

Summary
Type I interferon (IFN) is critical for controlling pathogen infection; however, its regulatory mechanisms in plasmacytoid cells (pDCs) still remain unclear. Here, we have shown that nucleic acid sensors cGAS-, STING-, MDA5-, MAVS-, or transcription factor IRF3-deficient mice produced high amounts of type I IFN-α and IFN-β (IFN-α/β) in the serum and were resistant to lethal plasmodium yoelii YM infection. Robust IFN-α/β production was abolished when gene encoding nucleic acid sensor TLR7, signaling adaptor MyD88, or transcription factor IRF7 was ablated or pDCs were depleted. Further, we identified SOCS1 as a key negative regulator to inhibit MyD88-dependent type I IFN signaling in pDCs. Finally, we have demonstrated that pDCs, cDCs, and macrophages were required for generating IFN-α/β-induced subsequent protective immunity. Thus, our findings have identified a critical regulatory mechanism of type I IFN signaling in pDCs and stage-specific function of immune cells in generating potent immunity against lethal YM infection.
Keywords: Type I IFN signaling, Innate immune response, Plasmacytoid dendritic cells, Malaria infection
Graphical Abstract
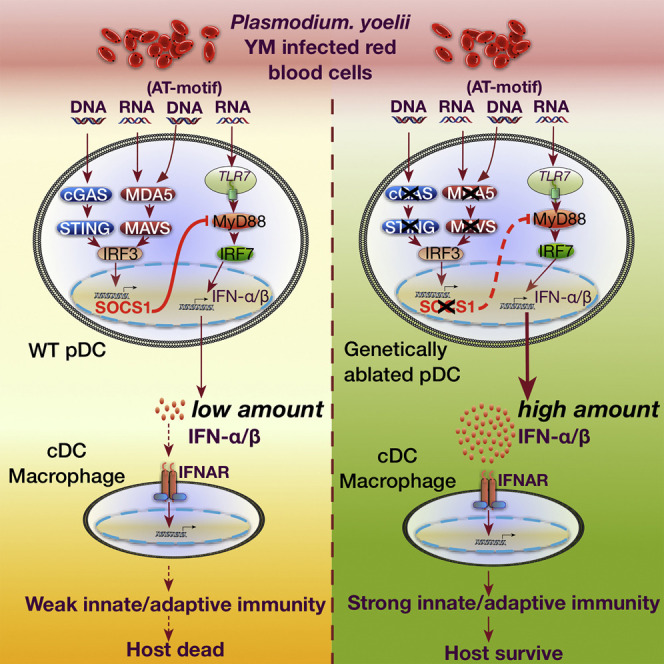
Highlights
-
•
cGAS functions as a DNA sensor in vivo for detecting malaria genomic DNA
-
•
STING- and MAVS-mediated signaling induces a negative regulator SOCS1 expression
-
•
SOCS1 inhibits MyD88-mediated type I IFN signaling in pDCs
-
•
Type I IFN produced by pDCs activates cDCs and macrophages for adaptive immunity
Malaria is a worldwide deadly infectious disease; how type I IFN signaling is regulated in response to malaria infection remains poorly understood. Yu et al. identify a cross-regulatory mechanism of two type I IFN signaling pathways in plasmacytoid DCs, which is critical for generating protective immunity against lethal malaria infection.
Introduction
Type I interferon (IFN) and NF-κB pathways play key roles in controlling disease pathogenesis (Goubau et al., 2013, Paludan and Bowie, 2013, Takeuchi and Akira, 2010). The innate immune response serves as the first line of defense against invading pathogens and relies on the recognition of pathogen-associated molecular patterns (PAMPs) by several classes of pattern-recognition receptors (PRRs), including Toll-like receptors (TLRs), nucleotide-binding domain and leucine-rich repeat containing gene family (NLRs), and RIG-I-like receptors (RLRs) (Goubau et al., 2013, Paludan and Bowie, 2013, Takeuchi and Akira, 2010). Activation of most TLRs leads to the recruitment of a common adaptor molecule, MyD88, which acts on a series of downstream signaling molecules to trigger the NF-κB pathway (Goubau et al., 2013, Hayden and Ghosh, 2008, Paludan and Bowie, 2013), whereas stimulation of TLR7 and TLR9 in specialized plasmacytoid dendritic cells (pDCs) can trigger robust MyD88-dependent IFN regulatory factor (IRF) 7 transcription factor-mediated type I IFN signaling and produce large amounts of IFN-α and IFN-β (IFN-α/β) in response to viral infection (Liu, 2005, Wang et al., 2011). By contrast, activation of cytosolic RNA sensors (RIG-I and MDA5) recruits the mitochondrial antiviral-signaling protein (MAVS) adaptor protein, while stimulation of DNA sensors recruits stimulator of IFN genes (STING), which both result in triggering IRF3-mediated type I IFN signaling (Goubau et al., 2013, Paludan and Bowie, 2013, Sun et al., 2013, Takeuchi and Akira, 2010). Innate immune responses must be tightly regulated to maintain immune balance, but how these signaling pathways are regulated in vivo, particularly in response to malaria infection, remains unresolved.
Malaria is a deadly infectious disease and is responsible for up to 0.5 million deaths each year (Miller et al., 2013). Lack of effective vaccines against malaria has been one of the major limiting factors in controlling the disease (Riley and Stewart, 2013). The incomplete understanding of the malaria-strain-specific response and regulatory mechanisms impedes our ability to develop effective malaria vaccines (Langhorne et al., 2008, Riley and Stewart, 2013, Stevenson and Riley, 2004). Recent studies show that the liver stage of infection can induce type I IFN signaling activation, either through the cytosolic PRRs, MDA5-MAVS pathway (Liehl et al., 2014), or multiple mechanisms (Miller et al., 2014). After the liver stage of infection and expansion, merozoites are released into the blood and infect red blood cells (iRBCs), which activate several key signaling pathways, including NF-κB, type I IFN, and the inflammasome (Gazzinelli et al., 2014, Miller et al., 2014, Riley and Stewart, 2013, Wu et al., 2014). However, the role of TLRs and type I IFN in malaria blood-stage infection is unclear or remain controversial (Baccarella et al., 2013, Gowda et al., 2012, Haque et al., 2014, Liehl and Mota, 2012, Sharma et al., 2011, Togbe et al., 2007, Wu et al., 2010). Therefore, we have used a lethal malaria plasmodium yoelii YM (YM for short) strain as a model to probe the molecular signaling pathways and mechanisms of host immune response during blood stage infection.
In this study, we identified cGAS as a key DNA sensor that triggers the type I IFN signaling pathway after YM infection. cGAS-, STING-, MAVS-, or MDA5-deficient mice were resistant to lethal YM infection compared with wild-type (WT) mice. High serum amounts of IFN-α/β in these genetically deficient mice were abolished upon depletion of pDC or ablation of Tlr7, Myd88, and Irf7. Our experiments further revealed that the STING- and MAVS-mediated type I IFN pathway induced SOCS1 expression, which potently inhibited MyD88-dependent type I IFN signaling in pDCs. Furthermore, we showed that early robust IFN-α/β production by pDCs in genetically ablated mice during the first 24 hr of YM infection subsequently activated cDCs and macrophages, which, in turn, induced T- and B-cell-mediated adaptive immune responses against malaria infection.
Results
Mice Deficient in cGAS, STING, MDA5, MAVS, or IRF3 Are Resistant to YM Infection
Since the role of TLRs in defending against different malaria species remains controversial (Baccarella et al., 2013, Gowda et al., 2012, Togbe et al., 2007), we first investigated whether TLRs and their downstream signaling molecules play a role in controlling lethal YM infection. We infected Tlr2 −/−, Tlr3 −/−, Tlr4 −/−, Tlr7 −/−, Tlr9 −/−, Ticam1 −/− (coding for TRIF), Myd88 −/−, and WT mice by intraperitoneal (i.p.) injection of YM-iRBCs. We found that WT and genetically deleted mice died within 8 days after infection (Figures 1 A–1C and S1A–S1D). Notably, Tlr7 −/− and Myd88 −/− mice showed increased parasitemia and died sooner after infection than WT mice, suggesting that TLR7 and MyD88 may play a role in the control of parasitemia and host mortality.
Figure 1.

Mice Deficient in cGAS, STING, MDA5, MAVS, or IRF3, but Not in TLR Signaling Molecules, Are Protected from YM Infection
(A–C) Daily parasitemias and mortality rates of WT (black lines) and TLR-deficient mice (red lines) after YM (0.5 × 106 iRBCs) infection: Tlr7−/− (A), Tlr9−/− (B), and Myd88−/− (C).
(D–I) Daily parasitemias and mortality rates of WT (black lines) and indicated deficient mice (red lines) after YM infection: Mb21d1−/− (D), Tmem173gt (E), Ifih1−/− (F), Mavs−/− (G), Irf3−/− (H), and Irf7−/− (I) mice.
(J) Quantitative analysis of IFN-β mRNA in STING stably expressing 293T cells transfected with Flag-Mb21d1, Zbp1, Ddx41, or Ifi203 followed by YM gDNA stimulation for 18 hr.
(K) Quantitative analysis of IFN-β mRNA in RAW264.7 cells transfected with Mb21d1-, Zbp1-, Ddx41-, Ifi203-specific siRNA, or scramble siRNA for 48 hr, followed by YM gDNA stimulation for 6 hr.
(L) RAW264.7 cells were transfected with Mb21d1-specific siRNA or scramble siRNA, followed by YM gDNA stimulation for 24 hr. Cell supernatants were collected for ELISA analysis.
(M) WT and indicated deficient mice were infected with YM. Serum amounts of IFN-α/β, IL-6, and IFN-γ were determined by ELISA.
Data are representative of three independent experiments and are plotted as the mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 versus corresponding control. NS, not significant.
See also Figure S1.
To test whether RNA and DNA sensor-mediated type I IFN signaling plays a role in host immune responses to YM infection, we infected Mb21d1 −/− (coding for cGAS), Tmem173 gt (coding for STING), Ifih1 −/− (coding for MDA5), and Mavs −/− mice with YM. We found that these genetically ablated mice remarkably reduced parasitemia compared with WT mice. Tmem173 gt, Ifih1 −/−, and Mavs −/− mice were completely resistant to YM infection, whereas Mb21d1 −/− mice showed partial protection (Figures 1D–1G). Furthermore, YM infection was completely cleared in Ifih1 −/−, Mavs −/−, and Tmem173 gt mice within 4 weeks (Figure S1F). However, mice with ablation of the Ddx58 gene (coding for RIG-I) showed no difference in malaria parasitemia and host death, compared with WT mice (Figure S1E). IRF3 is a key factor for stimulating type I IFN signaling for IFN-β production in almost all cell types, whereas IRF7 is responsible for activating type I IFN signaling to produce IFN-α/β only in pDCs (Liu, 2005, Wang et al., 2011). Infection of Irf3 −/− and Irf7 −/− mice with YM revealed that Irf3 −/− mice, but not Irf7 −/− mice, remarkably reduced parasitemia and were resistant to YM infection (Figures 1H and 1I). Collectively, these results suggest that activation of cGAS-STING and MDA5-MAVS-mediated IRF3-dependent type I IFN signaling leads to a lethal phenotype of YM infection.
Identification of cGAS as a Malaria DNA Sensor and Robust Production of IFN-α/β in cGAS-, STING-, MDA5-, or MAVS-Deficient Mice
To understand how cGAS-, STING-, MDA5-, and MAVS-deficient mice were resistant to YM infection, we first determined whether cGAS and MDA5 functioned as DNA or RNA sensors for detecting YM genomic DNAs (gDNAs) or RNAs. We found that both purified YM gDNA and RNA could induce IFN-β mRNA expression in RAW264.7 cells (Figure S1G), which is in agreement with other studies showing that malaria RNA can be detected by a MDA5 sensor (Liehl et al., 2014, Wu et al., 2014). As expected, in vitro stimulation with YM gDNA or RNA showed impaired or completely abolished activation of IFN-β mRNA and protein in bone-marrow-derived conventional DCs (cDCs) from MDA5-, MAVS-, and STING-deficient mice compared to cDCs from WT mice (Figure S1H). Although STING has been shown to play an important role in the host response to AT-rich malarial DNA (Sharma et al., 2011), it is not clear which DNA sensors are responsible for recognizing YM gDNA and activating STING-mediated type I IFN signaling. To this end, we transfected 293T-STING stable cells with plasmids containing the gene encoding one of four putative DNA sensors (cGAS, DAI, DDX41, or IFI16) followed by YM gDNA stimulation. Ectopic expression of Mb21d1, but not Zbp1, Ddx41, or Ifi203, induced IFN-β mRNA expression after YM gDNA treatment (Figure 1J). Consistently, silencing of Mb21d1, but not Zbp1, Ddx41, or Ifi203, by specific siRNAs in RAW264.7 cells markedly reduced IFN-β mRNA and protein after YM gDNA treatment (Figures 1K, 1L, and S1I), suggesting that cGAS functions as a DNA sensor for detecting YM gDNA and inducing STING-mediated type I IFN signaling.
Previous studies showed that type I IFN signaling inhibits type II IFN (IFN-γ) or adaptive immune responses upon malaria and viral infection (Haque et al., 2011, Palomo et al., 2013, Teijaro et al., 2013, Teles et al., 2013, Wilson et al., 2013). To this end, we examined the serum amounts of IFN-α/β, IL-6, and IFN-γ in WT mice during YM infection and found that these cytokines were increased and peaked at 24 hr post infection and then completely disappeared in WT mice (Figure S1J). However, the serum amounts of IFN-α/β, IL-6, and IFN-γ in MDA5-, MAVS-, STING-, and cGAS-deficient mice were much higher than in WT mice at 24 hr after YM infection (Figure 1M), suggesting that MDA5, MAVS, STING, or cGAS deficiency enhances type I IFN production in vivo after YM infection.
Robust Production of IFN-α/β Is Required for Generating Immunity against YM Infection
To determine which cytokines are responsible for generating protective immunity against YM infection, we injected WT mice with exogenous recombinant IFN-α/β, IFN-γ, or IL-6 and then tested their ability to inhibit parasitemia and reduce host mortality after YM infection. WT mice treated with IFN-α/β after YM infection had markedly decreased parasitemia and host mortality, but treatment with IL-6 or IFN-γ failed to do so (Figure 2 A). Since the production of IFN-α/β in serum peaked at 24 hr after YM infection and then disappeared, we asked whether the timing of cytokine treatment was important for protective immunity. We performed similar experiments with cytokine injections at different time points and found that the amounts of IFN-α/β-mediated protection were the highest at 18 hr after YM infection, intermediate at 32 hr, and the lowest at 48 hr (Figure 2B). These results suggest that timing of IFN-α/β administration or in vivo production is critical for inducing protective immunity against YM infection.
Figure 2.

Type I IFN and Blockade of IFNAR Determine the Fate of YM-Infected Mice
(A) Parasitemias and mortality rates of WT mice infected with YM, followed by i.v. administration of recombinant mouse IFN-α/β, IL-6, IFN-γ, or control BSA protein at 18 hr post infection.
(B) Parasitemias and mortality rates of WT mice infected with YM, followed by i.v. administration of recombinant mouse IFN-α/β at 18, 32, or 48 hr post infection.
(C and D) Parasitemias (C) and mortality rates (D) of WT, Ifih1−/−, Mavs−/−, and Tmem173gt mice treated with anti-IFNAR1 blocking antibody at days −1, 2, 4, and 6 (500 μg), followed by infection with YM (2 × 106 iRBCs).
Data are representative of three independent experiments and are plotted as the mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 versus corresponding control. NS, not significant. † denotes mouse death.
See also Figure S2.
Next, we sought to determine the molecular mechanisms of how an early robust burst of IFN-α/β can generate potent protective immune responses. We hypothesized that IFN-α/β may initiate a cascade of signaling events to generate potent immune responses. As the first step, we asked whether IFN-α/β-mediated activation of IFNAR (IFN-α/β receptor) is required for controlling parasitemia and host mortality. We blocked type I IFN signaling in Ifih1 −/−, Mavs −/−, and Tmem173 gt mice by administering anti-IFNAR1 antibody. Although anti-IFNAR1 treatment did not have much effect on the production of IFN-α/β, IFN-γ, and IL-6 in Mavs −/− mice (Figure S2A), the Ifih1 −/−, Mavs −/−, and Tmem173 gt mice treated with anti-IFNAR1 antibody with a single injection at 1 day before infection (i.e., day −1) or four injections at days −1, 2, 4, and 6 showed increased parasitemias and died around day 15 (Figures 2C, 2D, and S2B), suggesting that the IFN-α/β-mediated downstream signaling pathway is critical in controlling parasitemia and host mortality. To further test whether IFN-α/β-induced immunity requires IFNAR and its downstream signaling, we infected Ifnar1 −/− mice with YM and found comparable low amounts of IFN-α/β, IL-6, and IFN-γ in serum compared with those in WT mice (Figure S2C). Whereas WT mice treated with exogenous recombinant IFN-α/β became resistant to YM infection, Ifnar1 −/− mice with recombinant IFN-α/β did not reduce parasitemia and mortality (Figure S2D), indicating that IFNAR is required for IFN-α/β-mediated protective immunity against YM infection.
Role of TLR7-MyD88-IRF7 Type I IFN Signaling in Robust Production of IFN-α/β
Next, we sought to identify the key signaling pathways that are responsible for the robust production of IFN-α/β in Ifih1 −/−, Mavs −/−, and Tmem173 gt mice at 24 hr after YM infection. We first asked whether ablation of the Tlr7, Myd88, or Irf7 gene could reduce IFN-α/β production and reverse the resistant phenotypes of Ifih1 −/−, Mavs −/−, and Tmem173 gt mice to YM infection. To test this possibility, we generated Ifih1 −/−:Myd88 −/−, Mavs −/−:Myd88 −/−, and Tmem173 gt:Myd88 −/− mice and then infected them, along with WT, Ifih1 −/−, Mavs −/−, and Tmem173 gt mice, with YM. As expected, serum amounts of IFN-α/β were very high in Ifih1 −/−, Mavs −/−, and Tmem173 gt mice. However, we could not detect any IFN-α/β in the sera of Ifih1 −/−:Myd88 −/−, Mavs −/−:Myd88 −/−, and Tmem173 gt:Myd88 −/− mice or in the sera of Myd88 −/− mice (Figures 3 A, S3A, and S3B), suggesting a critical role for MyD88 in the rapid production of high amounts of IFN-α/β in response to YM infection. Consistent with these data, MyD88 ablation sensitized Ifih1 −/−, Mavs −/−, and Tmem173 gt mice to high parasitemia and host lethality (Figures 3B, S3C, and S3D). These results suggest that the rapid production of high amounts of IFN-α/β in the Ifih1 −/−, Mavs −/−, and Tmem173 gt mice after YM infection is MyD88 dependent.
Figure 3.

Requirement and Regulation of TLR7-MyD88-IRF7 in Type I IFN-Mediated Protection of Mice from YM Infection
(A) Serum amounts of IFN-α/β in WT, Myd88−/−, Mavs−/−, and Mavs−/−:Myd88−/− mice at 0, 18, and 24 hr after YM infection.
(B) Daily YM parasitemias and mortality rates of WT, Myd88−/−, Mavs−/−, and Mavs−/−:Myd88−/− mice after YM infection.
(C) Serum amounts of IFN-α/β in WT, Tlr7−/−, Tmem173gt, and Tmem173gt:Tlr7−/− mice at 0, 24, and 48 hr after YM infection.
(D) Serum amounts of IFN-α/β in WT and indicated deficient mice at 24 hr after YM infection.
(E–F) Daily YM parasitemias and mortality rates of WT and indicated deficient mice after YM infection.
Data are plotted as the mean ± SD and are representative of three independent experiments. ∗∗p < 0.01, ∗∗∗p < 0.001 versus corresponding control. ND, not detected.
See also Figure S3.
Using a similar strategy, we showed that serum amounts of IFN-α/β were very high in Tmem173 gt and Irf3 −/− mice but undetectable in the sera of Tlr7 −/−, Irf7 −/−, Tmem173 gt:Tlr7 −/−, and Irf3 −/−:Irf7 −/− mice (Figures 3C and 3D). TLR7 or IRF7 ablation converted the resistant phenotypes of Tmem173 gt and Irf3 −/− mice to the sensitive phenotypes of Tmem173 gt:Tlr7 −/− and Irf3 −/−:Irf7 −/− mice after YM infection (Figures 3E and 3F). Similar results were observed in Mavs −/−:Tlr7 −/− and Ifih1 −/−:Tlr7 −/− mice (Figures S3E and S3F). By contrast, we did not observe any difference in IFN-α/β production, parasitemia, and host death between Ifih1 −/− and Mavs −/− mice versus Ifih1 −/−:Tlr9 −/− and Mavs −/−:Tlr9 −/− mice (Figures S3G and S3H), indicating that TLR9 is not required for cytokine production and protective immunity. Taken together, these results suggest that TLR7-MyD88-IRF7 molecules constitute key components of MyD88-dependent type I IFN signaling, which has been shown to operate in only pDC for production of large amounts of IFN-α/β after viral infection (Liu, 2005, Wang et al., 2011).
pDCs Are the Major Source of Type I IFN Cytokine Production
To determine a role of pDCs in malaria infection, we first tested whether pDCs could be preferentially targeted by YM infection. To this end, we infected WT mice with YM and isolated cDCs, macrophages, and pDCs at 18 hr post infection to detect malaria 18S rRNA by PCR with malaria-specific primers. We found that malaria 18S rRNA could be detected in pDCs, but not in macrophages and cDCs (Figure S4A), suggesting that only pDCs are involved in detecting YM infection within the first 18 hr. To directly demonstrate that pDCs are the major source of type I IFN cytokine production, we depleted pDCs in WT, Ifih1 −/−, Mavs −/−, and Tmem173 gt mice by injection of mPDCA-1 antibody at 12 hr before and 12 hr after YM infection. Depletion of pDCs significantly reduced serum amounts of IFN-α/β in WT and deficient mice compared with control antibody treatment (Figure 4 A). Similar results were obtained in Mb21d1 −/− mice (Figure S4B), suggesting that IFN-α/β are primarily produced by pDCs. Importantly, pDC depletion by mPDCA-1 treatment markedly increased parasitemias and mortality in Ifih1 −/−, Mavs −/−, and Tmem173 gt mice infected with YM compared with control antibody-treated mice (Figures 4B and 4C). These results clearly suggest that pDCs are responsible for the early and rapid production of type I IFN in the Ifih1 −/−, Mavs −/−, and Tmem173 gt mice, which, in turn, activates type I IFN downstream signaling for potent innate immunity against YM infection.
Figure 4.

Requirement of Plasmacytoid DCs in Type I IFN-Mediated Protection of Mice from YM Infection
(A–C) Depletion of pDCs cell population in WT, Ifih1−/−, Mavs−/−, and Tmem173gt mice by administration of anti-mPDCA-1 antibody at 12 hr before and 12 hr after YM infection. Serum amounts of IFN-α/β collected at 24 hr after infection in WT and deficient mice untreated or treated with anti-mPDCA-1 are shown in (A). Daily YM parasitemias and mortality rates are shown in (B and C).
(D and E) WT, WT:BDCA2-DTR, Mavs−/−, and Mavs−/−:BDCA2-DTR mice were treated with DT as indicated at 1 day before and 1, 3, and 5 days after YM infection. Serum amounts of IFN-α/β collected at 24 hr after infection are shown in (D). Daily YM parasitemias and mortality rates are shown in (E).
Data are plotted as the mean ± SD and are representative of three independent experiments. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 versus corresponding control. NS, not significant.
See also Figure S4.
To further define the role of pDCs in type I IFN production and host protection after YM infection, we genetically ablated pDCs in vivo by generating Mavs −/−:BDCA2-DTR mice, as previously described for BDCA2-DTR mice (Swiecki et al., 2010). Upon diphtheria toxin (DT) administration, pDCs were depleted in Mavs −/−:BDCA2-DTR mice, but not in Mavs −/− mice. These treated mice were then infected with YM. We found that treatment of Mavs −/−:BDCA2-DTR mice with DT markedly reduced IFN-α/β compared with DT-treated Mavs −/− mice (Figure 4D). Importantly, DT-treated Mavs −/−:BDCA2-DTR mice were sensitive to YM infection compared with DT-treated Mavs −/− mice, which remained resistant to YM infection (Figure 4E). These results suggest that pDCs are required for robust production of type I IFN cytokines and host protective immunity against YM infection.
SOCS1 Is Induced by STING- and MAVS-Mediated Type I Signaling and Inhibits Type I IFN Signaling through MyD88
To further elucidate the mechanisms by which ablation of MDA5, MAVS, or STING markedly enhanced MyD88-dependent type I IFN production in pDCs, we hypothesized that activation of STING- and MAVS-mediated type I IFN signaling by YM may induce expression of negative regulators, which, in turn, may inhibit MyD88-dependent type I IFN signaling activation in pDCs. To test this possibility, we examined the expression pattern of several negative regulators in splenocytes from YM-infected WT and deficient mice, and found that the mRNA amounts of Socs1, Socs3, Inpp5d (coding for SHIP1), and Inppl1 (coding for SHIP2) were markedly increased in WT but were little or low in MDA5-, MAVS-, and STING-deficient splenocytes (Figures 5 A and S5A). Downregulation of Socs1 was also observed in cGAS-deficient mice (Figure S5B). By contrast, we did not observe appreciable changes in the expression of other potential negative regulators, such as Otud5, Gsk3b, Nlrc3, Pcbp2, Ube3c, and Rnf5, between YM-infected WT and deficient mice (Figure S5A). To further assess the expression of these negative regulators in pDCs, we purified pDCs from Flt3L-induced bone-morrow-derived DCs of WT and deficient mice by flow cytometry (Figure S5C) and then stimulated them with YM RNA or gDNA. SOCS1 was the only negative regulator showing a marked increase in gene expression in WT pDCs but significantly lower gene expression in Ifih1 −/−, Mavs −/−, and Tmem173 gt pDCs (Figures 5B and 5C). No appreciable differences in expression of other negative regulators Socs3, Inpp5d, and Inppl1 were observed between WT and deficient pDCs after YM RNA or gDNA stimulation (Figure S5D). To further substantiate these findings, we purified pDCs, macrophages, and cDCs from YM-infected WT and Mavs −/− mice (18 hr after infection) and then directly assessed SOCS1 expression in these cells, and we found that SOCS1 mRNA abundance in pDCs from WT mice were significantly higher than pDCs from Mavs −/− mice (Figure 5D). Notably, we did not detect any difference in expression of SOCS1 in macrophages and cDCs from WT and Mavs −/− mice (Figure 5D). Importantly, SOCS1 mRNA abundance was inversely correlated with IFN-α/β mRNA and protein expression in pDCs from YM-infected WT and Mavs −/− mice (Figures 5E and 5F). Similar results were obtained using purified pDCs, cDCs, and macrophages treated in vitro with YM-iRBCs (Figures S5E and S5F).
Figure 5.

SOCS1 Is a Key Negative Regulator Induced by STING- and/or MAVS-Mediated Signaling and Inhibits Type I IFN Signaling through MyD88 in pDCs
(A) Expression of negative regulators Socs1 in the spleens of WT and deficient (Ifih1−/−, Mavs−/−, and Tmem173gt) mice at the indicated time points after YM infection. RNAs from splenocytes were isolated and used for expression analysis.
(B and C) Expression of Socs1 in pDCs of WT and deficient (Ifih1−/−, Mavs−/−, and Tmem173gt) mice after YM RNA (B) or gDNA (C) stimulation.
(D) WT and Mavs−/− mice were infected with YM for 18 hr. pDCs, cDCs, and macrophages were isolated and purified from infected mice and used for analysis of Socs1 mRNA expression by qPCR.
(E and F) IFN-α/β expression at the mRNA (E) and protein (F) amounts in pDCs, cDCs, or macrophages from WT and Mavs−/− mice after overnight culture.
(G) SOCS1 interacts with MyD88 and IRAK1. 293T cells were transfected with HA-Socs1 and Flag-Irak1, Flag-Irak4, or Flag-Myd88. Immunoprecipitation was performed with an anti-Flag, followed by immunoblotting with an anti-HA or anti-Flag antibody. Whole-cell lysates were immunoblotted for protein expression.
Data are plotted as the mean ± SD and are representative of three independent experiments. ∗∗p < 0.01, ∗∗∗p < 0.001 versus corresponding control. NS, not significant. ND, not detected.
See also Figures S5 and S6.
Next, we determined how SOCS1 inhibits MyD88-dependent type I IFN signaling. Although previous studies showed that SOCS1 inhibits the NF-κB signaling pathway by interacting with IRAK1 (Nakagawa et al., 2002), the involvement of SOCS1 in MyD88-mediated type I IFN signaling is not known. To determine whether SOCS1 directly interacts with MyD88, we performed co-immunoprecipitation of 293T cells expressing Socs1 plus Myd88, Irak1, or Irak4, and we found that SOCS1 directly interacted with MyD88 (Figure 5G), in addition to IRAK1. To determine whether STING-, MDA5-, and MAVS-induced SOCS1 are responsible for inhibition of MyD88-dependent type I IFN production in pDCs, we silenced Socs1, Socs3, Inpp5d, and Inppl1 in WT pDCs by siRNA transfection (Figure S6A) and then determined IFN-α/β production after YM RNA and gDNA stimulation. Only silencing of Socs1 resulted in a significant increase in IFN-α/β mRNA, as well as IFN-β protein after YM RNA and gDNA stimulation (Figures S6B and S6C), suggesting that STING- or MAVS-mediated type I IFN signaling induces SOCS1 expression, which, in turn, inhibits MyD88-dependent type I IFN production in pDCs.
SOCS1-Mediated Inhibition of Type I IFN Signaling Is pDC Specific and MyD88 Dependent
Next, we sought to determine the role of SOCS1 in inhibiting MyD88-mediated type I IFN production and protective immunity in vivo. Due to neonatal mortality and the complex inflammatory pathology of Socs1 −/− mice, the in vivo role of SOCS1 in response to malaria infection could not be addressed. Hence, we first silenced the Socs1 gene in WT mice with Socs1-specific siRNA (Figures 6 A and S6D). As expected, silencing of Socs1 increased IFN-γ production through its inhibitory effect on JAK1. We also found increased serum amounts of IFN-α/β compared with scrambled siRNA-treated WT mice after YM infection (Figure 6B). Importantly, silencing of Socs1 resulted in reduction of parasitemia and increased survival of WT mice after YM infection compared with control mice (Figure 6C). To further confirm these observations, we genetically ablated the Socs1 gene in WT bone marrow cells using the CRISPR/Cas9 system, and then we generated chimeric mice (Figures S6E and S6F). SOCS1-deficient chimeric mice produced high amounts of IFN-α/β, reduced parasitemia, and increased survival after YM infection (Figures S6G and S6H). These results suggest that silencing or genetic deletion of Socs1 relieves its inhibition of the MyD88-dependent type I IFN signaling pathway, thus leading to the control of parasitemia and host survival.
Figure 6.

SOCS1 Is Responsible for Inhibition of Myd88-Dependent Type I IFN in pDCs
(A–C) C57BL/6 mice were treated with Socs1-specific or scramble siRNA for 48 hr, followed by YM (0.5 × 106 iRBCs) infection. Diagram of experiment procedure are shown in (A). Serum amounts of IFN-α/β and IFN-γ in Socs1-specific or scrambled siRNA-treated mice at 24 hr after YM infection are shown in (B). Daily YM parasitemias and mortality rates are shown in (C).
(D and E) WT mice were treated with scramble siRNA, Socs1-specific siRNA, or Socs1-specific siRNA with anti-mPDCA-1 antibody at 24 hr before infection, followed by YM (0.5 × 106 iRBCs) infection. Serum amounts of IFN-α/β collected at 24 hr after infection are shown in (D). Daily YM parasitemias and mortality rates are shown in (E).
(F) WT and Myd88−/− mice were treated with Socs1-specific or scramble siRNA for 48 hr, followed by YM (0.5 × 106 iRBCs) infection. Daily YM parasitemias and mortality rates are shown.
Data are plotted as the mean ± SD and are representative of three independent experiments. ∗∗p < 0.01, ∗∗∗p < 0.001 versus corresponding control. NS, not significant.
See also Figure S6.
To provide direct evidence that SOCS1-mediated inhibition of MyD88-dependent type I IFN signaling is pDC-specific, we silenced Socs1 in WT mice at 24 hr before infection with or without pDC depletion using anti-mPDCA-1. We showed that the high serum amounts of IFN-α/β observed in Socs1-silenced mice were abolished upon pDC depletion (Figure 6D). Consistently, pDC depletion in Socs1-silenced mice also increased parasitemia and host mortality after infection compared with Socs1-silenced mice without pDC depletion (Figure 6E). These results suggest that SOCS1-mediated inhibition of MyD88-dependent type I IFN signaling is pDC specific.
Because SOCS1 can inhibit MyD88-dependent type I IFN signaling (as presented in this study) and JAK1 in STAT signaling (a downstream IFN-stimulated signaling pathway for IFN-γ production) (Figure S6I), we determined whether SOCS1-mediated type I IFN signaling inhibition is MyD88-dependent. As expected, Socs1-silenced mice reduced parasitemia and increased survival after YM infection. By contrast, Myd88 −/− mice with Socs1 silencing did not show reduced parasitemia or increased survival compared with those treated with scrambled siRNA (Figure 6F). Consistently, we could not detect any IFN-α/β in Myd88 −/− mice regardless of Socs1 silencing, indicating that SOCS1-mediated type I IFN signaling inhibition is MyD88 dependent (Figure S6J). Taken together, these results suggest that SOCS1, which is induced by the STING and/or MAVS pathways in response to YM infection, mainly inhibits MyD88-dependent type I IFN signaling and host survival.
pDCs, Macrophages, and cDCs Are Required in a Stage-Specific Manner for Generating IFN-α/β-Induced Protective Immunity against YM Infection
To understand how the early burst of type I IFN-α/β production in pDCs induces subsequent protective immunity and whether macrophages and cDCs are required for inducing potent immunity against YM infection, we depleted specific cell populations in different phases and then determined their contribution to cytokine production, parasitemia, and host survival (Figure 7 A). We found that depletion of pDCs with anti-mPDCA-1 in Mavs −/− mice during the cytokine production phase (24 hr before infection) markedly reduced IFN-α/β production; however, depletion of pDCs after 24 hr of infection had no effect on IFN-α/β production compared with mice treated with a control antibody (Figure 7B). Consistently, Mavs −/− mice treated with anti-mPDCA-1 during the cytokine production phase had increased parasitemia and host mortality but had no change in parasitemia and host survival when treated with anti-mPDCA-1 in the effector phase compared with control antibody-treated mice (Figure 7C). Collectively, these results clearly demonstrate the importance of pDCs in the production of IFN-α/β within the first 24 hr, but not after 24 hr of infection, for the protective immunity.
Figure 7.

Stage-Specific Role of pDCs, Macrophages, and cDCs in Generating IFN-α/β-Induced Protective Immunity
(A) Schematic figure to show cell depletion at different stages of IFN-α/β cytokine production after YM infection.
(B and C) Depletion of pDCs in Mavs−/− mice by anti-mPDCA-1 antibody administrated at 24 hr before or 24 hr after YM infection. Serum amounts of IFN-α/β collected at 24 hr after infection are shown in (B). Daily YM parasitemias and mortality rates are shown in (C).
(D and E) Depletion of macrophages in Tmem173gt mice by clodronate (700 μg/injection) administered 2 days before or 1 day after YM infection. Control liposome served as a control. Serum amounts of IFN-α/β collected at 24 hr after YM infection in Tmem173gt mice untreated (control liposome) or treated with clodronate at the indicated times are shown in (D). Parasitemias and mortality rates are shown in (E).
(F and G) WT chimeric mice were irradiated and transplanted with bone marrow cells of Mavs−/−:Zbtb46-DTR mice, then untreated or treated with DT as indicated at 4 days before or 1 day after YM infection. Serum amounts of IFN-α/β collected at 24 hr after infection are shown in (F), and daily YM parasitemias and mortality rates are shown in (G).
(H and I) WT and Rag2−/− mice were infected with YM, followed by i.v. administration of recombinant mouse IFN-α/β at 18 hr post infection. Parasitemias at day 5 and day 7 are shown in (H). Daily YM parasitemias and mortality rates are shown in (I).
(J and K) Intracellular staining of IFN-γ were measured by flow cytometry in splenocytes of WT and Mavs−/− mice at day 10 post YM infection, followed by stimulation with crude antigens (YM iRBCs). Statistical analysis is shown in (J). Meanwhile, splenocytes from YM-infected WT and Mavs−/− mice were cultured with crude antigens (iRBCs) overnight, and cell supernatants were collected for ELISA analysis (K).
(L) Malaria specific IgG in serum from WT and Mavs−/− mice at day 10 post YM infection, evaluated by ELISA.
Data are representative of three independent experiments and are plotted as the mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 versus corresponding control. NS, not significant. † denotes mouse death.
See also Figure S7.
To determine the importance of macrophages in cytokine production, parasitemia, and host mortality, we depleted macrophages with clodronate at 2 days before or 1 day after YM infection (Figure S7A). Macrophage depletion in Tmem173 gt mice did not affect serum amounts of IFN-α/β, regardless of depletion at 2 days before or 1 day after YM infection (Figure 7D). However, macrophage depletion during the effector phase (at 1 day post infection) markedly increased parasitemia and host mortality compared with a liposome control or treatment at 2 days before infection (Figure 7E), suggesting a critical role of macrophages during the effector phase, but not during the cytokine production phase. To determine the relative role of cDCs in anti-malaria immunity, we generated Mavs −/−:Zbtb46-DTR (zinc figure transcription factor-driven DTR expression in cDCs) mice. As expected in a previous study (Meredith et al., 2012), DT injection into Zbtb46-DTR bone marrow chimeras resulted in specific depletion of cDCs in 12 hr and maintained depletion for 5 days. Thus, we injected DT into Mavs −/−:Zbtb46-DTR chimeric mice at 4 days before or at 1 day post infection. We found that there was no change in the serum amounts of IFN-α/β when DT was injected either before or after YM infection (Figure 7F), suggesting that cDCs are not responsible for the IFN-α/β production during the early phase (24 hr) of infection. However, Mavs −/−:Zbtb46-DTR mice treated with DT after 24 hr of infection showed increased parasitemia and died sooner after infection compared to untreated mice or mice treated with DT at 4 days before infection (Figure 7G), suggesting that cDCs are required for generating protective immunity during the effector phase. Consistent with these results, we also found that malaria 18S rRNA could be detected in pDCs, but not in macrophages or cDCs, at 18 hr post infection. However, malaria 18S rRNA could be detected in macrophages and cDCs of Mavs −/− mice at day 3 post infection (Figure S7B), suggesting that pDCs are the major cell population that sense and detect YM infection within the first 24 hr, whereas macrophages and cDCs are involved in the detection of YM infection at later time points for the induction of adaptive immunity. Taken together, these results suggest that pDCs, but not macrophages or cDCs, are critically required for the production of IFN-α/β in the first 24 hr after YM infection, whereas macrophages and cDCs are required for IFN-α/β-induced immunity against YM after 24 hr post infection.
Next, we determined whether B and T cells are required for IFN-α/β-induced immunity against YM, in particular for clearing malaria in the late phase of infection. To test this possibility, we infected WT and Rag2-deficient mice, which lack B and T cells, with YM in the presence or absence of exogenous murine recombinant IFN-α/β. Although cytokine-treated WT and Rag2 −/− mice demonstrated reduced parasitemia compared with untreated WT and Rag2 −/− mice, we found no differences in the percent of parasitemia between WT and Rag2 −/− mice either in untreated groups or cytokine-treated groups in the first 5 days after infection (Figure 7H). However, after 7 days post infection, parasitemia in cytokine-treated Rag2 −/− mice markedly increased to the amounts observed in untreated WT or untreated Rag2 −/− mice (Figures 7H and 7I). WT untreated, Rag2 −/− untreated, and Rag2 −/− cytokine-treated mice died within 8 days after YM infection, whereas cytokine-treated WT mice maintained significantly lower parasitemia than the other three groups and survived beyond 8 days (Figure 7I), suggesting that B and T cells are required for generating adaptive immunity to control parasitemia and clear parasites at a later time. To further understand cellular and molecular basis of YM-induced adaptive immunity in WT and Mavs −/− mice, we infected WT and Mavs −/− mice with 0.1 × 106 YM iRBCs (10 times lower dosage to keep WT mice alive) and isolated splenocytes at day 10 post infection, then we stimulated with crude antigen (P.y YM-iRBC) for T and B cells function analysis. Our results showed that the percentage of splenic IFN-γ+ CD4+ and IFN-γ+ CD8+ cells from Mavs −/− mice were much higher than those from WT mice (Figure 7J). Consistently, IFN-γ protein amounts in the supernatant of splenocytes after overnight culture with crude antigens and concentrations of malaria-specific immunoglobulin G (IgG) in serum were significantly higher in Mavs −/− mice than in WT mice (Figures 7K and 7L), suggesting that YM-infected Mavs −/− mice developed much stronger adaptive (B and T cell) immune responses than WT mice.
Discussion
Our study shows that cGAS functions as a DNA sensor for recognizing malaria gDNA and triggers STING-mediated type I IFN signaling. STING-deficient mice were more resistant to YM infection than were cGAS-deficient mice, suggesting that other DNA sensors might function in recognizing malarial genomic DNA. For RNA sensing, MDA5, but not RIG-I, is responsible for detecting malaria RNA and triggers MAVS-mediated type I IFN signaling. It is known that RIG-I recognizes RNA with a triphosphate (PPP) moiety and 5′ blunt-ended 20 nucleotides, whereas MDA5 recognizes long dsRNA (1–2 kb) (Goubau et al., 2013). These structural and length requirements of dsRNA may explain why malaria RNA interacts with MDA5, but not with RIG-I, to trigger downstream MAVS-dependent type I IFN signaling. Although cGAS-STING- and MDA5-MAVS-mediated type I IFN signaling is important and operates in all cell types, including macrophages and cDCs, we did not detect YM malaria 18S rRNA or observe IFN-β and SOCS1 expression in macrophages and cDCs within the first 18–24 hr post infection. Depletion of macrophages or cDCs did not affect the production of IFN-α/β at 24 hr post infection, further suggesting that macrophages and cDCs are not involved the production of IFN-α/β in the early stage of YM infection.
To understand high serum amounts of IFN-α/β in Ifih1 −/−, Mavs −/−, Tmem173 gt, or Mb21d1 −/− mice, we have provided several lines of evidence to support our conclusion: (1) Ifih1 −/− :Myd88 −/−, Mavs −/− :Myd88 −/−, or Tmem173 gt :Myd88 −/− mice failed to produce any IFN-α/β and become susceptible to YM infection, compared with Ifih1 −/−, Mavs −/−, and Tmem173 gt mice; (2) depletion of pDCs by a specific antibody or genetic ablation markedly reduced the production of IFN-α/β, suggesting that pDCs are the major sources for production of these cytokines. Thus, our results provide direct evidence that MyD88-dependent type I IFN signaling in pDCs plays a critical role in the early production of IFN-α/β and protection against malaria infection.
To identify the mechanisms and sequential order of immune responses induced by IFN-α/β-producing pDCs, we depleted pDCs, macrophages, cDCs, or T cells at different times and determined their roles in the production of IFN-α/β, parasitemia, and host survival. Our results clearly demonstrate the requirement for pDCs in the production of IFN-α/β during the first 24 hr of infection, but not after that time. Conversely, depletion of macrophages or cDCs during the first 24 hr of infection affected neither IFN-α/β production nor parasitemia or host mortality in this YM model. However, depletion of macrophages or cDCs after 24 hr post infection resulted in increased parasitemia and host mortality, suggesting that these cell types participated in anti-malaria immunity, which is consistent with previous results showing that classic DCs are required for inducing T cell response (Voisine et al., 2010). Although B cells and T cells are not required for IFN-α/β production and parasitemia control during the first 5–6 days post infection, they are required for the control of parasitemia and host mortality at later times (>6 days), as suggested by previous studies using cerebral malaria (Ball et al., 2013, Haque et al., 2014).
We have shown that TLR7, but not TLR9, was essential for IFN-α/β production in pDCs. YM RNA activates TLR7 and recruits the downstream adaptor MyD88 to trigger IRF7-mediated type I IFN signaling pathway. However, previous studies show that mice deficient with STING, TBK1, IRF3 and IRF7, or IFNAR1 expression are resistant to P. berghei ANKA infection (Haque et al., 2014, Sharma et al., 2011), suggesting that type I IFN impairs host anti-malaria responses to ANKA. To explain why type I IFN plays opposing roles in host immune responses to different malaria strain infection, we examined the timing and amounts of IFN-α/β production in response to two lethal Plasmodium strains (P. yoelii YM and P. berghei ANKA) and found that the timing and amounts of IFN-α/β production are different in WT mice in response to YM or ANKA infection: low amount of IFN-α/β only at day 1 after YM infection versus large amounts of IFN-α/β at day 4 after ANKA infection (Haque et al., 2014). We propose that early robust production or administration of type I IFN (18–24 hr post infection) is essential to induce innate and adaptive immunity against malaria. By contrast, late time production of type I IFN (3–4 days after ANKA infection) impairs host anti-malaria immune responses. Thus, different malaria strains have evolved to evade host immune system by either inhibiting type I IFN production at the early infection phase (in YM strain) or shifting type I IFN production to the late time (4 days post infection) (in ANKA strain). Similar results have been obtained in LCMV infection or other pathogens (O’Connell et al., 2004, Teles et al., 2013, Wilson et al., 2013). In these cases, persistent production of type I IFN even at day 9 post LCMV infection is observed, which, in turn, induces the expression of negative regulators, such as IL-10 and PD-L1, to inhibit adaptive immune response. Our study showed that robust type I IFN production (only at day 1) produced by pDCs activates cDCs and macrophages to induce adaptive immune response for the clearance of YM infection. Our notion is also supported by a recent study, showing that administration of type I IFN at an early time (6–12 hr post-infection) after SARS-CoV infection promotes host innate and adaptive immune response. By contrast, delayed type I IFN production promotes lung immunopathology with diminished survival and impairs anti-viral immunity (Channappanavar et al., 2016). Thus, early robust production of type I IFN (18–24 hr post infection) promotes innate and adaptive immune responses, while delayed type I IFN production (3–4 days post infection) impairs innate and adaptive immune responses. The timing and amounts of type I IFN production may be dependent upon cell tropism (targeting different cell types during infection) and distinct strain-specific pathogenesis (Gun et al., 2014, Wu et al., 2014). Further studies are needed to understand the molecular mechanisms responsible for these opposing roles of type I IFN in malaria and viral infection.
Our study identified SOCS1 as a key negative regulator, which interacted with MyD88 and inhibited MyD88-dependent type I IFN signaling. A recent study shows a role for STING in negatively regulating inflammatory responses of macrophages in systemic lupus erythematosus (SLE) through upregulation of A20, SOCS1, and SOCS3 (Sharma et al., 2015), but its role in MyD88-dependent type I IFN signaling is not known in this disease model. We further showed that inactivation of Ifih1, Tmem173, or Mavs markedly reduced Socs1 expression in vitro and in vivo and increased IFN-α/β mRNA and protein expression. Silencing or genetic deletion of Socs1 in YM-infected WT mice markedly reduced parasitemia and host mortality. The predominant role of SOCS1 is to regulate MyD88-dependent type I IFN signaling in pDCs because the observed increase in IFN-α/β expression after siRNA-mediated Socs1 silencing disappeared when pDCs were depleted, suggesting that Socs1 silencing mainly affects MyD88-dependent type I IFN signaling in pDCs. Based on these findings, we propose a working model to illustrate how type I IFN is regulated in response to YM infection (Figure S7C). Activation of cGAS-STING and MDA5-MAVS by YM infection triggers IRF3-mediated type I IFN signaling in pDCs, which produce low amounts of IFN-α/β and activate negative regulator SOCS1 in WT pDCs. SOCS1 then inhibits MyD88-dependent type I IFN signaling pathway, which produces more than 100-fold higher cytokine amounts than those produced by STING- and MAVS-mediated IRF3-dependent type I IFN signaling pathway. By contrast, SOCS1 expression is markedly reduced in Ifih1 −/−, Mavs −/−, and Tmem173 gt pDCs, allowing the activation of MyD88-dependent type I IFN signaling. Thus, lethal YM infection activates two distinct type I IFN signaling pathways in pDCs. YM takes advantage of the cross-inhibition of TLR7-MyD88-mediated type I IFN signaling by cGAS-STING- and MDA5-MAVS-mediated type I IFN through upregulation of Socs1 in WT mice to avoid host innate immune response. Silencing or genetic ablation of genes, such as Socs1 or those in the STING- and MAVS-mediated type I IFN signaling pathway, relieves SOCS1-suppressive effects on TLR7-MyD88-mediated type I IFN signaling and permits robust production of IFN-α/β in the first 24 hr post infection, then activates downstream signaling pathways via IFNAR in other innate immune cells, such as macrophages and cDCs, for priming adaptive immunity mediated by B and T cells to clear YM infection at later times. Thus, our findings not only identify a critical regulatory mechanism between different type I IFN signaling pathways in pDCs, as well as a critical role of stage-dependent type I IFN production and specific contribution of different immune cells for developing protective immunity, but also provide potential therapeutic targets for the development of safe and effective malaria vaccines.
Experimental Procedures
Malaria Parasites and Mice
The parasite YM has been previously described (Li et al., 2011). For YM infection, 1 × 106 iRBCs (otherwise indicated specifically in the figure legend) suspended in 200 μL PBS from the donor mice were injected i.p. into experimental mice. All mouse-related procedures were performed according to experimental protocols approved by the Animal Care and Welfare Committee at Houston Methodist Research Institute and in accordance with NIH-approved animal study protocol LMVR-11E. See Supplemental Experimental Procedures for more details of this and following sections.
Antibody Treatments
To block type I IFN receptor, anti-mouse IFN-α/β receptor antibodies, in the amount of 500 μg in PBS at day 0, 2, 4, and 250 μg at day 6 after infection, were injected i.p. into WT and deficient mice. To deplete pDCs, pDC-depleting functional-grade mAb (anti-mPDCA-1 IgG) and the corresponding isotype control IgG served as control and were purchased from Miltenyi Biotec, and two i.p. injections of antibody (250 μg/mouse) per mouse were administered 12 hr prior and after YM infection.
Isolation and Purification of Immune Cell Populations
Bone marrow cells were isolated from the tibia and femur and cultured in RPMI1640 medium with 10% FBS, 1% penicillin-streptomycin, 55 μM β-mercaptoethanol, and 10% L929 conditioned media containing macrophage-colony stimulating factor for 5 days for BMDMs, 20 ng/mL murine GM-CSF and 10 ng/ml IL-4 for 6–8 days for cDCs, and 200 ng/mL Flt3L for pDCs. pDCs were further purified by flow cytometry analysis gating on CD11b−B220+CD11c+ cell population.
RNAi-Mediated Silencing in Mice
In vivo ready siRNAs were mixed with invivofectamine 2.0 liposomes (Invitrogen) following the manufacturer’s instructions and injected intravenously (i.v.) in a volume of 100 μL at a dose of 5 mg/kg. Mice were infected with YM (0.5 × 106 iRBCs) 48 hr after siRNA treatment.
RNA Preparation and qPCR
Total RNA was harvested from splenic tissue or stimulated cells using the TRIzol reagent, and the complimentary cDNA was generated using reverse transcriptase III. Real-time PCR was carried out using the ABI Prism 7000 analyzer using the SYBR GreenER qPCR Super Mix Universal and specific primers.
Diphtheria Toxin Treatment
Ifih1 −/−, Mavs −/−, and Tmem173 gt mice were crossed with BDCA2-DTR transgenic mice to generate Ifih1 −/−: BDCA2-DTR, Mavs −/−:BDCA2-DTR and Tmem173 gt:BDCA2-DTR mice, respectively, and then treated with diphtheria toxin (DT) i.p. at dose of 100–120 ng/mouse. pDCs were depleted by DT injection at 1 day before and 1, 3, and 5 days after YM infection. For cDC depletion, bone marrow chimeras were reconstituted for at least 6–8 weeks after lethal irradiation (950 cGy) and i.v. transferred with 10 × 106 bone marrow cells from Mavs −/−:Zbtb46-DTR mice and then injected with DT at a dose of 2.5 ng per gram of body weight.
ELISA
Mouse serum or cell supernatants were collected at the indicated time after infection or stimulation and subjected to analysis with commercial ELISA kits for mouse IFN-α, IFN-β (PBL Biomedical Laboratories), IL-6, or IFN-γ (eBioscience) following the manufacturer’s instructions.
Statistical Analysis
All analyses were performed using GraphPad Prism version 5.0 (GraphPad Software, La Jolla, CA). Data are presented as means ± SD unless otherwise stated. Statistical significance of differences between two groups was assessed by unpaired Student’s t tests and a p value of <0.05 was considered significant.
Author Contributions
X.Y., B.C., and M.W. performed the experiments. P.T., X.D., Q.L., P.L., C.X., and J.L. provided assistance or technique support in some experiments. J.W. and X.-Z.S. provided malaria strains and some experimental assistance. X.Y., H.Y.W., X.-Z.S., and R.-F.W. performed data analysis and wrote the manuscript. R.-F.W. supervised the entire project and designed experiments.
Acknowledgments
We thank Marco Colonna (Washington University School of Medicine) for providing Tlr9 −/− mice, Richard A. Flavell (Yale University) for Tlr7 −/− mice, Kate Fitzgerald (University of Massachusetts Medical School) and Tadatsugo Taniguchi (The University of Tokyo) for Irf3 −/−:Irf7 −/− mice, Michael Gale (University of Washington) and Wenxin Wu (University of Oklahoma Health Science Center) for Ddx58 −/− mice, and Skip Virgin (Washington University at St. Louis) for Mb21d1 −/− mice, Bo Ning for art drawing, and Jana S. Burchfield for editing the manuscript. X.Y. and B.C. were partially supported by the China Scholar Council. This work was supported, in part, by grants (R01CA101795 and DA030338) from the NCI and NIDA, NIH to R.-F.W. and by the Division of Intramural Research at the National Institute of Allergy and Infectious Diseases (NIAID).
Published: October 25, 2016
Footnotes
Supplemental Information includes Supplemental Experimental Procedures and seven figures and can be found with this article online at http://dx.doi.org/10.1016/j.immuni.2016.10.001.
Contributor Information
Xin-zhuan Su, Email: xsu@niaid.nih.gov.
Rong-Fu Wang, Email: rwang3@houstonmethodist.org.
Supplemental Information
References
- Baccarella A., Fontana M.F., Chen E.C., Kim C.C. Toll-like receptor 7 mediates early innate immune responses to malaria. Infect. Immun. 2013;81:4431–4442. doi: 10.1128/IAI.00923-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball E.A., Sambo M.R., Martins M., Trovoada M.J., Benchimol C., Costa J., Antunes Gonçalves L., Coutinho A., Penha-Gonçalves C. IFNAR1 controls progression to cerebral malaria in children and CD8+ T cell brain pathology in Plasmodium berghei-infected mice. J. Immunol. 2013;190:5118–5127. doi: 10.4049/jimmunol.1300114. [DOI] [PubMed] [Google Scholar]
- Channappanavar R., Fehr A.R., Vijay R., Mack M., Zhao J., Meyerholz D.K., Perlman S. Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in SARS-CoV-Infected Mice. Cell Host Microbe. 2016;19:181–193. doi: 10.1016/j.chom.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazzinelli R.T., Kalantari P., Fitzgerald K.A., Golenbock D.T. Innate sensing of malaria parasites. Nat. Rev. Immunol. 2014;14:744–757. doi: 10.1038/nri3742. [DOI] [PubMed] [Google Scholar]
- Goubau D., Deddouche S., Reis e Sousa C. Cytosolic sensing of viruses. Immunity. 2013;38:855–869. doi: 10.1016/j.immuni.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowda N.M., Wu X., Gowda D.C. TLR9 and MyD88 are crucial for the development of protective immunity to malaria. J. Immunol. 2012;188:5073–5085. doi: 10.4049/jimmunol.1102143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gun S.Y., Claser C., Tan K.S., Rénia L. Interferons and interferon regulatory factors in malaria. Mediators Inflamm. 2014;2014:243713. doi: 10.1155/2014/243713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haque A., Best S.E., Ammerdorffer A., Desbarrieres L., de Oca M.M., Amante F.H., de Labastida Rivera F., Hertzog P., Boyle G.M., Hill G.R., Engwerda C.R. Type I interferons suppress CD4+ T-cell-dependent parasite control during blood-stage Plasmodium infection. Eur. J. Immunol. 2011;41:2688–2698. doi: 10.1002/eji.201141539. [DOI] [PubMed] [Google Scholar]
- Haque A., Best S.E., Montes de Oca M., James K.R., Ammerdorffer A., Edwards C.L., de Labastida Rivera F., Amante F.H., Bunn P.T., Sheel M. Type I IFN signaling in CD8- DCs impairs Th1-dependent malaria immunity. J. Clin. Invest. 2014;124:2483–2496. doi: 10.1172/JCI70698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden M.S., Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- Langhorne J., Ndungu F.M., Sponaas A.M., Marsh K. Immunity to malaria: more questions than answers. Nat. Immunol. 2008;9:725–732. doi: 10.1038/ni.f.205. [DOI] [PubMed] [Google Scholar]
- Li J., Pattaradilokrat S., Zhu F., Jiang H., Liu S., Hong L., Fu Y., Koo L., Xu W., Pan W. Linkage maps from multiple genetic crosses and loci linked to growth-related virulent phenotype in Plasmodium yoelii. Proc. Natl. Acad. Sci. USA. 2011;108:E374–E382. doi: 10.1073/pnas.1102261108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liehl P., Mota M.M. Innate recognition of malarial parasites by mammalian hosts. Int. J. Parasitol. 2012;42:557–566. doi: 10.1016/j.ijpara.2012.04.006. [DOI] [PubMed] [Google Scholar]
- Liehl P., Zuzarte-Luís V., Chan J., Zillinger T., Baptista F., Carapau D., Konert M., Hanson K.K., Carret C., Lassnig C. Host-cell sensors for Plasmodium activate innate immunity against liver-stage infection. Nat. Med. 2014;20:47–53. doi: 10.1038/nm.3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.J. IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu. Rev. Immunol. 2005;23:275–306. doi: 10.1146/annurev.immunol.23.021704.115633. [DOI] [PubMed] [Google Scholar]
- Meredith M.M., Liu K., Darrasse-Jeze G., Kamphorst A.O., Schreiber H.A., Guermonprez P., Idoyaga J., Cheong C., Yao K.H., Niec R.E., Nussenzweig M.C. Expression of the zinc finger transcription factor zDC (Zbtb46, Btbd4) defines the classical dendritic cell lineage. J. Exp. Med. 2012;209:1153–1165. doi: 10.1084/jem.20112675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller L.H., Ackerman H.C., Su X.Z., Wellems T.E. Malaria biology and disease pathogenesis: insights for new treatments. Nat. Med. 2013;19:156–167. doi: 10.1038/nm.3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J.L., Sack B.K., Baldwin M., Vaughan A.M., Kappe S.H. Interferon-mediated innate immune responses against malaria parasite liver stages. Cell Rep. 2014;7:436–447. doi: 10.1016/j.celrep.2014.03.018. [DOI] [PubMed] [Google Scholar]
- Nakagawa R., Naka T., Tsutsui H., Fujimoto M., Kimura A., Abe T., Seki E., Sato S., Takeuchi O., Takeda K. SOCS-1 participates in negative regulation of LPS responses. Immunity. 2002;17:677–687. doi: 10.1016/s1074-7613(02)00449-1. [DOI] [PubMed] [Google Scholar]
- O’Connell R.M., Saha S.K., Vaidya S.A., Bruhn K.W., Miranda G.A., Zarnegar B., Perry A.K., Nguyen B.O., Lane T.F., Taniguchi T. Type I interferon production enhances susceptibility to Listeria monocytogenes infection. J. Exp. Med. 2004;200:437–445. doi: 10.1084/jem.20040712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palomo J., Fauconnier M., Coquard L., Gilles M., Meme S., Szeremeta F., Fick L., Franetich J.F., Jacobs M., Togbe D. Type I interferons contribute to experimental cerebral malaria development in response to sporozoite or blood-stage Plasmodium berghei ANKA. Eur. J. Immunol. 2013;43:2683–2695. doi: 10.1002/eji.201343327. [DOI] [PubMed] [Google Scholar]
- Paludan S.R., Bowie A.G. Immune sensing of DNA. Immunity. 2013;38:870–880. doi: 10.1016/j.immuni.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley E.M., Stewart V.A. Immune mechanisms in malaria: new insights in vaccine development. Nat. Med. 2013;19:168–178. doi: 10.1038/nm.3083. [DOI] [PubMed] [Google Scholar]
- Sharma S., DeOliveira R.B., Kalantari P., Parroche P., Goutagny N., Jiang Z., Chan J., Bartholomeu D.C., Lauw F., Hall J.P. Innate immune recognition of an AT-rich stem-loop DNA motif in the Plasmodium falciparum genome. Immunity. 2011;35:194–207. doi: 10.1016/j.immuni.2011.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S., Campbell A.M., Chan J., Schattgen S.A., Orlowski G.M., Nayar R., Huyler A.H., Nündel K., Mohan C., Berg L.J. Suppression of systemic autoimmunity by the innate immune adaptor STING. Proc. Natl. Acad. Sci. USA. 2015;112:E710–E717. doi: 10.1073/pnas.1420217112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson M.M., Riley E.M. Innate immunity to malaria. Nat. Rev. Immunol. 2004;4:169–180. doi: 10.1038/nri1311. [DOI] [PubMed] [Google Scholar]
- Sun L., Wu J., Du F., Chen X., Chen Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiecki M., Gilfillan S., Vermi W., Wang Y., Colonna M. Plasmacytoid dendritic cell ablation impacts early interferon responses and antiviral NK and CD8(+) T cell accrual. Immunity. 2010;33:955–966. doi: 10.1016/j.immuni.2010.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi O., Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- Teijaro J.R., Ng C., Lee A.M., Sullivan B.M., Sheehan K.C., Welch M., Schreiber R.D., de la Torre J.C., Oldstone M.B. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science. 2013;340:207–211. doi: 10.1126/science.1235214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teles R.M., Graeber T.G., Krutzik S.R., Montoya D., Schenk M., Lee D.J., Komisopoulou E., Kelly-Scumpia K., Chun R., Iyer S.S. Type I interferon suppresses type II interferon-triggered human anti-mycobacterial responses. Science. 2013;339:1448–1453. doi: 10.1126/science.1233665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togbe D., Schofield L., Grau G.E., Schnyder B., Boissay V., Charron S., Rose S., Beutler B., Quesniaux V.F., Ryffel B. Murine cerebral malaria development is independent of toll-like receptor signaling. Am. J. Pathol. 2007;170:1640–1648. doi: 10.2353/ajpath.2007.060889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voisine C., Mastelic B., Sponaas A.M., Langhorne J. Classical CD11c+ dendritic cells, not plasmacytoid dendritic cells, induce T cell responses to Plasmodium chabaudi malaria. Int. J. Parasitol. 2010;40:711–719. doi: 10.1016/j.ijpara.2009.11.005. [DOI] [PubMed] [Google Scholar]
- Wang Y., Swiecki M., McCartney S.A., Colonna M. dsRNA sensors and plasmacytoid dendritic cells in host defense and autoimmunity. Immunol. Rev. 2011;243:74–90. doi: 10.1111/j.1600-065X.2011.01049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson E.B., Yamada D.H., Elsaesser H., Herskovitz J., Deng J., Cheng G., Aronow B.J., Karp C.L., Brooks D.G. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science. 2013;340:202–207. doi: 10.1126/science.1235208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X., Gowda N.M., Kumar S., Gowda D.C. Protein-DNA complex is the exclusive malaria parasite component that activates dendritic cells and triggers innate immune responses. J. Immunol. 2010;184:4338–4348. doi: 10.4049/jimmunol.0903824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J., Tian L., Yu X., Pattaradilokrat S., Li J., Wang M., Yu W., Qi Y., Zeituni A.E., Nair S.C. Strain-specific innate immune signaling pathways determine malaria parasitemia dynamics and host mortality. Proc. Natl. Acad. Sci. USA. 2014;111:E511–E520. doi: 10.1073/pnas.1316467111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.