

Abstract
The cytosolic helicase retinoic acid-inducible gene-I (RIG-I) initiates immune responses to most RNA viruses by detecting viral 5′-triphosphorylated RNA (pppRNA). Although endogenous mRNA is also 5′-triphosphorylated, backbone modifications and the 5′-ppp-linked methylguanosine (m7G) cap prevent immunorecognition. Here we show that the methylation status of endogenous capped mRNA at the 5′-terminal nucleotide (N1) was crucial to prevent RIG-I activation. Moreover, we identified a single conserved amino acid (H830) in the RIG-I RNA binding pocket as the mediator of steric exclusion of N1-2′O-methylated RNA. H830A alteration (RIG-I(H830A)) restored binding of N1-2′O-methylated pppRNA. Consequently, endogenous mRNA activated the RIG-I(H830A) mutant but not wild-type RIG-I. Similarly, knockdown of the endogenous N1-2′O-methyltransferase led to considerable RIG-I stimulation in the absence of exogenous stimuli. Studies involving yellow-fever-virus-encoded 2′O-methyltransferase and RIG-I(H830A) revealed that viruses exploit this mechanism to escape RIG-I. Our data reveal a new role for cap N1-2′O-methylation in RIG-I tolerance of self-RNA.
Keywords: mRNA, cap, 2′O-methyl, 5′-triphosphate RNA, immune recognition of RNA, virus, RIG-I, MTr1, innate immune tolerance mechanism
Graphical Abstract
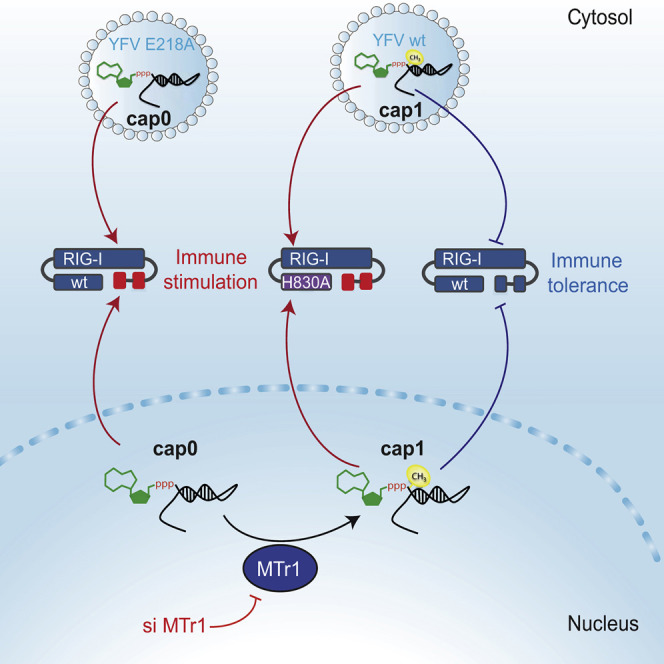
Highlights
-
•
N1-2′O-methylation is crucial to block RIG-I activation by viral and self RNA
-
•
Self-RNA exclusion is singularly governed by the conserved H830 in RIG-I
-
•
H830A alteration leads to indiscriminate recognition of endogenous RNA by RIG-I
-
•
Cellular N1-2′O-methyltransferase knockdown renders endogenous RNA stimulatory
The cytosolic receptor RIG-I initiates immune responses against most RNA viruses by detecting viral RNA. Schlee and colleagues report that a conserved amino acid in the RNA binding pocket prevents recognition of endogenous RNA bearing a N1-2′O-methyl group as a marker of “self” and that flaviviruses exploit this tolerance mechanism for immunoescape.
Introduction
Most highly pathogenic and emerging viruses are RNA genome-based viruses, giving rise to zoonotic and epidemic diseases (e.g., influenza) or causing viral hemorrhagic fever (yellow fever, dengue fever, Lassa fever, Ebola disease) (Bray, 2008). The first barrier against invasion of RNA viruses is a cytosolic innate intracellular defense response that is present in all cell types and is initiated by activation of the viral RNA-sensing innate immune receptors RIG-I (retinoic acid-inducible gene 1I) and MDA5 (melanoma differentiation-associated protein 5) in the cytosol (Gitlin et al., 2006, Kato et al., 2006, Loo et al., 2008). Upon sensing of viral RNA, RIG-I or MDA5 induces type I interferon (IFN) secretion leading to upregulation of antiviral IFN-induced proteins in the infected and neighboring cells, which inhibits virus replication or protects against new infection. Further downstream events attract immune cells and trigger the adaptive immune response.
Discrimination of a few viral RNA molecules from the abundant host RNA in the cytosol occurs by detection of unusual structures or modifications of the viral RNA. The structural features leading to recognition by MDA5 have remained elusive. By contrast, because of high-resolution structures of RIG-I/ligand complexes, the mechanism of RNA recognition by RIG-I is well understood (Civril et al., 2011, Jiang et al., 2011, Kowalinski et al., 2011, Lu et al., 2010, Luo et al., 2011, Wang et al., 2010). RIG-I is activated by double-stranded 5′-triphosphorylated RNA (ppp-dsRNA) (Hornung et al., 2006, Pichlmair et al., 2006, Schlee and Hartmann, 2010, Schlee et al., 2009). Like most viral RNAs, endogenous mRNA and RNA polymerase III transcripts are also 5′-triphosphorylated. However, mRNAs of eukaryotes possess a cap structure consisting of a triphosphorylated 5′ end that is 5′-5′ linked to a guanosine methylated at N7 (m7G) (Figure 1 A). The m7G cap is essential for eIF4a-dependent mRNA-dependent protein translation from yeast to vertebrates. 2′O-methyl modifications at N1 and N2 are conserved features of mRNA cap structures of higher eukaryotes (Banerjee, 1980) but haven’t been implicated in mRNA translation control so far (Bélanger et al., 2010). A number of differences between eukaryotic RNA and microbial RNAs have been described that explain the discrimination of self and non-self by RIG-I. Apart from the presence of a 5′-triphosphate moiety in fully base-paired regions in various microbial RNA species, this also includes certain modifications in eukaryotic RNAs that exert an inhibitory function on otherwise stimulatory RNA molecules. This includes backbone and nucleobase modifications in eukaryotic RNAs, a well as m7G cap modification of eukaryotic mRNAs (Hornung et al., 2006, Pichlmair et al., 2006). The relative contribution of these activating or inhibiting signatures in putative RIG-I ligands remains to be determined, especially in light of the fact that in vitro transcription was used to study the impact of the latter modifications (Goubau et al., 2014, Schlee et al., 2009, Schmidt et al., 2009; see Supplemental Experimental Procedures). In the present study, by using synthetic pppRNA, we re-assessed the impact of m7G on RIG-I stimulation and analyzed also the influence of single modifications (including G methylation at N7 and 2′O-methylation of the N1 and N2 position of the RNA) on the interaction with RIG-I. We found that a m7G cap alone could only partially reduce RIG-I stimulation at low RIG-I ligand concentrations, whereas the 2′O-methylation of the 5′-terminal nucleotide (N1) entirely abrogated RIG-I activation even at high ligand concentrations. Structure-guided mutational analysis led to the identification of a highly conserved histidine (H830) in the RNA binding domain of RIG-I, which is dispensable for RIG-I activation but mediates steric exclusion of N1-methylated RNA. We furthermore provide evidence that endogenous RNA stimulated RIG-I in the absence of endogenous cap1 2′O-methylation or if H830 was mutated. Furthermore, we demonstrated that RNA viruses exploited this tolerance mechanism to avoid recognition by RIG-I. The 100% conservation of this “licensing” histidine position in RIG-I among vertebrates and even sea anemone RIG-I highlights a fundamental role of this self-RNA tolerance mechanism in evolution. As such, this is the first study showing the biological relevance of endogenous cap 2′O-methylation and the corresponding enzymes for immune tolerance of self-RNA, a pathway that is relevant for any cell type in the body.
Figure 1.

2′O-Methylation at N1 Position of RNA Critically Determines the Abolition of RIG-I Activation
(A) The chemical structure of cap structures as contained in eukaryotes’ mRNA is presented (m7GpppNmNm). Important structural features are labeled. The cap2 structure, which occurs only in higher eukaryotes’ mRNA, consists of a G, 5′-5′ triphosphate linked to N1, with methylation at N7 of the G residue. N1 and N2 are 2′O-methylated.
(B and C) Chloroquine-treated human PBMCs were stimulated with the indicated synthetic RNA oligonucleotides (see also Table S1 and Figure S2) at concentrations of 2.5 nM and 5 nM (B) or a dose titration was performed (C). Before stimulation, RNA oligonucleotides were hybridized with the complementary RNA (AsGA) to blunt-ended double-stranded RNA. IFN-α production was analyzed 20 hr after stimulation. Data from four donors are depicted as mean values + SEM.
(D) Murine bone-marrow-derived dendritic cells from MDA5- or RIG-I-deficient or wild-type mice were stimulated with indicated RNA ligands (50 nM) and murine IFN-α was determined by ELISA (linear range limit: 80 pg/ml) 20 hr after transfection. One representative experiment out of two is shown. Error bars indicate SD.
Results
2′O-Methylation at the N1 Position of RNA Critically Determines the Abolition of RIG-I Activation
Capping of mRNA is required for effective ribosomal translation. Three forms of cap structures are known: cap0 is a N7-methyl guanosine (m7G) linked to the gamma phosphate of the 5′-triphosphate of RNA (m7GpppNN; N = nucleotide; Figure 1A); cap1 is identical to cap0 except for an additional 2′O-methyl group at the N1 position (m7GpppNmN); and cap2 carries a second 2′O-methyl group at the N2 position (m7GpppNmNm). Although the cap N7-methyl guanosine in cap structures is well known to be required for effective ribosomal translation, the impact of the 2′O-methyl groups at N1 and N2 on translation is unclear. So far, it has been described that replication of (+)ssRNA viruses lacking the viral N1-2′O-methyl transferase is restricted by the type I IFN-induced, pppRNA binding protein IFIT1 (Abbas et al., 2013, Daffis et al., 2010, Habjan et al., 2013, Pichlmair et al., 2011). To study the impact of the different components of cap structures on immunorecognition of RNA by RIG-I, we established methods to generate well-characterized 24-mer RNA oligonucleotides with identical sequence and containing a 5′-triphosphate with additional single or combined features of the cap0/1/2 structures (e.g., with or without guanosine cap, N7-methylated guanosine cap, 2′O-methyl at N1 or N2; see Figures S2A–S2E and Table S1). These RNA oligonucleotides were hybridized with complementary RNA (antisense RNA oligonucleotide: AsGA; Table S1) resulting in short double-stranded blunt-end RNA oligonucleotides (ppp-dsRNA). The double-stranded RNA oligonucleotides were transfected into human peripheral blood mononuclear cells (PBMCs) in the presence of chloroquine, which eliminates endosomal TLR-mediated recognition. In this setting, IFN-α induction by short ppp-dsRNA is exclusively mediated by RIG-I (Schlee et al., 2009, Wang et al., 2010). We found that 5′ppp5′-linked G (GpppGA) did not impair RIG-I activation (Figure 1B), whereas N7-methylation of the 5′ppp5′-linked G (cap0 = m7GpppGA; Figure 1B) only partially reduced IFN-α induction at the concentrations used (2.5 nM). In dose response experiments, complete inhibition of RIG-I activation by m7G cap was seen at concentrations below 0.5 nM (Figure 1C). By contrast, a single 2′O-methyl group at N1 (pppGmA) completely abolished RIG-I activation at all RNA concentrations tested (up to 15.8 nM; Figures 1B and 1C). The combination of the 2′O-methyl group at N1 and m7G as in cap1 (m7GpppGmA; Figure 1B) also showed no activity. However, 2′O-methylation at N2 with and without m7G (m7GpppGAm, pppGAm) only partially reduced the RIG-I activity (Figure 1B). These data reveal a dominant role of the 2′O-methyl group at N1 for the inability of RIG-I to sense capped pppRNA. Stimulation of bone-marrow-derived dendritic cells (BM-DCs) deficient for Ifih1 (encoding MDA5) or Ddx58 (encoding RIG-I) with ligands comprising different cap structures confirmed the exclusive detection of capped pppRNA by RIG-I and the absolute inhibitory effect of 2′O-methylation (Figure 1D).
The Amino Acid H830 in the RNA Binding Domain of RIG-I Is Responsible for Steric Exclusion of N1-2′O-Methylated cap1 RNA
RIG-I is composed of two N-terminal signaling adaptor domains (CARD), a helicase domain (DECH), and the C-terminal domain (CTD), which was identified to harbor the pppRNA binding site (Cui et al., 2008, Takahasi et al., 2008). The CTD possesses a basic binding cleft with ppp-dsRNA-CTD interactions at amino acids F853 (5′-terminal base pair stacking), K858, H847, K861, K888 (ppp binding), K907 (internucleotide phosphate binding), and H830 (contact to 2′OH of N1) (Lu et al., 2010, Wang et al., 2010). We mutated selected amino acids of the binding pocket of RIG-I to alanine and expressed the full-length RIG-I constructs in HEK293blue cells, which lack RIG-I activity, if not primed by type I IFN. The IP10 gene is known to be directly activated by RIG-I (Brownell et al., 2014). IP10 secretion was studied as a surrogate parameter of RIG-I activation after stimulation with synthetic ppp-dsRNA or ppp-ssRNA (Figure 2 A). The highly conserved amino acids F853, K861, K888, and K907 (Figure S1), which are part of the basic ppp-dsRNA binding cleft of the CTD domain, were essential for RIG-I activation by ppp-dsRNA (Figure 2A). Although the highly conserved H830 is in contact with 2′OH of N1, the H830A mutation did not decrease but instead increased ppp-dsRNA-induced RIG-I stimulation (Figure 2A). Immunoblot analysis confirmed that this increase was not due to higher expression of the mutated RIG-I(H830A) (Figure 2B). Indeed, its 100% conservation across all known vertebrate species and sea anemone RIG-I (Figure S1) points to a key function of H830 in RIG-I biology. Because H830 is not required for RIG-I activation and given its position in proximity to N1, we speculated that H830 might be involved in a steric interference of the 2′O-methyl group (trans-C3′-oriented) through its side chain (Figure 2C; Wang et al., 2010). To address this hypothesis, we performed a set of experiments in which we compared wild-type (WT) RIG-I to RIG-I(H830A) by using different pppRNA ligands. Indeed, in contrast to RIG-I(WT), the RIG-I(H830A) mutant was still activated by ppp-dsRNA containing 2′O-methyl at N1 (pppGmA) and showed considerable activation in the presence of a complete cap1 structure (m7GpppGmA) (Figure 2D, black bars).
Figure 2.

Immune Tolerance of 2′O-Methylated cap1 Structures Is Mediated by H830 in the ppp-dsRNA Binding Cleft of RIG-I
(A) Wild-type or indicated full-length human RIG-I mutants (see also Figure S1) were overexpressed for 12 hr in HEK293blue cells and stimulated with 5 nM synthetic 5′-ppp-ss or dsRNA. IP10 production was analyzed 20 hr after stimulation. Data from three independent experiments are depicted as mean values + SEM.
(B) Immunoblot of RIG-I(WT) and indicated RIG-I mutants overexpressed in HEK293blue cells for 48 hr.
(C) Interaction of H830 with 2′OH of N1 as revealed from crystal structure (Wang et al., 2010) of CTD-ppp-dsRNA complex is displayed.
(D) RIG-I(WT) or RIG-I(H830A) was overexpressed in HEK293blue cells and stimulated with indicated synthetic ppp-dsRNA ligands (5 nM). IP10 production was analyzed 20 hr after stimulation. Data from three independent experiments are depicted as mean values ± SEM, normalized on RIG-I(WT)+pppGA (100% = 9 ng/ml IP10 in average).
(E–G) Homogenous interaction assay (AlphaScreen) of purified RIG-I(WT) or RIG-I(H830A) protein with indicated synthetic ligands. Protein concentrations were kept constant, and ligand concentrations were titrated. Titration plots (E and F) and apparent dissociation constants (Kd in G) are shown. AlphaScreen units are proportional to RIG-I-ligand complex concentrations. One representative experiment of two is shown.
Error bars indicate SD. Purity of RIG-I(WT) and RIG-I(H830A) proteins are shown in Figure S2F.
By using a homogenous ligand interaction assay, we analyzed the binding affinity of RIG-I(WT) and RIG-I(H830A) to the different ppp-RNAs (Figures 2E–2G). Consistent with reduced RIG-I-stimulating activity, binding of pppGmA to RIG-I(WT) was 4.5-fold diminished in comparison to pppGA, and 5′ppp5′-linked m7G (m7GpppGA) also inhibited binding but to a lower extent (∼2-fold) (Figures 2E and 2G). The binding of cap1-bearing dsRNA (m7GpppGmA) to RIG-I(WT) was lower than the detection limit of the assay (Figures 2E and 2G; Kd > 1,000 nM).
Similar to RIG-I(WT), the cap0 structure (m7GpppGA) reduced binding of RIG-I(H830A) 2-fold, demonstrating that H830 does not interfere with the m7G cap. In contrast to RIG-I(WT) and consistent with the functional activity, binding of RIG-I(H830A) to pppRNA with and without 2′O-methylation at N1 was nearly equal (Figures 2F and 2G). In line with this result, RNA with a complete cap1 structure (m7GpppGmA) bound to RIG-I(H830A) to an extent comparable to the non-2′O-methylated cap0-bearing RNA (m7GpppGA) (Figures 2F and 2G).
Of note, pppGmA did still demonstrate some residual binding to RIG-I(WT) in the Alphascreen assay although the assay cannot resolve the binding site. One possible explanation for this residual interaction with RIG-I(WT) could be non-productive binding as reported previously (Marq et al., 2011). Here, RIG-I binding was reported for ligands that could not activate RIG-I or induce type I IFN. In particular, dsRNA with an overhanging 5′-ppp-nucleotide (ppp(+1nt)-dsRNA) was observed to be a weak competitor for blunt ppp-dsRNA (Marq et al., 2011). To test whether N1-2′O-methylation has similar effects as a 5′-ppp-overhang, we performed a competition assay as described previously (Marq et al., 2011). We transfected ppp-dsRNA as pppGA hybridized to asGA (pppGA+asGA) mixed with a 24-fold excess of asGA (single-stranded control, non-binder), pppGmA+asGA (blunt N1-2′O-methylated), or pppGA+asGA(−1) (ppp(+1nt)-dsRNA, non-productive binder) (Figure S2F; Marq et al., 2011). Although ppp(+1nt)-dsRNA, as previously published, reduced RIG-I stimulation by ppp-dsRNA 2-fold, no competitive inhibition via N1-2′O-methylated ppp-dsRNA (pppGmA) could be detected. Thus, unlike ppp(+1nt)-dsRNA, pppGmA binding does not seem to be sufficient to block binding of pppGA in this setting.
Altogether, the results provide strong evidence that H830 impairs accurate binding of N1-2′O-methylated RNA via steric hindrance, leading to immune ignorance of 2′O-methylated pppRNA.
H830 and cap1 N1-2′O-Methylation of Self RNA Prevent Immunorecognition of Endogenous RNAs
The high conservation of H830 in RIG-I and its function in interference with accurate binding of 5′-triphosphate RNA carrying a 2′O-methyl group at N1 suggest the existence of self-RNA molecules that depend on N1 methylation to evade recognition by RIG-I. Although base-paired 5′-triphosphate RNA is required for RIG-I stimulation, GpppNN-RNA intermediates with such RIG-I ligand properties might occur, causing some degree of immune stimulation in the absence of 2′O-methylation (or in the presence of the mutated form of RIG-I that accepts 2′O-methylation). Indeed, we found that long-term expression of RIG-I(H830A) in HEK293blue cells lacking RIG-I activity led to a significant IP10 response in the absence of exogenous RIG-I ligand (Figure 3 A, black bars). To show that IP10 induction is induced by endogenous RNA species, we overexpressed RIG-I-CTD-FLAG in HEK293blue cells and purified endogenous RNA bound to immunoprecipitated RIG-I-CTD-FLAG (Figure 3B, left). RIG-I-bound endogenous RNA stimulated the RIG-I(H830A) mutant but not RIG-I(WT) (Figure 3B, right). The RIG-I-stimulating activity of isolated endogenous RNA was partially sensitive to alkaline phosphatase (AP), which hydrolyzes free phosphates, and to treatment with tobacco acid pyrophosphatase (TAP), which hydrolyzes the triphosphate bridge of mRNA caps (Figure S3A). However, only the combined treatment abolished RIG-I stimulation, indicating the presence of stimulatory capped and uncapped pppRNA species (Figure 3B). It was reported that mRNA cap1 2′O-methylation occurs in the nucleus and is performed by the endogenous cap1 methyltransferase MTr1 (Bélanger et al., 2010, Perry and Kelley, 1976). To study the impact of diminished 2′O-methylation of endogenous RNA transcripts, we knocked down hMTr1 in primary human fibroblasts (Figures 3C and S3B). Intriguingly, inhibition of endogenous cap methylation induced a substantial type I IFN response 72 hr after hMTr1 knock down (Figure 3C). Correspondingly, siRNA-mediated knock-down of MTr1 induced IFN-β mRNA in primed A549 cells (Figure 3D). By contrast, RIG-I-deficient A549 cells did not upregulate IFN-β upon depletion of MTr1 (Figures 3D and S3C). A similar scenario held true in HEK293blue cells silenced for hMTr1 (Figures 3E and S3D) and lacking significant endogenous RIG-I activity. Expression of RIG-I(WT) in combination with knockdown of hMTr1 (Figure 3E, right) led to increased IP10 production as compared to the control setting (Figure 3E, left). Overexpression of RIG-I(H830A) by itself also led to an increased IP10 response, yet additional silencing of hMTr1 did not considerably augment RIG-I(H830A)-induced IP10 levels (Figure 3E, right). Altogether these results indicated that hMTr1 activity was required for an active suppression of endogenous RNA recognition by RIG-I.
Figure 3.

H830 and cap1 2′O-Methylation of RNA Prevent Recognition of Endogenous RNAs
(A) RIG-I(WT) and RIG-I(H830A) were expressed in HEK293blue cells in the absence of exogenous RIG-I stimuli and IP10 was monitored at indicated times (24, 48, 72 hr) after transfection. Data from three independent experiments with technical duplicates are depicted as mean values + SEM.
(B) Flag-tagged RIG-I-CTD was overexpressed in HEK293blue cells. Endogenous RNA binding to immune-precipitated RIG-I-CTD was extracted and used for stimulation of RIG-I(WT)- and RIG-I(H830A)-expressing HEK293blue cells. Before stimulation, RNAs were treated with tobacco acid pyrophosphatase (TAP) or alkaline phosphatase (AP) or left untreated (nt). IP10 induction values 20 hr after stimulation from three independent experiments with technical duplicates are depicted as mean values + SEM. TAP hydrolyzes and inactivates free and capped, AP-only free triphosphate (ppp). Right panel: Ethidium bromide-stained agarose gel of RIG-I(WT)-CTD-bound RNAs. Untreated (nt), treated with TAP, or AP.
(C) Fibroblasts isolated from human nasal conchas were transfected with control siRNA (control siRNA pool) or siRNA against hMTr1 (siRNA hMTr1 pool) and harvested after 70 hr. RNA was isolated and IFN-β mRNA induction relative to TBP-1 mRNA expression was determined by real-time PCR. One representative of two experiments in triplicates is shown. Error bars indicate SD.
(D) IFN-β mRNA induction by siRNAs against hMTr1 in wild-type (WT) or RIG-I-deficient (Ddx58−/−) A549 cells were treated with indicated siRNAs, primed with 1,000 U/ml IFN-α, and assessed for IFN-β mRNA induction by qPCR 72 hr after siRNA transfection. Data from three independent experiments with technical duplicates are depicted as mean values + SEM.
(E) IP10 induction in RIG-I(WT)- or RIG-I(H830A)-expressing HEK293blue cells 72 hr after treatment with control siRNA (control siRNA pool) or siRNA against hMTr1 (siRNA hMTr1 pool); linear range limit, 31 pg/ml.
Data from three independent experiments with technical duplicates are depicted as mean values ± SEM (B, D, E). For statistics, two-way ANOVA and Bonferroni post-test was applied: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. Knockdown efficiencies were determined by immunoblot (Figure S3A) of hMTr1 and real-time PCR (Figures S3B and S3C).
Yellow Fever Virus cap1 2′O-Methyltransferase Activity Impairs Recognition by RIG-I
Viruses have evolved numerous mechanisms that allow capping and methylation of viral RNA in the cytosol. Flaviviruses (positive ssRNA) express the NS5 protein, which performs RNA polymerization as well as guanine N7 and ribose 2′O-methylations (Figure S4; Zhou et al., 2007). To examine whether cytosolic RNA viruses use N1-2′O-methylation to suppress type I IFN induction by RIG-I, we constructed a yellow fever virus replicon (YFVR-WT) and abrogated 2′O-methylation activity of NS5 by mutation of NS5-E218 to A (YFVR-E218A) (Zhou et al., 2007); replication was monitored by renilla luciferase activity encoded by YFVR (Figure S4B). YFVR-E218A (no cap1 methylation of RNA) replication was strongly impaired in A549 cells as compared to the YFVR-WT (Figure 4 A, WT versus 218). By contrast, no difference in replication of both YFV replicons was found in Vero cells, which lack type I IFN genes (Figure 4B, WT versus 218). Likewise, when A549 or Vero cells were infected with whole yellow fever virus particles (Figures 4C and 4D), the NS5-E218A mutation impaired virus production in A549 cells to a much higher extent (>60-fold reduction, Figure 4C) than in Vero cells (<5-fold reduction, Figure 4D). These data indicate that cap1 methylation is involved in the immune escape of yellow fever virus from a type I IFN-dependent host restriction system. To further strengthen this hypothesis, we used RIG-I-deficient and STAT1-deficient A549 cells to assess the influence on YFV replication (Figure 4E). Intriguingly, YFV-E218A reached the same virus titer in RIG-I-deficient A549 cells as the wild-type virus. Similar to Vero cells, the replication of YFV-E218A was not impaired in STAT1-deficient A549 cells. Viral replication was reflected by induction of the type I IFN response (Figure 4F). As determined 8 hr after infection, YFV-E218A induced a 4-fold higher IFIT1 expression than wild-type YFV. By contrast, no considerable induction of IFIT1 was detected in RIG-I- and STAT1-deficient A549 cells. The data demonstrate that RIG-I is essential for type I IFN induction by YFV in A549 cells. Furthermore, these data clearly show that the ability of YFV to methylate RNA (lost with the mutation E218A) reduces immunorecognition of the virus by RIG-I and enhances viral replication. In HEK293blue cells lacking endogenous RIG-I expression, YFVR-WT and YFVR-E218A induced IP10 only when RIG-I was expressed (Figure 4G). Analogous to cells with reduced endogenous MTr1 (Figure 3E), YFVR-E218A lacking the viral N1-2′O-methyltransferase activity induced a 3-fold higher IP10 level than YFVR-WT in RIG-I(WT)-expressing cells (Figure 4G, RIG-I WT blue versus orange). On the other hand, RIG-I(H830A), ignoring 2′O-methylation status, induced equal amounts of IP10 in response to YFVR-WT and YFVR-E218A (Figure 4G), up to the same level as the response to YFVR-E218A RNA in RIG-I(WT) transfected cells.
Figure 4.

YFV cap1 2′O-Methyltransferase Prevents Immune Recognition
(A and B) Immune-competent A549 cells (A) or type I IFN gene-deficient Vero cells (B) were transfected with YFV replicon (YFVR) RNA or YFVR-E218A RNA-deficient for viral cap1 2′O-methyltransferase activity (see also Figure S4A). Replication was monitored by replicon-derived luciferase activity. Average of two experiments in technical duplicates is shown. Error bars indicate SD.
(C and D) A549 cells (C) or Vero cells (D) were infected with whole yellow fever virus particles YFV-WT or YFV-E218A (MOI 0.01). Virus production was quantified by plaque assay in BHK cells 24, 48, or 72 hr after infection. One representative of two experiments in technical duplicates is shown. Error bars indicate range.
(E) Wild-type, RIG-I-deficient (Ddx58−/−), or STAT1-deficient A549 cells were infected with YFV-WT or YFV-E218A (MOI 0.01) and virus production was quantified as in (C) 72 hr after infection.
(F) Wild-type, RIG-I-deficient (Ddx58−/−), or STAT1-deficient A549 cells were infected with YFV-WT or YFV-E218A (MOI 1) and IFIT1 mRNA was measured 8 hr after infection by RT-PCR.
(E and F) Average values of two experiments in technical duplicates are shown; error bars indicate SEM.
(G) Untransfected (no RIG-I, mock), RIG-I(WT)-, or RIG-I(H830A)-expressing HEK293blue cells were transfected with YFVR-WT or YFVR(218) replicon RNA. The mean values of four experiments in technical duplicates is shown, error bars: SEM 100% = 895 ng/ml IP10 in average, linear range limit: 31 pg/ml.
(E–G) For statistics, two-way ANOVA and Bonferroni post-test were applied: ∗∗p < 0.01, ∗∗∗p < 0.001.
Altogether these results demonstrated that cap1 2′O-methylation of YFV RNA constitutes a viral subversion mechanism to evade RIG-I recognition.
Discussion
Posttranscriptional modification is key for the discrimination of foreign from self nucleic acids. Using synthetic modified ppp-RNA, we analyzed the influence of individual cap modifications including N7 methylation and 2′O-methylation. We found that masking the 5′ RNA triphosphate by the non-methylated 0G-cap alone did not influence RIG-I stimulation. Additional N7-methylation of 0GpppNN-RNA, resulting in a cap0 structure (m7GpppNN-RNA), reduced RIG-I activation, but only partially. By contrast, 2′O-methylation at the penultimate nucleotide (N1) as found in vertebrate mRNA cap1 structures (m7GpppNmN-RNA) abrogated RIG-I activation completely.
Because not only viral RNA but also abundant cytosolic self-RNA molecules harbor a triphosphate at the 5′ end, the distinction of self from viral RNA is molecularly challenging. The detection system needs to provide high sensitivity for few viral RNA copies and immune tolerance toward highly abundant endogenous RNA species. Based on our previous structural data (Wang et al., 2010), we are now able to provide for the first time the molecular mechanism of how an innate immune receptor discriminates 2′O-methylation of self RNA, as the RIG-I RNA binding pocket prevents binding of N1-2′O-methylated RNA.
We found that sufficient self-tolerance necessitates several levels of self-RNA labeling and that methylations are the decisive modifications to prohibit recognition by RIG-I. By using chemically synthesized RNA, we are now able to dissect single modifications independently and quantify their contributions. Non-methylated 0G cap alone did not affect RIG-I stimulation. This is in accordance with the ppp-dsRNA/RIG-I crystal structure (Lu et al., 2010, Wang et al., 2010) from which it can be predicted that a 0G cap does not sterically interfere with binding to RIG-I. By contrast, N7-methylation (m7G cap) impaired RIG-I stimulation at low ligand concentrations but not to a full extent. In previous studies, we found a higher inhibitory impact of the methylguanidine (m7G) cap0 structure, which can be explained by the in vitro transcription process used to generate these ligands (Hornung et al., 2006, Pichlmair et al., 2006) (see Supplemental Experimental Procedures for details). Additional N7-methylation introduces a positive charge into the cap, which weakens the interaction with the basic (positively charged) pppRNA-binding cleft. Furthermore, m7G mediates binding to eIF4A (Fechter and Brownlee, 2005), potentially competing with binding to RIG-I. Both effects can reduce RIG-I activation but are not sufficient to mediate the complete ignorance of self 5′-triphosphate RNA by RIG-I. Our data show that complete self-tolerance is accomplished by a single 2′O-methyl group at the 5′-terminal residue (N1), a characteristic molecular feature of higher eukaryotes’ mRNA (Banerjee, 1980). This immune tolerance is mediated by a single highly conserved amino acid, H830, in the pppRNA-binding domain of RIG-I. The responsible histidine is present in all known species that express RIG-I and was therefore deemed to be involved in RNA binding (Lu et al., 2010, Wang et al., 2010). Mutation of H830 to alanine restored the binding of and stimulation by 2′O-methyl pppRNA while the stimulatory activity of non-methyl pppRNA remained unaffected, highlighting an exclusive immune regulatory function of H830. N1-2′O-methylated and non-methylated ppp-dsRNAs or m7Gppp-dsRNA bind with considerably different affinities to wild-type RIG-I but with similar affinities to mutant RIG-I(H830A), indicating a direct steric effect of H830 on N1-2′O-methylated RNA. An inactive ligand (pppGmA) still showed residual but non-productive RIG-I binding. The discrepancy between binding (only 5-fold reduction) and biological activity (no RIG-I activation at any concentration) might appear contradictory at first view. However, an assay that simply measures binding affinity can resolve neither the site nor the orientation of protein/ligand interaction. Previously, non-productive binding to RIG-I of dsRNA with competitive, ppp(+1nt)-dsRNA, and non-competitive, blunt OH-dsRNA, ligands has been reported (Marq et al., 2011). Our competition assay revealed that N1-2′O-methylated dsRNA is a much weaker competitive inhibitor than ppp(+1nt)-dsRNA (if at all) for pppGA binding. Thus, pppGmA has a similar profile to the blunt OH-dsRNA sequences used by Marq et al. (2011). These sequences can bind RIG-I but are nonetheless incapable of inhibiting pppGA binding or inducing type I IFN. In retrospect, because both blunt OH-dsRNA and pppGmA cannot properly interact with the lysine-rich binding cleft, the similarities observed between these two classes of dsRNA are completely in line with our other data. Although we demonstrate that pppGmA is sterically hindered by H830, OH-dsRNA clearly lacks the phosphates requisite for binding-cleft interaction. By contrast, ppp(+1nt)-dsRNA should be able to interact with the designated binding cleft (K888, K858, K861, H830, K907), and it could be that this is essential to its ability to act as a competitive, non-productive ligand. Nonetheless, from our data we cannot conclude whether the detected pppGmA/RIG-I interaction results from an inaccurate binding as a result from a distorted helix orientation within the binding cleft or whether interaction is mediated by other, unspecific contacts. Whatever the nature of this interaction, it does not succeed in RIG-I activation.
In addition, it must be emphasized that the in vivo N1-methylation of mRNA does not occur in an isolated fashion without m7G-capping. In fact, MTr1 requires m7G-capped RNA as a substrate. Moreover, in this physiological situation, these two modifications are strongly synergistic. m7G-capping mediates a reduction in type I IFN induction for RIG-I(WT) and RIG-I(H830A) alike, and additional N1-methylation of m7G-capped dsRNA (m7GpppGmA) completely abrogates the interaction with RIG-I(WT). Thus, in vivo, it is MTr1-mediated N1-methylation that renders endogenous mRNA completely immunologically inert.
Because RIG-I is exclusively stimulated by base-paired pppRNA ends (Schlee et al., 2009), the existence of endogenous RIG-I ligands is not self-evident. Although we did not identify the RNA species responsible for RIG-I activation, our results demonstrate that endogenous RNA species exist, which can be detected in the absence of N1-2′O-methylation or the RIG-I H830 side chain. Phosphatase treatment of endogenous RNA revealed that in the cell both capped and uncapped RNA molecules exist that stimulate mutant RIG-I(H830A). Base-paired RNA structures can be a result of intra- or intermolecular base pairing or could be formed by a recently described endogenous RNA replication mechanism (Kapranov et al., 2010). The fact that stimulation of RIG-I(H830A) by endogenous RNA was relatively weak indicates that the concentration of stimulatory RIG-I ligands is either low or that endogenous RIG-I ligands present a suboptimal ligand structure. However, its long-term immune stimulatory effects could lead to severe autoinflammatory effects in vivo.
Our results on the YFV-encoded 2′O-methyltransferase demonstrate that viruses employ 2′O-methylation of N1 to escape recognition by RIG-I: the 2′O-methyltransferase deletion mutant YFVR-E218A of the viral replicon led to decrease of replication in A549 cells but not in Vero cells, which are known to have deleted IFN genes. Accordingly, the replication of the mutated virus YFV-E218A was strongly impaired in A549 cells but not in Vero- or STAT1-deficient A549 cells, which have a defect in type I IFN signaling. Intriguingly, viral titers of YFV-E218A and YFV-WT were equal in RIG-I-deficient A549 cells. IFIT1 mRNA (as an indicator of type I IFN induction) was induced more strongly by YFV-E218A than YFV-WT. Of note, no IFIT1 induction by YFV-WT or YFV-E218A occurred in absence of RIG-I in A549 cells.
Analogous to our YFV 2′O-methyltransferase mutant YFV-E218A, Daffis et al. (2010) analyzed the flavivirus WNV including a mutant lacking 2′O-methyltransferase activity (WNV-E218A). They showed that the type I IFN-induced protein IFIT1 mediates repression of WNV-E218A replication in the brain (in vivo) and in macrophages in vitro (Daffis et al., 2010). As revealed by their experiments, the type I IFN-induced IFIT1 protein could act only downstream of MAVS-dependent type I IFN-inducing pathways, which in turn requires prior detection of the viral RNA via either RIG-I or MDA5 (Daffis et al., 2010). In a follow-up study, the same group observed that Ifit −/− mice still survive subcutaneous WNV-E218A infection at a viral titer that is lethal for wild-type WNV (Szretter et al., 2012), suggesting 2′O-methylation-sensitive immune mechanisms beyond IFIT1. Altogether, this is in line with our results that RIG-I is a 2′O-methylation-sensitive receptor of viral RNA and that IFIT1 expression is induced by RIG-I and therefore cannot act without prior upregulation by type I IFN-inducing receptors like RIG-I.
Züst et al. (2011) observed that a mutant of the (+)ssRNA coronavirus MHV lacking viral N1-2′O-methyltransferase activity showed enhanced IFN-β induction and suppressed virus replication in murine macrophages and that both effects were MDA5 dependent. However, MDA5 deficiency neither led to restoration of WNV-E218A infectivity in primary cells nor rescued WNV-E218A virulence in mice (Szretter et al., 2012). In addition, a viral mRNA has been identified, which stimulated MDA5, when expressed under the control of an RNA polymerase-II-controlled promoter that produces N1-2′O-methylated mRNA (cap1 structure) (Luthra et al., 2011), which suggests that N1-2′O-methylation does not generally impair MDA5 engagement. None of the above studies examined RIG-I-deficient cells. In the light of our results and the above mentioned data (Daffis et al., 2010, Szretter et al., 2012), we conclude that IFIT1 acts as type I IFN-induced effector downstream of type I IFN-inducing receptors (e.g., RIG-I and MDA5) to sequester viral mRNA from translation (Habjan et al., 2013). Because no direct impact of N1-2′O-methylation on binding of viral RNA to MDA5 was shown, still an indirect effect remains possible. Because coronavirus recognition in murine macrophages is known to require MDA5 (Roth-Cross et al., 2008), it is conceivable that stimulation via MDA5 is required for type I IFN-dependent upregulation of IFIT1, which then sequesters non-2′O-methyl RNA, thereby inhibiting translation of viral proteins, which suppress recognition of RNA or interfere with type I IFN-inducing pathways. In this study, we provide molecular evidence for a direct physical interaction of RIG-I and RNA, which is sterically impaired by 2′O-methyl at the N1 position, leading to abolishment of RIG-I activation.
In conclusion, with this study we identified the highly conserved biological function of the N1-2′O-methylation of capped RNA, and we resolved the molecular mechanism that allows the distinction of self versus foreign RNA by RIG-I. We provide evidence that endogenous stimulatory RNA molecules exist at sufficient numbers to allow RIG-I activation and that the proper function of the endogenous 2′O-methyltransferase prevents RIG-I-driven autoinflammation; furthermore, that viruses mimic the molecular mechanism of self-tolerance by introducing a viral 2′O-methyltransferase. To our knowledge, RIG-I is the first example of an innate immune receptor that adapted its structure to tolerate self-RNA (that is actively labeled self in the nucleus) rather than adapting its structure to detect a specific foreign molecule. Thus, like the adaptive immune system, innate immunity employs active mechanisms that secure tolerance toward self while potently responding to foreign molecular pattern. Our study emphasizes endogenous RNA methyltransferases (N7 and N1/2-2′O-methyltransferases) as the crucial safeguards for maintenance of immune tolerance of self-RNA.
Experimental Procedures
Preparation of Capped RNA
Triphosphorylated RNA oligonucleotides were chemically synthesized as described (Goldeck et al., 2014). Internal methylation occurred by incorporation of methylated nucleotides during chemical synthesis. Capping (addition of 5′ 7mG) occurred by incubation of pppRNA with vaccina virus capping enzyme (Epicenter), GTP, and S-Adenosyl methionine (SAM) according to the manufacturers’ protocol (Figure S2). Preparation of “GpppGA” occurred in absence of SAM. Uncapped pppRNA was eliminated by treatment with 5′-polyphosphatase (Epicenter) and Terminator Nuclease (Epicenter) (Figure S2). Polyacrylamide gel electrophoresis (PAGE) and MALDI-ToF mass spectrometry (Metabion) was performed as described (Figure S2; Schlee et al., 2009).
Cell Culture
Human PBMCs were isolated as described (Schlee et al., 2009). For stimulation, 4 × 106 cells (PBMCs) were cultured in 96-well plates. The PBMC studies were approved by the local ethics committee (Ethikkommission der Medizinischen Fakultät Bonn) according to the ICH-GCP guidelines. Written informed consent was provided by voluntary blood donors.
To inhibit TLR7/8 activity, cells were pre-incubated with 2.5 μg/ml chloroquine for 30 min. Cells were kept in RPMI 1640 (10% FCS, 1.5 mM L-Glu, 100 U/ml penicilin, 100 μg/ml streptomycin). For transfection, nucleic acids were complexed with Lipofectamine(LF) 2000 (Life Technologies). HEK293blue cells (Invivogen) are reporter cells for type 1 IFN. By incident, as demonstrated (Figures 2A and 2D), they show negligible RIG-I background activity. Murine BM-DCs were generated by culturing murine bone marrow cells for 7 days with GM-CSF.
Detection of Cytokines
IFN-α and IP10 levels were analyzed with commercial ELISA assay kits. In our hands the linear range limit for the IP-10 ELISA (BD Biosciences) was 31 pg/ml and for the IFN-α ELISA (eBioscience) was 8 pg/ml.
Generation of RIG-I Mutants
Mutagenesis of full-length RIG-I was performed as described (Wang et al., 2010). (Primers listed in Table S1.) Constructs were confirmed by sequencing. Equal protein expression of RIG-I mutants was confirmed by immunoblot with an antibody against Flag (Sigma) (Figure 2B).
Protein Purification and Analysis
(His6)-Flag-tagged RIG-I(WT) and RIG-I(H830A) were transiently overexpressed in HEK293blue cells and lysed in a CHAPS-containing lysis buffer (150 mM NaCl, 50 mM Tris/HCl [pH 7.4], 2 mM MgCl2, 1 mM DTT, 1% CHAPS) including protease inhibitor cocktail (Roche). The lysate was incubated overnight at 4°C with anti-FLAG beads (Sigma). Anti-FLAG beads were washed subsequently with lysis buffer and high salt wash buffer (300 mM NaCl, 50 mM Tris/HCl [pH 7.4], 5 mM MgCl2, 1 mM DTT, 0.1% CHAPS). RIG-I-FLAG was eluted by addition of FLAG-peptide (300 μg/ml) solution to the beads. The purity and concentration of recombinant RIG-I-derivates was determined by SDS-PAGE/Coomassie blue stain.
Alpha Screen RIG-I-Binding Assay
The binding affinity of RNA for (His6)-FLAG-tagged RIG-I and RIG-I(H830A) was determined by an amplified luminescent proximity homogenous assay (AlphaScreen; Perkin Elmer). In this assay purified HF-RIG-I was incubated with concentrations of biotinylated RNA for 1 hr at 37°C in buffer (50 mM KCl, 5 mM HEPES [pH 7.0], 3 mM MgCl2, 0.5 mM DTT, 0.01% Tween, 1% BSA) and subsequently incubated for 30 min at 25°C with HF-RIG-I-binding Nickel Chelate acceptor beads (Perkin-Elmer) and biotin-RNA-binding Streptavidine donor beads (Perkin Elmer).
Pull-Down of Endogenous RNA by RIG-I-CTD
For the affinity purification of endogenous RNA, a RNA pull-down was conducted using the RNA-binding domain CTD. A RIG-I-CTD expression plasmid containing a (His6)-Flag-Tag was generated via PCR cloning. RIG-I-CTD was transiently overexpressed in HEK293blue cells and lysed concordant to the protein purification protocol (see above). The lysate was incubated 2 hr at 4°C with anti-FLAG beads (Sigma). The beads were washed stringently with lysis buffer. Trizol/chloroform extraction of the bound RNA from the RIG-I-CTD and treatment with alkaline phosphatase (AP; Fermentas) and tobacco acid pyrophosphatase (TAP; Epicenter) was conducted according to manufacturers’ instructions.
siRNA-Mediated Knockdown Experiments
For RNAi/RIG-I mutant experiments, 15 × 103 HEK293blue cells were seeded per well (96-well format) and 50 ng RIG-I expression plasmid (WT or H830A) per well was transfected with Genejuice (Merck). After 12 hr, 100 nM siRNA against sihMTr1 or control (siGenome SMART pool, Dharmacon) was transfected with LF2000. WT or Ddx58 −/− A549 cells were seeded at 30 × 103/well (48-well format) and transfected (RNAiMAX, Life Technologies) with siRNAs against hMTr1 or luciferase (target site + 3′dTdT: siLuc: cauaaggcuaugaagagauac, sihMTr1#1: gugaaggauuggguaaaua, sihMTr1#2: ggaaaugagcgauuggaug, Biomers). Efficient knockdown was confirmed via qPCR (Figure S3).
Construction of Yellow Fever Virus Constructs and Quantification of Replication
The construction of YFV wild-type and NS5 E218A mutant virus and replicons (Figure S4) expressing renilla luciferase is described in the Supplemental Experimental Procedures. In brief, replication-competent YFVR- or YFV-RNA were generated by in vitro transcription. Replication of YFV-replicons was monitored by renilla-Luciferase assay 24 hr after transfection. Virus was generated by electroporation of BHK-J cells with YFV-RNA. Virus titers were determined by BHK-J cell plaque assays as described (Kümmerer and Rice, 2002).
Generation of Knockout Cell Lines
A549 cells were transfected with LF2000 with 200 ng of a CAS9-gRNA expression plasmid targeting RIG-I (5′-GGGTCTTCCGGATATAATCC(TGG)-3′) or STAT1 (5′-CAGGAGGTCATGAAAACGGA(TGG)-3′). Knockout clones were confirmed by functional testing, immunoblot, and Sanger sequencing.
Author Contributions
Conceptualization, M.S., J.L., T.Z., C.S.-W., A.K.B., V.H., and G.H.; Methodology, M.S., C.S.-W., A.K.B., B.M.K., R.K., S.W., T.Z., J.L., M.G., A.-M.H., T.S., J.L.S.-B., J.-P.S., and A.R.; Formal Analysis, A.-M.H., A.K.B., M.S., and C.S.-W.; Investigation, M.S., C.S.-W., A.K.B., A.-M.H., T.Z., M.G., B.M.K., and S.W.; Writing – Original Draft, M.S., C.S.-W., V.H., E.B., A.K.B., and G.H.; Writing – Review & Editing, E.B., M.S., V.H., J.L., W.B., A.K.B., C.C., C.D., and G.H.; Funding Acquisition, M.S., G.H., V.H., W.B., C.C., E.B., C.D., B.M.K.; Resources, J.L., M.G., T.S., J.L.S.-B., J.-P.S., A.R., B.M.K., R.K., and C.C.; Supervision, M.S., J.L., B.M.K., C.D., C.C., W.B., V.H., and G.H.
Acknowledgments
We thank Janett Wieseler for excellent technical assistance and Cristina A. Hagmann and Jasper v.d. Boorn for critically reading the manuscript. We are also thankful to Charles M. Rice for providing pACNR/YF 17D. B.M.K., C.C., G.H., M.S., V.H., and W.B. are supported by the Deutsche Forschungsgemeinschaft (http://www.dfg.de; SFB670 to M.S., G.H., V.H., and W.B., DFG SCHL1930/1-1 to M.S., SFB704 to G.H. and W.B., SFB832 and KFO177 to G.H. and C.C., and KU1201/4-1 to B.M.K.). E.B. is supported by the University Hospital of Bonn (BONFOR). A.K.B., E.B., G.H., M.S., V.H., and W.B. are supported by the DFG Excellence Cluster ImmunoSensation. C.D., G.H., V.H., and W.B. are supported by the German Center of Infectious Disease (DZIF). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. C.S.-W., G.H., and V.H. are co-founders and shareholders of the Rigontec GmbH.
Published: July 14, 2015
Footnotes
Supplemental Information includes four figures, one table, and Supplemental Experimental Procedures and can be found with this article online at http://dx.doi.org/10.1016/j.immuni.2015.06.015.
Supplemental Information
References
- Abbas Y.M., Pichlmair A., Górna M.W., Superti-Furga G., Nagar B. Structural basis for viral 5′-PPP-RNA recognition by human IFIT proteins. Nature. 2013;494:60–64. doi: 10.1038/nature11783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee A.K. 5′-terminal cap structure in eucaryotic messenger ribonucleic acids. Microbiol. Rev. 1980;44:175–205. doi: 10.1128/mr.44.2.175-205.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bélanger F., Stepinski J., Darzynkiewicz E., Pelletier J. Characterization of hMTr1, a human Cap1 2′-O-ribose methyltransferase. J. Biol. Chem. 2010;285:33037–33044. doi: 10.1074/jbc.M110.155283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray M. Highly pathogenic RNA viral infections: challenges for antiviral research. Antiviral Res. 2008;78:1–8. doi: 10.1016/j.antiviral.2007.12.007. [DOI] [PubMed] [Google Scholar]
- Brownell J., Bruckner J., Wagoner J., Thomas E., Loo Y.M., Gale M., Jr., Liang T.J., Polyak S.J. Direct, interferon-independent activation of the CXCL10 promoter by NF-κB and interferon regulatory factor 3 during hepatitis C virus infection. J. Virol. 2014;88:1582–1590. doi: 10.1128/JVI.02007-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civril F., Bennett M., Moldt M., Deimling T., Witte G., Schiesser S., Carell T., Hopfner K.P. The RIG-I ATPase domain structure reveals insights into ATP-dependent antiviral signalling. EMBO Rep. 2011;12:1127–1134. doi: 10.1038/embor.2011.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui S., Eisenächer K., Kirchhofer A., Brzózka K., Lammens A., Lammens K., Fujita T., Conzelmann K.K., Krug A., Hopfner K.P. The C-terminal regulatory domain is the RNA 5′-triphosphate sensor of RIG-I. Mol. Cell. 2008;29:169–179. doi: 10.1016/j.molcel.2007.10.032. [DOI] [PubMed] [Google Scholar]
- Daffis S., Szretter K.J., Schriewer J., Li J., Youn S., Errett J., Lin T.Y., Schneller S., Zust R., Dong H. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature. 2010;468:452–456. doi: 10.1038/nature09489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fechter P., Brownlee G.G. Recognition of mRNA cap structures by viral and cellular proteins. J. Gen. Virol. 2005;86:1239–1249. doi: 10.1099/vir.0.80755-0. [DOI] [PubMed] [Google Scholar]
- Gitlin L., Barchet W., Gilfillan S., Cella M., Beutler B., Flavell R.A., Diamond M.S., Colonna M. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc. Natl. Acad. Sci. USA. 2006;103:8459–8464. doi: 10.1073/pnas.0603082103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldeck M., Tuschl T., Hartmann G., Ludwig J. Efficient solid-phase synthesis of pppRNA by using product-specific labeling. Angew. Chem. Int. Ed. Engl. 2014;53:4694–4698. doi: 10.1002/anie.201400672. [DOI] [PubMed] [Google Scholar]
- Goubau D., Schlee M., Deddouche S., Pruijssers A.J., Zillinger T., Goldeck M., Schuberth C., Van der Veen A.G., Fujimura T., Rehwinkel J. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5′-diphosphates. Nature. 2014;514:372–375. doi: 10.1038/nature13590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habjan M., Hubel P., Lacerda L., Benda C., Holze C., Eberl C.H., Mann A., Kindler E., Gil-Cruz C., Ziebuhr J. Sequestration by IFIT1 impairs translation of 2'O-unmethylated capped RNA. PLoS Pathog. 2013;9:e1003663. doi: 10.1371/journal.ppat.1003663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung V., Ellegast J., Kim S., Brzózka K., Jung A., Kato H., Poeck H., Akira S., Conzelmann K.K., Schlee M. 5′-Triphosphate RNA is the ligand for RIG-I. Science. 2006;314:994–997. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- Jiang F., Ramanathan A., Miller M.T., Tang G.Q., Gale M., Jr., Patel S.S., Marcotrigiano J. Structural basis of RNA recognition and activation by innate immune receptor RIG-I. Nature. 2011;479:423–427. doi: 10.1038/nature10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapranov P., Ozsolak F., Kim S.W., Foissac S., Lipson D., Hart C., Roels S., Borel C., Antonarakis S.E., Monaghan A.P. New class of gene-termini-associated human RNAs suggests a novel RNA copying mechanism. Nature. 2010;466:642–646. doi: 10.1038/nature09190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H., Takeuchi O., Sato S., Yoneyama M., Yamamoto M., Matsui K., Uematsu S., Jung A., Kawai T., Ishii K.J. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- Kowalinski E., Lunardi T., McCarthy A.A., Louber J., Brunel J., Grigorov B., Gerlier D., Cusack S. Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell. 2011;147:423–435. doi: 10.1016/j.cell.2011.09.039. [DOI] [PubMed] [Google Scholar]
- Kümmerer B.M., Rice C.M. Mutations in the yellow fever virus nonstructural protein NS2A selectively block production of infectious particles. J. Virol. 2002;76:4773–4784. doi: 10.1128/JVI.76.10.4773-4784.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo Y.M., Fornek J., Crochet N., Bajwa G., Perwitasari O., Martinez-Sobrido L., Akira S., Gill M.A., García-Sastre A., Katze M.G., Gale M., Jr. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J. Virol. 2008;82:335–345. doi: 10.1128/JVI.01080-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C., Xu H., Ranjith-Kumar C.T., Brooks M.T., Hou T.Y., Hu F., Herr A.B., Strong R.K., Kao C.C., Li P. The structural basis of 5′ triphosphate double-stranded RNA recognition by RIG-I C-terminal domain. Structure. 2010;18:1032–1043. doi: 10.1016/j.str.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo D., Ding S.C., Vela A., Kohlway A., Lindenbach B.D., Pyle A.M. Structural insights into RNA recognition by RIG-I. Cell. 2011;147:409–422. doi: 10.1016/j.cell.2011.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luthra P., Sun D., Silverman R.H., He B. Activation of IFN-γ; expression by a viral mRNA through RNase L and MDA5. Proc. Natl. Acad. Sci. USA. 2011;108:2118–2123. doi: 10.1073/pnas.1012409108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marq J.B., Hausmann S., Veillard N., Kolakofsky D., Garcin D. Short double-stranded RNAs with an overhanging 5′ ppp-nucleotide, as found in arenavirus genomes, act as RIG-I decoys. J. Biol. Chem. 2011;286:6108–6116. doi: 10.1074/jbc.M110.186262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry R.P., Kelley D.E. Kinetics of formation of 5′ terminal caps in mRNA. Cell. 1976;8:433–442. doi: 10.1016/0092-8674(76)90156-2. [DOI] [PubMed] [Google Scholar]
- Pichlmair A., Schulz O., Tan C.P., Näslund T.I., Liljeström P., Weber F., Reis e Sousa C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science. 2006;314:997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- Pichlmair A., Lassnig C., Eberle C.A., Górna M.W., Baumann C.L., Burkard T.R., Bürckstümmer T., Stefanovic A., Krieger S., Bennett K.L. IFIT1 is an antiviral protein that recognizes 5′-triphosphate RNA. Nat. Immunol. 2011;12:624–630. doi: 10.1038/ni.2048. [DOI] [PubMed] [Google Scholar]
- Roth-Cross J.K., Bender S.J., Weiss S.R. Murine coronavirus mouse hepatitis virus is recognized by MDA5 and induces type I interferon in brain macrophages/microglia. J. Virol. 2008;82:9829–9838. doi: 10.1128/JVI.01199-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlee M., Hartmann G. The chase for the RIG-I ligand--recent advances. Mol. Ther. 2010;18:1254–1262. doi: 10.1038/mt.2010.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlee M., Roth A., Hornung V., Hagmann C.A., Wimmenauer V., Barchet W., Coch C., Janke M., Mihailovic A., Wardle G. Recognition of 5′ triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity. 2009;31:25–34. doi: 10.1016/j.immuni.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt A., Schwerd T., Hamm W., Hellmuth J.C., Cui S., Wenzel M., Hoffmann F.S., Michallet M.C., Besch R., Hopfner K.P. 5′-triphosphate RNA requires base-paired structures to activate antiviral signaling via RIG-I. Proc. Natl. Acad. Sci. USA. 2009;106:12067–12072. doi: 10.1073/pnas.0900971106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szretter K.J., Daniels B.P., Cho H., Gainey M.D., Yokoyama W.M., Gale M., Jr., Virgin H.W., Klein R.S., Sen G.C., Diamond M.S. 2′-O methylation of the viral mRNA cap by West Nile virus evades ifit1-dependent and -independent mechanisms of host restriction in vivo. PLoS Pathog. 2012;8:e1002698. doi: 10.1371/journal.ppat.1002698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahasi K., Yoneyama M., Nishihori T., Hirai R., Kumeta H., Narita R., Gale M., Jr., Inagaki F., Fujita T. Nonself RNA-sensing mechanism of RIG-I helicase and activation of antiviral immune responses. Mol. Cell. 2008;29:428–440. doi: 10.1016/j.molcel.2007.11.028. [DOI] [PubMed] [Google Scholar]
- Wang Y., Ludwig J., Schuberth C., Goldeck M., Schlee M., Li H., Juranek S., Sheng G., Micura R., Tuschl T. Structural and functional insights into 5′-ppp RNA pattern recognition by the innate immune receptor RIG-I. Nat. Struct. Mol. Biol. 2010;17:781–787. doi: 10.1038/nsmb.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Ray D., Zhao Y., Dong H., Ren S., Li Z., Guo Y., Bernard K.A., Shi P.Y., Li H. Structure and function of flavivirus NS5 methyltransferase. J. Virol. 2007;81:3891–3903. doi: 10.1128/JVI.02704-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Züst R., Cervantes-Barragan L., Habjan M., Maier R., Neuman B.W., Ziebuhr J., Szretter K.J., Baker S.C., Barchet W., Diamond M.S. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat. Immunol. 2011;12:137–143. doi: 10.1038/ni.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.