

Abstract

Practical relevance A number of systemic diseases are associated with neurological deficits. Most systemic diseases that impact on the nervous system result in multifocal neurological signs; however, isolated deficits can also be observed. This article reviews the clinical signs, pathophysiology, diagnosis, treatment and prognosis of four important systemic diseases with neurological consequences: feline infectious peritonitis, toxoplasmosis, hypertension and hepatic encephalopathy.
Clinical challenges Early recognition of systemic signs of illness in conjunction with neurological deficits will allow for prompt diagnosis and treatment. While neurological examination of the feline patient can undoubtedly be challenging, hopefully the accompanying articles in this special issue will enable the clinician to approach these cases with more confidence.
Evidence base The veterinary literature contains numerous reports detailing the impact of systemic disease on the nervous system. Unfortunately, very few references provide detailed descriptions of large cohorts of affected cats. This review summarises the literature underpinning the four key diseases under discussion.
Diseases with neurological consequences
There are a number of systemic disorders in cats that can result in neurological consequences. In most such disorders, signs of systemic disease overshadow the neurological deficits. In some systemic disorders, neurological signs dominate the clinical picture.
Four important systemic diseases with neurological consequences are feline infectious peritonitis (FIP), toxoplasmosis, hypertension and hepatic encephalopathy (HE).
Feline infectious peritonitis
Feline infectious peritonitis is a fatal viral infection of wild and domestic Felidae worldwide that is caused by a mutant variant of a ubiquitous feline enteric coronavirus (FECV). 3 The clinical syndrome of immune-mediated vasculitis and pyogranulomatous inflammation that has become known as FIP was first described in 1963. 4 Later a viral etiology, feline infectious peritonitis virus (FIPV), was identified and classified as a coronavirus. 5 Although it was initially believed that FIPV and FECV were distinct entities, it is now known that FIPV develops out of spontaneous mutation of FECV within an infected cat. 6 Therefore, the antigenicity and genome of FIPV is nearly identical to FECV but FIPV possesses a very different pathogenicity. 3 Throughout the remainder of this article, the term feline coronavirus (FCoV) will be used when referring to all feline coronaviral infections, and the terms FECV and FIPV will be used to denote specifics regarding enteric infection and FIPV infections, respectively.
Distributed worldwide, FCoV is a single-stranded RNA virus belonging to the genus Coronavirus of the family Coronaviridae. Consequently, FIPV infection represents a disease with worldwide significance. While the systemic consequences of FIPV infection are clearly significant, the focus of the following discussion is on the impact of FIPV on the nervous system.
Clinical work-up
Assessment of the feline patient with suspected neurological disease requires with a thorough neurological examination aimed at reaching an accurate neuroanatomic diagnosis. Only then can an appropriate differential diagnosis list be formulated — that is, a systematic and prioritized list of disease processes. The specifics of the neurological examination of cats are presented in a separate article in this special issue (doi:10.1016/j.jfms.2009.03.002) and are also reviewed elsewhere.1,2
Knowledge of the epidemiology of FCoV infection is important in order to understand the pathogenesis of FIPV. Feline coronavirus infection is virtually endemic, with studies revealing that:
Approximately 50% of cats in the United States and Europe have antibodies against FCoV; 3
In Australia, the seroprevalence in owned cats is approximately 34%; 7
In the UK, 82% of show cats, 53% of breeding cats and 15% of single-cat homes have anti-FCoV antibodies; 8
In Italy, the seroprevalence is 82% in cats from breeding colonies, shelters and homes; 9
In Switzerland, similarly 80% of breeding cats, and 50% of free-roaming cats, test positive for anti-FCoV antibodies. 10
The significance of this worldwide distribution relates to the fact that FIPV is the result of spontaneous mutation of FECV, which means that cats worldwide are susceptible to developing FIP. 6 Despite this, approximately 5% of cats in multicat homes and a smaller percentage of cats in single-cat homes develop FIP.3,11,12 Notably, it is young cats and immunosuppressed cats that are most susceptible to developing FIP. In addition, certain purebreed cats, specifically the Birman, Ragdoll, Bengal, Rex, Abyssinian and Himalayan breeds, have a greater risk of developing FIP, which suggests a genetic influence on susceptibility. 13
Transmission of FCoV is through infected fecal material via the orofecal route, leading to enteric infection. Enteric FCoV infection typically results in inappetence and/or mild gastrointestinal signs such as vomiting and diarrhea. Infected cats shed FECV for up to 10 months post infection; thereafter infected cats shed virus intermittently or continuously, serving as chronic carriers and thereby perpetuating reinfection of other individuals. 14 As with all coronaviruses, FECV undergoes a high rate of mutation. The degree of mutation, and therefore the development of a mutation leading to FIP, appear greater in susceptible individuals as well as in individuals with a high viral load.15,16 In part, this may explain the fact that more than 50% of cats with FIP are under a year of age. 3
Pathogenesis
The initial step in the pathogenesis of FIP is the mutation of FCoV, in the process of which the virus gains the ability to replicate within macrophages. Once this occurs, the virus can be disseminated throughout the body. Ultimately, FIP is an immune complex disease that is a consequence of virus or viral antigens complexed with antiviral antibodies. 16 After distribution by macrophages, the virus may enter tissue and replicate, resulting in attraction of neutrophils and macrophages and leading to granuloma formation. 16 Alternatively antigen-antibody complexes may adhere to the vasculature, fix complement and attract macrophages, resulting in granuloma formation. 17 Regardless of the exact mechanism, the combination of macrophage infiltration and complement fixation leads to the release of inflammatory cytokines and vasoactive amines; these cause vasculitis resulting in pyogranulomatous inflammation and exudation of protein-rich fluid from capillaries.
Clinical signs
Clinical signs of FIP are often vague (Fig 1), with affected cats displaying non-specific systemic signs including intermittent fever, lethargy, anorexia, vomiting, diarrhea and weight loss. Physical examination may disclose abdominal effusion, abdominal lymphadenopathy, and irregular and asymmetrical kidneys.
Fig 1.
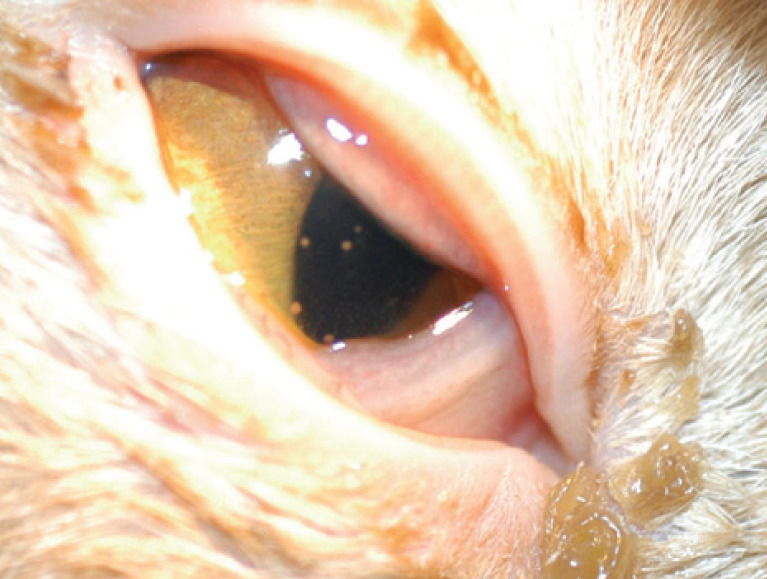
Right eye of a cat with FIP. Note the keratoprecipitates secondary to anterior uveitis on the endothelial surface of the cornea
Neurological signs.
Neurological signs may be observed in approximately 30% of affected cats,18–21 either alone or in conjunction with systemic signs. Most cats with neurological signs have non-effusive FIP and typically display multifocal neurological signs. 20 Abnormal mental state and behavior, seizures, ataxia, deficits in cranial nerve function, and varying degrees of tetra- or paraparesis are among the signs seen.18–21 Affected cats often demonstrate signs consistent with central vestibular disease including head tilt, vestibular ataxia, abnormal nystagmus and postural reaction deficits. 20
Pathology
Grossly, the meninges may appear thickened and opaque, especially those overlying the ventral aspect of the brain. 18 Occasionally, no gross abnormalities are observed. In general, lesions are surface-oriented, distributed over the external leptomeningeal and ependymal surfaces of the nervous system, with the caudal and ventral aspects of the brain being most severely affected. 18 Histologically, typical pyogranulomatous inflammatory infiltrates are observed. Lesions comprise meningitis, ependymitis and periventriculitis consisting primarily of lymphocytes, macrophages, and varying numbers of plasma cells forming perivascular cuffs. 18 Subependymal periventricular inflammatory infiltrate may obstruct the mesencephalic aqueduct leading to obstructive hydrocephalus. Similarly, obstruction of the central canal of the spinal cord may lead to hydromyelia. Occasionally, infiltrate extends into the superficial neuropil and cranial nerve roots. 18
Neurological signs may be observed in approximately 30% of cats with FIP, either alone or in conjunction with systemic signs.
Treatment of feline infectious peritonitis
Since FIP is an immune-mediated disease process, therapy has been directed at immunosuppression and/or immunomodulation with the goal of providing symptomatic care. Immunosuppressive therapy using corticosteroids (prednisone at 2–4 mg/kg/day) may allow mildly affected cats to maintain an acceptable quality of life for weeks to months. In addition to corticosteroids, a wide array of drugs including chemotherapy agents (cyclophosphamide and melphalan), an antiviral (ribavrin), a thromboxane synthetase inihibitor (ozagrel hydrochloride) and a variety of immunomodulating drugs (promodulin, human interferon-a, Propionibacterium acnes, and feline interferon-ω) have been investigated. 36
The interpretation of results from most studies has been hindered by a lack of control groups and the difficulty in establishing a definitive diagnosis of FIP in treated cats. 36 Still, most therapeutic trials have failed and, disappointingly, an effective treatment regime remains elusive.
Diagnosis
Hematology in affected cats usually reveals a normocytic, normochromic, nonregenerative anemia, leukocytosis consisting of a neutrophilia, and lymphopenia. 22 Approximately 50% of cats with the effusive form and 70% of cats with the dry form have increased serum proteins, primarily comprising a hyperglobulinemia. 23 Protein electrophoresis discloses a polyclonal gammopathy, mainly involving the γ-globulins. 24 Other biochemical changes may be observed depending on the severity of involvement of other organ systems including abnormal liver enzyme, bilirubin, blood urea nitrogen and creatinine levels. 25
State of the art review
‘A review of feline infectious peritonitis virus infection: 1963–2008’ by Dr Niels Pedersen, of the University of California, Davis, was published in the April 2009 issue of J Feline Med Surg doi:10.1016/j.jfms.2008.09.008
Although common clinicopathologic abnormalities in affected cats have been defined, changes in routine hematology and biochemical evaluations are often non-specific. Consequently, establishing a definitive ante mortem diagnosis of FIP is extremely challenging. 26 Definitive diagnosis can only be achieved through histopathological identification of pyogranulomatous inflammation within tissue coupled with identification of the virus. 16 Viral identification in tissue samples can be performed using immunohistochemistry or through PCR testing. However, there are several tests that may help support a presumptive ante mortem diagnosis. Importantly, interpretation of results from such tests should be evaluated in conjunction with clinical signs and results of other diagnostics. Taken outside the context of signs and other clinicopathologic data, most tests are unable to provide a definitive diagnosis.
When present, effusions should be analyzed. Typically, effusion from an affected cat should be consistent with a modified transu-date. 3 Cats with neurological signs should undergo magnetic resonance imaging (MRI) of the brain. This may disclose hydro-cephalus. 18 Additionally, T2-weighted and T2-weighted FLAIR images may reveal periventricular hyperintensities consistent with periventriculitis. 18 Although these findings are not pathognomonic, MRI of the brain should be pursued in order to eliminate the potential that clinical signs may be a consequence of a disease process other than FIPV.
Analysis of cerebrospinal fluid (CSF) often reveals increased protein content (50–350 mg/dl) with a pleocytosis consisting of neutrophils, lymphocytes and macrophages.20,27,28
The evaluation of FCoV antibody titers (often erroneously referred to as an ‘FIP titer’) in blood and other fluids has been extensively studied. However, despite years of investigation, caution should be exercised when interpreting FCoV antibody titers in blood and effusions as high titers can be observed in healthy cats and low to absent titers in affected cats. 3 Feline coronavirus antibody titers can be measured in CSF and may correlate with the presence of neurological FIPV. 18 Cerebrospinal fluid FCoV antibody titers correlate with serum FCoV antibody titers but, most importantly, elevated CSF FCoV antibody titers may also be observed in cats affected by neurological diseases other than FIP. 29
KEY POINTS: FELINE INFECTIOUS PERITONITIS
Approximately 30% of cats with FIPV infection demonstrate neurological signs such as abnormal mentation, seizures, vestibular dysfunction, and varying degrees of para- and tetraparesis.
Definitive diagnosis requires observation of pyogranulomatous inflammation and identification of virus in tissue. A presumptive diagnosis is made by utilizing a variety of clinicopathologic data including imaging, biochemical analysis, measurement of serum α-glycoprotein, FCoV antibodies, and PCR testing.
FIPV is fatal and treatment is mainly palliative.
While PCR assays can be performed on blood and effusions in affected cats, they are unable to distinguish between the mutated FCoV causing FIP, and the non-pathogenic FCoV. 30 In addition, healthy cats can be viremic with FCoV. 31 Therefore, PCR identification of virus in blood or effusions does not provide a definitive diagnosis. 32 While its application in CSF has not been studied, PCR identification of virus in CSF may allow a definitive diagnosis of FIP.
Measurement of serum α1-acid glypoprotein (AGP), an acute phase protein that increases during inflammation, has been used in the diagnosis of FIP.33,34 In cats with signs and clinicopathologic data highly suggestive of FIP, elevation in serum AGP provides strong supportive evidence of FIPV infection. 33 However, AGP may also increase in other conditions associated with inflammation, such as feline immunodeficiency virus (FIV) infection, or in cats with a high viral load of FECV, which may limit its potential as a diagnostic tool for FIP.34,35
Prognosis
Unfortunately, the prognosis for cats with FIP is grave as all affected cats succumb to the disease. To date, no therapy has been shown to alter the eventuality of humane euthanasia or death of affected cats.
Toxoplasmosis
Toxoplasmosis is caused by an obligate intracellular protozoan parasite, Toxoplasma gondii. The definitive hosts are the domestic cat and other Felidae. Many mammals can become infected with T gondii and serve as intermediate hosts; however, fecal shedding of infective oocysts occurs only in cats.
Systemic and ocular toxoplasmosis have been well described in cats. The emphasis in the following discussion is on central nervous system (CNS) toxoplasmosis.
Life cycle and transmission
There are three methods of transmission of T gondii: fecal-oral, ingestion of tissue cysts, and congenital. Reproduction of the organism can involve both a sexual and asexual phase. The asexual phase occurs in many mammals and birds, which serve as intermediate hosts. As the definitive host, the sexual phase of the life cycle can only occur in cats and it does so within the intestinal tract. As a result, unsporulated oocysts, which are non-infectious, are passed in the feces. These oocysts require 1–5 days for sporulation to occur, at which point they become infectious. Ingestion of sporulated oocysts by another cat begins another cycle.
With the exception of finding T gondii in tissue, no single test provides a definitive diagnosis.
Toxoplasma gondii also displays an extraintestinal life cycle. After infectious oocysts have been ingested, the organism is capable of penetrating the wall of the intestinal tract and disseminating to multiple organs. Within these other organs, asexual reproduction occurs, giving rise to tissue cysts — bradyzoites (so named given their slow replication) and tachyzoites (in which replication is rapid). Ingestion of bradyzoites in tissue is probably responsible for the majority of infections. In cats it can result in intestinal replication, while ingestion of bradyzoites by other animals can only lead to extraintestinal infection. Ingestion of infectious organism during gestation can also lead to congenital infection. 37
Cysts that form as a result of extraintestinal infection are likely to persist for life.38,39 Encysted bradyzoites are the most probable source of continual release of antigen and reactivation of infection. 38 Reactivation of infection is thought to occur secondarily to immunosuppression. Cats infected with FIV appear to be predisposed to the development of acute toxoplasmosis. 40 There appears to be a high seroprevalence of T gondii infection in cats co-infected with FIV.41,42 The significance of this relationship is unknown as the sero-prevalence of T gondii infection in FIV-infected cats is similar to that in the general population.39,43 Immunosuppression as a result of ciclosporin therapy has also led to acute toxoplasmosis.44,45 With the availability of renal transplantation in cats, the role of immunosuppression in reactivation of infection and the development of clinical disease has gained importance.46–48
Clinical signs
Clinical infection with T gondii is not common in cats. Infections can be considered acute or chronic. 49 Acute toxoplasmosis typically affects younger cats. The most common clinical signs are anorexia, lethargy, fever, dyspnea and sudden death.49–52 Chronic toxoplasmosis typically affects older cats and manifests over weeks to months. Signs are similar to acute infection and may include vomiting, diarrhea, anorexia, weight loss, fever and icterus.42,49–51
Clinical signs reflecting organ involvement include lymphadenopathy, myocardial disease, pancreatitis, hepatitis, anterior uveitis and chorioretinitis.41,42,49–51,53,54
Neurological signs
Central nervous system involvement occurs in almost all clinically affected cats. 50 Neurological signs typically reflect a multifocal distribution and include hypothermia, behavioral changes, seizures, ataxia, blindness, anisocoria, torticollis, vestibular disease, muscle hyperesthesia, and paresis/paralysis.21,42,50,55–58 Central nervous system signs may or may not occur in conjunction with systemic disease. Occasionally, cats may present with signs consistent with focal disease such as seizures or paralysis related to spinal cord disease.55,56,59
Diagnosis
The diagnosis of clinical toxoplasmosis can be challenging. Hematologic findings are nonspecific, often consisting of non-regenerative anemia, neutrophilic leukocytosis, lymphocytosis and monocytosis.41,42,50 Biochemical abnormalities generally reflect organ involvement and include azotemia, elevation in liver enzymes, hyperbilirubinemia and hyper-proteinemia.41,42,50 Thoracic radiographs may show a diffuse interstitial to bronchial pattern in which infiltrate may coalesce into areas of patchy alveolar patterns (Fig 2).49–51,60
Fig 2.

(a) Lateral radiograph of the thorax of a cat presenting with tachypnea and a cough, showing a diffuse bronchointerstitial pattern. (b) Cytology of an endotracheal wash sample. Multiple T gondii tachyzoites are present extracellularly (arrows) and intracellularly in a reactive macrophage. Courtesy of Dr E Rozanski, Cummings School of Veterinary Medicine, Tufts University, North Grafton, MA, USA
Cerebrospinal fluid analysis typically reveals a mild lymphocytic pleocytosis predominantly, although other cell types may be observed; protein may be elevated to up to 149 mg/dl. 39 Neutrophilic pleocytosis has also been reported. 58
With the exception of identifying T gondii in tissue, no single test provides a definitive diagnosis. A presumptive diagnosis is based on a combination of clinical signs, evidence of recent or active infection (gained via serology for immunoglobulins or immune complexes, or PCR), exclusion of other disease processes, and response to therapy. 39
Serology for the detection of IgG and IgM anti-T gondii antibodies is widely used. 61 After experimental inoculation, an IgM response is detected in 1–3 weeks and an IgG response in 2–4 weeks. Immunoglobulin M responses peak within 3 weeks and persist for 3–16 weeks. In cats co-infected with FIV, there is a delayed conversion from an IgM to an IgG response. 62 Unfortunately, an IgM response does not necessarily correlate with active disease as occasionally IgM responses can be detected in clinically normal cats with chronic infection. Likewise, a single high IgG response does not predict active disease, as IgG responses can last up to 6 years. 63 A rising titer is strongly suggestive of active disease, however, and maximal titers are reached within 2–3 weeks. 63 In practice, given the insidious nature of the disease many cats have reached maximal immune responses by the time they are examined by a veterinarian, making documentation of a rising titer difficult. 61
Theoretically, identification of an immune response in the CNS, an immunoprivileged site, would suggest infection. However, immunoglobulins may extravasate from the blood into the CSF in other inflammatory diseases that disrupt the blood-brain barrier. Defining a ratio between serum and CSF IgG responses may help eliminate the possibility of passive cross over of antibodies secondary to another disease that compromises the integrity of the blood-brain barrier. A serum: CSF IgG response > 1 suggests local CNS production of immunoglobulin. In experimental oral inoculation, cats remain clinically normal yet develop a detectable IgG response in the CSF 4–12 weeks post inoculation and again 8–16 weeks after secondary exposure. 64 Importantly, an IgG response in the CSF can occur after exposure to killed tachyzoites in previously infected cats. 65 Therefore, observation of an IgG response in CSF does not necessarily document infection. 65
Experimental inoculation does not result in an IgM response in the CSF. 64 Potentially, therefore, detection of an IgM response in the CSF may be indicative of active disease, but this is yet to be confirmed.
KEY POINTS: TOXOPLASMOSIS
Neurological signs reflect a multifocal distribution in the CNS and include behavioral changes, seizures, ataxia, blindness, anisocoria, torticollis, vestibular disease, muscle hyperesthesia and paresis/paralysis.
Diagnosis requires cautious interpretation of serum and CSF IgG/IgM titers and PCR identification.
Clindamycin hydrochloride is the antibiotic of choice for the treatment of toxoplasmosis.
Treatment of toxoplasmosis
The treatment of choice for cats with clinical toxoplasmosis is clindamycin hydrochloride at 12–25 mg/kg divided per day.39,42 Clindamycin is almost completely absorbed after oral administration and achieves high concentrations in most tissues, including the lung. 68 Concentrations in CSF are low;69,70 however, the concentration in the brain may be higher given the lipophilic nature of the drug. 71 Clindamycin is well tolerated, with only minimal side effects (eg, vomiting and diarrhea) reported at dosages two and a half to four times the recommended dosage. 72 Parenteral formulations can be used in animals unable to receive oral medication or those experiencing gastrointestinal toxicity.
Reports of successful treatment are rare, which may reflect the difficulty of establishing a definitive diagnosis. Systemic clinical signs typically show improvement within 24–48 h of initiation of treatment. 39 Cats with systemic or ocular disease treated with antibiotic therapy may achieve clinical remission; however, recurrence of signs is likely as antibiotic therapy is unlikely to eliminate the organism entirely. 42
Although PCR assays have not been performed on CSF for the detection of T gondii, PCR assays have been utilized in the aqueous humor, another immunoprivileged site.66,67 Toxoplasma gondii can be identified in the aqueous humor of cats with uveitis using PCR; however, the organism can also be detected in the aqueous humor of clinically normal cats that have naturally been exposed to T gondii. 66 Consequently, PCR detection of T gondii in aqueous humor does not provide definitive proof of active disease. A similar interpretation of PCR analysis of CSF is likely.
Prognosis
Unfortunately, the prognosis is poor for cats displaying neurological signs or severe respiratory disease as most will succumb to the disease.21,56–58,60,73 Despite this, cats with focal CNS toxoplasmosis may achieve long term remission. 59
Hypertension
Since the initial description of systemic hypertension in cats, 74 the impact of hypertension systemically and on the nervous system has become increasingly recognized. A testament to this is a recent consensus statement from the American College of Veterinary Internal Medicine that has established guidelines for identification, evaluation and management of hypertension in dogs and cats. 75 In healthy cats, normal systemic blood pressure, which is often reported as a systolic measurement, is 118–162 mmHg.75–77 The wide range in the reported normal values is likely to reflect a lack of standardization in technique and equipment used to measure blood pressure. 75 Many factors affect blood pressure measurement including recording device, cuff size and operator skill, as well as patient factors such as size and demeanor of the cat. Although increasing age was found to be associated with increased blood pressure in one study, other reports have found no effect of age on blood pressure.77–79
Hypertension has been defined as a sustained increase in systolic blood pressure ≥ 160–170 mmHg. 75 Although the prevalence of hypertension in cats has not been accurately established, one study documented hypertension in 2% of healthy cats. 80 In cats referred for evaluation of disease associated with hypertension, or animals with clinical signs compatible with hypertension, a 30% prevalence was found. 79
Hypertension can be divided into three categories: white coat, secondary and idiopathic.
White coat hypertension is an artefactual increase in blood pressure that develops secondarily to excitement or anxiety, and is likely to be the result of activation of the sympathetic nervous system. 81 It is observed in cats, and results in a median increase in systolic blood pressure of 17.6 mmHg ± 1.5 mmHg. 81 While white coat hypertension does not cause a clinical problem, it does complicate definitive documentation of clinically relevant hypertension.
Secondary hypertension develops as a consequence of systemic disease. By far the most common causes of secondary hypertension are renal disease (chronic renal disease and renal transplantation) and hyperthyroidism.78,80,82–86 Other causes include primary hyperaldosteronism, pheochromocytoma, diabetes mellitus and erythropoietin treatment.74,87–89
Idiopathic hypertension is defined as the presence of hypertension in the absence of other disease conditions that can lead to hypertension. It has also been referred to as primary or essential hypertension. Although hypertension in cats is most commonly classified as secondary, approximately 20% of cats with hypertension are considered idiopathic.82,89
Pathology
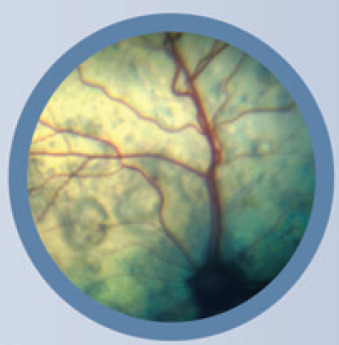
Chronic systemic hypertension has a variety of pathological consequences that collectively are referred to as end-organ or target organ damage. Important target organ damage is observed in the kidneys, eyes, heart and nervous system. 90 In the kidney, this leads to an accelerated decline in renal function, proteinuria and death. Hypertension can exist in animals at any stage of renal disease, and may be seen in non-azotemic animals. 75 In the eye, hypertension leads to hypertensive retinopathy and choroidopathy (Fig 3). Exudative retinal detachment, retinal hemorrhage, multifocal retinal edema and tortuosity of the retinal vessels may be observed, and commonly result in blindness.74,82,83,89,91–93 In the heart, hypertension may result in cardiomegaly and left ventricular hypertrophy.74,83,89,94 A systolic murmur, gallop rhythm and congestive heart failure may be observed.82,89 In the nervous system, hypertension may result in a hypertensive encephalopa-thy.83,85,86,89,95 Two studies have variously documented neurological signs in 29% and 46% of cats with hypertension.83,89
Fig 3.
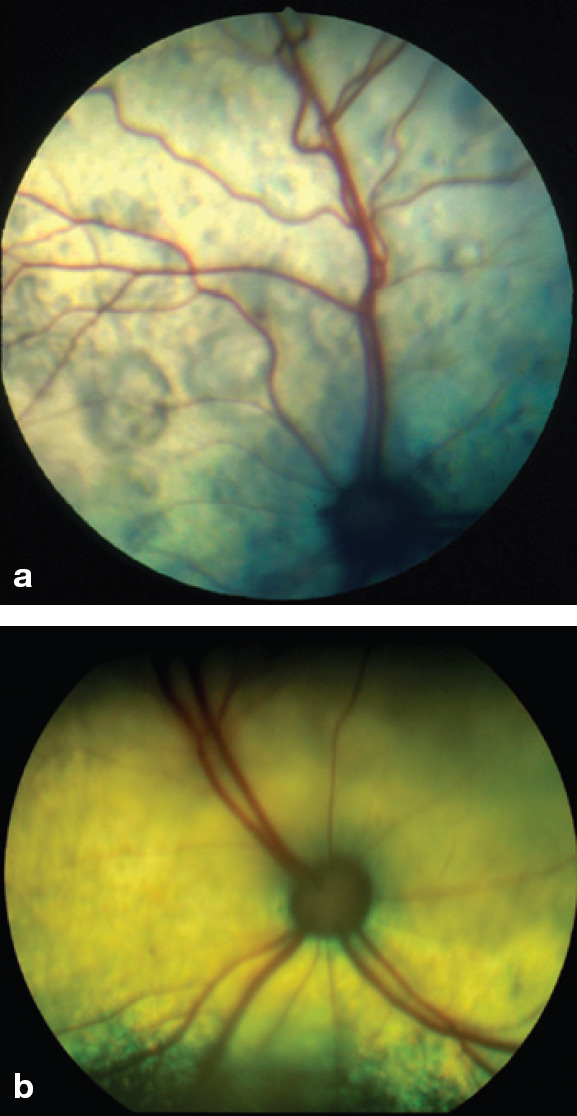
Images of the fundus in cats with hypertensive retinopathy. (a) The retinal blood vessels are engorged and tortuous. There are multifocal areas of subretinal effusion throughout the tapetum. (b) The dorsal tapetum is decreased in reflectivity secondary to subretinal effusion and focal areas of retinal detachments. There is an area of subretinal effusion at the 6 o'clock position.
Courtesy of Dr David Gould, Davies Veterinary Specialists, Hertfordshire, UK
Clinical signs
Since hypertension in most cats can be categorized as secondary, clinical signs typically reflect the underlying disease process. Consequently, affected cats often demonstrate signs relating to renal disease or hyperthyroidism, given the high prevalence of hypertension with these disorders.
Neurological signs.
Neurological signs typically reflect intracranial disease. Seizures, changes in mentation (obtundation, stupor or coma), and vestibular dysfunction (head tilt, vestibular ataxia and abnormal nystagmus) are the most common neurological manifestations of hypertension.83,85,86,89,95 Other neurological signs associated with hypertension include blindness, weakness, ataxia, behavioral changes, tremors, sudden collapse, flexion of the neck, episodes of dragging the pelvic limb(s), and decorticate posture of the thoracic limbs.83,85,86,89,95
Pathophysiology
Although the pathophysiology underlying hypertensive encephalopathy remains unclear, it is thought to involve the development of vasogenic edema, which predominantly affects the white matter.96,97 With acute hypertension, the autoregulatory capacity of the brain vasculature may be exceeded, leading to hyperperfusion, breakdown of the blood-brain barrier, and cerebral edema.96,98 In experimental acute hypertension in cats, gross findings include coning of the vermis of the cerebellum, cerebellar herniation into the foramen magnum (Fig 4), rostral displacement of the colliculi, and widening and flattening of the cerebral gyri, all of which reflect raised intracranial pressure. 95 Microscopically, the consequences of edema are observed such as marked pallor of the cerebral white matter, accentuation of the separation between axons and myelin sheaths, and widening of the perivascular space. 95 In chronic hypertension, brain vasculature may be chronically vasoconstricted leading to hypertrophy and hyperplasia of the smooth muscle. 89 As a result, fibrous changes develop, allowing leakage of plasma which ultimately causes degeneration of the vasculature predisposing to ruptures and microhemorrhages. 89 Multifocal cerebral arteriosclerosis with hemorrhages has been observed in cats with spontaneous hypertension. 83
Fig 4.
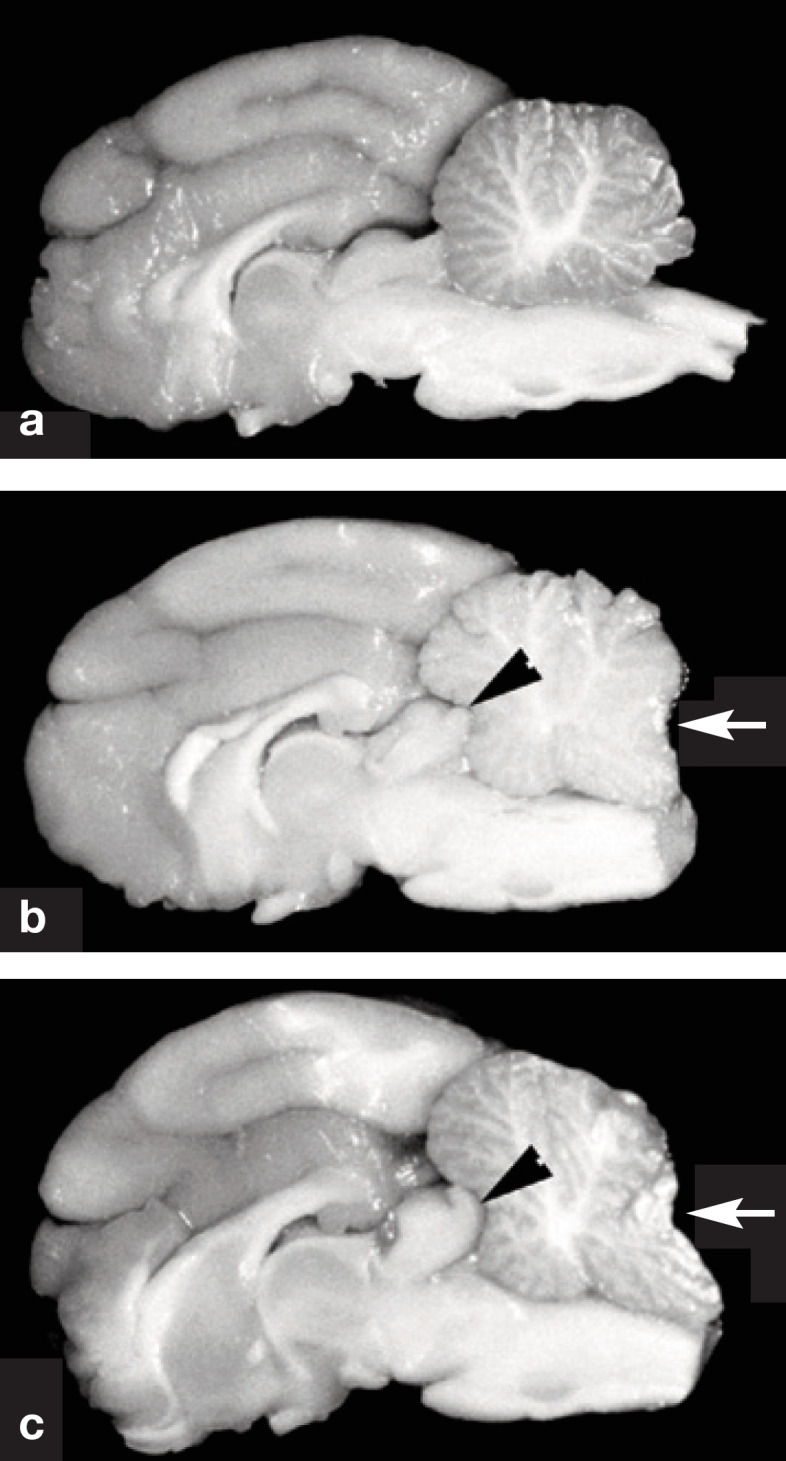
Gross specimens of the brain of cats in longitudinal section. (a) Normal cat. (b) Cat with moderately severe hypertension. The cerebral gyri are swollen resulting in compression in the rostral aspect of the cerebellum (arrowhead). Note the coning of the cerebellar vermis, which is a consequence of herniation of vermis through the foramen magnum (arrow). (c) Cat with severe hypertension. Similar features are noted as in (b). The cerebrum appears rounded, and there is severe coning and caudal displacement of the cerebellar vermis (arrow). Courtesy of Dr C Brown, University of Georgia, Athens, GA, USA
Diagnosis
A presumptive diagnosis of hypertensive encephalopathy is relatively straightforward and requires the documentation of hypertension (systolic blood pressure ≥ 160–170 mmHg) with contemporaneous neurological signs. The gold standard for blood pressure measurement is invasive intra-arterial monitoring. However, this is often not feasible in clinical practice. 99 Consequently, indirect blood pressure monitoring is used most commonly. 99 Accurate and reliable indirect blood pressure measurements can be performed using Doppler flow ultrasonography and oscillometry. 99 Minimal, mild, moderate and severe risk categories for target organ damage have been defined based on blood pressure recordings (see below). 75
Ideally, measurement of blood pressure should be performed in a quiet environment, with the cat first allowed time to acclimatize to minimize the effect of white coat hypertension. The same experienced, trained personnel should perform the measurements to ensure reliability and eliminate potential operator error. Between three and five recordings should be taken to demonstrate consistency. Often the firs is disregarded as the animal acclimatizes to the procedure.
Interpretation of blood pressure recording

Treatment of hypertension
Unless animals are showing evidence of target organ damage, or are at severe risk of developing target organ damage (see box on page 401), there is no requirement for immediate therapeutic intervention. Instead, repeated blood measurements over a period of time, combined with identification and treatment of any potential underlying disease process leading to hypertension, may be all that is needed to control blood pressure.
In cats that remain hypertensive despite control of an underlying disease process, or those with idiopathic hypertension in the mild to moderate risk category for target organ damage, the decision to pursue hypertensive therapy requires a dedicated owner as treatment is generally lifelong and involves frequent re-evaluations.
In animals displaying signs consistent with hypertensive encephalopathy, prompt intervention should be pursued. The treatment of choice for hypertension in cats is amlodipine besylate, a calcium channel blocker.82,99,101–103 A dose of 0.625–1.25 mg/cat orally once to twice daily reliably reduces blood pressure with minimal risk of causing hypotension.82,101–104 Furthermore, treatment with amlodipine is not associated with increases in blood urea nitrogen and creatinine in cats with chronic renal failure.103,104
In cats in which amlodipine is ineffective at controlling hypertension, adjunctive therapy with the β1 selective β-blocker, atenolol, may be instituted at 6.25–12.5 mg/cat PO q 12–24 h. 105 Alternatively, the angiotensin-converting enzyme inhibitor, benazepril, at 0.25–0.5 mg/kg PO q 12–24 h can be used. 106 However, benazepril therapy is associated with only a small but significant reduction in blood pressure in cats with chronic renal failure. 106
In acute hypertension, subcutaneous hydralazine (1.0–2.5 mg/cat) has been effective at reducing blood pressure without significant risk of hypotension. 84 While parenteral hypotensive medications may be preferable in the setting of severe hypertensive encephalopathy, the use of such medications requires continuous, direct arterial blood pressure measurement and is associated with a significant risk of hypotension.
In cats with severe neurological dysfunction that do not respond to a reduction in blood pressure, treatment for raised intracranial pressure due to brain edema may be warranted. This entails diuretic therapy with mannitol (0.5 to 2 g/kg IV over 10–15 mins often combined with furosemide 0.7 mg/kg IV), or other hypertonic agents. 107 Note, however, that diuretic therapy should not be used until blood pressure has normalized, as these agents can transiently increase blood pressure. 107
Identification of hypertension should prompt investigation for an underlying disease process. A complete blood count, biochemistry profile and urinalysis should be performed in all hypertensive cats. In cats older than 5 years of age, serum thyroxine level should also be measured. When indicated, endocrinological testing for Cushing's disease or diabetes mellitus should be performed. In cats with suspected or confirmed renal disease, quantification of a proteinuria should be performed. Thoracic radiographs should be obtained to assess cardiovascular structures, and abdominal ultrasonography should be performed to assess renal structure and identify any concurrent disease. Echocardiography is warranted in cats with a murmur, gallop rhythm or other signs consistent with cardiac disease. In all cats with hypertension, echocardiography allows assessment of any secondary cardiac changes.
In cats with severe neurological dysfunction MRI may be warranted. In addition to assessing CNS pathology related to hypertension, MRI allows exclusion of other disease processes that can produce similar neurological signs. Given the potential for raised intracranial pressure and brain herniation in hypertensive encephalopathy, caution should be exercised prior to advanced imaging; the requirement for general anesthesia can lead to deterioration or death in animals with severe raised intracranial pressure. In humans with hypertensive encephalopathy, MRI of the brain discloses hyperintensities in the white matter of the parietal and occipital lobes of the cerebrum on T2-weighted images. 96 Less frequently, similar findings may be observed in the brainstem. 100 Magnetic resonance imaging in hypertensive cats has not been studied; however, given the gross and microscopic changes observed in affected cats, similar findings would be expected.
Prognosis
Unfortunately, control of hypertension does not appear to have a significant effect on survival time.82,83,108,109 However, amlodipine does seem to reduce the degree of proteinuria in cats with renal disease, and a reduction in proteinuria appears to have a positive effect on survival time.108,109
KEY POINTS: HYPERTENSION
The most common neurological signs associated with hypertension include seizures and vestibular dysfunction.
Diagnosis involves accurate documentation of increased systolic blood pressure in a cat demonstrating compatible neurological signs.
Amlodipine besylate is the drug of choice to treat hypertensive cats.
Hepatic encephalopathy
Hepatic encephalopathy is the clinical syndrome of abnormal neurological function caused by portosystemic shunting, with or without intrinsic liver disease. 110 As a result, HE can develop in cats with acquired or congenital liver disorders. By far the most common cause of HE in cats is portosystemic shunting of blood secondary to a congenital vascular anomaly. 111
Clinical signs
Regardless of the cause of the underlying liver disease, the clinical signs of HE are similar and can be divided into systemic and neurological signs. Affected cats often display intermittent clinical signs that may be associated with eat-ing. 112 Cats with portosystemic shunts are generally small in stature, fail to thrive and grow, and lose weight.112–115 Pytalism is a common clinical sign, occurring in approximately 75% of cats.112,116–120 Other, less common clinical signs include gastrointestinal signs such as decreased appetite, anorexia, pica, vomiting, diarrhea or constipation.112,113,117 Cats may demonstrate polydipsia, and polyuria, pollakiuria and stranguria may occur as a consequence of cystic calculi.112,113,117 Affected cats may have copper-coloured irises. 121
Neurological signs
In general, neurological examination findings reflect a prosencephalic lesion localization, and typically comprise abnormal behavior, abnormal mentation and seizures. Affected cats may display aggression.112–120,122,123 Other abnormal behaviors include aimless pacing and wandering, head pressing and collapse. Cats may appear obtunded.112–120,122,123 Constant or episodic blindness may also be observed.112–120,122,123
Pathophysiology
Although a complete understanding of the mechanisms underlying HE remains elusive, it is clear that the pathophysiology involved is multifactorial. Despite numerous potential factors, ammonia remains key in the development of HE. 124 Ammonia is produced by bacteria in the gastrointestinal tract, primarily the colon, as a consequence of protein metabolism. 125 It is also produced by the gastrointestinal cells as a result of metabolism of glutamine, the main cellular energy source for the epithelium. 126 A further source of ammonia is the kidneys, during states of hypokalemia or alkalosis. 126
Normally, the liver efficiently removes ammonia from the portal vasculature, ultimately converting it to urea. In animals with hepatic failure or portosystemic shunts, hyperammonemia may develop. However, the severity of the neurological signs does not always correlate with the degree of hyperammonemia. 127 In fact, blood ammonia levels may be normal in cats with HE. 127 This relates to a greater rate of uptake of ammonia in the CNS in HE, leading potentially to a high CNS ammonia level in the face of a normal blood ammonia level. 128
By far the most common cause of hepatic encephalopathy in cats is portosytemic shunting of blood as the result of a congenital vascular anomaly.
Although HE is a syndrome of neuronal dysfunction, the neuropathological consequences of increased CNS ammonia are played out in the astrocyte. 129 Normally, CNS ammonia undergoes energy-dependent metabolism to glutamine by the astrocytes. 130 With increased CNS ammonia, astrocytes become overwhelmed, leading to energy depletion. 110 Additionally, the increased concentration of glutamine in astrocytes may act as an osmotic stress, leading to cell swelling. 110 Increased numbers of swollen astrocytes — referred to as Alzeheimer type II astrocytes — is the only structural change observed microscopically in the brain in HE. 131 As a consequence of cellular edema, neurotransmitter processing in the astrocytes is affected, resulting in upregulation of neuronal benzodiazepine receptors and the production of neurosteroids which increase γ-aminobutyric acid (GABA) neurotransmission and thereby ultimately affect neuronal function. 110 Ammonia may also have a direct toxic effect on neurons. 110
While ammonia remains a focal point in the pathogenesis of HE, other factors are also involved. Mercaptans are formed during the degradation of sulfur-containing amino acids. These substances exert a neurotoxic effect through inhibition of ATPase activity, thereby potentiating the effect of ammonia. 132 Short and medium chain fatty acids are derived from bacterial metabolism of carbohydrates or from incomplete β-oxidation of long chain fatty acids in the liver. 127 Like mercaptans, these molecules may inhibit energy metabolism as well as inhibiting urea cycle enzymes in the liver. 126 A putative role for mercaptans, short and long chain fatty acids is unknown, but they may act synergistically with ammonia. 127
Hepatic encephalopathy may develop through an imbalance of inhibitory (GABA) and excitatory (glutamate) neurotrans-mitters. 133 There is evidence to implicate excessive GABAergic tone in HE, which would result in global inhibition of neurological function. 133 In humans with HE, there are increased GABA concentrations in the CNS leading to excessive GABAergic tone. 134 In addition, there may also be increased concentrations of endogenous benzodiazepines in the CNS.127,133 Benzodiazepines also bind to the GABA receptor, potentiating the effect of GABA. 126
Treatment of hepatic encephalopathy
Treatment should be directed at the underlying cause of HE as well as controlling the clinical signs of HE. One of the primary aims is to reduce blood ammonia levels. For animals with mild to moderately severe clinical signs, which are capable of taking oral medications, lactulose should be administered at a starting dose of 1 ml PO q 8–12 h. 116 The dosage is adjusted based on stool consistency and response. In more severely affected cats, lactulose can also be administered per rectum. Prior to rectal administration, warm water enemas should be performed to remove fecal material. Lactulose is a non-aborbable disaccharide that undergoes extensive metabolism by colonic bacteria, first to constituent monosaccharides and then to volatile fatty acids. 140 Ultimately, lactulose decreases the production/absorption of ammonia. 140 This is accomplished in several ways: namely, by decreasing the colonic luminal pH, leading to conversion of ammonia (NH3) to ammonium (NH4+), trapping it intraluminally; decreasing transit time through the osmotic cathartic effect of lactulose; and interfering with intestinal absorption of glutamine, thereby decreasing the production of ammonia. 141
Antibiotic therapy is often combined with lactulose administration. Antibiotics with activity against ureaseproducing bacteria are effective at reducing ammonia production. Neomycin is an oral aminoglycoside antibiotic that undergoes minimal systemic absorption. It is administered at 20 mg/kg PO q 8 h. 116 Despite the limited systemic absorption, systemic concentrations capable of causing side effects are possible. 116 Metronidazole is also effective at reducing urease-producing microbes, and is administered at 10 mg/kg PO q 12 h. 116 Reduced hepatic metabolism in animals with liver disease may result in an increased incidence of neurotoxicity. 142 Alternatively, ampicillin or amoxicillin-clavulanate can be administered. In severely affected animals, antibiotics (ampicillin, amoxicillin or metronidazole) should be administered parenterally.
There are several precipitating factors that can lead to HE, and these should be identified and corrected in the individual animal. Correction of dehydration, hypoglycemia and hypokalemia is imperative. Animals with clinical signs suggestive of gastrointestinal bleeding, such as melena, anorexia and vomiting, should be treated with H2-blockers. In moderately to severely affected animals, food should be withheld until therapy results in significant improvements. Once a clinically significant improvement is obtained, affected animals should be fed a diet restricted in protein, with limited aromatic amino acids and short chain fatty acids. 114 Commercial diets designed for animals with liver disease are available. Alternatively, diets designed for renal failure can be utilized in animals with HE.

The severity of the neurological signs does not always correlate with the degree of hyperammonemia. In fact, blood ammonia levels may be normal in cats with HE.
False neurotransmitters may also play a role in HE. 126 In liver disease, the production of branched chain amino acids is reduced. 127 Branched and aromatic amino acids compete for the same transporter into the CNS. 126 As a consequence of reduced branched chain amino acids, there may be a relative increase in the aromatic amino acids; these so-called false neurotransmitters may act like GABA and other inhibitory neurotransmitters. 135 Ultimately, the clinical signs of HE may be a result of global inhibition of neurotransmission.
Diagnosis
A complete blood count, biochemistry and urinalysis should be performed in all animals with clinical signs suggestive of HE. On hematology, microcytosis may be present. 112 Biochemical abnormalities may include low blood urea nitrogen, increased liver enzymes, decreased total protein and albumin concentrations, and hypocholesterolemia. 112 Urinalysis may disclose hyposthenuria and ammonium urate crystals. 112 A presumptive diagnosis can be made by documenting altered liver function in the setting of clinical signs consistent with HE. An elevated fasting blood ammonia level helps confirm the clinical suspicion. In order to provide accurate results, blood samples should be placed in a heparinized tube and transferred to the laboratory on ice for immediate testing. Red blood cells contain large amounts of ammonia; hence hemolysis may result in falsely elevated blood ammonia levels. Alternatively, a presumptive diagnosis of HE can be made by demonstrating altered liver function through fasting bile acid stimulation testing and on the basis of response to therapy.
A suspected or confirmed diagnosis of HE should prompt investigations for congenital or acquired portosystemic shunting. Portosystemic shunts are most commonly diagnosed by ultrasonography or per rectal portal scintigraphy using 99mtechnetium pertechnetate.136–139 Positive contrast portography also can be performed; however, this necessitates general anesthesia and laparotomy. 113
Prognosis
The prognosis for animals with HE is dependent on the underlying liver disease. With treatment, most animals experience an improvement in the clinical signs related to HE. Severely affected animals may not respond to therapy, however. Animals with increased blood ammonia tend to respond better to treatment than those with normal blood ammonia.
KEY POINTS: HEPATIC ENCEPHALOPATHY
Neurological signs reflect prosencephalic dysfunction and include abnormal mentation and seizures.
Diagnosis relies on demonstrating abnormal liver function, compatible neurological signs, and response to therapy.
Treatment is directed at reducing serum ammonia concentrations.
References
- 1.Garosi L. Lesion localization and differential list. In: Platt SR, Olby NJ, eds. BSAVA manual of canine and feline neurology. 3rd edn. Gloucester: British Small Animal Veterinary Association, 2004: 24–34. [Google Scholar]
- 2.Lorenz MD, Kornegay JN. Neurological history and examination. In: Handbook of veterinary neurology. 4th edn. St Louis, MO: Saunders Elsevier, 2004: 3–44. [Google Scholar]
- 3.Hartmann K. Feline infectious peritonitis. Vet Clin North Am Small Anim Pract 2005; 35: 39–79, vi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Holzworth J. Some important disorders of cats. Cornell Vet 1963; 53: 157–60. [PubMed] [Google Scholar]
- 5.O'Reilly KJ, Fishman B, Hitchcock LM. Feline infectious peritonitis: Isolation of a coronavirus. Vet Rec 1979; 104: 348. [DOI] [PubMed] [Google Scholar]
- 6.Vennema H, Poland A, Foley J, Pedersen NC. Feline infectious peritonitis viruses arise by mutation from endemic feline enteric coronaviruses. Virology 1998; 243: 150–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bell ET, Toribio J, White JD, Malik R, Norris JM. Seroprevalence study of feline coronavirus in owned and feral cats in Sydney, Australia. Aust Vet J 2006; 84: 74–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sparkes AH, Gruffydd-Jones TJ, Harbour DA. Feline coronavirus antibodies in UK cats. Vet Rec 1992; 131: 223–24. [DOI] [PubMed] [Google Scholar]
- 9.Pratelli A. Comparison of serologic techniques for the detection of antibodies against feline coronaviruses. J Vet Diagn Invest 2008; 20: 45–50. [DOI] [PubMed] [Google Scholar]
- 10.Kummrow M, Meli ML, Haessig M, et al. Feline coronavirus serotypes 1 and 2: Seroprevalence and association with disease in Switzerland. Clin Diagn Lab Immunol 2005; 12: 1209–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Addie D, Jarrett J. Feline coronavirus antibodies in cats. Vet Rec 1992; 131: 202–3. [DOI] [PubMed] [Google Scholar]
- 12.Pedersen NC. Serologic studies of naturally occurring feline infectious peritonitis. Am J Vet Res 1976; 37: 1449–53. [PubMed] [Google Scholar]
- 13.Pesteanu-Somogyi LD, Radzai C, Pressler BM. Prevalence of feline infectious peritonitis in specific cat breeds. J Feline Med Surg 2006; 8: 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Foley JE, Poland A, Carlson J, Pedersen NC. Patterns of feline coronavirus infection and fecal shedding from cats in multiple-cat environments. J Am Vet Med Assoc 1997; 210: 1307–12. [PubMed] [Google Scholar]
- 15.Foley JE, Poland A, Carlson J, Pedersen NC. Risk factors for feline infectious peritonitis among cats in multiple-cat environments with endemic feline enteric coronavirus. J Am Vet Med Assoc 1997; 210: 1313–18. [PubMed] [Google Scholar]
- 16.Pedersen NC. An overview of feline enteric coronavirus and infectious peritonitis virus infections. Feline Pract 1995; 23: 7–22. [Google Scholar]
- 17.Nafe LA. Topics in feline neurology. Vet Clin North Am Small Anim Pract 1984; 14: 1289–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Foley JE, Lapointe JM, Koblik P, Poland A, Pedersen NC. Diagnostic features of clinical neurologic feline infectious peritonitis. J Vet Intern Med 1998; 12: 415–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Foley JE, Leutenegger C. A review of coronavirus infection in the central nervous system of cats and mice. J Vet Intern Med 2001; 15: 438–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kline KL, Joseph RJ, Averill DR. Feline infectious peritonitis with neurologic involvement: Clinical and pathological findings in 24 cats. J Am Anim Hosp Assoc 1994; 30: 111–18. [Google Scholar]
- 21.Negrin A, Lamb CR, Cappello R, Cherubini GB. Results of magnetic resonance imaging in 14 cats with meningoencephalitis. J Feline Med Surg 2007; 9: 109–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paltrinieri S, Grieco V, Comazzi S, Cammarata Parodi M. Laboratory profiles in cats with different pathological and immunohistochemical findings due to feline infectious peritonitis (FIP). J Feline Med Surg 2001; 3: 149–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sparkes AH, Gruffydd-Jones TJ, Harbour DA. An appraisal of the value of laboratory tests in the diagnosis of feline infectious peritonitis. J Am Anim Hosp Assoc 1994; 30: 345–50. [Google Scholar]
- 24.Potkay S, Bacher JD, Pitts TW. Feline infectious peritonitis in a closed breeding colony. Lab Anim Sci 1974; 24: 279–89. [PubMed] [Google Scholar]
- 25.Wolf AM. Feline infectious peritonitis, part 2. Feline Pract 1997; 25: 24–8. [Google Scholar]
- 26.Pedersen NC. Feline infectious peritonitis and feline enteric coronavirus infections. I. Feline enteric coronaviruses. Feline Pract 1983; 13: 5–20. [Google Scholar]
- 27.Rand JS, Parent J, Percy D, Jacobs R. Clinical, cerebrospinal fluid, and histological data from twenty-seven cats with primary inflammatory disease of the central nervous system. Can Vet J 1994; 35: 103–10. [PMC free article] [PubMed] [Google Scholar]
- 28.Foley JE, Rand C, Leutenegger C. Inflammation and changes in cytokine levels in neurological feline infectious peritonitis. J Feline Med Surg 2003; 5: 313–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boettcher IC, Steinberg T, Matiasek K, Greene CE, Hartmann K, Fischer A. Use of anti-coronavirus antibody testing of cerebrospinal fluid for diagnosis of feline infectious peritonitis involving the central nervous system in cats. J Am Vet Med Assoc 2007; 230: 199–205. [DOI] [PubMed] [Google Scholar]
- 30.Herrewegh A, de Groot R, Cepica A, Egberink H, Horzinek M, Rottier P. Detection of feline coronavirus RNA in feces, tissues, and body fluids of naturally infected cats by reverse transcriptase PCR. J Clin Microbiol 1995; 33: 684–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gunn-Moore DA, Gruffydd-Jones TJ, Harbour DA. Detection of feline coronaviruses by culture and reverse transcriptase-polymerase chain reaction of blood samples from healthy cats and cats with clinical feline infectious peritonitis. Vet Microbiol 1998; 62: 193–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gamble D, Lobbiani A, Gramegna M, Moore L, Colucci G. Development of a nested PCR assay for detection of feline infectious peritonitis virus in clinical specimens. J Clin Microbiol 1997; 35: 673–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paltrinieri S, Giordano A, Tranquillo V, Guazzetti S. Critical assessment of the diagnostic value of feline alpha1-acid glycoprotein for feline infectious peritonitis using the likelihood ratios approach. J Vet Diagn Invest; 19: 266–72. [DOI] [PubMed] [Google Scholar]
- 34.Duthie S, Eckersall PD, Addie DD, Lawrence CE, Jarrett O. Value of agrl-acid glycoprotein in the diagnosis of feline infectious peritonitis. Vet Rec 1997; 141: 299–303. [DOI] [PubMed] [Google Scholar]
- 35.Paltrinieri S, Metzger C, Battilani M, Pocacqua V, Gelain ME, Giordano A. Serum [alpha]1-acid glycoprotein (AGP) concentration in non-symptomatic cats with feline coronavirus (FCoV) infection. J Feline Med Surg 2007; 9: 271–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hartmann K, Ritz S. Treatment of cats with feline infectious peritonitis. Vet Immunol Immunopathol 2008; 123: 172–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dubey JP, Lappin MR, Thulliez P. Diagnosis of induced toxoplasmosis in neonatal cats. J Am Vet Med Assoc 1995; 207: 179–85. [PubMed] [Google Scholar]
- 38.Dubey JP. Toxoplasmosis in cats. Feline Pract 1986; 16: 12–6, 8–45. [Google Scholar]
- 39.Dubey JP, Lappin MR. Toxoplasmosis and neosporosis. In: Greene CE, ed. Infectious diseases of the dog and cat. 3rd edn. St Louis, MO: Saunders Elsevier, 2006: 754–75. [Google Scholar]
- 40.Davidson MG, Rottman JB, English RV, Lappin MR, Tompkins MB. Feline immunodeficiency virus predisposes cats to acute generalized toxoplasmosis. Am J Pathol 1993; 143: 1486–97. [PMC free article] [PubMed] [Google Scholar]
- 41.O'Neil SA, Lappin MR, Reif JS, Marks A, Greene CE. Clinical and epidemiological aspects of feline immunodeficiency virus and Toxoplasma gondii coinfections in cats. J Am Anim Hosp Assoc 1991; 27: 211–20. [Google Scholar]
- 42.Lappin MR, Greene CE, Winston S, Toll SL, Epstein ME. Clinical feline toxoplasmosis: Serological diagnosis and therapeutic management of 15 cases. J Vet Intern Med 1989; 3: 139–43. [DOI] [PubMed] [Google Scholar]
- 43.Lappin MR, Greene CE, Prestwood AK, Dawe DL, Marks A. Prevalence of Toxoplasma gondii infection in cats in Georgia using enzyme-linked immunosorbent assays for IgM, IgG, and antigens. Vet Parasitol 1989; 33: 225–30. [DOI] [PubMed] [Google Scholar]
- 44.Barrs VR, Martin P, Beatty JA. Antemortem diagnosis and treatment of toxoplasmosis in two cats on cyclosporin therapy. Aust Vet J 2006; 84: 30–5. [DOI] [PubMed] [Google Scholar]
- 45.Last RD, Suzuki Y, Manning T, Lindsay D, Galipeau L, Whitbread TJ. A case of fatal systemic toxoplasmosis in a cat being treated with cyclosporin A for feline atopy. Vet Dermatol 2004; 15: 194–98. [DOI] [PubMed] [Google Scholar]
- 46.Bernsteen L, Gregory CR, Aronson LR, Lirtzman RA, Brummer DG. Acute toxoplasmosis following renal transplantation in three cats and a dog. J Am Vet Med Assoc 1999; 215: 1123–26. [PubMed] [Google Scholar]
- 47.Adin CA, Gregory CR, Kyles AE, Cowgill L. Diagnostic predictors of complications and survival after renal transplantation in cats. Vet Surg 2001; 30: 515–21. [DOI] [PubMed] [Google Scholar]
- 48.Kadar E, Sykes JE, Kass PH, Bernsteen L, Gregory CR, Kyles AE. Evaluation of the prevalence of infections in cats after renal transplantation: 169 cases (1987–2003). J Am Vet Med Assoc 2005; 227: 948–53. [DOI] [PubMed] [Google Scholar]
- 49.Petrak M, Carpenter J. Feline toxoplasmosis. J Am Vet Med Assoc 1965; 146: 728–34. [PubMed] [Google Scholar]
- 50.Dubey JP, Carpenter JL. Histologically confirmed clinical toxoplasmosis in cats: 100 cases (1952–1990). J Am Vet Med Assoc 1993; 203: 1556–66. [PubMed] [Google Scholar]
- 51.Meier H, Holzworth J, Griffiths RC. Toxoplasmosis in the cat; fourteen cases. J Am Vet Med Assoc 1957; 131: 395–414. [PubMed] [Google Scholar]
- 52.Dubey JP, Johnstone I. Fatal neonatal toxoplasmosis in cats. J Am Anim Hosp Assoc 1982; 18: 461–67. [Google Scholar]
- 53.Dubey JP, Mattix ME, Lipscomb TP. Lesions of neonatally induced toxoplasmosis in cats. Vet Pathol 1996; 33: 290–95. [DOI] [PubMed] [Google Scholar]
- 54.Simpson KE, Devine BC, Gunn-Moore D. Suspected toxoplasma-associated myocarditis in a cat. J Feline Med Surg 2005; 7: 203–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Goncalves R, Platt SR, Francisco J. Clinical and magnetic resonance imaging findings in 92 cats with clinical signs of spinal cord disease. J Feline Med Surg 2009; 11: 53–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Heidel JR, Dubey JP, Blythe LL, Walker LL, Duimstra JR, Jordan JS. Myelitis in a cat infected with Toxoplasma gondii and feline immunodeficiency virus. J Am Vet Med Assoc 1990; 196: 316–18. [PubMed] [Google Scholar]
- 57.McConnell JF, Sparkes AH, Blunden AS, Neath PJ, Sansom J. Eosinophilic fibrosing gastritis and toxoplasmosis in a cat. J Feline Med Surg 2007; 9: 82–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Singh M, Foster DJ, Child G, Lamb WA. Inflammatory cerebrospinal fluid analysis in cats: Clinical diagnosis and outcome. J Feline Med Surg 2005; 7: 77–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pfohl JC, Dewey CW. Intracranial Toxoplasma gondii granuloma in a cat. J Feline Med Surg 2005; 7: 369–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Feeney DA, Sautter JH, Lees GE. An unusual case of acute disseminated toxoplasmosis in a cat. J Am Anim Hosp Assoc 1981; 17: 311–14. [Google Scholar]
- 61.Lappin MR. Feline toxoplasmosis: Interpretation of diagnostic test results. Semin Vet Med Surg (Small Anim) 1996; 11: 154–60. [DOI] [PubMed] [Google Scholar]
- 62.Lappin MR, Marks A, Greene CE, et al. Effect of feline immunodeficiency virus infection on Toxoplasma gondii-specific humoral and cell-mediated immune responses of cats with serologic evidence of toxoplasmosis. J Vet Intern Med 1993; 7: 95–100. [DOI] [PubMed] [Google Scholar]
- 63.Dubey JP, Lappin MR, Thulliez P. Long-term antibody responses of cats fed Toxoplasma gondii tissue cysts. J Parasitol 1995; 81: 887–93. [PubMed] [Google Scholar]
- 64.Muñana KR, Lappin MR, Powell CC, Cooper CM, Chavkin MJ. Sequential measurement of Toxoplasma gondii-specific antibodies in the cerebrospinal fluid of cats with experimentally induced toxoplasmosis. Prog Vet Neurol 1995; 6: 27–31. [Google Scholar]
- 65.Lappin MR, Chavkin MJ, Munana KR, Cooper CM. Feline ocular and cerebrospinal fluid Toxoplasma gondii-specific humoral immune responses following specific and nonspecific immune stimulation. Vet Immunol Immunopathol 1996; 55: 23–31. [DOI] [PubMed] [Google Scholar]
- 66.Burney DP, Chavkin MJ, Dow SW, Potter TA, Lappin MR. Polymerase chain reaction for the detection of Toxoplasma gondii within aqueous humor of experimentally-inoculated cats. Vet Parasitol 1998; 79: 181–86. [DOI] [PubMed] [Google Scholar]
- 67.Lappin MR, Burney DP, Dow SW, Potter TA. Polymerase chain reaction for the detection of Toxoplasma gondii in aqueous humor of cats. Am J Vet Res 1996; 57: 1589–93. [PubMed] [Google Scholar]
- 68.Harari J, Lincoln J. Pharmacologic features of clindamycin in dogs and cats. J Am Vet Med Assoc 1989; 195: 124–25. [PubMed] [Google Scholar]
- 69.Davidson MG, Lappin MR, Rottman JR, et al. Paradoxical effect of clindamycin in experimental, acute toxoplasmosis in cats. Antimicrob Agents Chemother 1996; 40: 1352–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Brown SA, Zaya MJ, Dieringer TM, et al. Tissue concentrations of clindamycin after multiple oral doses in normal cats. J Vet Pharmacol Ther 1990; 13: 270–77. [DOI] [PubMed] [Google Scholar]
- 71.Papich MG, Riviere JE. Chloramphenicol and derivatives, macrolides, lincosamides, and miscellaneous antimicrobials. In: Adams HR, ed. Veterinary pharmacology and therapeutics. 8th edn. Ames: Iowa State University Press, 2001: 868–97. [Google Scholar]
- 72.Greene CE, Lappin MR, Marks A. Effect of clindamycin on clinical, hematological, and biochemical parameters in clinically healthy cats. J Am Anim Hosp Assoc 1992; 28: 323–26. [Google Scholar]
- 73.Holzworth J. Encephalitic toxoplasmosis in a cat. J Am Vet Med Assoc 1954; 124: 313–16. [PubMed] [Google Scholar]
- 74.Morgan RV. Systemic hypertension in four cats: Ocular and medical findings. J Am Anim Hosp Assoc 1986; 22: 615–21. [Google Scholar]
- 75.Brown S, Atkins C, Bagley R, et al. Guidelines for the identification, evaluation, and management of systemic hypertension in dogs and cats. J Vet Intern Med 2007; 21: 542–58. [DOI] [PubMed] [Google Scholar]
- 76.Brown SA, Langford K, Tarver S. Effects of certain vasoactive agents on the long-term pattern of blood pressure, heart rate, and motor activity in cats. Am J Vet Res 1997; 58: 647–52. [PubMed] [Google Scholar]
- 77.Mishina M, Watanabe T, Fujii K, Maeda H, Wakao Y, Takahashi M. Non-invasive blood pressure measurements in cats: Clinical significance of hypertension associated with chronic renal failure. J Vet Med Sci 1998; 60: 805–8. [DOI] [PubMed] [Google Scholar]
- 78.Kobayashi DL, Peterson ME, Graves TK, Lesser M, Nichols CE. Hypertension in cats with chronic renal failure or hyperthyroidism. J Vet Intern Med 1990; 4: 58–62. [DOI] [PubMed] [Google Scholar]
- 79.Sparkes AH, Caney SM, King MC, Gruffydd-Jones TJ. Inter- and intraindividual variation in Doppler ultrasonic indirect blood pressure measurements in healthy cats. J Vet Intern Med 1999; 13: 314–18. [DOI] [PubMed] [Google Scholar]
- 80.Bodey AR, Sansom J. Epidemiological study of blood pressure in domestic cats. J Small Anim Pract 1998; 39: 567–73. [DOI] [PubMed] [Google Scholar]
- 81.Belew AM, Barlett T, Brown SA. Evaluation of the white-coat effect in cats. J Vet Intern Med 1999; 13: 134–42. [DOI] [PubMed] [Google Scholar]
- 82.Elliott J, Barber PJ, Syme HM, Rawlings JM, Markwell PJ. Feline hypertension: Clinical findings and response to antihypertensive treatment in 30 cases. J Small Anim Pract 2001; 42: 122–29. [DOI] [PubMed] [Google Scholar]
- 83.Littman MP. Spontaneous systemic hypertension in 24 cats. J Vet Intern Med 1994; 8: 79–86. [DOI] [PubMed] [Google Scholar]
- 84.Kyles AE, Gregory CR, Wooldridge JD, et al. Management of hypertension controls postoperative neurologic disorders after renal transplantation in cats. Vet Surg 1999; 28: 436–41. [DOI] [PubMed] [Google Scholar]
- 85.Gregory CR, Mathews KG, Aronson LR, Ilkiw JE, LeCouteur RA, Aldrich J. Central nervous system disorders after renal transplantation in cats. Vet Surg 1997; 26: 386–92. [DOI] [PubMed] [Google Scholar]
- 86.Mathews KG, Gregory CR. Renal transplants in cats: 66 cases (1987–1996). J Am Vet Med Assoc 1997; 211: 1432–36. [PubMed] [Google Scholar]
- 87.Cowgill LD, James KM, Levy JK, et al. Use of recombinant human erythropoietin for management of anemia in dogs and cats with renal failure. J Am Vet Med Assoc 1998; 212: 521–28. [PubMed] [Google Scholar]
- 88.Flood SM, Randolph JF, Gelzer ARM, Refsal K. Primary hyperaldosteronism in two cats. J Am Anim Hosp Assoc 1999; 35: 411–16. [DOI] [PubMed] [Google Scholar]
- 89.Maggio F, DeFrancesco TC, Atkins CE, Pizzirani S, Gilger BC, Davidson MG. Ocular lesions associated with systemic hypertension in cats: 69 cases (1985–1998). J Am Vet Med Assoc 2000; 217: 695–702. [DOI] [PubMed] [Google Scholar]
- 90.Bartges JW, Willis AM, Polzin DJ. Hypertension and renal disease. Vet Clin North Am Small Anim Pract 1996; 26: 1331–45. [DOI] [PubMed] [Google Scholar]
- 91.Turner JL, Brogdon JD, Lees GE, Greco DS. Idiopathic hypertension in a cat with secondary hypertensive retinopathy associated with a high-salt diet. J Am Anim Hosp Assoc 1990; 26: 647–51. [Google Scholar]
- 92.Stiles J, Polzin DJ, Bistner SI. The prevalence of retinopathy in cats with systemic hypertension and chronic renal failure or hyperthyroidism. J Am Anim Hosp Assoc 1994; 30: 564–72. [Google Scholar]
- 93.Sansom J, Barnett KC, Dunn KA, Smith KC, Dennis R. Ocular disease associated with hypertension in 16 cats. J Small Anim Pract 1994; 35: 604–11. [Google Scholar]
- 94.Henik RA, Stepien RL, Bortnowski HB. Spectrum of M-mode echocardiographic abnormalities in 75 cats with systemic hypertension. J Am Anim Hosp Assoc 2004; 40: 359–63. [DOI] [PubMed] [Google Scholar]
- 95.Brown CA, Munday JS, Mathur S, Brown SA. Hypertensive encephalopathy in cats with reduced renal function. Vet Pathol 2005; 42: 642–49. [DOI] [PubMed] [Google Scholar]
- 96.Hinchey J, Chaves C, Appignani B, et al. A reversible posterior leuko-encephalopathy syndrome. N Engl J Med 1996; 334: 494–500. [DOI] [PubMed] [Google Scholar]
- 97.Uchino M, Haga D, Nomoto J, Mito T, Kuramitsu T. Brainstem involvement in hypertensive encephalopathy: A report of two cases and literature review. Eur Neurol 2007; 57: 223–26. [DOI] [PubMed] [Google Scholar]
- 98.Johansson BB. The blood-brain barrier and cerebral blood flow in acute hypertension. Acta Med Scand Suppl 1983; 678: 107–12. [DOI] [PubMed] [Google Scholar]
- 99.Acierno MJ, Labato MA. Hypertension in renal disease: Diagnosis and treatment. Clin Tech Small Anim Pract 2005; 20: 23–30. [DOI] [PubMed] [Google Scholar]
- 100.Bartynski WS. Posterior reversible encephalopathy syndrome, part 1: Fundamental imaging and clinical features. Am J Neuroradiol 2008; 29: 1036–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Snyder PS. Amlodipine: A randomized, blinded clinical trial in 9 cats with systemic hypertension. J Vet Intern Med 1998; 12: 157–62. [DOI] [PubMed] [Google Scholar]
- 102.Snyder PS, Sadek D, Jones GL. Effect of amlodipine on echocardiographic variables in cats with systemic hypertension. J Vet Intern Med 2001; 15: 52–6. [DOI] [PubMed] [Google Scholar]
- 103.Henik RA, Snyder PS. Treatment of systemic hypertension in cats with amlodipine besylate. J Am Anim Hosp Assoc 1997; 33: 226–34. [DOI] [PubMed] [Google Scholar]
- 104.Mathur S, Syme H, Brown CA, et al. Effects of the calcium channel antagonist amlodipine in cats with surgically induced hypertensive renal insufficiency. Am J Vet Res 2002; 63: 833–39. [DOI] [PubMed] [Google Scholar]
- 105.Brown SA, Henik RA. Diagnosis and treatment of systemic hypertension. Vet Clin North Am Small Anim Pract 1998; 28: 1481–94, ix. [DOI] [PubMed] [Google Scholar]
- 106.Brown SA, Brown CA, Jacobs G, Stiles J, Hendi RS, Wilson S. Effects of the angiotensin converting enzyme inhibitor benazepril in cats with induced renal insufficiency. Am J Vet Res 2001; 62: 375–83. [DOI] [PubMed] [Google Scholar]
- 107.Bagley RS. Pathophysiologic sequelae of intracranial disease. Vet Clin North Am Small Anim Pract 1996; 26: 711–33. [PubMed] [Google Scholar]
- 108.Jepson RE, Elliott J, Brodbelt D, Syme HM. Effect of control of systolic blood pressure on survival in cats with systemic hypertension. J Vet Intern Med 2007; 21: 402–9. [DOI] [PubMed] [Google Scholar]
- 109.Syme HM, Markwell PJ, Pfeiffer D, Elliott J. Survival of cats with naturally occurring chronic renal failure is related to severity of proteinuria. J Vet Intern Med 2006; 20: 528–35. [DOI] [PubMed] [Google Scholar]
- 110.Jalan R, Shawcross D, Davies N. The molecular pathogenesis of hepatic encephalopathy. Int J Biochem Cell Biol 2003; 35: 1175–81. [DOI] [PubMed] [Google Scholar]
- 111.Maddison JE. Medical management of chronic hepatic encephalopathy. In: Bonagura JD, Kirk RW, eds. Kirk's current veterinary therapy XII small animal practice. Philadelphia: WB Saunders, 1995: 743–49. [Google Scholar]
- 112.Center SA, Magne ML. Historical, physical examination, and clinicopathologic features of portosystemic vascular anomalies in the dog and cat. Semin Vet Med Surg (Small Anim) 1990; 5: 83–93. [PubMed] [Google Scholar]
- 113.Birchard SJ, Sherding RG. Feline portosystemic shunts. Compend Contin Educ Pract Vet 1992; 14: 1295–301. [Google Scholar]
- 114.Levy JK, Bunch SE, Komtebedde J. Feline portosystemic vascular shunts. In: Bonagura JD, Kirk RW, eds. Kirk's current veterinary therapy XII small animal practice. Philadelphia: WB Saunders, 1995: 743–49. [Google Scholar]
- 115.Schunk CM. Feline portosystemic shunts. Semin Vet Med Surg (Small Anim) 1997; 12: 45–50. [PubMed] [Google Scholar]
- 116.White RN, van Hijfte Forster MA, Petrie G, Lamb CR, Hammond RA. Surgical treatment of intrahepatic portosystemic shunts in six cats. Vet Rec 1996; 139: 314–17. [DOI] [PubMed] [Google Scholar]
- 117.VanGundy TG, Boothe HW, Wolf A. Results of surgical management of feline portosystemic shunts. J Am Anim Hosp Assoc 1990; 26: 55–62. [Google Scholar]
- 118.Scavelli TD, Hornbuckle WE, Roth L, et al. Portosystemic shunts in cats: Seven cases (1976–1984). J Am Vet Med Assoc 1986; 189: 317–25. [PubMed] [Google Scholar]
- 119.Levesque DC, Oliver JE, Jr, Cornelius LM, Mahaffey MB, Rawlings CA, Kolata RJ. Congenital portacaval shunts in two cats: Diagnosis and surgical correction. J Am Vet Med Assoc 1982; 181: 143–45. [PubMed] [Google Scholar]
- 120.Berger B, Whiting PG, Breznock EM, Bruhl-Day R, Moore PF. Congenital feline portosystemic shunts. J Am Vet Med Assoc 1986; 188: 517–21. [PubMed] [Google Scholar]
- 121.Lipscomb VJ, Jones HJ, Brockman DJ. Complications and long-term outcomes of the ligation of congenital portosystemic shunts in 49 cats. Vet Rec 2007; 160: 465–70. [DOI] [PubMed] [Google Scholar]
- 122.Brockman DJ, Brown DC, Holt DE. Unusual congenital portosystemic communication resulting from persistence of the extrahepatic umbilical vein. J Small Anim Pract 1998; 39: 244–48. [DOI] [PubMed] [Google Scholar]
- 123.Ware WA, Montavon P, DiBartola SP, Couto CG. Atypical portosystemic shunt in a cat. J Am Vet Med Assoc 1986; 188: 187–88. [PubMed] [Google Scholar]
- 124.Mas A. Hepatic encephalopathy: From pathophysiology to treatment. Digestion 2006; 73 (Suppl 1): 86–93. [DOI] [PubMed] [Google Scholar]
- 125.Hoyumpa AM, Jr, Schenker S. Perspectives in hepatic encephalopathy. J Lab Clin Med 1982; 100: 477–87. [PubMed] [Google Scholar]
- 126.Gerber T, Schomerus H. Hepatic encephalopathy in liver cirrhosis: Pathogenesis, diagnosis and management. Drugs 2000; 60: 1353–70. [DOI] [PubMed] [Google Scholar]
- 127.Maddison JE. Hepatic encephalopathy. Current concepts of the pathogenesis. J Vet Intern Med 1992; 6: 341–53. [DOI] [PubMed] [Google Scholar]
- 128.Lockwood AH, McDonald JM, Reiman RE, et al. The dynamics of ammonia metabolism in man. Effects of liver disease and hyperammonemia. J Clin Invest 1979; 63: 449–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Albrecht J, Jones EA. Hepatic encephalopathy: Molecular mechanisms underlying the clinical syndrome. J Neurol Sci 1999; 170: 138–46. [DOI] [PubMed] [Google Scholar]
- 130.Berl S, Takagaki G, Clarke DD, Waelsch H. Metabolic compartments in vivo. Ammonia and glutamic acid metabolism in brain and liver. J Biol Chem 1962; 237: 2562–69. [PubMed] [Google Scholar]
- 131.Hooper PT. Spongy degeneration in the central nervous system of domestic animals. Part III: Occurrence and pathogenesis of hepatocerebral disease caused by hyperammonaemia. Acta Neuropathol 1975; 31: 343–51. [DOI] [PubMed] [Google Scholar]
- 132.Zieve L, Doizaki WM, Zieve J. Synergism between mercaptans and ammonia or fatty acids in the production of coma: A possible role for mercaptans in the pathogenesis of hepatic coma. J Lab Clin Med 1974; 83: 16–28. [PubMed] [Google Scholar]
- 133.Hardy RM. Pathophysiology of hepatic encephalopathy. Semin Vet Med Surg (Small Anim) 1990; 5: 100–6. [PubMed] [Google Scholar]
- 134.Baraldi M, Zeneroli ZL. Experimental hepatic encephalopathy: Changes in the binding of gamma-aminobutyric acid. Science 1982; 216: 427–29. [DOI] [PubMed] [Google Scholar]
- 135.James JH, Ziparo V, Jeppsson B, Fischer JE. Hyperammonaemia, plasma aminoacid imbalance, and blood-brain aminoacid transport: A unified theory of portal-systemic encephalopathy. Lancet 1979; 2: 772–75. [DOI] [PubMed] [Google Scholar]
- 136.Daniel GB, Bright R, Ollis P, Shull R. Per rectal portal scintigraphy using 99mtechnetium pertechnetate to diagnose portosystemic shunts in dogs and cats. J Vet Intern Med 1991; 5: 23–7. [DOI] [PubMed] [Google Scholar]
- 137.Tillson DM, Winkler JT. Diagnosis and treatment of portosystemic shunts in the cat. Vet Clin North Am Small Anim Pract 2002; 32: 881–99, vi–vii. [DOI] [PubMed] [Google Scholar]
- 138.d'Anjou MA, Penninck D, Cornejo L, Pibarot P. Ultrasonographic diagnosis of portosystemic shunting in dogs and cats. Vet Radiol Ultrasound 2004; 45: 424–37. [DOI] [PubMed] [Google Scholar]
- 139.Koblik PD, Hornof WJ. Transcolonic sodium pertechnetate Tc 99m scintigraphy for diagnosis of macrovascular portosystemic shunts in dogs, cats, and potbellied pigs: 176 cases (1988–1992). J Am Vet Med Assoc 1995; 207: 729–33. [PubMed] [Google Scholar]
- 140.Morgan M, Blei A, Grüngreiff K, et al. The treatment of hepatic encephalopathy. Metab Brain Dis 2007; 22: 389–405. [DOI] [PubMed] [Google Scholar]
- 141.van Leeuwen PA, van Berlo CL, Soeters PB. New mode of action for lactulose. Lancet 1988; 1: 55–6. [DOI] [PubMed] [Google Scholar]
- 142.Taboada J. Medical management of animals with portosystemic shunts. Semin Vet Med Surg (Small Anim) 1990; 5: 107–19. [PubMed] [Google Scholar]