

Abstract
The severe acute respiratory syndrome-associated coronavirus (SARS-CoV) infected more than 8,000 people across 29 countries and caused more than 900 fatalities. Based on the concept of chemical genetics, we screened 50,240 structurally diverse small molecules from which we identified 104 compounds with anti-SARS-CoV activity. Of these 104 compounds, 2 target the SARS-CoV main protease (Mpro), 7 target helicase (Hel), and 18 target spike (S) protein-angiotensin-converting enzyme 2 (ACE2)-mediated viral entry. The EC50 of the majority of the 104 compounds determined by SARS-CoV plaque reduction assay were found to be at low micromolar range. Three selected compounds, MP576, HE602, and VE607, validated to be inhibitors of SARS-CoV Mpro, Hel, and viral entry, respectively, exhibited potent antiviral activity (EC50 < 10 μM) and comparable inhibitory activities in target-specific in vitro assays.
Introduction
The severe acute respiratory syndrome-associated coronavirus (SARS-CoV) recently emerged as the causative agent of an endemic atypical pneumonia. Within a year, SARS-CoV infected more than 8,000 people across 29 countries and cost more than 900 human lives [1]. Lack of knowledge of the novel coronavirus SARS-CoV and the absence of efficacious therapeutic agents were the main reasons for the failure to manage the outbreak of SARS effectively. After the causative agent of the devastating disease was identified by us and others 2, 3, 4, the genome of SARS-CoV was decoded rapidly by several groups 5, 6, 7. Subsequently, reverse genetics with SARS-CoV cDNA was accomplished [8], and ACE2 was identified as a functional receptor for the virus [9], highlighting the rapid responses of the scientific community to this previously unknown global pathogen.
Advancements in synthetic organic chemistry, molecular biology, and informatics have made possible the use of large collections of small molecules (chemical libraries) to investigate protein/chemical interactions in vitro and in vivo 10, 11, 12, 13. The term “chemical genetics” has been coined to signify the use of chemicals to perturb systematically, and thus determine, the function of proteins in the same way that mutations are used in classical genetics 14, 15, 16. We decided to dissect the pathogenic pathways of the SARS-CoV using chemical genetics. We hypothesized that by using forward chemical genetics 15, 16, in which small molecules that induce altered phenotypes in cells or organisms are identified and their cellular targets will then be determined subsequently, we will be able to isolate novel small-molecule compounds perturbing the biological pathways that are essential for the pathogenesis of SARS-CoV.
Recent findings demonstrate that the first step in SARS-CoV infection is mediated by S protein association with ACE2 [9]. After being internalized into the target cells, SARS-CoV undergoes a very rapid replication cycle through a series of concerted transcriptional, translational, posttranslational, and proteolytic processing events, leading to maturation and release of infective viral particles into the culture supernatant 17, 18, 19. We speculated that the various biological pathways involved in viral pathogenesis could be perturbed by small molecules using chemical genetic approaches. To examine the feasibility of employing chemical genetic approaches in SARS-CoV research, we acquired a chemical library (ChemBridge Corporation) of 50,240 structurally diverse small-molecule compounds that vary in functional groups and charges. As the SARS-CoV replicates effectively in Vero cells (African green monkey kidney cell line) and full cytopathic effects (CPE) of the infected cells can be observed within 96 hr postinfection, Vero cell CPE was used as a phenotypic indicator of successful viral infection in a cell-based assay to screen for small-molecule compounds that perturb the infectivity of the virus. The employment of high-throughput screening (HTS) technologies to generate a collection of structurally diverse small-molecule compounds perturbing the pathogenesis of the SARS-CoV will lay down the foundation to dissecting the molecular basis of viral infections using chemical genetics.
Results and Discussion
Phenotype-Based HTS
In a primary screening (at 20 μg/ml of each compound), we identified 1003 “hits” (a hit rate of 2%) that protected Vero cells from SARS-CoV-induced CPE. When the hits were rearrayed and the concentration of selected compounds was lowered to 10 μg/ml for secondary screening, 104 compounds retained consistent protective effects against SARS-CoV-induced CPE in Vero cells (Figure 1) . Further evaluation by quantitative plaque reduction assays demonstrated that the EC50 (median effective concentration) of the selected compounds were below 10 μg/ml, with 78 compounds having an EC50 below 2 μg/ml. For subsequent studies, the concentrations of selected compounds were converted to molar units for more precise comparison of their biological activities. The TC50 (median toxic concentration) of selected compounds was determined to be >50 μM by MTT (3-[4,5-dimethylth-iazol-2-yl]-2,5-diphenyltetrazolium bromide) assay. To test our hypothesis that the 104 selected compounds represent diverse molecular blockers of various biological pathways crucial for SARS-CoV infectivity, we screened for molecules targeting viral entry, transcription, and proteolytic processing, the three major processes essential for successful viral replication in the host.
Figure 1.

Isolation of Biologically Active Small-Molecule Inhibitors of SARS-CoV in a Phenotype-Based Screen
A schematic illustration of major processes involved in the phenotype-based screen is shown. Enlarged images of Vero cells from a typical 384-well tissue culture plate used in screening are also included to indicate the criteria for hit selection. Only those compounds that fully protected the Vero cells from SARS-CoV-induced CPE were selected as hits.
Compounds Targeting SARS-CoV Mpro
SARS-CoV Mpro is believed to play a major role in proteolytic processing of the viral polyproteins and is regarded as a prime target for anti-SARS-CoV drug development 20, 21. The recently available crystal structure of the SARS-CoV Mpro makes it a suitable target for structure-based rational drug design [22]. We cloned the SARS-CoV Mpro in an E. coli expression system and obtained purified enzyme to examine if any of the 104 selected compounds targets this vital component of the virus. High-performance liquid chromatography (HPLC)-based assays were used to monitor the in vitro cleavage activity of the viral Mpro. The purified SARS-CoV Mpro cleaved efficiently and specifically a synthetic peptide with the sequence H2N-TSAVLQ↓SGFRKW-COOH mimicking the putative autolytic cleavage site (the cleavage site is indicated with a ↓) of the N-terminal part of Mpro [22]. Screening of the 104 compounds at a concentration of 20 μg/ml identified two candidates as potential inhibitors of the SARS-CoV Mpro. One of the compounds (designated MP576) displayed potent inhibitory activity with an IC50 (median inhibitory concentration) of 2.5 μM (Figure 2A) and an EC50 of 7 μM in the Vero cell-based SARS-CoV plaque reduction assay (Figure 2B). Furthermore, MP576 can be docked favorably into the active site of SARS-CoV Mpro (Figure 2C), consistent with the in vitro cleavage data indicating that MP576 is a novel nonpeptide inhibitor of SARS-CoV Mpro. Since SARS-CoV Mpro shares a similar structural fold with serine proteases 21, 23, we tested the inhibitory activity of MP576 on human neutrophil elastase (HNE). Using azocasein as the substrate, we demonstrated that 100 μM of MP576 failed to inhibit the proteolytic activity of HNE (Figure 2D), suggesting that MP576 is not a general protease inhibitor.
Figure 2.
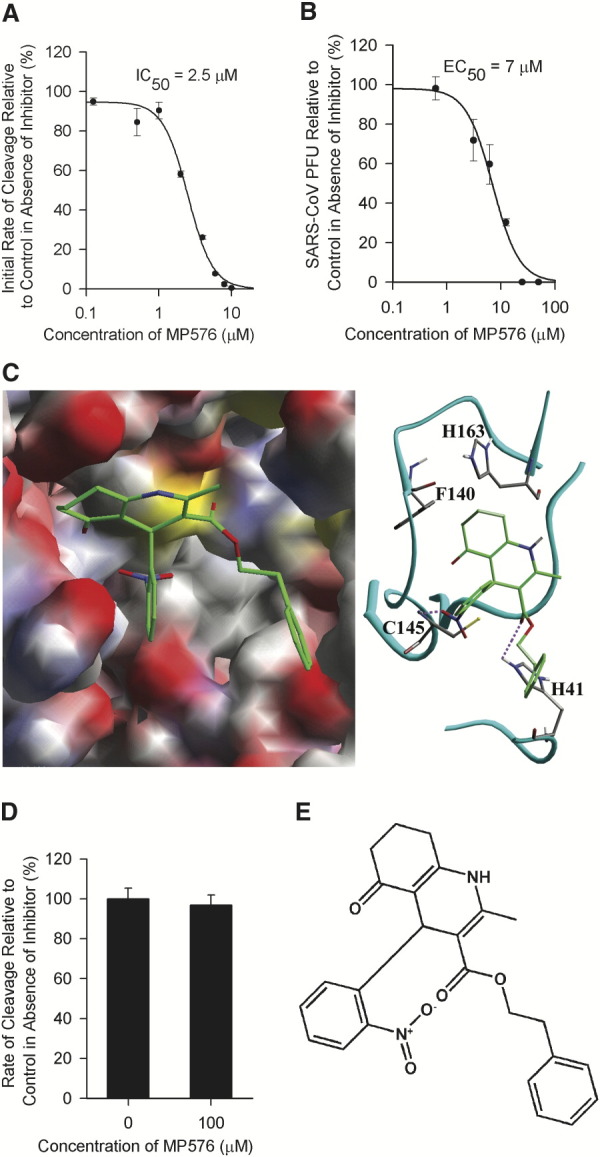
MP576 Targets SARS-CoV Mpro
(A) MP576 inhibited in vitro cleavage activity of SARS-CoV Mpro with an IC50 of 2.5 μM. Synthetic peptide H2N-TSAVLQSGFRKW-COOH was used as the substrate in the assay.
(B) MP576 inhibited SARS-CoV plaque formation with an EC50 of 7 μM.
(C) MP576 is predicted to occupy the active site of SARS-CoV Mpro (left) and hydrogen bonds with Cys145 N and His41 Nϵ2 (right). The active site of SARS-CoV Mpro is shown as a surface model (left) and the backbone trace (light blue) of SARS-CoV is shown, along with the side chains of residues crucial for substrate binding and catalysis (right). MP576 carbons are shown in green. Predicted hydrogen bonds are shown as dashed lines.
(D) MP576 (100 μM) did not inhibit human neutrophil elastase (HNE). Azocasein was used as the substrate in the assay.
(E) Chemical structure of MP576 (3-quinolinecarboxylic acid, 1,4,5,6,7,8-hexahydro-2-methyl-4-(2-nitrophenyl)-5-oxo-, 2-phenylethyl ester).
Compounds Targeting SARS-CoV Hel
To identify compounds that interfere with viral components essential for transcription and replication, we selected the SARS-CoV Hel, which we had previously cloned and characterized [24], for our assay system. It was demonstrated that the SARS-CoV NTPase/helicase had an unstimulated ATPase activity that could be stimulated by the addition of polynucleotides, in particular dT24. As the functional conformation of the helicase is likely to be the nucleic acid-bound state, we initially screened the 104 active compounds against the polynucleotide-stimulated ATPase activity of SARS-CoV Hel at a concentration of 20 μg/ml. Seven compounds exhibited inhibitory activity. One of the compounds (designated HE602) exhibited substantial inhibitory activity at 2 μg/ml. HE602 was found to strongly inhibit the polynucleotide-stimulated ATPase activity of SARS-CoV Hel with an IC50 of 6.9 μM (Figure 3A) , but barely inhibited the unstimulated ATPase activity even at a concentration of 200 μM (Figure 3B). In the helicase assay, a duplex DNA was prepared incorporating a 5′ overhang that is required for SARS-CoV helicase activity. The shorter strand of the duplex DNA incorporated a 32P label so that differential migration of the duplex or single-stranded oligonucleotide could be easily observed following polyacrylamide gel electrophoresis. It can be clearly seen that both 200 and 20 μM concentrations of HE602 inhibited the helicase activity of the SARS-CoV Hel, while 2 and 0.2 μM concentrations did not inhibit the helicase activity (Figure 3C). It was only possible to perform a qualitative assay with this system, but these results are fully consistent with the IC50 value that we measured using our quantitative ATPase assay. Furthermore, HE602 inhibited the viral plaque formation in Vero cell with an EC50 of 6 μM (Figure 3D). The effective inhibition of only the stimulated ATPase activity together with its ability to inhibit helicase activity indicates that HE602 has distinct parallels with the pharmacological profiles of inhibitors targeted against the herpes simplex virus (HSV) helicase-primase protein 23, 25.
Figure 3.

HE602 Targets SARS-CoV Hel
(A) HE602 inhibited polynucleotide-stimulated ATPase activity of SARS-CoV Hel.
(B) HE602 (200 μM) did not inhibit significantly the nonstimulated ATPase activity.
(C) Inhibition of SARS-CoV Hel helicase activity by HE602 observed by polyacrylamide electrophoresis of labeled duplex and single-stranded DNA. The positions of the 32P-labeled DNA partial duplex and released single strand are indicated to the left of the polyacrylamide gel (the star indicates the position of the 32P label).
(D) HE602 inhibited SARS-CoV plaque formation with an EC50 of 6 μM.
(E) Chemical structure of HE602 (3-Benzyloxy-N-(4-chloro-3-oxazolo[4,5-b]pyridine-2-yl-phenyl)benzamide).
Compounds Targeting S Protein-ACE2-Mediated Viral Entry
We used a SARS-CoV pseudotype virus assay to identify compounds that block S protein-ACE2-mediated cellular entry of the SARS-CoV. Briefly, 293T cells expressing ACE2 [9] were infected with SARS-CoV pseudovirus carrying the HIV-1 backbone and a luciferase reporter gene. The viral entry level was quantified by measuring luciferase activity at 72 hr postinfection. Of the 104 compounds, 18 inhibited the entry of pseudovirus into the engineered 293T cells. One compound (designated VE607) inhibited pseudovirus entry with an EC50 of 3 μM (Figure 4A) and inhibited SARS-CoV plaque formation with an EC50 of 1.6 μM (Figure 4B). The viral entry-blocking property of VE607 was confirmed by using modified plaque reduction assay that indicates if the compound is involved in interfering with the viral entry process; by excluding the inhibitor in the early stage of the infection when viral entry (0–2 hr postinfection) is crucial, we demonstrated that the protective effects of VE607 against viral plaque formation were greatly reduced (Figure 4C). The viral entry-blocking activity of VE607 is SARS-CoV specific, as it did not protect Vero cells from poliovirus plaque formation (Figure 4D).
Figure 4.

VE607 Blocks S Protein-ACE2-Mediated SARS-CoV Entry
(A) VE607 blocked SARS-CoV S protein pseudotype HIV-1 virus infection of 293T cells expressing ACE2 with an EC50 of 3 μM.
(B) VE607 inhibited SARS-CoV plaque formation with an EC50 of 1.6 μM.
(C) VE607 (10 μM) became ineffective when excluded from the plaque reduction assay at stage crucial for viral entry (0–2 hr postinfection).
(D) VE607 (50 μM) did not inhibit poliovirus plaque formation.
(E) Chemical structure of VE607 (1-[3-(2-Hydroxyl-3-piperidin-1-yl-propoxy)-phenoxy]-3-piperidin-1-ylpropan-2-ol).
Implications and Prospects
We set out to examine the feasibility of establishing a HTS-based chemical genetic platform to dissect the molecular mechanisms of SARS-CoV pathogenesis. We first used forward chemical genetics to isolate a group of small-molecule compounds perturbing the pathogenicity of SARS-CoV in Vero cells from a diverse chemical library. After screening 50,240 compounds, 104 fulfilled the criteria of being protective against SARS-CoV infection in cell culture without apparent cytotoxicity. The structures of the active compounds varied (unpublished data), suggesting that this collection of small molecules may modulate different biological pathways involved in viral propagation. To examine if this selected reservoir of biologically active compounds could be used to probe essential pathways involved in SARS-CoV infection, three pathways (viral entry, transcription/replication, and proteolytic processing of viral polyproteins) speculated to be crucial for SARS-CoV infection were chosen as testing candidates. By selecting without bias these important molecular targets/pathways and subsequent successful identification of their molecular blockers from the 104 compounds obtained by cell-based HTS, we can speculate that this reservoir of biologically active compounds covers most, if not all, biological pathways crucial to SARS-CoV infection. The specific inhibitory activities of MP576, HE602, and VE607 in various in vitro assays are summarized in Table 1 . Though the inhibitory roles of MP576, HE602, VE607 and other active compounds are indisputable, the precise mode of inhibition of these novel inhibitors is currently under investigation. In addition, one has to be cautioned that the ultimate validation of their targets will be the selection of chemical-resistant viruses and subsequent identification of genetic mutations conferring the resistant phenotype.
Table 1.
Specificity of MP576, HE602, and VE607
| Compound | SARS-CoV Mpro Inhibition (IC50) | SARS-CoV Hel Inhibition (IC50) | SARS-CoV Entry Inhibition (EC50) | Cytotoxicity on Vero Cells (TC50) |
|---|---|---|---|---|
| MP576 | 2.5 μM | >50 μM | >50 μM | >50 μM |
| HE602 | >50 μM | 6.9 μM | >50 μM | >50 μM |
| VE607 | >50 μM | >50 μM | 3 μM | >50 μM |
The highest concentration of compounds used in the assays was 50 μM. The specific conditions of each assay employed were stated in Experimental Procedures.
Since CPE in Vero cells was used as a phenotypic indicator of successful viral infection, one can expect that the compounds identified in our study could target the virus, the host, or both. Some compounds may also have multiple effects; these compounds will be selected as hits by our screening system as the sum of the biological activities of that compound leads to the protection of Vero cells from SARS-CoV-induced CPE. Screening the 104 compounds against other common RNA viruses indicates that the majority (>85%) of the compounds are SARS-CoV specific while ∼3% have broad spectrum activity against all viral candidates tested (R.Y.K. and K.-Y.Y., unpublished data). From this observation, one may speculate that the majority of the compounds selected from a SARS-CoV-based screen will target pathways that are unique to SARS-CoV infection, while a small portion of the compounds may target pathways common to other RNA viruses. Further studies on this pool of compounds should lead to better understanding of their mode of actions and the biological processes involved in viral infections.
While the classical genetic manipulations of the SARS-CoV are still at pioneering stage [8] and usually require prior knowledge of the genome, HTS-based chemical genetics offers an alternative approach to rapidly and systematically perturb the biological pathways involved in viral pathogenesis. As cell-permeable small molecules can rapidly interfere with the functions of their targets, compounds isolated from our chemical library should compose a biologically relevant platform for the dissection of the dynamic cellular processes involved in SARS-CoV infection. Furthermore, since no biologically active small-molecule inhibitor designed specifically for SARS-CoV Mpro, Hel, or S protein-ACE2-mediated entry has been described thus far, lead optimization of MP576, HE602, and VE607 should yield promising compounds with far superior anti-SARS-CoV activity.
Significance
Viral infection involves complex mechanisms and interactions of both the virus and the host. Phenotype-based screen in conjunction with high-throughput screening (HTS) technologies provides a biologically relevant platform for the investigation of various important cellular processes involved in viral infection. Based on the concept of chemical genetics, we have established a HTS platform to identify small-molecule compounds that will perturb the pathogenic pathways of SARS-CoV in Vero cells. After screening 50,240 structurally diverse small-molecule compounds against SARS-CoV infection in a cellular model, we have identified 104 compounds with potent anti-SARS-CoV activities. Of the 104 compounds isolated, 2 target the SARS-CoV main protease (Mpro), 7 target helicase (Hel), and 18 target spike (S) protein-angiotensin-converting enzyme 2 (ACE2)-mediated viral entry. Three selected compounds, MP576, HE602, and VE607, targeting SARS-CoV Mpro, Hel and viral entry, respectively, exhibited potent antiviral activities in cell-based assays and comparable inhibitory activities in target-specific in vitro assays. The successful identification of novel small molecules targeting SARS-CoV Mpro, Hel, and viral entry, demonstrates that compounds isolated from a phenotype-based screen of a large diverse chemical library provide a reservoir of biologically active small molecules perturbing the pathogenic pathways of the virus. Our study validates chemical genetics as a rapid and effective approach to tackle emerging diseases such as SARS and provides a reservoir of novel biologically active small molecules targeting major processes involved in SARS-CoV infection.
Experimental Procedures
Phenotype-Based HTS
The compounds were assayed at a final concentration of 20 μg/ml in 384-well microtitre plates (Greiner bio-one). Vero cells were seeded at 5 × 103 cells per well in complete Eagle's minimal essential medium (EMEM) (Invitrogen) supplemented with 5% heat-inactivated fetal bovine serum (FBS) (Invitrogen), with or without the addition of chemicals. One hundred TCID50 (50% tissue-culture infectious dose) of SARS-CoV strain HKU39849 was added subsequently to each well. Assay plates were incubated at 37°C in 5% CO2 for 96 hr. CPE of the infected cells were scored 96 hr postinfection using a Leica DMIL inverted microscope equipped with DC300F digital imaging system (Leica Microsystems). Experiments were carried out in duplicates. Compounds that exerted full protection (no CPE observed) were selected as hits. The cytotoxicity of selected compounds was determined by MTT (3-[4,5-dimethylth-iazol-2-yl]-2,5-diphenyltetrazolium bromide) assay (Roche) according to manufacturer's instructions.
Virus Plaque Reduction Assay
Twenty-four-well tissue culture plates (TPP, Switzerland) with a confluent monolayer of Vero cells (1 × 105 cells per well) in EMEM with 1% FBS were prepared. One hundred plaque forming units (PFU) of SARS-CoV were added to each well with or without the addition of chemicals. After being incubated for 2 hr at 37°C with 5% CO2, cell culture media and unbound viral particles were aspirated and 1 ml of overlay (1% low-melting agarose in EMEM with 1% FBS and appropriate concentrations of inhibitors) was added to each well immediately. Plates were further incubated for 48 hr under identical conditions. Cells were fixed by adding 1 ml of 10% formaldehyde. The agarose plugs were removed subsequently and cells stained with 0.5% crystal violet in 70% methanol and the viral plaques counted. The poliovirus (type 1) used in the study was a clinical isolate from a patient in Queen Mary Hospital, Hong Kong. SigmaPlot 8.0 (SPSS) was used for statistical analysis and graph plotting. Experiments were carried out in quadruplicate and repeated twice. Results are expressed as a percentage of controls in the absence of inhibitors. The mean value is shown with standard deviation (SD). Dose-response data are best fit to the logistic equation.
Purification of SARS-CoV Mpro and HPLC-Based Cleavage Assay
The coding sequence of SARS-CoV Mpro was cloned from SARS-CoV strain HKU39849 and inserted into plasmid pET28b (Novagen) and expressed as a His-tag fusion protein in E. coli BL21 cells (Novagen). Culture was induced with 0.5 mM isopropyl-β-D-thiogalactoside (IPTG) for 4 hr and cell pellet sonicated subsequently. After affinity purification by HiTrap Chelating column and cleaved with factor Xa (Amersham Biosciences) to release the N-terminal His tag, the authentic SARS-CoV Mpro was further purified using anion-exchange chromatography. Cleavage assays were carried out at 25°C in buffer containing 20 mM Tris-HCl (pH 7.3), 150 mM NaCl, 1% dimethyl sulphoxide (DMSO), with or without inhibitors. 200 nM purified SARS-CoV Mpro was preincubated with the buffer for 30 min at 25°C, and a synthetic substrate H2N-TSAVLQSGFRKW-COOH (SynPep) was added to a final concentration of 500 μM to initiate the reaction. The cleavage products were resolved by HPLC using a SOURCE 5RPC column (2.1 × 150 mm) (Amersham Biosciences) with a 20 min linear gradient of 10%–30% acetonitrile in 0.1% trifluoroacetic acid (TFA). The absorbance was determined at 215 or 280 nm, and peak areas were integrated to quantify the cleavage products. The initial rates of cleavage were determined under the condition that 5%–10% of the total substrate was cleaved. SigmaPlot 8.0 (SPSS) was used for statistical analysis and graph plotting. Experiments were carried out in triplicate and repeated twice. Results are expressed as a percentage of controls in the absence of inhibitors. The mean value is shown with SD. Dose-response data are best fit to the logistic equation.
Molecular Docking
The atomic coordinates of SARS-CoV Mpro were downloaded from Protein Data bank (PDB ID code 1UK4). The three-dimensional structure of compound MP576 was generated by Chem3D (CambridgeSoft). Hydrogen atoms were assigned using InsightII (Accelrys), and partial charge and potential were assigned by CHARMM force field. Autodock3.0.5 Lamarckian Genetic Algorithm [26] and Cerius2 LigandFit were used for docking. The binding free energy was obtained using Autodock3.0.5 [26]. The Sybyl Mol2 files for Autodock were prepared using InsightII. The Autodock default parameters were employed for docking, except for 0.35 Å grid point spacing. The docking energy grid 17.5Å3 was used for the compound, and the grids were centered at the active site.
HNE Activity Assay
200 nM of HNE (Calbiochem, CA) was preincubated at 25°C in 75 μl of 20 mM Tris-HCl, pH 7.3, 150 mM NaCl, for 30 min in the presence or absence of 100 μM of MP576. Azocasein (Sigma) was added to the mixture to a final concentration of 0.5% in 100 μl and incubated at 25°C for 60 min. The reaction was stopped by the addition of 250 μl 6% TFA. The mixture was centrifuged at 5000 × g for 15 min at 25°C. The absorbance at 366 nm was measure using a U-2800 spectrophotometer (Hitachi). Rate of cleavage was calculated according to published report [27]. SigmaPlot 8.0 (SPSS) was used for statistical analysis and graph plotting. Experiments were carried out in triplicate and repeated twice. Results are expressed as a percentage of controls in the absence of inhibitors. The mean value is shown with SD.
SARS-CoV Hel ATPase and Helicase Assays
SARS-CoV Hel was cloned and purified as described previously [24], incorporating an N-terminal His-T7 tag derived from pET32a (Novagen). ATPase assays were performed using a phosphomolybdate-malachite green assay to measure phosphate released as described previously [24]. dT24 at a concentration of 200 nM was used in the polynucleotide stimulated ATPase assay. For the helicase assay, 32P-labeled “released” oligo (5′-GGTGCAGCCGCAGCGGTGCTCG-3′) was thermally annealed to oligo “5T20” (5′-TTTTTTTTTTTTTTTTTTTTCGAGCACCGCTGCGGCTGCACC-3′) in buffer (25 mM HEPES, pH 7.4, 1 mM EDTA, 20 mM NaCl) by heating to 95°C for 5 min, followed by cooling to 20°C at 0.5°C–1°C/min. Purified SARS-CoV Hel protein (0.2 pmol) in 42 μl of helicase buffer (25 mM HEPES, pH 7.4, 10% glycerol, 1 mM DTT, 5 mM magnesium acetate) was incubated at 20°C with 1 μl of either DMSO, or inhibitor in DMSO (to the appropriate final concentration), for 5 min, then 32P-labeled d5T partial DNA duplex was added (0.2 pmol, 3.9 ng). The reaction was initiated by the addition of ATP to 0.2 mM (where applicable) and was incubated at 20°C for 6 min before quenching with 12 μl of SDS-loading buffer (2.5% SDS, 40 mM EDTA, 0.1% xylene cyanol, 0.05% bromophenol blue, 15% w/v Ficoll). 12 μl aliquots were immediately resolved on 15% polyacrylamide gels in 0.5× TBE (45 mM Tris base, 5 mM EDTA, 45 mM boric acid) at 20°C. For the positive control, 0.2 pmol of 32P-labeled 5T20 partial DNA duplex was denatured by heating to 95°C for 5 min, then flash cooling in ice water. Gels were analyzed by Phosphor-Imaging on a Storm 860 (Molecular Dynamics), using ImageQuaNT software. SigmaPlot 8.0 (SPSS) was used for statistical analysis and graph plotting. Experiments were carried out in triplicate and repeated twice. Results are expressed as a percentage of controls in the absence of inhibitors. The mean value is shown with SD. Dose-response data are best fit to the logistic equation.
Pseudotype Virus Entry Assay
SARS-CoV full-length S protein was used to pseudotype HIV-1 backbone carrying a luciferase reporter gene. The pseudotype virus was generated by cotransfecting 293T cells with an optimized S gene expression plasmid and a vector carrying HIV-1NL4-3Luc+ genome (L.Z. and D.D.H., unpublished data). The pseudotype virus was subsequently used to infect 293T ACE2 cells. When the inhibition assay was performed, the pseudotype virus was first incubated with the inhibitors for 30 min at 37°C before addition to the target cells. The infected cells were cultured for an additional 72 hr before measurement of luciferase activity. SigmaPlot 8.0 (SPSS) was used for statistical analysis and graph plotting. Experiments were carried out in triplicate and repeated twice. Results are expressed as a percentage of controls in the absence of inhibitors. The mean value is shown with SD. Dose-response data are best fit to the logistic equation.
Acknowledgements
We thank Julian Davies and Robert Shapiro for helpful discussions and critical reading of the manuscript. We also thank Jason Madar for database construction and maintenance. We are in debt to two anonymous reviewers for their valuable comments and suggestions. The work was supported by the the Vice-Chancellor SARS Fund, the HKU SARS Donation Fund, the HKU seed grant for basic research, the Infectious Disease Fund (Mr. William Benter), and the Research Fund for the Control of Infectious Diseases.
Published: September 17, 2004
References
- 1.World Health Organization (WHO). (www.who.int/csr/sars/en/).
- 2.Peiris J.S., Lai S.T., Poon L.L., Guan Y., Yam L.Y., Lim W., Nicholls J., Yee W.K., Yan W.W., Cheung M.T. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet. 2003;361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S., Urbani C., Comer J.A., Lim W. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 4.Drosten C., Gunther S., Preiser W., van der Werf S., Brodt H.R., Becker S., Rabenau H., Panning M., Kolesnikova L., Fouchier R.A. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 5.Rota P.A., Oberste M.S., Monroe S.S., Nix W.A., Campagnoli R., Icenogle J.P., Penaranda S., Bankamp B., Maher K., Chen M.H. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 6.Marra M.A., Jones S.J., Astell C.R., Holt R.A., Brooks-Wilson A., Butterfield Y.S., Khattra J., Asano J.K., Barber S.A., Chan S.Y. The genome sequence of the SARS-associated coronavirus. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 7.Zeng F.Y., Chan C.W., Chan M.N., Chen J.D., Chow K.Y., Hon C.C., Hui K.H., Li J., Li V.Y., Wang C.Y. The complete genome sequence of severe acute respiratory syndrome coronavirus strain HKU-39849 (HK-39) Exp. Biol. Med. (Maywood) 2003;228:866–873. doi: 10.1177/15353702-0322807-13. [DOI] [PubMed] [Google Scholar]
- 8.Yount B., Curtis K.M., Fritz E.A., Hensley L.E., Jahrling P.B., Prentice E., Denison M.R., Geisbert T.W., Baric R.S. Reverse genetics with a full-length infectious cDNA of severe acute respiratory syndrome coronavirus. Proc. Natl. Acad. Sci. USA. 2003;100:12995–13000. doi: 10.1073/pnas.1735582100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li W., Moore M.J., Vasilieva N., Sui J., Wong S.K., Berne M.A., Somasundaran M., Sullivan J.L., Luzuriaga K., Greenough T.C. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mayer T.U., Kapoor T.M., Haggarty S.J., King R.W., Schreiber S.L., Mitchison T.J. Small molecule inhibitor of mitotic spindle bipolarity identified in a phenotype-based screen. Science. 1999;286:971–974. doi: 10.1126/science.286.5441.971. [DOI] [PubMed] [Google Scholar]
- 11.Peterson R.T., Link B.A., Dowling J.E., Schreiber S.L. Small molecule developmental screens reveal the logic and timing of vertebrate development. Proc. Natl. Acad. Sci. USA. 2000;97:12965–12969. doi: 10.1073/pnas.97.24.12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kao R.Y., Jenkins J.L., Olson K.A., Key M.E., Fett J.W., Shapiro R. A small-molecule inhibitor of the ribonucleolytic activity of human angiogenin that possesses antitumor activity. Proc. Natl. Acad. Sci. USA. 2002;99:10066–10071. doi: 10.1073/pnas.152342999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuruvilla F.G., Shamji A.F., Sternson S.M., Hergenrother P.J., Schreiber S.L. Dissecting glucose signalling with diversity-oriented synthesis and small-molecule microarrays. Nature. 2002;416:653–657. doi: 10.1038/416653a. [DOI] [PubMed] [Google Scholar]
- 14.Mitchison T.J. Towards a pharmacological genetics. Chem. Biol. 1994;1:3–6. doi: 10.1016/1074-5521(94)90034-5. [DOI] [PubMed] [Google Scholar]
- 15.Schreiber S.L. Chemical genetics resulting from a passion for synthetic organic chemistry. Bioorg. Med. Chem. 1998;6:1127–1152. doi: 10.1016/s0968-0896(98)00126-6. [DOI] [PubMed] [Google Scholar]
- 16.Stockwell B.R. Chemical genetics: ligand-based discovery of gene function. Nat. Rev. Genet. 2000;1:116–125. doi: 10.1038/35038557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thiel V., Ivanov K.A., Putics A., Hertzig T., Schelle B., Bayer S., Weissbrich B., Snijder E.J., Rabenau H., Doerr H.W. Mechanisms and enzymes involved in SARS coronavirus genome expression. J. Gen. Virol. 2003;84:2305–2315. doi: 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- 18.Ng M.L., Tan S.H., See E.E., Ooi E.E., Ling A.E. Early events of SARS coronavirus infection in vero cells. J. Med. Virol. 2003;71:323–331. doi: 10.1002/jmv.10499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ng M.L., Tan S.H., See E.E., Ooi E.E., Ling A.E. Proliferative growth of SARS coronavirus in Vero E6 cells. J. Gen. Virol. 2003;84:3291–3303. doi: 10.1099/vir.0.19505-0. [DOI] [PubMed] [Google Scholar]
- 20.Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 21.Yan L., Velikanov M., Flook P., Zheng W., Szalma S., Kahn S. Assessment of putative protein targets derived from the SARS genome. FEBS Lett. 2003;554:257–263. doi: 10.1016/S0014-5793(03)01115-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z., Sun L., Mo L., Ye S., Pang H. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. USA. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crute J.J., Grygon C.A., Hargrave K.D., Simoneau B., Faucher A.M., Bolger G., Kibler P., Liuzzi M., Cordingley M.G. Herpes simplex virus helicase-primase inhibitors are active in animal models of human disease. Nat. Med. 2002;8:386–391. doi: 10.1038/nm0402-386. [DOI] [PubMed] [Google Scholar]
- 24.Tanner J.A., Watt R.M., Chai Y.B., Lu L.Y., Lin M.C., Peiris J.S., Poon L.L., Kung H.F., Huang J.D. The severe acute respiratory syndrome (SARS) coronavirus NTPase/helicase belongs to a distinct class of 5′ to 3′ viral helicases. J. Biol. Chem. 2003;278:39578–39582. doi: 10.1074/jbc.C300328200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kleymann G., Fischer R., Betz U.A., Hendrix M., Bender W., Schneider U., Handke G., Eckenberg P., Hewlett G., Pevzner V. New helicase-primase inhibitors as drug candidates for the treatment of herpes simplex disease. Nat. Med. 2002;8:392–398. doi: 10.1038/nm0402-392. [DOI] [PubMed] [Google Scholar]
- 26.Morris G.M., Goodsell D.S., Halliday R.S., Huey R., Hart W.E., Belew R.K., Olson A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998;19:1639–1662. [Google Scholar]
- 27.Hellberg A., Nowak N., Leippe M., Tannich E., Bruchhaus I. Recombinant expression and purification of an enzymatically active cysteine proteinase of the protozoan parasite Entamoeba histolytica. Protein Expr. Purif. 2002;24:131–137. doi: 10.1006/prep.2001.1548. [DOI] [PubMed] [Google Scholar]