

Abstract
Ubiquitin carboxyl-terminal hydrolases (UCHs) are implicated in the proteolytic processing of polymeric ubiquitin. The high specificity for the recognition site makes UCHs useful enzymes for in vitro cleavage of ubiquitin fusion proteins. In this work, an active C-terminal His-tagged UCH from Drosophila melanogaster (DmUCH) was produced as a secretory form in a recombinant strain of the methylotrophic yeast Pichia pastoris. The production of recombinant DmUCH by Muts strain was much higher than that by Mut+ strain, which was confirmed by Western blot analysis. When expression was induced at pH 6.0 in a BMMY/methanol medium, the concentration of recombinant DmUCH reached 210 mg l−1. With the (His)6-tag, the recombinant DmUCH was easily purified by Ni-NTA chromatography and 18 mg pure active DmUCH were obtained from 100 ml culture broth supernatant. Ubiquitin–magainin fusion protein was efficiently cleaved by DmUCH, yielding recombinant magainin with high antimicrobial activity. After removing the contaminants by Ni-NTA chromatography, recombinant magainin was purified to homogeneity easily by reversed-phase HPLC. Analysis of the recombinant magainin by ESI-MS showed that the molecular weight of the purified recombinant magainin was 2465 Da, which perfectly matches the mass calculated from the amino acid sequence. The result of mass spectrometry confirmed that the purified His-tagged DmUCH can recognize the ubiquitin–magainin fusion protein and cleave it at the carboxyl terminus of ubquitin precisely. Our results showed that P. pastoris is a robust system to express the secreted form of DmUCH.
Keywords: Ubiquitin carboxyl-terminal hydrolase, Pichia pastoris, Expression, Purification, Activity assay
In recent years, one strategy, which has been developed for the expression of heterologous gene in Escherichia coli, is to fuse the gene of interest downstream of the ubiquitin gene to produce a fusion protein [1], [2], [3], [4], [5], [6], [7], [8], [9], [10], [11], [12]. Ubiquitin-based tags have been used to not only increase expression levels but also to aid in protein folding. The technology has emerged as an alternative for the production, solubility and correct folding of otherwise intractable proteins. When fused to ubiquitin, insoluble proteins fold properly and become soluble [1], [2], [3], [4], [5], [6], [7], [8], [9], [10], [11], [12]. Furthermore, the ubiquitin tag can be removed using a group of enzymes called deubiquitinating enzymes that cleave the tail sequence following a Gly–Gly dipeptide in the ubiquitin-derived substrates [1], [13]. Deubiquitinating enzymes consist of at least two families: the UCH 2 (ubiquitin carboxyl-terminal hydrolase) family [13], [14], [15] and the UBP (ubiquitin-specific processing protease) family [16], [17], [18], [19], [20]. UBPs are thought to disassemble polyubiquitin chains, whereas UCHs are shown to hydrolyze bonds between small adducts and ubiquitin to generate free monomeric ubiquitin [1], [3].
More recently, the UCH has been shown to have a broad application in cleaving ubiquitin fusion proteins produced in E. coli [5], [6], [7], [10], [11], [12]. The enzyme is particularly suitable for this role because of its high degree of specificity, its tolerance to a wide range of reaction conditions, and the fact that its recognition sequence lies entirely at the amino-terminal side of the scissile bond [12]. This enzymatic activity of the UCH allows the release of the carboxyl-terminal fusion partners from fusion proteins without adding any unwanted amino acid residues to their amino termini. Thus, to cater for the applications, high yields of recombinant UCHs are needed. Recombinant UCHs have been reported in E. coli [12], [21], [22], in which a renaturation procedure is necessary. In addition, since E. coli is a prokaryotic expression system, it lacks post-translational modifications, such as proteolytic processing, folding and glycosylation that occur in a eukaryotic system. These limitations have prompted biotechnologists to seek new expression systems.
Recently, methylotrophic yeast, Pichia pastoris, has been developed as excellent host for the large-scale expression of proteins from different sources [23]. P. pastoris is known for its high-level expression of heterologous proteins and its tightly regulated alcohol oxidase1 (AOX1) gene promoter [24]. P. pastoris can be easily grown to high cell densities using defined minimal media and it is possible to introduce eukaryotic post-translational modifications. The techniques needed for molecular genetic manipulation are similar to those well established in Saccharomyces cerevisiae. Therefore, even very toxic proteins can be produced in large-scale in this system [25], [26], [27], [28].
In the present study, we report the expression, purification and characterization of the recombinant UCH from Drosophila melanogaster (DmUCH) with a 6xHis-tag at the carboxyl terminus in P. pastoris.
Materials and methods
Materials
All the restriction enzymes and immobilized metal ion affinity chromatography (IMAC) (Ni-NTA) resins were purchased from Qiagen (Germany). E. coli Top10F’was used for routine plasmid amplification. T4 DNA ligase and Taq DNA polymerase were from TaKaRa Biotechnology (Dalian, China). Plasmid pPICZα-A and P. pastoris GS115 were purchased from Invitrogen.
Gene and plasmid construction
First strand cDNA from the third instar D. melanogaster larvae was synthesized using the Trizol kit (Invitrogen)[28]. The cDNA encoding D. melanogaster ubiquitin carboxyl-terminal hydrolase (DmUCH) (GenBank Accession No. NM_057592) was amplified using the first strand cDNA as a template and a pair of gene specific primers designed based on the nucleotide sequence of DmUCH. The gene specific primers are: PF (5′-CCG CTC GAG AAA AGA ATG TTA ACC TGG ACG CCA CTT G-3′) and PR (5′-CAT GCG GCC GCT TGT TGT GCC GCG GTC AAG GCC-3′). In order to express the native N-terminus of DmUCH, an XhoI restriction site was introduced to allow in-frame cloning of the gene into the α-factor secretion signal of pPICZα-A expression vector and a nucleotides sequence encoding the KEX2 cleavage site was placed ahead of DmUCH. At the C-terminus, the stop codon was eliminated and replaced with a NotI restriction site, thus, the expressed DmUCH should ligate to the 6xHis-tag at the carboxyl terminus. PCR was performed as follows: preheating at 94 °C for 7 min, 30 cycles at 94 °C for 40 s, 55 °C for 40 s and 72 °C for 50 s, followed by an elongation at 72 °C for 7 min. A 690 bp fragment was recovered from the gel by using the E.Z.N.A Gel Extraction Kit, and cloned into a pMD18-T vector using the Original TA cloning Kit (TaKaRa, Japan). The nucleotide sequence of the DmUCH was confirmed by sequencing. The pMD18-DmUCH plasmid was digested with XhoI and NotI sequentially, and the DNA fragment was gel-purified and then cloned into the P. pastoris expression vector pPICZα-A (Fig. 1 ). The constructed vector, pPICZα-DmUCH, contains the complete DmUCH structural gene downstream the α-factor sequence, and was used to transform the competent E. coli Top10F′. The plasmid DNA was prepared and the insert was confirmed by sequencing.
Fig. 1.

The schematic representation of the vector used for the expression of DmUCH.
Transformation of P. pastoris and selection of transformants
Pichia pastoris GS115 strain was transformed with SalI-linearized pPICZα-DmUCH by electroporation following the manufacturer’s instructions (Invitrogen). All Zeocine-resistant colonies were replica-plated onto MMH plates (1.34% YNB, 0.5% methanol, 1.61 μM biotin, 0.004% histidine, 1.5% agar) and MDH plates (same compositions as in MMH but with 2% glucose for 0.5% methanol) to determine the methanol-utilizing phenotypes. After 3–4 days of incubation, the Mut+ phenotypes grew normally on both MMH and MDH plates, whereas the Muts phenotypes grew very slowly on MMH plates. Both Muts and Mut+ strains were used for shake-flask cultivation.
Shake-flask cultivation of P. pastoris
Ten positive colonies were grown in 100 ml of BMGYH (1% yeast extract, 2% peptone, 100 mM potassium phosphate, pH 6.0, 1.34% YNB, 1.61 μM biotin, 0.004% histidine, 1% glycerol) in 500 ml shaking flask until O.D600 = 5 at 30 °C. The cell culture was centrifuged and the pellet was suspended to an O.D600 of 1.0 in 20 ml of BMMYH (same as BMGYH but with 0.5% methanol for 1% glycerol). The resuspended culture was grown for 48 h by addition of 1.0% methanol every 24 h. Samples were withdrawn for the detection of the recombinant protein by SDS–PAGE and Western blot using the mouse anti-His (C-term) antibody [28].
Purification of the secreted DmUCH
One hundred milliliter of cultured medium supernatant was dialyzed overnight in 20 mM sodium phosphate buffer (pH 7.5) containing 300 mM NaCl (buffer A) and applied to a nickel chelating Sepharose column (1.6 × 10 cm) pre-equilibrated with the binding buffer (20 mM Na3PO4, 500 mM NaCl, 5 mM imidazole, pH 7.4). The column was washed with the binding buffer and the bound protein was eluted with a linear gradient of 5–500 mM imidazole in the buffer (20 mM Na3PO4, 500 mM NaCl, pH 7.4) at 1 ml/min. The DmUCH fractions were pooled and dialyzed overnight against a buffer containing 50 mM potassium phosphate, 50 mM NaCl and 50% glycerol, pH 8.0. The purity of the protein sample was determined by 12% SDS–PAGE and Western blot. The protein concentration of the recombinant enzyme was determined using the Bio-Rad dye agent with BSA as a standard [29].
Preparation of the ubiquitin–magainin fusion protein
The expression plasmid (pQE-ubiquitin–magainin) harboring the sequence encoding the 6xHis-ubquitin-magainin was constructed according to the procedures reported previously [2]. The resulting expression plasmids were transformed into E. coli strain M15 (pREP4) for recombinant protein expression. Cells were grown in LB-broth supplemented with 100 μg/ml ampicillin and 25 μg/ml kanamycillin at 37 °C. Overnight cultures were diluted 1:100, grown to O.D600 = 0.8, and then induced for protein expression by addition of isopropylthiogalactoside (IPTG) to a final concentration of 0.4 mM. Protein expression was carried out for 5 h at 30 °C. The cell pellets were thawed on ice and resuspended in 50 mM Tris–HCl (pH 7.0) containing 500 mM NaCl. Resuspended cells were lysed by sonication on ice for 20 s for four times. Cell debris was removed by centrifugation at 12,000g for 30 min at 4 °C. The fusion protein (ubquitin-magainin) was purified from the supernatant by a HisTrap HP (5 ml) column (Amersham Biosciences, Sweden) pre-equilibrated with the binding buffer (20 mM Na3PO4, 500 mM NaCl, 5 mM imidazole, pH 7.4). Purified 6xHis-ubquitin-magainin was concentrated and stored at −20 °C in 50 mM Tris, pH 7.5, 0.5 mM EDTA, 5 mM DTT and 50% glycerol.
Analysis of enzymatic activity of DmUCH
The purified DmUCH (0.2 μg) was incubated with the fusion protein ubiquitin–magainin (20 μg) in 50 μl assay buffer (50 mM HEPES/NaOH, pH 7.8, 0.5 mM EDTA, 1 mM DTT, 0.1 mg/ml ovalbumin) at 37 °C for various time periods. Aliquots were withdrawn at different time intervals and the reactions were terminated by boiled for 10 min, and the progress of the reaction was monitored by Tricine–SDS–PAGE [30] and antibacterial activity assay using agar diffusion method [31].
Purification and analysis of recombinant magainin
The reaction mixture was first applied to the HisTrap HP (5 ml) column to remove the His-tagged ubiquitin, His-tagged DmUCH and undigested fusion proteins. The flow-through fraction was filtrated using the Amicon ultrafiltration device. One hundred milliliters of the filtrate were collected, including the proteins below 10.0 kDa. The filtrate was applied to the semi-preparative reversed-phase HPLC on a C18 column (250 × 4.6 mm, 5 μm 300 Å), pre-equilibrated in 0.1% TFA and 18% acetonitrile. The bound protein was eluted with a linear gradient of acetonitrile (18–45%, v/v) in 0.1% TFA at 1.35% per minute. The flow rate was 1.0 ml/min and the absorbance of the elutant was monitored at 280 nm. The purity and integrity of magainin eluted from the reverse-phase HPLC was determined by Tricine–SDS–PAGE and electrospray ionization mass spectrometry .
Results
Influence of methanol utilization phenotype of the host on DmUCH production
After 3–4 days of incubation, the Mut+ phenotypes grew normally on both MMH and MDH plates, whereas the Muts phenotype grew very slowly on MMH plates and the high zeocine-resistant positive transformants were selected on MDH plates. In order to screen the high-level expression stains, we investigated the expression of recombinant DmUCH-His by both Muts and Mut+ strains in shake flasks. Western blot analysis revealed that the DmUCH-His level expressed in the Muts strain was much higher than in the Mut+ strain. Thus, the Muts strain was selected for suspension culture in shake flasks.
Expression and purification of DmUCH
The P. pastoris clone that produced highest DmUCH was selected for subsequent large-scale protein expression with the optimal conditions (pH 6.0 and 120 h methanol induction). A major recombinant DmUCH of about 29 kDa was observed after 48 h induction, and its concentration increased up to 210 mg/l after 120 h induction (Fig. 2 ). Western blot analysis revealed that the DmUCH was specifically recognized by mouse anti-His (C-term) antibody, and the high expression of the protein was on day 5 (Fig. 2). With the 6xHis-tag at the C-terminus of DmUCH, recombinant DmUCH was easily purified to homogeneity using chelating Sepharose chromatography, which appeared as a single protein band around 29 kDa on SDS–PAGE (Fig. 3 ). The apparent molecular weight (∼29 kDa) of recombinant DmUCH is consistent with the one calculated from the deduced amino acid sequence. About 18 mg of DmUCH-His was purified from 100 ml of culture medium.
Fig. 2.
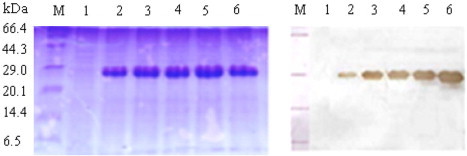
Analyses of recombinant DmUCH in the culture media by SDS–PAGE and Western blot. Samples of culture media were removed at 24 h intervals after methanol induction and separated by 12% SDS–PAGE. Proteins were either stained with Coomassie blue (A) or transferred to a nitrocellular (PVDF) membrane and then identified by immunoblotting using mouse anti-His (C-term) antibody. Lane 1, sample from DmUCH/6His -expressing P. pastoris prior to methanol induction; Lanes 2–6, samples from DmUCH/6His-expressing P. pastoris after 1–5 days of methanol induction; lane M, polypeptide SDS–PAGE molecular weight markers.
Fig. 3.

SDS–PAGE analysis of the purified recombinant DmUCH/6His Lane M, polypeptide SDS–PAGE molecular weight markers; Lane 1, purified recombinant DmUCH/6His.
Enzymatic activity of recombinant DmUCH
Ubiquitin–magainin was incubated with the recombinant DmUCH. The time-course release of magainin from the fusion proteins was monitored by antimicrobial assay using agar diffusion method. Samples were collected from the reaction solutions at various time intervals and assayed for antibacterial activity. The antibacterial activity reached the maximum after 60 min of cleavage by DmUCH (Fig. 4 ). Tricine–SDS–PAGE analysis indicated that nearly all fusion proteins were completely cleaved, resulting in the cleavage products of ubiquitin and magainin at the expected size of 8.6 and 2.5 kDa, respectively, (Fig. 5 A). These results indicated that the purified recombinant DmUCH can recognize the ubiquitin–magainin fusion protein and cleave the fusion protein precisely.
Fig. 4.

Time course cleavage of the ubiquitin–magainin fusion protein by DmUCH/6His. Aliquots of the reaction mixture were withdrawn at different time intervals after incubation and the progress of the reaction was monitored by the antibacterial activity assay using the agar diffusion method. Antibacterial activity was assayed by measuring the zones of growth inhibition in thin agar plates with E. coli K12D31 as described by Hultmark et al. [31]. Experiments were repeated three times, the antibacterial activity reached the maximum after 60 min of cleavage by DmUCH and the diameter of the clearing zone was 1.1 ± 0.05 cm. ck, the inhibition zone with PBS (negative control); 1–6, the inhibition zones of the reaction mixture after incubation for 10, 20, 30, 40, 50 and 60 min.
Fig. 5.

SDS–PAGE analysis of the cleavage products from the ubiquitin–magainin fusion protein by DmUCH/6His for 1 h at 37 °C(A) and Tricine–SDS–PAGE analysis of the purified recombinant magainin (B). Lanes M1 and M2, polypeptide SDS–PAGE molecular weight markers; Lane 1, the reaction mixture after incubation for 60 min; Lane 2, the reaction mixture after incubation for 30 min; Lane 3, the reaction mixture before incubation; Lane 4, purified recombinant magainin.
Purification and analysis of recombinant magainin
The reaction mixtures containing the ubiquitin–magainin fusion protein and recombinant DmUCH were applied to the HisTrap HP (5 ml) column. After removing the 6xHis-UBI, DmUCH and undigested fusion protein by chelating Sepharose chromatography, recombinant magainin was easily purified to homogeneity by ultrafiltration and reverse-phase HPLC chromatography. Analysis of the purified magainin by Tricine–SDS–PAGE revealed that the molecular mass of magainin is about 2.5 kDa, consistent with the theoretical molecular mass of 2465 Da (Fig. 5B). Analysis of the recombinant magainin by Mass spectrometry showed a single, non-dispersed signal (Fig. 6 ). The average mass of the molecular ion [M+4H]4+ is 618.05 Da, which perfectly matches the mass (2465 Da) calculated from the amino acid sequence. The result of mass spectrometry confirmed that the purified DmUCH-His can recognize the ubiquitin–magainin fusion protein and cleave it at the carboxyl terminus of ubquitin precisely.
Fig. 6.
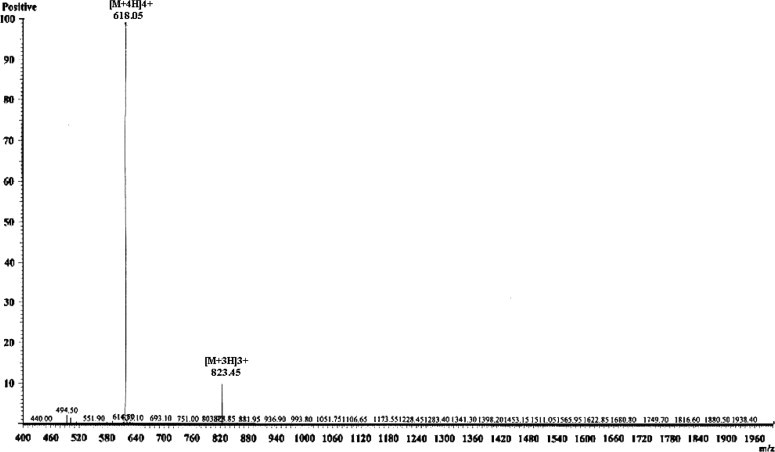
ESI-MS analysis of the purified recombinant magainin.
Discussion
Recently, ubiquitin and SUMO (small ubiquitin modifying protein) have been fused to the N-terminus of several proteins, including matrix metalloprotease X (MMP13), green fluorescent protein (GFP) and SARS-CoV 3CL protease, as fusion partners to enhance expression and solubility of target proteins [3], [5], [8]. In our laboratory, we have developed an efficient E. coli-based expression system to express antibacterial peptides fused to the poly-histidine-tagged ubiquitin to enable a simple one-step purification of the fusion antibacterial peptides by immobilized metal affinity chromatography (IMAC)[2]. In order to obtain the active antibacterial peptides from the ubiquitin fusion proteins, the ubiquitin C-terminal hydrolases (UCHs) that can cleave the ubiquitin molecule at the C-terminal Gly76 to release ubiquitin and active antibacterial peptides are needed. Therefore, in the present study, we report the high-level expression and secretion of recombinant ubiquitin C-terminal hydrolase of D. melanogaster (DmUCH) in the methylotrophic yeast, P. pastoris.
In our laboratory, the gene encoding DmUCH was inserted into pQE-30 and expressed in E. coli M15. But most of recombinant DmUCH have been expressed as inclusion bodies, and low-yield functional recombinant DmUCH was obtained after refolding the inclusion bodies. Recombinant DmUCH was successfully secreted into the culture supernatants by P. pastoris, and with the 6xHis-tag at the C-terminus of DmUCH, recombinant DmUCH was purified to homogeneity by an affinity chromatography (IMAC). Enzymatic activity assays indicated that the recombinant DmUCH had high activity to cleave the ubiquitin–magainin fusion proteins and released the magainin that had activity against E. coli K12D31.
Fusing a partner to the N-terminus of target proteins/peptides for expression, solubility and purification has been reported [32], [33], [34], [35]. But the fusion partner must be removed to obtain functional peptides. This can be achieved by introducing a protease recognition site, including tobacco etch virus (Tev) protease [36], factor Xa, or thrombin protease, in the fusion proteins[35]. However, cleavage of the fusion proteins with proteases may result in recombinant peptides with an unauthentic N-terminus [37], [38]. Furthermore, cleavage of the fusion proteins is rarely complete, which reduces the yield. In addition, proteases may nonspecifically cleave the fusion proteins. The ubiquitin and SUMO fusion proteins also require proteases to remove the tags. However, the ubiquitin C-terminal hydrolases (UCHs), which are members of the cysteine protease superfamily, are distinct from other proteases in that they recognize the tertiary structure of the SUMO or ubiquitin. When the target protein is fused directly to the C-terminus of SUMO or ubiquitin, removal of the SUMO or ubiquitin will not add extra residues to the N-terminus of the target protein and therefore native-like recombinant proteins can be obtained [8], [39]. In the present study, when the ubiquitin–magainin was treated with recombinant DmUCH, accumulation of recombinant magainin was observed with the reaction times. The ESI-MS analysis showed that the molecular weight of the purified magainin was 2465 Da, which is almost identical to the mass calculated from the amino acid sequence. These results indicated that the purified recombinant DmUCH can recognize and cleave the ubiquitin–magainin fusion precisely with high activity to release the intact magainin with a native N-terminus.
His-tags are the most widely used affinity tags which may have a positive effect in the biochemical properties of the target proteins. Purification of His-tagged proteins is based on the chelating metal ions as affinity ligands. The use of short histidine stretches or His-tags, typically as affinity tags at either the N-terminus or C-terminus, enables the purification of the desired protein from the crude extract in a single step by IMAC (immobilized metal ion affinity chromatography) [40], [41], [42]. The most widely used IMAC supports are either nickel nitrilotriacetic acid (Ni-NTA) resins or different chelating Sepharose matrices. Importantly, the binding specificity enables the purification of proteins under both native and denaturing conditions [41]. Now, numerous proteins and peptides have been purified using the His-tags, and several therapeutic candidates are in clinical studies [43]. His-tags have also been used for purification of proteins using the expanded bed adsorption [44], [45]. In the present study, the (His)6-tag has been attached to the C-terminus of DmUCH and to the N-terminus of ubiquitin–magainin fusion protein, and the fusion proteins were easily purified by Ni-NTA. The (His)6-tag also facilitates purification of recombinant magainin after the ubiquitin–magainin was cleaved by DmUCH for 60 min, because the fusion protein, UBI and UCH, but not the released magainin, all contain a His-tag and can be removed by Ni-NTA, and the magainin is the only peptide remained in the flow-through.
Many substrates, including recombinant ubiquitin fusions and synthetic ubiquitn fusions, have been used to test the enzymatic activity of UCHs [5], [6], [7], [10], [12]. However, the procedures are very complex and the expense for synthetic fusions is very expensive. In the present study, the recombinant 6xHis-ubiquitin–magainin fusion protein was used as a substrate to analyze the activity of recombinant DmUCH. The cleavage of ubiquitin–magainin to ubiquitin and magainin by the purified recombinant DmUCH was first monitored by the high antibacterial activity of magainin released from the fusion protein using the agar diffusion method. Release of magainin was then confirmed by Tricine–SDS–PAGE and ESI-MS analyses. The agar diffusion method, which requires small volumes of cleavage mixtures, is simple and easy to manipulate, thus, it would be a versatile and efficient method to assay the activity of DmUCH.
We showed in this study that the ubiquitin carboxyl–terminal hydrolase from D. melanogaster (DmUCH) can be heterologously expressed in P. pastoris using the secretion signal of yeast α-mating factor. The recombinant DmUCH was expressed at high-levels (up to 210 mg/l) in the culture medium and easily purified by a single step affinity chromatography. The recombinant DmUCH exhibited high enzymatic activity. The successful expression and purification of the active DmUCH in P. pastoris will provide the opportunity to elucidate its structure in relation to its mechanism of action and offer an enzyme source for cleavage of ubiquitin fusion proteins in near future.
Acknowledgments
This work was supported by a grant from the National Basic Research and Development Program of China 973 Project (2006CB102005), the Special Scientific Research Fund for Commonwealth Trade of China (200803005) and the National Postdoctoral Foundation of China (20070410240).
Footnotes
Abbreviations used: UCH, ubiquitin carboxyl terminal hydrolase; AOX1, alcohol oxidase1; IPTG, isopropyl-β-d-thiogalactoside; IMAC, immobilized metal ion affinity chromatography; SDS–PAGE, sodium dodecyl sulfate–polyacrylamide gel electrophoresis; DmUCH, ubiquitin carboxyl-terminal hydrolase from D. melanogaster.
References
- 1.Baker R.T. Protein expression using ubiquitin fusion and cleavage. Current Opinion in Biotechnology. 1996;7:541–546. doi: 10.1016/s0958-1669(96)80059-0. [DOI] [PubMed] [Google Scholar]
- 2.Xu X.X., Jin F.L., Yu X.Q., Ren S.X., Hu J., Zhang W.Q. High-level expression of the recombinant hybrid peptide cecropinA(1–8)-magainin2(1–12) with an ubiquitin fusion partner in Escherichia coli. Protein Expression and Purification. 2007;55:175–182. doi: 10.1016/j.pep.2007.04.018. [DOI] [PubMed] [Google Scholar]
- 3.Butt T.R., Edavettal S.C., Hall J.P., Mattern M.R. SUMO fusion technology for difficult-to-express proteins. Protein Expression and Purification. 2005;43:1–9. doi: 10.1016/j.pep.2005.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Assadi-Porter M.F., Patry S., Markley J.L. Efficient and rapid protein expression and purification of small high disulfide containing sweet protein brazzein in E. coli. Protein Expression and Purification. 2008;58:263–268. doi: 10.1016/j.pep.2007.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zuo X., Mattern M.R., Tan R., Li S., Hall J., Sterner D.E., Shoo J., Tran H., Lim P., Sarafianos S.G., Kazi L., Navas-Martin S., Weiss S.R., Butt T.R. Expression and purification of SARS coronavirus proteins using SUMO-fusions. Protein Expression and Purification. 2005;42:100–110. doi: 10.1016/j.pep.2005.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mildner A.M., Paddock D.J., LeCureux L.W., Leone J.W., Anderson D.C., Tomasselli A.G., Heinrikson R.L. Production of chemokines CTAPIII and NAP/2 by digestion of recombinant ubiquitin–CTAPIII with yeast ubiquitin C-Terminal hydrolase and human immunodeficiency virus protease. Protein Expression and Purification. 1999;16:347–354. doi: 10.1006/prep.1999.1081. [DOI] [PubMed] [Google Scholar]
- 7.Butt T.R., Jonnalagadda S., Monia B.P., Sternberg E.J., Marsh J.A., Stadel J.M., Ecker D.J., Crooke S.T. Ubiquitin fusion augments the yield of cloned gene products in Escherichia coli. Proceedings of the National Academy of Sciences of the United States of America. 1989;86:2540–2544. doi: 10.1073/pnas.86.8.2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Malakhov M.P., Malakhova O.A., Drinker M., Weeks S., Butt T.R. SUMO fusion and SUMO-specific proteases for efficient expression and purification of proteins. Journal of Structural and Functional Genomics. 2004;5:75–86. doi: 10.1023/B:JSFG.0000029237.70316.52. [DOI] [PubMed] [Google Scholar]
- 9.Koken M.H., Odijk H.H., van Duin M., Fornerod M., Hoeijmakers J.H. Augmentation of protein production by a combination of the T7 RNA polymerase system and ubiquitin fusion: overproduction of the human DNA repair protein, ERCC1, as an ubiquitin fusion protein in Escherichia coli. Biochemical and Biophysical Research Communications. 1993;195:643–653. doi: 10.1006/bbrc.1993.2094. [DOI] [PubMed] [Google Scholar]
- 10.Lee E.K., Hwang J.H., Shin D.Y., Kim D.I., Yoo Y.J. Production of recombinant amyloid-β peptide 42 as an ubiquitin extension. Protein Expression and Purification. 2005;40:183–189. doi: 10.1016/j.pep.2004.12.014. [DOI] [PubMed] [Google Scholar]
- 11.Pilon A.L., Yost P., Chase T.E., Lohnas G.L., Bentley W.E. High-level expression and efficient recovery of ubiquitin fusion proteins from Escherichia coli. Biotechnology Progress. 1996;12:331–337. doi: 10.1021/bp9600187. [DOI] [PubMed] [Google Scholar]
- 12.Catanzariti A.M., Soboleva T.A., Jans D.A., Board P.G., Baker R.T. An efficient system for high-level expression and easy purification of authentic recombinant proteins. Protein Science. 2004;13:1331–1339. doi: 10.1110/ps.04618904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller H.I., Henzel W.J., Ridgway J.B., Kuang W.J., Chisholm V., Liu C.C. Cloning and expression of a yeast ubiquitin–protein cleaving activity in Escherichia coli. Biotechnology. 1989;7:698–704. [Google Scholar]
- 14.Woo S.K., Lee J.I., Park I.K., Yoo Y.J., Cho C.M., Kang M.-S., Ha D.B., Tanaka K., Chung C.H. Multiple ubiquitin C-terminal hydrolases from chick skeletal muscle. Journal of Biological Chemistry. 1995;270:18766–18773. doi: 10.1074/jbc.270.32.18766. [DOI] [PubMed] [Google Scholar]
- 15.Seliger B., Fedorushchenko A., Brenner W., Ackermann A., Atkins D., Hanash S., Lichtenfels R. Ubiquitin COOH-terminal hydrolase 1: a biomarker of renal cell carcinoma associated with enhanced tumor cell proliferation and migration. Clinical Cancer Research. 2007;13:27–37. doi: 10.1158/1078-0432.CCR-06-0824. [DOI] [PubMed] [Google Scholar]
- 16.Tobias J.W., Varshavsky A. Cloning and functional analysis of the ubiquitin-specific protease gene UBP1 of Saccharomyces cerevisiae. Journal of Biological Chemistry. 1991;266:12021–12028. [PubMed] [Google Scholar]
- 17.Baek K.H., Mondoux M.A., Jaster R., Levin E.F., D’Andrea A.D. DUB-2A, a new member of the DUB subfamily of hematopoietic deubiquitinating enzymes. Blood. 2001;98:636–642. doi: 10.1182/blood.v98.3.636. [DOI] [PubMed] [Google Scholar]
- 18.Malakhov M.P., Malakhova O.A., Kim K.I., Ritchie K.J., Zhang D.-E. UBP43 (USP18) specifically removes ISG15 from conjugated proteins. Journal of Biological Chemistry. 2002;277:9976–9981. doi: 10.1074/jbc.M109078200. [DOI] [PubMed] [Google Scholar]
- 19.Sloper-Mould K.E., Eyre H.J., Wang X.-W., Sutherland G.R., Baker R.T. Characterization and chromosomal localization of USP3, a novel human ubiquitin-specific protease. Journal of Biological Chemistry. 1999;274:26878–26884. doi: 10.1074/jbc.274.38.26878. [DOI] [PubMed] [Google Scholar]
- 20.Yan N., Doelling J.H., Falbel T.G., Durski A.M., Vierstra R.D. The ubiquitin-specific protease family from arabidopsis. AtUBP1 and 2 are required for the resistance to the amino acid analog canavanine. Plant Physiology. 2000;124:1828–1843. doi: 10.1104/pp.124.4.1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Franklin K., Layfield R., Landon M., Ramage R., Brown A., Love S., Muir T., Urquhart K., Bownes M., Mayer R.J. Capillary electrophoresis assay for ubiquitin carboxyl-terminal hydrolases with chemically synthesized ubiquitin–valine as substrate. Analytical Biochemistry. 1997;247:305–309. doi: 10.1006/abio.1997.2099. [DOI] [PubMed] [Google Scholar]
- 22.Yu H.A., Kim S.G., Kim E.J., Lee W.J., Kim D.O., Park K., Park Y.C., Seo J.H. Characterization of ubiquitin C-terminal hydrolase 1 (YUH1) from Saccharomyces cerevisiae expressed in recombinant Escherichia coli. Protein Expression and Purification. 2007;56:20–26. doi: 10.1016/j.pep.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 23.Cereghino J.L., Cregg J.M. Heterologous protein expression in the methylotrophic yeast Pichia pastoris. FEMS Microbiology Reviews. 2000;24:45–66. doi: 10.1111/j.1574-6976.2000.tb00532.x. [DOI] [PubMed] [Google Scholar]
- 24.Sreekrishna K., Brankamp R.G., Kropp K.E., Blankenship D.T., Tsay J.-T., Smith P.L., Wierschke J.D., Subramaniam A., Birkenberger L.A. Strategies for optimal synthesis and secretion of heterologous proteins in the methylotrophic yeast Pichia pastoris. Gene. 1997;190:55–62. doi: 10.1016/s0378-1119(96)00672-5. [DOI] [PubMed] [Google Scholar]
- 25.Li L., Wang J.X., Zhao X.F., Kang C.J., Liu N., Xiang J.H., Li F.H., Sueda S., Kondo H. High level expression, purification, and characterization of the shrimp antimicrobial peptide, Ch-penaeidin, in Pichia pastoris. Protein Expression and Purification. 2005;39:144–151. doi: 10.1016/j.pep.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 26.Almeida M.S., Cabral K.S., de Medeiros L.N., Valente A.P., Almeida F.C.L., Kurtenbach E. cDNA cloning and heterologous expression of functional cysteine-rich antifungal protein Psd1 in the yeast Pichia pastoris. Archives of Biochemistry and Biophysics. 2001;395:199–207. doi: 10.1006/abbi.2001.2564. [DOI] [PubMed] [Google Scholar]
- 27.Cabral K.M.S., Almeida M.S., Valente A.P., Almeida F.C.L., Kur-tenbach E. Production of the active antifungal pisum sativum defensin 1 (Psd1) in Pichia pastoris: overcoming the inefficiency of the STE13 protease. Protein Expression and Purification. 2003;31:115–122. doi: 10.1016/s1046-5928(03)00136-0. [DOI] [PubMed] [Google Scholar]
- 28.Jin F.L., Xu X.X., Zhang W.Q., Gu D.X. Expression and characterization of a housefly cecropin gene in the methylotrophic yeast, Pichia pastoris. Protein Expression and Purification. 2006;49:39–46. doi: 10.1016/j.pep.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 29.Ausubel F.M., Brent R., Kingston R.E., Moore D.D., Seidman J.G., Smith J.A., Struhl K. Protocols in molecular biology. Trends in Cell Biology. 1996;6:366–367. [Google Scholar]
- 30.Schacgger H., Jagow G.V. Tricine–sodium dodecyl sulfate–polyacryl-amide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Analytical Biochemistry. 1987;166:368–379. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- 31.Hultmark D., Engstrom A., Bennich H. Insect immunity: isolation and structure of cecropin d and four minor antibacterial components from cecropia pupa. European Journal of Biochemistry. 1982;127:207–217. doi: 10.1111/j.1432-1033.1982.tb06857.x. [DOI] [PubMed] [Google Scholar]
- 32.Rao X.C., Li S., Hu J.C., Jin X.L., Hu X.M., Huang J.J., Chen Z.J., Zhu J.M., Hu F.Q. A nove lcarrier molecule for high-level expression of peptide antibiotics in Escherichia coli. Protein Expression and Purification. 2004;36:11–18. doi: 10.1016/j.pep.2004.01.020. [DOI] [PubMed] [Google Scholar]
- 33.Feng X.J., Wang J.H., Shan A.S., Teng D., Yang Y.L., Yao Y., Yang G.P., Shao Y.Y., Liu S., Zhang F. Fusion expression of bovine lactoferricin in Escherichia coli. Protein Expression and Purification. 2006;47:10–117. doi: 10.1016/j.pep.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 34.Skosyrev V.S., Kulesskiy E.A., Yakhnim A.V., Temirov Y.V., Vinokurov L.M. Expression of the recombinant antibacterial peptide sarcotoxin IA in Eschericha coli cells. Protein Expression and Purification. 2003;28:350–356. doi: 10.1016/s1046-5928(02)00697-6. [DOI] [PubMed] [Google Scholar]
- 35.Hara S., Yamakawa M. Production in Escherichia coli of moricin a novel type antibacterial peptide from the silkworm, Bombyx mori. Biochemical and Biophysical Research Communications. 1996;22:664–669. doi: 10.1006/bbrc.1996.0461. [DOI] [PubMed] [Google Scholar]
- 36.Carrington J.C., Cary S.M., Parks T.D., Dougherty W.G. A second proteinase encoded by a plant polyvirus genome. EMBO Journal. 1989;8:365–370. doi: 10.1002/j.1460-2075.1989.tb03386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jenny R.J., Mann K.G., Lundblad R.L. A critical review of the methods for cleavage of fusion proteins with thrombin and factor Xa. Protein Expression and Purification. 2003;31:1–11. doi: 10.1016/s1046-5928(03)00168-2. [DOI] [PubMed] [Google Scholar]
- 38.Prouty W., Goldberg A. Effects of protease inhibitors on protein breakdown in Escherichia coli. Journal of Biological Chemistry. 1972;247:3341–3352. [PubMed] [Google Scholar]
- 39.Bachmair A., Finley D., Varshavsky A. In vivo half-life of a protein is a function of its amino-terminal residue. Science. 1986;234:179–186. doi: 10.1126/science.3018930. [DOI] [PubMed] [Google Scholar]
- 40.Arnau J., Lauritzen C., Petersen G.E., Pedersen J. Current strategies for the use of affinity tags and tag removal for the purification of recombinant proteins. Protein Expression and Purification. 2006;48:1–13. doi: 10.1016/j.pep.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 41.Porath J. Immobilized metal ion affinity chromatography. Protein Expression and Purification. 1992;3:263–281. doi: 10.1016/1046-5928(92)90001-d. [DOI] [PubMed] [Google Scholar]
- 42.Hage D.S. Affinity chromatography: a review of clinical applications. Clinical Chemistry. 1999;45:593–615. [PubMed] [Google Scholar]
- 43.Chester K., Pedley B., Tolner B., Violet J., Mayer A., Sharma S., Boxer G., Green A., Nagl S., Begent R. Engineering antibodies for clinical applications in cancer. Tumor Biology. 2004;25:91–98. doi: 10.1159/000077727. [DOI] [PubMed] [Google Scholar]
- 44.Abdullah N., Chase H.A. Removal of poly-histidine fusion tags from recombinant proteins puri ed by expanded bed adsorption. Biotechnology and Bioengineering. 2005;92:501–513. doi: 10.1002/bit.20633. [DOI] [PubMed] [Google Scholar]
- 45.Tan Y.P., Ling T.C., Tan W.S., Yuso K., Tey B.T. Purification of recombinant nucleocapsid protein of Newcastle disease virus from unclarified feedstock using expanded bed adsorption chromatography. Protein Expression and Purification. 2006;46:14–121. doi: 10.1016/j.pep.2005.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]