

Abstract
Viruses, which are among the simplest infective pathogens, can produce characteristic endocrine manifestations in infected patients. In addition to the classic modification of the host endocrine system by either direct or indirect destruction of the endocrine organs and/or effects exerted by systemic production of inflammatory and/or stress mediators, recent progress in molecular virology and endocrinology has revealed that virus-encoded molecules might alter the host endocrine-signaling systems by affecting extracellular and/or intracellular signal transduction and hormone sensitivity of host target tissues. Here, we provide a brief overview of such viral-mediated modulation of host endocrine signaling systems. We propose that virus-encoded molecules and the signaling systems they influence are potential therapeutic targets for the treatment of disorders that are associated with some viral infections.
Introduction
Viruses are among the simplest infective pathogens on earth. Viruses cause a variety of pathological conditions of occasionally pandemic proportions, and constitute potential major threats to the human population [1]. Even after the development of vaccines, which have been tremendously effective in controlling many viral infections, viruses are still among the most common human pathogens, sometimes causing life-threatening diseases [1]. The latter is evident from recent epidemics, such as acquired immunodeficiency syndrome (AIDS), caused by the human immunodeficiency virus type-1 (HIV-1), and severe acute respiratory syndrome (SARS), caused by the SARS corona virus 2, 3.
Replication and survival of viruses inside host cells are dependent on the cellular machinery of the host. Viruses might usurp the regulation of and change host-cell functions for their benefit. Indeed, through molecules encoded by viral genetic material, whose expression is synchronized with specific phases of the viral life cycle, viruses increase the chance of their own replication and survival. Virus-induced alterations of host-cell functions cause a broad spectrum of cellular damage, from a transient, mild change of function to neoplastic transformation, apoptosis and necrosis. Recent research has revealed that viruses, through the molecules they encode, modulate endocrine signaling systems in the host to cause characteristic changes in both endocrine organs and hormone target tissues.
Here, we provide a brief overview of recent progress in understanding virus-induced modifications of endocrine signaling systems and of the resultant pathological states in the host. On the basis of this understanding, potential new therapeutic targets for the development of anti-viral disease agents are envisioned.
Virus-induced modifications of host endocrine systems: the classic view
Generally, entry of viruses into and infection of host tissues evokes several levels of host reactions that might be associated with pathological manifestations. Thus, infection of host tissues might damage infected organs and tissues directly. By contrast, viruses themselves and tissues that are infected with viruses might activate the host immune reaction, leading to the development of inflammatory lesions and/or the destruction of infected tissues [4]. In addition, host immune responses induced against viruses and infected tissues might damage uninfected tissues, partly because of the similarity between viral antigens and those of host cells (molecular mimicry), and because of a systemic, generalized inflammatory reaction that results in dysfunction of multiple organs [5].
Viral-induced inflammation and viremia also activate the systemic stress response, in which the hypothalamic–pituitary–adrenal (HPA) axis has a major role. Secreted glucocorticoids, the end-effectors of the HPA axis, protect peripheral organs from inflammation-induced tissue damage by suppressing an overshoot of the host immune reaction and by rendering tissues resistant to toxic inflammatory agents 6, 7, 8. Thus, viral infections classically induce the host reactions shown in Box 1 and Figure 1 . Examples of classic host responses to viral infections are also described in Table 1 .
Box 1. Virus-induced alterations of the host endocrine system.
-
•
Activation of the HPA axis/stress system indirectly, as a result of a general inflammatory response to the systemic viral infection and secretion of mediators of inflammation with stress system-stimulating activity.
-
•
Damage of virus-infected endocrine cells by viral replication, proliferation and assembly.
-
•
Damage of virus-infected endocrine organs by activation of the immune reaction against these organs.
-
•
Damage against uninfected endocrine organs through an autoimmune mechanism.
-
•
Alteration of hormonal activity and/or hormone secretion by viral gene products.
Figure 1.
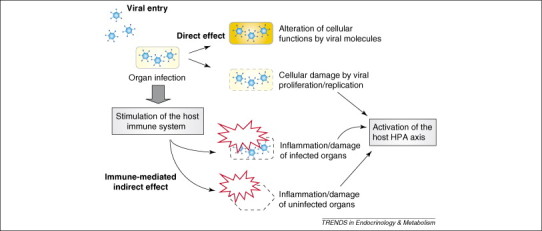
Virus-mediated modification of host endocrine organs. Viruses damage infected cells directly by proliferating inside cells, and can modulate cellular functions through molecules that are encoded by their genetic material. Viral infection might also trigger an inflammatory response, with consequent damage of both infected and uninfected tissues. Virus-induced tissue inflammation might increase the production of systemic mediators of inflammation, which, in turn, might stimulate the host HPA axis/stress system.
Table 1.
Viruses that influence host endocrine functions
| Mode of action | Virus type | Virus name | Affected organs and tissues | Diseases and symptoms | Refs |
|---|---|---|---|---|---|
| Activation of the HPA axis/stress system | Any viruses that cause systemic viremia and inflammation | Entire body, including the HPA axis/stress system | Sickness syndrome manifestations observed during a systemic inflammatory response, such as fever, malaise and appetite loss | [8] | |
| Direct destruction of endocrine organs | ssRNA virus | Mumps virus | Testis, ovary | Impairment of fertility (rare), low testosterone levels | [60] |
| ssRNA virus | Hantaan and Puumala viruses (hemorrhagic fever with renal syndrome) | Pituitary gland | Hypopituitarism | [61] | |
| dsDNA virus | Herpes simplex virus 1 and 2 (neonatal infection) | Adrenal gland, pituitary gland | Adrenal and pituitary inflammation | [62] | |
| ssRNA retrovirus | HIV-1 | Adrenal gland, pituitary gland, thyroid gland, testis, ovary | Adrenal insufficiency, hypopituitarism (rare) | [63] | |
| Autoimmune-mediated organ destruction | ssRNA virus | Hepatitis C virus (HCV) | Thyroid gland | Chronic thyroiditis (with interferon γ) | [64] |
| ssRNA virus | Rubella virus | Pancreatic islet cells, pituitary gland, adrenal gland, thyroid gland | Multiple endocrine organ failure | [65] | |
| ssRNA viruses | Mumps, Rubella and Coxsackie B viruses | Pancreatic islet cells | Insulin-dependent diabetes mellitus | [66] | |
| ssRNA viruses | Influenza, Coxsackie B virus, enteroviruses | Thyroid gland | Subacute thyroiditis | [67] | |
| dsDNA virus | Epstein Barr virus | Pancreatic islet cells | Insulin-dependent diabetes mellitus | [66] | |
Modification of host endocrine signaling systems by virus-encoded molecules: new mechanisms
Recent accumulation of knowledge about eukaryotic and viral genes has expanded our understanding of the roles of both host- and virus-encoded molecules in the regulation of gene expression and function. Numerous cell-surface molecules and nuclear hormone receptors (NRs) that transduce the signals of circulating hormones in the cytoplasm and/or the nucleus have been identified and their biological functions have been elucidated. In parallel, it has become evident that some viruses cause pathological changes because they encode molecules that affect host signaling pathways directly 9, 10 and these actions sometimes cause serious clinical problems (Table 2 ) 10, 11. Indeed, viruses have developed some of their unique, highly sophisticated molecules through the mutational selection of protein isoforms that increase the chance of their replication and/or survival inside infected cells, and their propagation from cell-to-cell and host-to-host.
Table 2.
Viruses and viral proteins that might alter the host endocrine signaling systems
| Virus type | Virus name | Virus-encoded molecule | Affected organs or tissues | Diseases and manifestations | Refs |
|---|---|---|---|---|---|
| ssRNA virus | Mumps virus | Nucleocapsid transcript (MVNT) | Bone and osteoclast | Paget's disease of bone (hypersensitivity to vitamin D) | [40] |
| ssRNA virus | SARS corona virus | Lung | Hypersensitivity to ANG II | [3] | |
| Lymphocytic choriomeningitis virus (LCMV) | Not determined | Pituitary gland (somatotroph) | Dwarfism in the rat (growth hormone/Pit-1) | [68] | |
| dsDNA virus | Adenovirus | E1A | Respiratory tract, stomach, intestine, urinary bladder | Possible hyposensitivity to steroid hormones, inhibition of p300/CBP and CtBP activity | 33, 45 |
| dsDNA virus | Molluscum contagiosum virus | MC013L | Skin | Molluscum contagiosum | [69] |
| Hyposensitivity to glucocorticoids and vitamin D | |||||
| ssRNA retrovirus | HIV-1 | Vpr and Tat | T cells, fat, liver and muscle | Hypersensitivity to glucocorticoids, immunosuppression, muscle wasting, lipodystrophy, insulin resistance, dyslipidemia, diabetes mellitus type 2 | [10] |
| Kaposi sarcoma | |||||
| gp120 | CNS | AIDS-related dementia and growth retardation | 12, 13 | ||
| Nef | Thyroid gland | Grave's disease | [15] | ||
| ssRNA retrovirus | HTLV-1 | Tax | T cells | Hypercalcemia (PTHrP) | [11] |
These viral molecules regularly influence crucial steps in host signaling systems and, thus, dramatically change cellular functions towards conditions that are beneficial to the replication, survival and/or propagation of the viruses that encode them. Sometimes, these viral molecules mimic functions of host proteins. For example, HIV-1-encoded gp120 molecules, which are located on the surface of the viral particle and have a major role in the entry of viruses into target cells, demonstrate amino acid sequence similarity to the growth hormone-releasing hormone (GHRH) receptor of the host and suppress the activation of this receptor by GHRH. This phenomenon might contribute to growth retardation of HIV-1-infected children [12]. This viral molecule is also similar to vasoactive intestinal peptide, which interacts with CD4, a receptor for HIV-1, to facilitate its entry into T cells 13, 14. The amino acid sequence of the HIV-1 Nef protein resembles a portion of the thyroid-stimulating hormone receptor, and this cross-reactivity might contribute to the development of Grave's disease in HIV-1-infected patients [15].
In addition to their extracellular actions, many viral molecules act inside infected cells to modulate intracellular host signaling systems, including transcriptional regulation of target genes by hormones. These molecules are intended primarily to benefit viral replication, but, frequently, they alter the intracellular milieu, disturbing cellular functions as a ‘side-effect’ and causing specific, recognizable, pathological conditions 10, 16. In the following sections, examples of extracellular and intracellular modifications of host endocrine signaling systems caused by viral molecules are discussed.
PTH-related polypeptide (PTHrP)-mediated hypercalcemia and the human T-cell leukemia (lymphotrophic) virus type-1 (HTLV-1)
Human T-cell leukemia (lymphotrophic) virus type 1 (HTLV-1), the single-stranded RNA virus of the Retroviridae family that specifically infects CD4-positive T cells, is a causative agent of adult T-cell leukemia (ATL) and HTLV-associated myelopathy [11]. ATL patients infected with HTLV-1 may also develop hypercalcemia because of aberrant expression of parathyroid hormone (PTH)-related protein (PTHrP) [17]. PTHrP is a 141 amino acid protein that shares significant N-terminal sequence homology with PTH [18]. In humans, PTHrP is present in many normal and neoplastic tissues, whereas PTH is present only in the parathyroid gland [18]. Leukemic cells from ATL patients, as well as lymphocytes from asymptomatic HTLV-1-positive carriers, contain high levels of PTHrP; the secreted protein exerts biological effects that are similar to those of PTH because PTHrP binds to the PTH receptor and stimulates downstream cellular events [18]. Hence, PTHrP, like PTH, increases serum Ca2+ concentrations by stimulating bone turnover and enhancing Ca2+ absorption from the kidney and the gut [18].
Accumulating evidence indicates that the HTLV-1-encoded Tax protein stimulates PTHrP synthesis in HTLV-1-infected lymphocytes by activating the PTHrP promoter [11]. Tax is a nuclear phosphoprotein of either 37 or 40 kDa that is required for transactivation of the HTLV-1 LTR and the transformation of T cells [19]. Tax does not bind DNA directly but transactivates numerous cellular genes by interacting physically with host transcription factors [19]. Tax appears to stimulate the promoter activity of the gene that encodes PTHrP by communicating with multiple transcription factors, including activator protein 1 (AP1), AP2, Est1 and SP1 [11]. Tax forms a ternary complex with Est1 and SP1, and their binding to the PTHrP promoter strongly stimulates its transcriptional activity [20]. Aggravated hypercalcemia, however, is observed most often in end-stage ATL when Tax expression is relatively low [11]. Mice infected experimentally with Tax-defective HTLV-1 have high levels of PTHrP and develop hypercalcemia [21]. Thus, as yet unknown viral factors appear to contribute to the development of HTLV-1-associated hypercalcemia in addition to Tax.
Alteration of glucocorticoid and insulin sensitivity in HIV-1-infection: implications for AIDS-related lipodystrophy and insulin-resistance syndrome
HIV-1 infection is associated with profound immunosuppression, muscle wasting and, occasionally, the AIDS-related lipodystrophy and insulin-resistance syndrome. The latter is characterized by a sometimes striking phenotype and marked metabolic disturbances, especially after addition of potent anti-HIV-1 treatment regimens, which have significantly improved patients’ life expectancy and allowed sufficient time for the syndrome to develop [10]. Patients with this syndrome have a combination of regional lipodystrophy and characteristic redistribution of adipose tissue with accumulation of visceral, breast and nuchal fat and loss of subcutaneous fat 10, 22. The AIDS-related lipodystrophy and insulin-resistance syndrome is accompanied by profound dyslipidemia and either carbohydrate intolerance or overt diabetes mellitus [10]. As the phenotypic and metabolic manifestations of the AIDS-related lipodystrophy and insulin-resistance syndrome are reminiscent of the typical phenotype of chronic glucocorticoid excess or Cushing syndrome, this syndrome was referred to initially as a Pseudo-Cushing state [10].
Anti-HIV-1 drugs, such as nucleotide reverse transcriptase and protease inhibitors, appear to contribute to the development of this syndrome, based on the evidence that the majority of AIDS patients develop this syndrome after taking such compounds 22, 23. However, some patients develop the characteristic features of the syndrome before treatment, which indicates that either HIV-1 infection itself induces these pathological changes or that it increases the vulnerability of patients to the facilitation of expression of this syndrome by anti-viral drugs 24, 25. In this context, anti-HIV-1 drugs might exacerbate already present, smoldering or subclinical lipodystrophy and insulin-resistance that is masked by illness-associated wasting, in agreement with the evidence that the drug effects listed above do not explain the complete clinical picture of AIDS-related lipodystrophy and insulin resistance 10, 26.
Because the clinical picture of AIDS-related lipodystrophy and insulin-resistance syndrome overlaps with that of Cushing syndrome, the adrenal function of patients with these syndromes has been examined, however, their HPA axis is normal [27]. Moreover, glucocorticoid receptors (GRs) are present at normal concentrations on peripheral leukocytes and the affinity of these receptors for dexamethasone is similar to that of controls. Therefore, biochemical hypercortisolism is not likely to be a major cause of immunosuppression, muscle wasting and AIDS-related lipodystrophy and insulin resistance. Rather, either localized or tissue-specific hypersensitivity to glucocorticoids might be involved in these phenomena [10]. Furthermore, hypersensitivity to glucocorticoids might explain the increased vulnerability of AIDS patients to HHV-8 infections and the development of Kaposi sarcoma [28].
In agreement with these findings, one of the HIV-1 proteins, Vpr, which is a 96-amino acid virion-associated accessory protein that has multiple functions (including influencing transcriptional activity and arresting the cell cycle), increases the effects of GR stimulation by several fold, functioning as a nuclear receptor coactivator in cooperation with a host cell coactivator complex containing p300 or its homolog CREB-binding protein (CBP) 29, 30, 31, 32. These proteins regulate many signal transduction cascades mediated by transcription factors, including NRs, CRE-binding protein (CREB), AP-1, nuclear factor-κB (NF-–B) and the signal transducers and activators of transcription [33]. p300 and CBP have intrinsic histone acetyltransferase activity and attract other histone acetyltransferase coactivators, such as the p300/CBP-interacting factor (p/CAF) and p160-type nuclear receptor coactivator proteins to the transcription initiation site [33]. Thus, p300/CBP act as regulatory ‘platforms’ for the transcription of numerous host genes, regulating transcriptional activity of many trans-acting factors and NRs (Figure 2 ). Vpr contains a NR coactivator box. This is similar to those on p160 and p300/CBP NR coactivators, potentiating the effect of ligand-bound GR and p160. Vpr penetrates the cell membrane easily to exert its biological effects even when added in the media surrounding the cells [34], thus, its effects might extend to tissues that are not infected with HIV-1.
Figure 2.
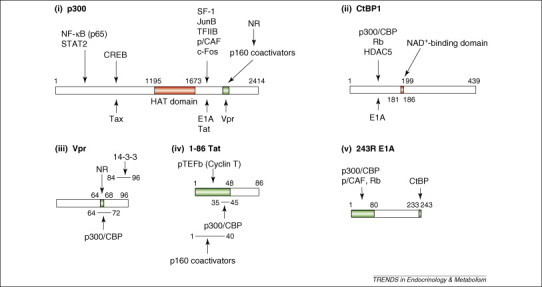
Linearized diagrams of (i) p300, (ii) CtBP1, (iii) Vpr, (iv) Tat, and (v) E1A and their mutual-interaction domains. Numerous transcription factors, transcriptional regulators and viral molecules bind the transcriptional coactivator p300 or its homolog CBP. Binding sites of p160 NR coactivators and Vpr overlap, and both bind NRs and p300 or CBP. Thus, Vpr mimics the host p160 NR coactivators and enhances NR transcriptional activity. p300/CBP facilitates attraction of transcription factors, cofactors and general transcription complexes by loosening the histone–DNA interaction through acetylation of histone tails by its HAT domain. The N-terminal portion of CtBP1 interacts physically with HDAC5 and Rb, which repress transcription. CtBP1 regulates interaction with its binding partners by sensing NADH levels through its NAD+-binding domain. E1A binds to the C-terminal portion of p300 and associates physically with the N-terminal portion of CtBP1 through its C-terminal end. The HAT domain of p300 and the NAD+-binding domain of CtBP1 are indicated in red. This figure is based on Refs 16, 33, 45. Abbreviations: CREB, CRE-binding protein; HAT, histone acetyltransferase; HDAC5, histone deacetylases 5; NF-κB, nuclear factor-κB; NAD, nicotinamide adenine dinucleotide; NR, nuclear hormone receptor; p/CAF, p300/CBP-associating factor; pTEFb, positive-acting transcription elongation factor b; Rb, retinoblastoma protein; SF-1, steroidogenic factor-1; STAT2, signal transducer and activator of transcription 2; TFIIB, transcription factor IIB.
Another HIV-1 accessory protein, Tat, the most potent transactivator of the HIV-1-LTR, also potentiates GR-induced transcriptional activity moderately, possibly through accumulation of the positive-acting transcription elongation factor b (pTEFb) complex, which is comprised of the cyclin-dependent kinase 9 and its partner molecule cyclin T, on glucocorticoid-responsive promoters [35]. Because Tat, like Vpr, circulates in blood and exerts its actions as either an auto/paracrine or an endocrine factor by penetrating the cell membrane [36], it is possible that Tat modulates tissue sensitivity to glucocorticoids irrespective of whether a cell is infected with HIV-1. Concomitantly with Vpr, Tat might induce tissue hypersensitivity to glucocorticoids that might contribute to viral proliferation indirectly, by suppressing activity of the host immune system and altering the host metabolic balance, both functions governed by glucocorticoids [10].
Vpr reduces tissue sensitivity to insulin by potentiating the actions of glucocorticoids as well as by modulating the transcriptional activity of insulin (Figure 3 ) 37, 38. Insulin uses the forkhead transcription factors (FoxOs) to control gene induction. Baseline, unphosphorylated FoxOs are active, reside in the nucleus and bind to their responsive sequences in the promoter region of insulin-responsive genes. By contrast, insulin activates Akt kinase, which phosphorylates specific serine and threonine residues in FoxOs to render them inactive [39]. Indeed, once FoxOs are phosphorylated at specific residues, they lose their transcriptional activity, by binding to and translocating into the cytoplasm with proteins of the 14–3-3 family [39]. We have found that Vpr moderately inhibits insulin-induced translocation of FoxO3a into the cytoplasm through inhibiting its association with 14–3-3 [37]. Based on these in vitro findings, Vpr might be a key viral factor that induces lipodystrophy, insulin resistance and hyperlipidemia by interfering with and/or modulating cellular activities, such as transactivation of NRs and insulin [26].
Figure 3.
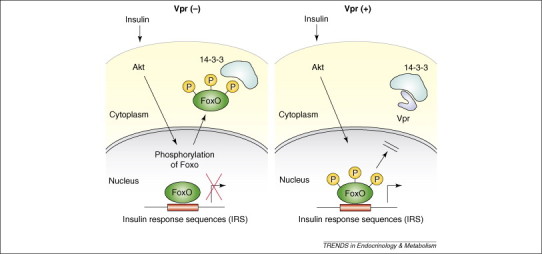
Vpr antagonizes the negative effect of insulin on FoxOs by retaining these transcription factors in the nucleus through inhibition of the interaction of FoxOs with proteins 14–3-3. Insulin induces cytoplasmic translocation of FoxO transcription factors by phosphorylating them and creating 14–3-3-binding sites. Vpr inhibits binding of 14–3-3 to phosphorylated FoxOs, thereby inhibiting the negative effect of insulin on FoxOs. These transcription factors thus remain in the nucleus and constitutively stimulate the transcriptional activity of their responsive gene promoters 26, 37, 38.
Unpublished information indicates that induction of insulin resistance by Vpr might be compounded by the ability of the viral protein to interfere with signal transduction by another NR, peroxisome proliferation receptor γ.*
Paget's disease of bone and the measles virus
Paget's disease of bone is a chronic, focal, skeletal disorder that causes bone deformities and fractures as a result of uncoupled bone remodeling [40]. The primary abnormality of this disease resides in the osteoclasts, which demonstrate increased capacity for bone resorption and hypersensitivity to vitamin D [40]. Immunocytochemical studies have shown that osteoclasts from patients with Paget's disease of bone contain paramyxoviral-like nuclear inclusions that cross-react with antibodies to measles virus, respiratory syncytial virus and canine distemper virus nucleocapsid antigen [40]. Nucleocapsid transcripts of measles virus (MVNP), the negative single-strand RNA virus of the Paramyxoviridae family, have also been detected in osteoclasts from patients with Paget's disease of bone [41]. Indeed, forced expression of MVNP in normal osteoclast precursor cells enhances formation of mature osteoclasts that possess many characteristics of those in patients with Paget's disease, including increased sensitivity to vitamin D [40]. Transgenic expression of MVNP induces bone lesions similar to those found in Paget's disease [42]. Osteoclasts that express MVNP demonstrate increased levels of TAFII-17, which is one of the components of the general transcriptional complexes that are attracted to the ligand-activated vitamin D receptor [40]. Thus, it is possible that Paget's disease of bone might be caused, in part, by the measles virus, through its encoded molecule MVNP increasing the sensitivity of osteoclasts to vitamin D.
Possible alteration of endocrine system by Adenovirus E1A protein
Adenoviruses are the double strand DNA viruses of the Adenoviridae family. This group of viruses causes illness of the respiratory system, such as common cold syndrome, pneumonia, croup and bronchitis, as well as illnesses of other organs, such as gastroenteritis, conjunctivitis and cystitis [43]. Adenoviruses encode the E1A protein, which is expressed just after infection and is necessary for the transcriptional regulation of adenovirus-encoded genes [43]. In addition to viral genes, E1A regulates the transcriptional activity of several host genes through interaction with the host transcriptional integrator p300 and its homologous molecule CBP (Figure 2) 33, 44. In an in vitro system, E1A, in contrast to Vpr, blocks the actions of glucocorticoids on the transcriptional activity of genes, producing resistance to glucocorticoids [30].
E1A also interacts with the C-terminal tail-binding protein (CtBP), which functions as a transcriptional repressor of numerous transcription factors by communicating with class II histone deacetylases and other inhibitory molecules, such as the retinoblastoma protein (Rb) (Figure 2) [45]. E1A suppresses the activity of p300/CBP and CtBP by binding to functionally crucial domains 33, 45. Although there is no supportive clinical evidence, it is possible that adenovirus changes the peripheral action of circulating hormones, such as glucocorticoids and other bioactive molecules that activate NRs and directly regulate the transcriptional activity of their target genes, ultimately contributing to the pathological states observed in adenoviral infection.
Angiotensin-converting enzyme 2 and SARS virus
In 2003, the virus that caused a newly identified illness called SARS spread rapidly worldwide, causing almost 800 deaths 3, 46. SARS starts with flu-like symptoms, such as fever, myalgia, somnolence, gastrointestinal symptoms, cough and sore throat, and eventually develops into pneumonia with severe respiratory failure 3, 46. Mechanical ventilation is required in ∼10–20% of cases and the death rate is almost 10% 3, 46. The causative virus of this severe respiratory syndrome is the SARS coronavirus (SARS-CoV), a positive single-strand RNA virus of the Coronaviridae family [47]. Subsequently, the angiotensin-converting enzyme 2 (ACE2) was identified as a cellular receptor for the entry of this virus [48]. ACE2 was first cloned as a homolog of ACE in 2000 49, 50. Although it functions as a carboxypeptidase similar to ACE, it has different substrate specificity: ACE2 cleaves a single residue from angiotensin I (ANG I) and ANG II to generate the inactive forms angiotensin (1–9) and angiotensin (1–7), respectively, whereas ACE removes dipeptides from the C-terminus of ANG I to produce potent, bioactive ANG II (Figure 4 ) [51].
Figure 4.
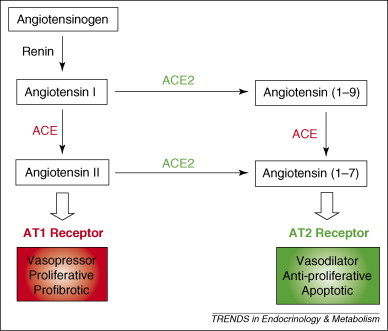
In the lung, ACE converts angiotensin I (ANG I) into ANG II, which acts as a vasopressor and a mitogen for smooth muscle cells and fibroblasts through the angiotensin 1 (AT1) receptor. By contrast, ACE2 [53] converts ANG I and ANG II into angiotensin (1–9) and angiotensin (1–7), respectively, which cause vasodilatation and apoptosis through the AT2 receptor 3, 51.
From mouse knockout studies, ACE2 is a negative regulator of the rennin–angiotensin system that counterbalances the function of ACE [52]. ANG II is a potent stimulator of vascular constriction, Na+ uptake in the renal tubules, aldosterone secretion from the adrenal glands and vasopressin secretion from the pituitary gland [53]. ANG II also has important roles in lung physiology and pathophysiology [51]. Indeed, ACE and ANG II function as promoting factors for acute lung injury, whereas ACE2 protects against lung injury [51]. As SARS-CoV infection downregulates ACE2 expression markedly in the lung and reduces circulating concentrations of ANG II [54], it is likely that SARS-CoV contributes to severe lung disease and other systemic complications through dysregulation of the renin−angiotensin system. This is in contrast to other coronaviruses that infect the lung epithelium like SARS-CoV but cause much milder diseases [51].
Conclusions: virus-induced modulation of host signaling systems as a new therapeutic target
Vaccines, which stimulate the host-immune reaction to protect the organism from the entry of viruses, are the most effective, first-line therapeutics against viral infection. This approach has been extremely useful in controlling epidemics, such as those caused by smallpox, poliomyelitis, measles, influenza, varicella and rubella viruses [1]. However, some viruses are resistant to vaccine development by conventional approaches. For example, so far, anti-HIV-1 vaccines have not been sufficiently effective to protect from infection by this virus, despite the tremendous efforts of the scientific community. This is possibly because of the high genetic instability of HIV-1, which changes the immunogenicity 55, 56.
Anti-viral agents are used because vaccines are either not available or have failed to protect us. Expansion of our understanding of virus-induced modifications of the host signaling systems, such as those discussed above, might prove highly beneficial for the development of new vaccines and novel anti-viral agents, based on knowledge of the potential indispensability of specific viral molecules for the entry, replication, and intracellular and extracellular survival and propagation of these viruses. Indeed, this approach has been followed successfully in the development of nucleoside analogs, such as acyclovir for the herpes simplex virus and ganciclovir for Cytomegalovirus, which specifically block transcription promoted by virus-encoded and virus-specific DNA polymerases [57]. Oseltamivir, a neuraminidase inhibitor that blocks the influenza virus-encoded neuraminidase, is effective in controlling infection by blocking the release of newly replicated viruses from the infected cells, a step in which this enzyme has a major role [58]. Thus, either vaccines or chemical compounds, including nucleotides (e.g. antisense DNA and siRNA) that inhibit the expression and/or function of specific viral molecules, are promising approaches for the treatment of viral illnesses. Indeed, vaccines against HIV-1 Tat are under development with promising effects [59]. We envision that future research to further elucidate the effects of viral molecules on the host endocrine signaling systems will improve our ability to intercept viral infection and prevent virus infection-associated diseases that affect endocrine systems. and endocrine target organs.
Acknowledgements
This article was funded by the Intramural Research Program of the National Institute of Child Health and Human Development, National Institutes of Health.
Footnotes
S. Shrivastav et al. Abstract. HIV-1 Vpr binds and inhibits PPAR-γ: implications for HIV-associated insulin resistance and lipodystrophy. International Meeting of the Institute of Human Virology: a symposium on HIV/AIDS and related topics. Baltimore, September 2000.
References
- 1.Mack T. A different view of smallpox and vaccination. N. Engl. J. Med. 2003;348:460–463. doi: 10.1056/NEJMsb022994. [DOI] [PubMed] [Google Scholar]
- 2.Simon V. HIV/AIDS epidemiology, pathogenesis, prevention, and treatment. Lancet. 2006;368:489–504. doi: 10.1016/S0140-6736(06)69157-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Turner A.J. ACE2: from vasopeptidase to SARS virus receptor. Trends Pharmacol. Sci. 2004;25:291–294. doi: 10.1016/j.tips.2004.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wittek A.E., Quinnan G.V.J. Immunology of viral infections. In: Belshe R.B., editor. Textbook of Human Virology. 2nd edn. Mosby-Year Book; 1991. pp. 116–155. [Google Scholar]
- 5.Oldstone M.B. Molecular mimicry and immune-mediated diseases. FASEB J. 1998;12:1255–1265. doi: 10.1096/fasebj.12.13.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chrousos G.P. The hypothalamic-pituitary-adrenal axis and immune-mediated inflammation. N. Engl. J. Med. 1995;332:1351–1362. doi: 10.1056/NEJM199505183322008. [DOI] [PubMed] [Google Scholar]
- 7.Meduri G.U., Chrousos G.P. Effectiveness of prolonged glucocorticoid treatment in acute respiratory distress syndrome: the right drug, the right way? Crit. Care Med. 2006;34:236–238. doi: 10.1097/01.ccm.0000196088.75067.4c. [DOI] [PubMed] [Google Scholar]
- 8.Silverman M.N. Immune modulation of the hypothalamic-pituitary-adrenal (HPA) axis during viral infection. Viral Immunol. 2005;18:41–78. doi: 10.1089/vim.2005.18.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gallimore P.H., Turnell A.S. Adenovirus E1A: remodelling the host cell, a life or death experience. Oncogene. 2001;20:7824–7835. doi: 10.1038/sj.onc.1204913. [DOI] [PubMed] [Google Scholar]
- 10.Kino T. AIDS-related lipodystrophy/insulin resistance syndrome. Horm. Metab. Res. 2003;35:129–136. doi: 10.1055/s-2003-39072. [DOI] [PubMed] [Google Scholar]
- 11.Prager D. Hypercalcemia, parathyroid hormone-related protein expression and human T-cell leukemia virus infection. Leuk. Lymphoma. 1994;14:395–400. doi: 10.3109/10428199409049695. [DOI] [PubMed] [Google Scholar]
- 12.Mulroney S.E. HIV gp120 inhibits the somatotropic axis: a possible GH-releasing hormone receptor mechanism for the pathogenesis of AIDS wasting. Proc. Natl. Acad. Sci. U. S. A. 1998;95:1927–1932. doi: 10.1073/pnas.95.4.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lafont V. Perturbation of in vitro HIV pathogenic effects by peptides showing sequence similarities with the C2 conserved domain of gp120. Immunol. Lett. 1993;37:249–250. doi: 10.1016/0165-2478(93)90038-4. [DOI] [PubMed] [Google Scholar]
- 14.Sacerdote P. Vasoactive intestinal peptide 1–12: a ligand for the CD4 (T4)/human immunodeficiency virus receptor. J. Neurosci. Res. 1987;18:102–107. doi: 10.1002/jnr.490180117. [DOI] [PubMed] [Google Scholar]
- 15.Burch H.B. Nucleotide and amino acid homology between the human thyrotropin receptor and the HIV-1 Nef protein: identification and functional analysis. Biochem. Biophys. Res. Commun. 1991;181:498–505. doi: 10.1016/s0006-291x(05)81447-0. [DOI] [PubMed] [Google Scholar]
- 16.Kino T., Chrousos G.P. Glucocorticoid and mineralocorticoid resistance/hypersensitivity syndromes. J. Endocrinol. 2001;169:437–445. doi: 10.1677/joe.0.1690437. [DOI] [PubMed] [Google Scholar]
- 17.Watanabe T. Constitutive expression of parathyroid hormone-related protein gene in human T cell leukemia virus type 1 (HTLV-1) carriers and adult T cell leukemia patients that can be trans-activated by HTLV-1 tax gene. J. Exp. Med. 1990;172:759–765. doi: 10.1084/jem.172.3.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gensure R.C. Parathyroid hormone and parathyroid hormone-related peptide, and their receptors. Biochem. Biophys. Res. Commun. 2005;328:666–678. doi: 10.1016/j.bbrc.2004.11.069. [DOI] [PubMed] [Google Scholar]
- 19.Grassmann R. Molecular mechanisms of cellular transformation by HTLV-1 Tax. Oncogene. 2005;24:5976–5985. doi: 10.1038/sj.onc.1208978. [DOI] [PubMed] [Google Scholar]
- 20.Dittmer J. Interaction of human T-cell lymphotropic virus type I Tax, Ets1, and Sp1 in transactivation of the PTHrP P2 promoter. J. Biol. Chem. 1997;272:4953–4958. doi: 10.1074/jbc.272.8.4953. [DOI] [PubMed] [Google Scholar]
- 21.Richard V. Humoral hypercalcemia of malignancy: severe combined immunodeficient/beige mouse model of adult T-cell lymphoma independent of human T-cell lymphotropic virus type-1 tax expression. Am. J. Pathol. 2001;158:2219–2228. doi: 10.1016/S0002-9440(10)64694-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hadigan C. Metabolic abnormalities and cardiovascular disease risk factors in adults with human immunodeficiency virus infection and lipodystrophy. Clin. Infect. Dis. 2001;32:130–139. doi: 10.1086/317541. [DOI] [PubMed] [Google Scholar]
- 23.Jain R.G. Metabolic complications associated with antiretroviral therapy. Antiviral Res. 2001;51:151–177. doi: 10.1016/s0166-3542(01)00148-6. [DOI] [PubMed] [Google Scholar]
- 24.Graham N.M. Metabolic disorders among HIV-infected patients treated with protease inhibitors: a review. J. Acquir. Immune Defic. Syndr. 2000;25(Suppl. 1):S4–S11. doi: 10.1097/00042560-200010001-00002. [DOI] [PubMed] [Google Scholar]
- 25.Qaqish R.B. HIV-associated lipodystrophy syndrome. Pharmacotherapy. 2000;20:13–22. doi: 10.1592/phco.20.1.13.34667. [DOI] [PubMed] [Google Scholar]
- 26.Kino T., Chrousos G.P. Human immunodeficiency virus type-1 accessory protein Vpr: a causative agent of the AIDS-related insulin resistance/lipodystrophy syndrome? Ann. N. Y. Acad. Sci. 2004;1024:153–167. doi: 10.1196/annals.1321.013. [DOI] [PubMed] [Google Scholar]
- 27.Yanovski J.A. Endocrine and metabolic evaluation of human immunodeficiency virus-infected patients with evidence of protease inhibitor-associated lipodystrophy. J. Clin. Endocrinol. Metab. 1999;84:1925–1931. doi: 10.1210/jcem.84.6.5740. [DOI] [PubMed] [Google Scholar]
- 28.Guo W.X., Antakly T. AIDS-related Kaposi's sarcoma: evidence for direct stimulatory effect of glucocorticoid on cell proliferation. Am. J. Pathol. 1995;146:727–734. [PMC free article] [PubMed] [Google Scholar]
- 29.Kino T., Pavlakis G.N. Partner molecules of accessory protein Vpr of the human immunodeficiency virus type 1. DNA Cell Biol. 2004;23:193–205. doi: 10.1089/104454904773819789. [DOI] [PubMed] [Google Scholar]
- 30.Kino T. Human immunodeficiency virus type 1 (HIV-1) accessory protein Vpr induces transcription of the HIV-1 and glucocorticoid-responsive promoters by binding directly to p300/CBP coactivators. J. Virol. 2002;76:9724–9734. doi: 10.1128/JVI.76.19.9724-9734.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kino T. The HIV-1 virion-associated protein vpr is a coactivator of the human glucocorticoid receptor. J. Exp. Med. 1999;189:51–62. doi: 10.1084/jem.189.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mirani M. HIV-1 protein Vpr suppresses IL-12 production from human monocytes by enhancing glucocorticoid action: potential implications of Vpr coactivator activity for the innate and cellular immunity deficits observed in HIV-1 infection. J. Immunol. 2002;169:6361–6368. doi: 10.4049/jimmunol.169.11.6361. [DOI] [PubMed] [Google Scholar]
- 33.Goodman R.H., Smolik S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000;14:1553–1577. [PubMed] [Google Scholar]
- 34.Sherman M.P. HIV-1 Vpr displays natural protein-transducing properties: implications for viral pathogenesis. Virology. 2002;302:95–105. doi: 10.1006/viro.2002.1576. [DOI] [PubMed] [Google Scholar]
- 35.Kino T. Nuclear receptor coactivator p160 proteins enhance the HIV-1 long terminal repeat promoter by bridging promoter-bound factors and the Tat-P-TEFb complex. J. Biol. Chem. 2002;277:2396–2405. doi: 10.1074/jbc.M106312200. [DOI] [PubMed] [Google Scholar]
- 36.Fawell S. Tat-mediated delivery of heterologous proteins into cells. Proc. Natl. Acad. Sci. U. S. A. 1994;91:664–668. doi: 10.1073/pnas.91.2.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kino T. HIV-1 accessory protein Vpr inhibits the effect of insulin on the Foxo subfamily of forkhead transcription factors by interfering with their binding to 14–3-3 proteins: potential clinical implications regarding the insulin resistance of HIV-1-infected patients. Diabetes. 2005;54:23–31. doi: 10.2337/diabetes.54.1.23. [DOI] [PubMed] [Google Scholar]
- 38.Kino T. Vpr protein of human immunodeficiency virus type 1 binds to 14–3-3 proteins and facilitates complex formation with Cdc25C: implications for cell cycle arrest. J. Virol. 2005;79:2780–2787. doi: 10.1128/JVI.79.5.2780-2787.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barthel A. FoxO proteins in insulin action and metabolism. Trends Endocrinol. Metab. 2005;16:183–189. doi: 10.1016/j.tem.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 40.Roodman G.D., Windle J.J. Paget disease of bone. J. Clin. Invest. 2005;115:200–208. doi: 10.1172/JCI24281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Basle M.F. Measles virus RNA detected in Paget's disease bone tissue by in situ hybridization. J. Gen. Virol. 1986;67:907–913. doi: 10.1099/0022-1317-67-5-907. [DOI] [PubMed] [Google Scholar]
- 42.Kurihara N. Expression of measles virus nucleocapsid protein in osteoclasts induces Paget's disease-like bone lesions in mice. J. Bone Miner. Res. 2006;21:446–455. doi: 10.1359/JBMR.051108. [DOI] [PubMed] [Google Scholar]
- 43.Liu C. Adenoviruses. In: Belshe R.B., editor. Textbook of Human Virology. 2nd edn. Mosby-Year Book; 1991. [Google Scholar]
- 44.Brockmann D., Esche H. The multifunctional role of E1A in the transcriptional regulation of CREB/CBP-dependent target genes. Curr. Top. Microbiol. Immunol. 2003;272:97–129. doi: 10.1007/978-3-662-05597-7_4. [DOI] [PubMed] [Google Scholar]
- 45.Chinnadurai G. CtBP, an unconventional transcriptional corepressor in development and oncogenesis. Mol. Cell. 2002;9:213–224. doi: 10.1016/s1097-2765(02)00443-4. [DOI] [PubMed] [Google Scholar]
- 46.Sorensen M.D. Severe acute respiratory syndrome (SARS): development of diagnostics and antivirals. Ann. N. Y. Acad. Sci. 2006;1067:500–505. doi: 10.1196/annals.1354.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ksiazek T.G. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 48.Li W. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Donoghue M. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ. Res. 2000;87:E1–E9. doi: 10.1161/01.res.87.5.e1. [DOI] [PubMed] [Google Scholar]
- 50.Tipnis S.R. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 2000;275:33238–33243. doi: 10.1074/jbc.M002615200. [DOI] [PubMed] [Google Scholar]
- 51.Kuba K. Angiotensin-converting enzyme 2 in lung diseases. Curr. Opin. Pharmacol. 2006;6:271–276. doi: 10.1016/j.coph.2006.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Crackower M.A. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417:822–828. doi: 10.1038/nature00786. [DOI] [PubMed] [Google Scholar]
- 53.Hunyady L., Catt K.J. Pleiotropic AT1 receptor signaling pathways mediating physiological and pathogenic actions of angiotensin II. Mol. Endocrinol. 2006;20:953–970. doi: 10.1210/me.2004-0536. [DOI] [PubMed] [Google Scholar]
- 54.Kuba K. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005;11:875–879. doi: 10.1038/nm1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Levy J.A. HIV pathogenesis: knowledge gained after two decades of research. Adv. Dent. Res. 2006;19:10–16. doi: 10.1177/154407370601900104. [DOI] [PubMed] [Google Scholar]
- 56.McMichael A.J. HIV vaccines. Annu. Rev. Immunol. 2006;24:227–255. doi: 10.1146/annurev.immunol.24.021605.090605. [DOI] [PubMed] [Google Scholar]
- 57.De Clercq E. Antivirals for the treatment of herpesvirus infections. J Antimicrob Chemother. 1993;32(Suppl. A):121–132. doi: 10.1093/jac/32.suppl_a.121. [DOI] [PubMed] [Google Scholar]
- 58.Moscona A. Neuraminidase inhibitors for influenza. N. Engl. J. Med. 2005;353:1363–1373. doi: 10.1056/NEJMra050740. [DOI] [PubMed] [Google Scholar]
- 59.Fanales-Belasio E. HIV-1 Tat-based vaccines: from basic science to clinical trials. DNA Cell Biol. 2002;21:599–610. doi: 10.1089/104454902760330138. [DOI] [PubMed] [Google Scholar]
- 60.Adamopoulos D.A. Pituitary-testicular interrelationships in mumps orchitis and other viral infections. BMJ. 1978;1:1177–1180. doi: 10.1136/bmj.1.6121.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pekic S. Hypopituitarism as a late complication of hemorrhagic fever. Endocrine. 2005;26:79–82. doi: 10.1385/ENDO:26:2:079. [DOI] [PubMed] [Google Scholar]
- 62.Kimberlin D.W. Herpes simplex virus infections in neonates and early childhood. Semin. Pediatr. Infect. Dis. 2005;16:271–281. doi: 10.1053/j.spid.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 63.Kino T., Chrousos G.P. AIDS/HPA axis. In: Chrousos G.P., editor. Adrenal Physiology and Diseases. 2003. http://endotext.org/adrenal/index.htm [Google Scholar]
- 64.Ramos-Casals M. Therapeutic management of extrahepatic manifestations in patients with chronic hepatitis C virus infection. Rheumatology (Oxford) 2003;42:818–828. doi: 10.1093/rheumatology/keg299. [DOI] [PubMed] [Google Scholar]
- 65.Clarke W.L. Autoimmunity in congenital rubella syndrome. J. Pediatr. 1984;104:370–373. doi: 10.1016/s0022-3476(84)81097-5. [DOI] [PubMed] [Google Scholar]
- 66.Oldstone M.B. Molecular mimicry, microbial infection, and autoimmune disease: evolution of the concept. Curr. Top. Microbiol. Immunol. 2005;296:1–17. doi: 10.1007/3-540-30791-5_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tomer Y., Davies T.F. Infection, thyroid disease, and autoimmunity. Endocr. Rev. 1993;14:107–120. doi: 10.1210/edrv-14-1-107. [DOI] [PubMed] [Google Scholar]
- 68.Oldstone M.B. Viral persistence: parameters, mechanisms and future predictions. Virology. 2006;344:111–118. doi: 10.1016/j.virol.2005.09.028. [DOI] [PubMed] [Google Scholar]
- 69.Chen N. Selective inhibition of nuclear steroid receptor function by a protein from a human tumorigenic poxvirus. Virology. 2000;274:17–25. doi: 10.1006/viro.2000.0410. [DOI] [PubMed] [Google Scholar]