

Abstract
Bcr-Abl is a dysregulated tyrosine kinase whose mechanism of activation is unclear. Here, we demonstrate that, like c-Abl, Bcr-Abl is negatively regulated through its SH3 domain. Kinase activity, transformation, and leukemogenesis by Bcr-Abl are greatly impaired by mutations of the Bcr coiled-coil domain that disrupt oligomerization, but restored by an SH3 point mutation that blocks ligand binding or a complementary mutation at the intramolecular SH3 binding site defined in c-Abl. Phosphorylation of tyrosines in the activation loop of the catalytic domain and the linker between the SH2 and catalytic domains (SH2-CD linker) is dependent on oligomerization and required for leukemogenesis. These results suggest that Bcr-Abl has a monomeric, unphosphorylated state with the SH3 domain engaged intramolecularly to Pro1124 in the SH2-CD linker, the form that is sensitive to the inhibitor imatinib (STI-571). The sole function of the coiled-coil domain is to disrupt the autoinhibited conformation through oligomerization and intermolecular autophosphorylation.
Introduction
As the direct cause of chronic myeloid leukemia (CML), Bcr-Abl is a key therapeutic target. While the catalytic activity of the c-Abl tyrosine kinase is very tightly regulated in vivo (Pendergast, 2002), the chimeric Bcr-Abl protein has constitutively increased tyrosine kinase activity (Konopka and Witte, 1985). c-Abl is regulated through a complex mechanism that includes intramolecular inhibition via the SH3 Barila and Superti-Furga 1998, Brasher et al. 2001, Van Etten et al. 1995 and N-terminal domains (Pluk et al., 2002), regulatory tyrosine phosphorylation Brasher and Van Etten 2000, Dorey et al. 2001, and binding of cellular inhibitors Pendergast et al. 1991a, Wen and Van Etten 1997. Despite almost two decades of intensive research, the mechanism of Abl kinase dysregulation upon fusion with Bcr is not clearly understood. Recently, a small molecule inhibitor of the Abl kinase (imatinib mesylate, formerly STI-571) has been shown to be remarkably effective for induction of hematologic and cytogenetic remissions in CML patients (Kantarjian et al., 2002), but clinical resistance to imatinib treatment is a significant problem (Shah et al., 2002). A better understanding of the regulation of Bcr-Abl catalytic activity is essential for future rational drug development.
The N terminus of Bcr contains a region predicted to form an amphipathic α helix that tetramerizes as a peptide in vitro (McWhirter et al., 1993). The crystal structure of the Bcr coiled coil has recently been solved (Zhao et al., 2002), and the monomer forms a left-handed α helix characterized by a heptad repeat with apolar residues in the first (a) and fourth (d) positions of the repeat. Two monomers associate in an antiparallel dimer that stacks to form a tetramer, consistent with experimental observations. Deletion of the initial 63 amino acids of Bcr or insertion of proline-containing peptides into the α-helical domain generates Bcr-Abl fusion proteins with decreased in vivo tyrosine kinase activity and impaired induction of anchorage-independent growth in Rat1 fibroblasts (McWhirter et al., 1993). These results suggest that oligomerization of Bcr-Abl increases tyrosine kinase activity and may also mediate other events, such as crosslinking of F-actin (McWhirter and Wang, 1993), which may be essential for transformation and leukemogenesis.
By analogy to receptor tyrosine kinases that are dimerized by extracellular ligands (Schlessinger, 2000), a simple model suggests that fusion of Bcr activates the Abl kinase by dimerization-induced intermolecular autophosphorylation. Fibroblast transformation by Bcr-Abl lacking the coiled-coil domain can be restored by replacement with the leucine zipper domain from GCN4 (McWhirter and Wang, 1997) or a conditional oligomerization motif (K.M.S. and R.A.V., unpublished data), demonstrating that the transforming properties of Bcr-Abl, like c-Abl (Smith and Van Etten, 2001), can be activated by dimerization. Transformation by Bcr-Abl lacking the coiled-coil domain can also be partially restored by deletion of the SH3 domain (Maru et al., 1996). However, there is no direct evidence that oligomerization is the critical function of Bcr sequences in transformation and leukemogenesis by Bcr-Abl, and mechanistic roles for autophosphorylation and the SH3 domain in Bcr-Abl catalytic and biological activity have not been defined.
To clarify the role of the Bcr coiled-coil domain in regulating the catalytic and oncogenic activity of p210 Bcr-Abl, we generated several novel Bcr coiled-coil mutations that are predicted to abrogate oligomerization, as well as complementary point mutations in the SH3 domain, a putative SH3 binding site, and potential autophosphorylation sites in Abl. We examined the ability of these mutants to form oligomers in vivo, their capacity to transform fibroblasts and hematopoietic cell lines, and their proficiency to induce leukemia in a murine bone marrow retroviral transduction/transplantation model of CML.
Results
Generation of Bcr-Abl Coiled-Coil Domain Mutants
Bcr contains an N-terminal coiled-coil domain formed by amino acids 28 through 63 that is included in all Bcr-Abl fusion proteins found in Ph+ leukemia patients (p190, p210, and p230). To investigate the role of the Bcr coiled-coil domain in regulating catalytic activity, cellular transformation, and leukemogenesis by p210 Bcr-Abl, we generated a series of mutations that are predicted to disrupt the coiled-coil domain while leaving the extreme N terminus of Bcr-Abl intact. p210Δcc removes amino acids 31 to 69 and deletes the entire coiled-coil domain (Figure 1C) . p210F54P mutates phenylalanine at position 54 to proline (Figure 1G) and is similar to a proline-containing peptide insertion mutant described previously (McWhirter et al., 1993). The F54P mutation places a proline in the middle of the domain and is predicted to disrupt the coiled-coil by introducing a kink to the middle of the α helix. The third mutant, p210LZA (Figure 1L), has point mutations at the “a” and “d” positions across the coiled-coil domain, with Val28, Ile31, Leu35, Ile42, Leu45, Val49, Met56, Leu59, and Leu63 changed to alanine. These point mutations are predicted to conserve the helical structure of the coiled-coil but replace the residues essential for stabilization of the dimer structure. Similar alanine substitution mutations in the leucine zipper domain of murine coronavirus spike protein abolish oligomerization and biological activity (Luo et al., 1999).
Figure 1.

Structure of the Bcr-Abl Proteins Used in this Study
The functional domains of the p210 Bcr-Abl protein are depicted, including (from left to right) Bcr exons 1–13 including the coiled-coil (CC) oligomerization domain and Abl exons 2–11 including the SH3 domain with location of Pro1013, the SH2 domain, the SH2-CD linker region with Pro1124 and Tyr1127, kinase domain with location of activation loop Tyr1294, three nuclear localization signals, DNA and actin binding domains, nuclear export signal, and the location of the BstEII site in the cDNA.
Bcr-Abl Coiled-Coil Domain Mutations Impair Oligomerization In Vivo
Bcr-Abl is predicted to form oligomers through the coiled-coil domain. To assess oligomerization by the coiled-coil domain mutants, we generated additional constructs that were truncated at a C-terminal BstEII site: p210Δcc/ΔBX, p210F54P/ΔBX, and p210LZA/ΔBX (Figures 1D, 1H, and 1M). The full-length and truncated constructs were coexpressed in 293T cells and immunoprecipitated with the GEX5 antibody (C) that recognizes only the full-length protein (Figure 2A , lane 3). The antibody pEX2 (N) recognizes and immunoprecipitates both full-length and truncated p210 proteins (Figure 2A, lane 2). Strong coassociation of the truncated p210 protein was seen with full-length p210 (Figure 2A) whereas the truncated p210Δcc no longer associated with the full-length version. Likewise, coassociation of the full-length and truncated p210 proteins was lost in the presence of either the F54P or LZA mutations. Thus, in vivo oligomerization of p210 Bcr-Abl is dependent on an intact amphipathic coiled-coil domain.
Figure 2.

Oligomerization of Bcr-Abl In Vivo Requires an Intact Amphipathic Coiled-Coil Domain
(A) Whole-cell extracts (ext; lanes 1, 4, 7, 10, 13), anti-Abl N-terminal (anti-pEX2 [Konopka and Witte, 1985], N; lanes 2, 5, 8, 11, 14), and anti-Abl C-terminal (anti-GEX5 [Van Etten et al., 1995], C; lanes 3, 6, 9, 12, 15) immunoprecipitates from 293T cells coexpressing p210, p210Δcc, p210F54P, and p210LZA with their respective BstEII truncation mutants (ΔBX).
(B) Whole-cell extracts (ext; lanes 1, 4, 7), anti-Abl N-terminal (anti-pEX2, N; lanes 2, 5, 8), and anti-Abl C-terminal (anti-GEX5, C; lanes 3, 6, 9) immunoprecipitates from 293T cells coexpressing p210Δcc/P1013L, p210F54P/P1013L, and p210LZA/P1013L with their respective BstEII truncation mutants (ΔBX). Abl proteins were detected with a pan-reactive Abl antibody (8E9). Arrowheads indicate the positions of p210, p210ΔBX, and endogenous c-Abl.
Decreased In Vivo Tyrosine Kinase Activity of Bcr-Abl Coiled-Coil Deletion and Alanine Substitution Mutants Is Restored by Loss of SH3 Function
We next assessed the in vivo tyrosine kinase activity of the Bcr-Abl coiled-coil mutant proteins. We transduced interleukin-3 (IL-3)-dependent Ba/F3 hematopoietic cells with BCR-ABL retroviruses, selected G418-resistant populations in the presence of IL-3, and prepared extracts after IL-3 deprivation. The Bcr-Abl coiled-coil mutant proteins were consistently expressed at levels higher than wild-type p210 Bcr-Abl (Figure 3 , bottom panel), suggesting that oligomerization of Bcr-Abl may affect the stability of the protein in vivo, possibly because activated Abl proteins are more sensitive to proteasomal degradation (Echarri and Pendergast, 2001). Despite higher Bcr-Abl levels, cells expressing p210Δcc or p210LZA had greatly decreased levels of phosphotyrosine on cellular proteins compared with wild-type p210-expressing cells (Figure 3, middle panel). Although tyrosine phosphorylation of a 210 kDa species that likely represents Bcr-Abl was moderately decreased in these cells, use of an antibody that specifically recognizes phosphorylated Tyr1127 in Bcr-Abl demonstrated that the p210Δcc and p210LZA proteins were essentially unphosphorylated at this site (Figure 3, top panel). Phosphorylation at the homologous residue in c-Abl is known to stimulate Abl kinase activity (Brasher and Van Etten, 2000). Surprisingly, the in vivo catalytic activity of p210 was relatively unaffected by the F54P mutation. This appeared to be the consequence of residual oligomerization of this mutant protein, because substitution of alanines for the “a” and “d” coiled-coil residues of p210F54P resulted in very low kinase activity (Figure 3, compare p210F54P and p210F45P/LZA). These results demonstrate that oligomerization is required for dysregulation of Bcr-Abl kinase activity in vivo.
Figure 3.

Mutations in SH3 or SH2-CD Linker Rescue In Vivo Tyrosine Kinase Activity of Oligomerization-Defective Bcr-Abl
Anti-phospho-Abl (top panel), anti-phosphotyrosine (middle panel), and anti-Abl (bottom panel) immunoblots of lysates from parental Ba/F3 (mock) or G418-resistant cells expressing the indicated proteins and starved of IL-3 for 4 hr. The extracts were equally loaded as determined by endogenous c-Abl levels. The slightly elevated levels of phosphotyrosine in cells expressing p210Δcc or p210LZA compared to parental Ba/F3 cells is consistent with previous observations of low but detectable tyrosine kinase activity of Bcr-Abl coiled-coil domain deletion mutants McWhirter et al. 1993, Zhang et al. 2001.
Previous studies demonstrated that transformation of cytokine-dependent hematopoietic cell lines by Bcr-Abl lacking amino acids 1–63 can be partially rescued by deletion of the SH3 domain (Maru et al., 1996). To determine whether the in vivo tyrosine kinase activity of oligomerization-defective Bcr-Abl can be restored by loss of SH3 ligand binding function, we generated two additional sets of Bcr-Abl mutants. In the first set, proline 1013 was mutated to leucine to generate p210Δcc/P1013L, p210F54P/P1013L, and p210LZA/P1013L (Figures 1E, 1I, and 1N). This mutation, which corresponds to P131L in type Ib/IV c-Abl, has been shown to activate c-Abl kinase and transforming activity and disrupts SH3 function by eliminating proline-rich ligand binding (Van Etten et al., 1995). In a second set of constructs, proline 1124 was mutated to leucine to generate p210Δcc/P1124L, p210F54P/P1124L, and p210LZA/P1124L (Figures 1F, 1J, and 1O). The P1124L mutation lies in the linker region between the Abl SH2 and catalytic domains at the site of intramolecular SH3 binding defined in c-Abl, and mutation of the corresponding residue (P242) to alanine or leucine has also been shown to activate the catalytic and transforming activity of type Ib/IV c-Abl Barila and Superti-Furga 1998, Brasher et al. 2001. Addition of either the P1013L or P1124L mutations to p210Δcc or p210LZA increased levels of cellular phosphotyrosine to that of wild-type p210-expressing cells without grossly altering the spectrum of phosphorylated proteins (Figure 3), but had little effect on the high constitutive activity of p210F54P. These data demonstrate that loss of SH3 ligand binding function restores maximal in vivo catalytic activity to Bcr-Abl in the absence of oligomerization.
Bcr-Abl Coiled-Coil Deletion and Alanine Substitution Mutants Are Defective for Transformation of Fibroblasts
To determine the impact of oligomerization on Bcr-Abl transforming activity, we tested the ability of the coiled-coil mutants to induce the anchorage-independent growth of fibroblasts. NIH 3T3 cells were transduced with BCR-ABL retroviruses and plated in soft agar. Deletion of the minimal coiled-coil domain abrogated the ability of p210 Bcr-Abl to transform fibroblasts to anchorage-independent growth (Figure 4A) , consistent with previous observations McWhirter et al. 1993, Zhang et al. 2001 and indicating that retention of Bcr amino acids 1–30 does not influence transformation. The LZA mutation also significantly reduced transformation of NIH 3T3 fibroblasts. The p210F54P mutant transformed fibroblasts nearly as efficiently as wild-type p210, but mutation of the hydrophobic leucine zipper residues to alanine (p210F54P/LZA; see Figure 1K) abolished transformation, again suggesting that the F54P point mutation does not completely block oligomerization. When combined with either the P1013L or P1124L mutations, induction of anchorage-independent growth in fibroblasts was restored for both the Δcc and LZA mutants, while mutation of Pro1013 or Pro1124 had little effect on the robust transforming activity of p210F54P. Thus, point mutations in the Abl SH3 domain or its putative intramolecular ligand can compensate for inactivating mutations in the coiled-coil domain.
Figure 4.

Defective Fibroblast and Ba/F3 Cell Transformation by Bcr-Abl Coiled-Coil Mutants
(A) Soft agar colony formation by NIH 3T3 fibroblasts transduced with pMSCVneo retrovirus expressing the indicated protein, normalized to one proviral copy per cell as described in Experimental Procedures. Transformation by p210F54P/LZA was not normalized by proviral copy number, but this virus had a neomycin resistance titer equivalent to the other stocks. Note that the y axis is a logarithmic scale. Standard deviation is indicated by the bars. Defective fibroblast transformation by the p210Y1294F mutant has been reported previously (Pendergast et al., 1993).
(B) Proliferation of Ba/F3 cell populations expressing p210, p210Δcc, p210F54P, or p210LZA in the absence of IL-3. Viable cell counts of triplicate wells were determined at 24 hr intervals. Bars indicate standard deviations. The difference in cell number at 96 hr between p210 and either p210Δcc or p210LZA (asterisk) was significant (p = 0.01, unpaired t test).
(C) Proliferation of Ba/F3 cell populations expressing p210, p210Δcc/P1013L, p210F54P/P1013L, or p210LZA/P1013L.
(D) Proliferation of Ba/F3 cell populations expressing p210, p210Δcc/P1124L, p210F54P/P1124L, or p210LZA/P1124L.
To confirm that the SH3 point mutations do not affect oligomerization of the p210 coiled-coil mutants, we generated additional constructs that were truncated at the N-terminal BstEII site, p210Δcc/P1013L/ΔBX, p210F54P/P1013L/ΔBX, and p210LZA/P1013L/ΔBX. The full-length and truncated constructs were coexpressed in 293T cells and immunoprecipitated as described above. None of the coiled-coil/P1013L double mutant proteins coassociated in vivo (Figure 2B), arguing that rescue of fibroblast transformation activity by SH3 mutation is not due to restoration of Bcr-Abl oligomerization.
The c-Abl kinase is activated fully following sequential autophosphorylation at two sites: Tyr412 in the activation loop of the catalytic domain and Tyr245 in the linker region between the SH2 and catalytic domains (Brasher and Van Etten, 2000). Using tyrosine to phenylalanine substitution mutants, we examined the requirement for these sites for Bcr-Abl kinase activity and transformation. Mutation of either the homologous activation loop tyrosine (Tyr1294) or the SH2-CD linker tyrosine (Tyr1127) alone modestly decreased Bcr-Abl in vivo kinase activity, while a Y1127/1294F double mutant displayed even lower activity (Figure 3). Tyr to Phe mutation at either position was sufficient to abolish fibroblast transformation by Bcr-Abl (Figure 4A). Taken together, these results demonstrate that loss of either oligomerization or autophosphorylation results in defective fibroblast transformation by Bcr-Abl.
Bcr-Abl Coiled-Coil Deletion and Alanine Substitution Mutants Have Decreased Mitogenic Activity in Cytokine-Dependent Hematopoietic Cells
Previous studies suggested that the coiled-coil domain is absolutely required for abrogation of cytokine dependence in a hematopoietic cell line by Bcr-Abl (McWhirter et al., 1993). To examine the effects of Bcr-Abl oligomerization in hematopoietic cells, we transduced murine IL-3-dependent Ba/F3 cells with BCR-ABL retroviruses, selected for G418 resistance, and tested populations for growth in the absence of IL-3 (Figure 4B). Like p210 Bcr-Abl, the p210F54P construct was able to support rapid IL-3-independent growth. In contrast, p210Δcc and p210LZA induced greatly diminished proliferation under the same conditions. If followed for an additional 48 hr, cultures expressing p210Δcc or p210LZA reached confluence (data not shown), demonstrating that oligomerization of Bcr-Abl enhances IL-3-independent growth but is not absolutely required for transformation of these cells, in agreement with more recent work He et al. 2002, Zhang et al. 2001. Mutation of the SH3 domain (P1013L) or the SH2-CD linker (P1124L) in p210Δcc and p210LZA resulted in IL-3 independent growth that was comparable to that of cells expressing wild-type p210 (Figures 4C and 4D). As in fibroblasts, the loss of Bcr-Abl oligomerization can be compensated by mutations that impair the negative regulatory function of the Abl SH3 domain.
Bcr-Abl Coiled-Coil Deletion and Alanine Substitution Mutants Are Defective for Transformation of Primary Bone Marrow B-Lymphoid Progenitors
To assess transformation of primary bone marrow B-lymphoid progenitors by the Bcr-Abl coiled-coil mutants, freshly harvested bone marrow was transduced with BCR-ABL retroviruses. When plated under Whitlock-Witte conditions, p210 Bcr-Abl stimulates the outgrowth of immature B-lymphoid cells that are stroma dependent and not highly leukemogenic in mice (McLaughlin et al., 1987). We have modified this assay to include serial dilution and plating of the transduced bone marrow, which allows for a qualitative assessment of transformation (Figure 5) . p210 Bcr-Abl expression resulted in the rapid outgrowth of cultures initiated with as few as 3 × 103 cells under these conditions (Figure 5A), while only adherent stromal cells were seen in cultures of untransduced cells (Figure 5B). The coiled-coil deletion or alanine substitution mutants were completely defective in this assay, with no detectable growth even at the highest plating density (Figures 5C and 5F). Expression of p210F54P resulted in growth of some wells at higher plating densities, but growth of the lower dilutions was impaired relative to wild-type p210 (Figure 5I), while the p210F54P/LZA double mutant was completely inactive (Figure 5O). Transformation by all three coiled-coil domain mutants was significantly increased by secondary mutations in the SH3 or SH2-CD linker sites, particularly by the P1124L mutation, which restored transformation to the level of wild-type p210 in all cases. Expression of p210Y1294F resulted in modestly decreased cell growth relative to that induced by wild-type p210 (Figure 5M), while the Y1127F and Y1127/1294F mutants showed no growth at any cell plating density (Figures 5L and 5N). These results demonstrate that oligomerization and autophosphorylation are required for transformation of primary B-lymphoid cells by Bcr-Abl. As with fibroblasts and hematopoietic cell lines, oligomerization via the coiled-coil domain can be functionally replaced by SH3 or SH2-CD linker mutations.
Figure 5.
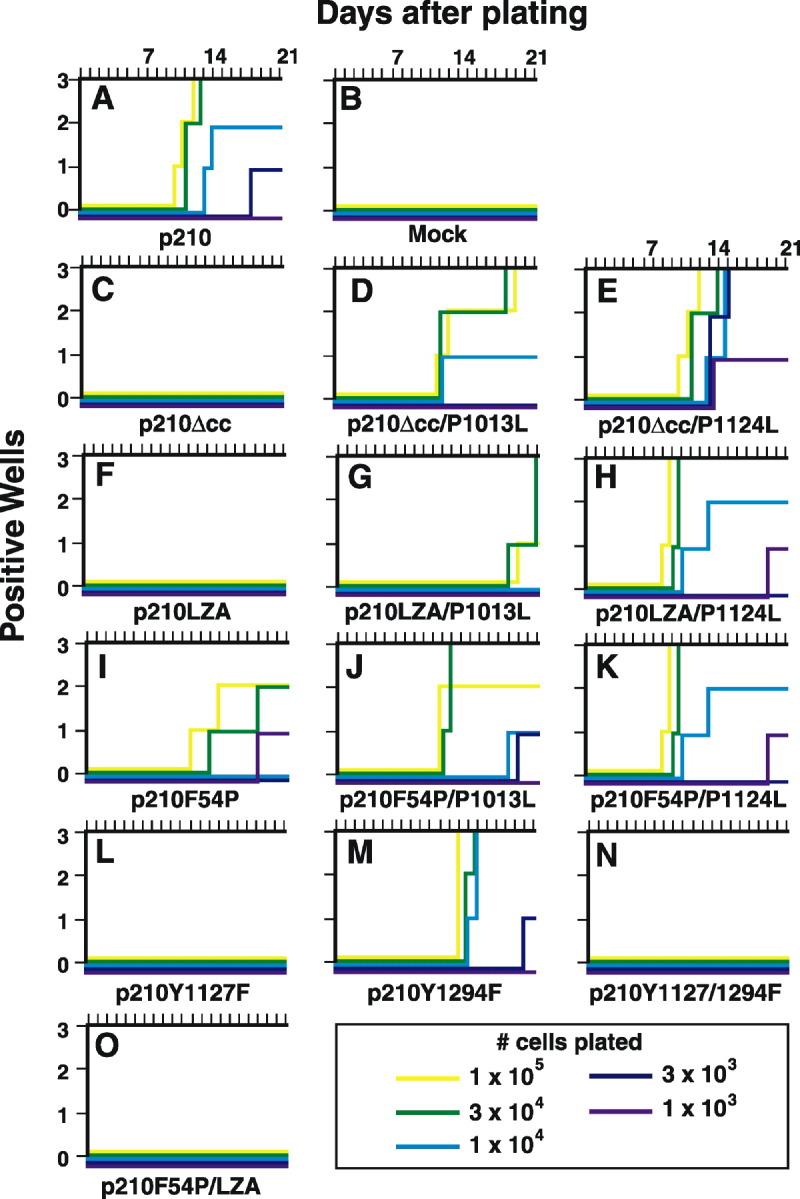
Bcr-Abl Coiled-Coil Deletion and Alanine Substitution Mutants Cannot Transform Primary Bone Marrow B-Lymphoid Progenitors
Balb/c bone marrow was transduced with the indicated BCR-ABL retrovirus and plated at indicated cell numbers per well in triplicate wells. Nontransduced cells were added to 106 total cells to provide stromal support. Wells were scored as positive when the viable nonadherent cell number reached 106/well. Although p210Y1294F was previously reported to be defective in this assay (Pendergast et al., 1993), this might be due to low virus titer in that study.
Oligomerization and Autophosphorylation Are Required for Induction of CML by Bcr-Abl
Transformation of fibroblasts and hematopoietic cell lines by Bcr-Abl mutants does not correlate well with their ability to induce leukemia in vivo (Van Etten, 2002), emphasizing the need to assess the role of Bcr-Abl oligomerization, SH3 function, and autophosphorylation in leukemogenesis. Hence, we tested the ability of the Bcr-Abl coiled-coil mutants to induce CML-like myeloproliferative disease in mice using the bone marrow retroviral transduction/transplantation model. In this assay, mice transplanted with p210-transduced bone marrow stem/progenitor cells from 5-fluorouracil-treated donors develop fatal myeloproliferative disease within 4 weeks posttransplantation (Figure 6) , characterized by peripheral blood leukocytosis with greatly increased neutrophils, hepatosplenomegaly, and infiltration of spleen, liver, and lungs with maturing myeloid cells (Li et al., 1999). In contrast, mice transplanted with marrow transduced with p210Δcc or p210LZA failed to develop CML-like disease, but instead succumbed to T-lymphoid leukemia/lymphoma (T-ALL) after a long latent period (Figures 6A and 6B), characterized by thymic enlargement and circulating CD4/CD8-positive cells (data not shown). Similar T-lymphoma occurs in recipients of marrow expressing Bcr-Abl lacking the entire Bcr N terminus or with a point mutation in the Grb2 binding site at Tyr177 He et al. 2002, Million and Van Etten 2000, Zhang et al. 2001, and results from transduction of T-lymphoid progenitors in the donor bone marrow. Recipients of marrow transduced with p210F54P developed rapidly fatal acute B-lymphoid leukemia (B-ALL) characterized by moderate splenomegaly, lymphadenopathy, normal thymus, and a hemorrhagic malignant pleural effusion that was the cause of death (Figure 6C). The same disease is induced by p210 Bcr-Abl with a point mutation in the SH2 domain that impairs catalytic activity (Roumiantsev et al., 2001). These results demonstrate that efficient oligomerization through the coiled-coil domain and maximal levels of Bcr-Abl kinase activity are required for induction of CML.
Figure 6.

Oligomerization and Autophosphorylation Are Required for Induction of CML by Bcr-Abl
Kaplan-Meier survival curve for recipients of marrow transduced with p210 Bcr-Abl and the indicated mutants. The number of individual mice in each arm is indicated. The p210 curve (solid black line) consists of 11 animals from three independent transplantation experiments. The disease phenotype of each animal is indicated by the shaded symbols. Mice that succumbed to two diseases simultaneously are indicated by the multi-shaded symbols. Significant differences in survival (Mantel-Cox test) were found between recipients of marrow transduced with p210 versus p210Δcc (p < 0.0001), p210Δcc versus p210ΔccP1013L (p = 0.0001), p210 versus p210LZA (p < 0.0001), p210LZA versus p210LZAP1124L (p = 0.026), p210 versus p2101294F (p < 0.0001), and p210 versus p210Y1127/1294F (p = 0.001).
The P1013L SH3 mutation rescued efficient induction of CML-like disease by p210Δcc (Figure 6A) with all recipients succumbing within 10 weeks posttransplant with leukocytosis due to maturing myeloid cells, hepatosplenomegaly, and pulmonary infiltrates and hemorrhage. These mice had normal lymph nodes and thymus and no evidence of B- or T-lymphoid malignancy based on histopathological and flow cytometric analysis (data not shown). This result is consistent with the induction of CML-like disease by p210 with deletions in both the coiled-coil and SH3 domains (Zhang et al., 2001) and implicates directly the ligand binding function of the SH3 domain in the rescue mechanism. The P1124L SH2-CD linker mutation also restored induction of CML-like disease by p210Δcc although less efficiently (Figure 6A). Likewise, mutation at either Pro1013 or Pro1124 restored induction of CML-like myeloproliferative disease by p210LZA (Figure 6B). Two recipients of p210LZA/P1013L-transduced marrow succumbed to myeloproliferative disease but with longer latency compared to mice receiving either p210- or p210Δcc/P1013L-transduced marrow, suggesting that the SH3 point mutation complements this particular coiled-coil domain mutation inefficiently, as it does for B-lymphoid transformation (Figure 5G). The P1124L mutation rescued p210LZA more effectively, with five of seven recipients developing CML-like disease and the remaining two animals developing T-ALL and histiocytic sarcoma (HS), another Bcr-Abl-induced hematologic malignancy that can develop in transplant recipients after long latent periods (Li et al., 1999). Interestingly, the P1013L mutation also partially rescued induction of myeloproliferative disease by p210F54P, with three of eight recipients developing typical CML-like disease and one succumbing to simultaneous CML and B-ALL. The remaining mice in this cohort had myeloproliferative disease with splenomegaly and peripheral blood leukocytosis, but eventually succumbed to T-lymphoma or HS. Together, these results argue that oligomerization of Bcr-Abl via the coiled-coil domain is required for induction of CML-like myeloproliferative disease in mice, but disruption of the SH3 domain ligand binding function or a complementary mutation in the intramolecular SH3 binding site can compensate for defects in oligomerization.
Mutation of the homolog of the major autophosphorylation site in c-Abl significantly attenuated leukemogenesis by p210 Bcr-Abl, with the majority of recipients of p210Y1294F-transduced marrow developing CML-like disease but only after a significant delay (Figure 6D). Single mutation of the homolog of the c-Abl secondary autophosphorylation site in the SH2-CD linker (Y1127F) had no effect on leukemogenesis, but p210 lacking both potential autophosphorylation sites (p210Y1127/1294F) was severely impaired for induction of CML-like disease, with the majority of mice lacking evidence of myeloproliferative disease and developing T-lymphoma (Figure 6D). These results demonstrate that two regulatory autophosphorylation sites that stimulate the catalytic activity of c-Abl by reversing an autoinhibited state are also required for efficient induction of CML by Bcr-Abl.
Discussion
The precise mechanism of dysregulation of c-Abl tyrosine kinase activity upon fusion of Bcr sequences to the Abl N terminus has been unclear despite intensive study. This deficiency in part reflected our limited understanding of the control of c-Abl catalytic activity, but several recent studies have begun to clarify this. A critical component of the c-Abl regulatory mechanism is shared with Src kinases: the Abl SH3 domain binds to a single proline (Pro242 in type Ib/IV c-Abl) in the SH2-CD linker that is conserved in Src family members. Mutations in SH3 that block ligand binding or mutation of the Pro242 SH3 binding site dysregulate c-Abl kinase activity in vivo and in vitro Barila and Superti-Furga 1998, Brasher et al. 2001, Van Etten et al. 1995. Enzymological studies of purified c-Abl have shown that Abl can undergo intermolecular autophosphorylation at two distinct regulatory tyrosines, Tyr412 and Tyr245 (Brasher and Van Etten, 2000). Phosphorylation of Tyr412 in the kinase domain activation loop stimulates Abl kinase activity and induces rapid phosphorylation at Tyr245, which may displace the SH3 domain from the adjacent Pro242 site, and results in a highly active enzyme with activity equivalent to SH3-mutated c-Abl. Physiological activation of c-Abl may involve auto- or trans-phosphorylation of Tyr412, perhaps by a Src family kinase (Plattner et al., 1999), followed by loss of self-inhibition by the SH3 domain.
Although the SH3 domains of Abl and Src carry out similar autoinhibitory functions, the two kinases differ in one important regulatory feature. A C-terminal phosphotyrosine in Src family kinases (Tyr527 in c-Src) functions as an intramolecular SH2 domain binding site (Sicheri et al., 1997) and is required for proper regulation of kinase activity. c-Abl is unphosphorylated in its inactive state (Van Etten et al., 1995) and, lacking this SH2-C-terminal interaction, employs other mechanisms to stabilize its inactive conformation. In the case of type Ib c-Abl, recent studies demonstrate that sequences N-terminal to the SH3 domain (the N-terminal “cap”) bind to the Abl catalytic domain (Pluk et al., 2002) and are required along with the N-terminal myristoyl group (Hantschel et al., 2003) for proper regulation of Abl kinase activity upon overexpression in vivo. The structural role of the Abl N terminus was revealed in a striking crystallographic study that demonstrated an interaction of the myristoyl group with the C-terminal lobe of the Abl catalytic domain (Nagar et al., 2003). Binding of the myristoyl group induces a conformational change that permits the docking of the SH2 domain with the noncatalytic face of the C-lobe in a fashion that closely resembles the inactive conformation of Src. Together, these studies suggest that there may be several routes to activation of c-Abl through displacement of either the myristoyl group, the SH3 domain, or the SH2 domain. However, there are several features of c-Abl that are unaccounted for by this model. When expressed at physiological levels, type Ia c-Abl is regulated despite the lack of a myristoyl group, as is a c-Abl Ib mutant lacking the myristoylation site and first 45 residues of the cap (Franz et al., 1989). In addition, there is evidence that c-Abl is also regulated in vivo by phosphoinositide lipids (Plattner et al., 2003) and protein inhibitors (Wen and Van Etten, 1997). Thus, further studies are necessary before we will have a complete understanding of c-Abl regulation.
Given these new insights into c-Abl regulation, how is Abl dysregulated upon fusion with Bcr? Relative to c-Abl, the Bcr-Abl fusion protein retains the Abl SH3 domain but lacks Abl first exon sequences and myristoylation and has gained the coiled-coil domain. A simple model suggested that oligomerization through the Bcr coiled-coil domain might constitutively dysregulate the Abl kinase. While deletion of the Bcr coiled-coil domain decreases kinase activity and transformation by Bcr-Abl (McWhirter et al., 1993), the underlying mechanism was not known. Using novel Bcr coiled-coil mutants, we have provided direct evidence that oligomerization of Bcr-Abl is the important kinase-activating function of the coiled-coil domain. The Bcr-Abl coiled-coil deletion and alanine substitution mutants failed to oligomerize, were defective for transformation of fibroblasts and primary B-lymphoid cells, and were unable to induce CML-like myeloproliferative disease in mice. The low but detectable in vivo tyrosine kinase of monomeric Bcr-Abl may reflect loss of a portion of the Abl N-terminal cap and the myristoyl group. The p210F54P mutant exhibited significant in vivo kinase and cell transforming activity that likely reflects residual oligomerization undetected by our stringent coimmunoprecipitation assay, because the p210F54P/LZA double mutant had low kinase activity and was completely defective for transformation. Phosphorylation of Bcr-Abl at two key regulatory tyrosines is both dependent on oligomerization and required for leukemogenesis, demonstrating that the critical event induced by Bcr-Abl oligomerization is likely to be intermolecular autophosphorylation.
Complete deletion of the SH3 domain can restore cell transformation by Bcr-Abl lacking the coiled-coil domain (Maru et al., 1996), but the mechanism involved was also unknown. We demonstrated that in vivo kinase activity, cellular transformation, and leukemogenesis by the Bcr-Abl coiled-coil deletion and alanine substitution mutants were restored by a point mutation (P1013L) in the Abl SH3 domain that disrupts proline-rich ligand binding or by a complementary mutation (P1124L) in the SH2-CD linker region at the site of intramolecular SH3 binding defined in c-Abl (Barila and Superti-Furga, 1998). Thus, Bcr-Abl is still autoregulated via its SH3 domain despite the lack of myristoylation, and cannot induce CML unless this inhibition is overcome through oligomerization and transphosphorylation or by SH3 mutation. It has been postulated that oligomerization of Bcr-Abl by the coiled-coil domain might contribute to the pathogenesis of CML through mechanisms other than direct effects on Bcr-Abl kinase activity, such as crosslinking of F-actin microfilaments (McWhirter and Wang, 1993). However, our results demonstrate that Bcr-Abl can induce CML-like disease independent of oligomerization when released from its autoinhibited state by other means.
Taken together, these observations suggest that an elegant and simple mechanism governs Bcr-Abl catalytic activity (Figure 7) . Our results argue that Bcr-Abl can assume an inactive state where the enzyme is monomeric and unphosphorylated. The primary consequence of oligomerization of Bcr-Abl is intermolecular autophosphorylation at Tyr1294 in the activation loop of the catalytic domain. As in c-Abl (Brasher and Van Etten, 2000), Tyr1294 phosphorylation may lead to secondary phosphorylation events, including phosphorylation of Tyr1127, the homolog of c-Abl Tyr245, which results in displacement of the SH3 domain from the SH2-CD linker. The order of phosphorylation may not be obligatory, as autophosphorylation of the linker tyrosine in both c-Abl and Bcr-Abl can occur when the activation loop tyrosine is mutated (Brasher and Van Etten, 2000; Figure 3). Enzymological studies of purified Bcr-Abl proteins should be helpful in confirming this model. While there are additional tyrosine phosphorylation sites in Bcr-Abl and c-Abl, our results suggest that Tyr1294 and Tyr1127 are the major sites that influence the regulation of catalytic activity in both enzymes. Other phosphorylation sites may contribute to leukemogenesis by Bcr-Abl independent of any role in autoregulation, as is the case with Tyr177, which forms a binding site for the SH2 domain of Grb2 and is required for induction of CML-like leukemia by Bcr-Abl (Million and Van Etten, 2000).
Figure 7.

A Model for Bcr-Abl Kinase Activation
The Bcr sequences, SH3, SH2, and catalytic domains of Abl are depicted. In its monomeric form, Bcr-Abl is unphosphorylated and the Abl SH3 domain interacts with the linker region between the SH2 and catalytic domains. This form of the enzyme binds efficiently to imatinib (shown in orange). Upon oligomerization, a primary phosphorylation event at Tyr1294 is rapidly followed by secondary phosphorylation at Tyr1127, which disrupts the SH3-linker interaction and results in full catalytic activity. In the absence of oligomerization via the coiled-coil domain, kinase and transformation activities can be restored by mutations in the SH3 domain that disrupt ligand binding or by complementary mutation of the SH3 ligand in the SH2-CD linker region. For simplicity, other phosphorylation sites including Tyr177 in Bcr are not shown.
There are several features of Bcr-Abl regulation yet unaccounted for by this model. The Abl SH2 domain seems to play a positive role in activation of Bcr-Abl catalytic activity, as deletions or point mutations in SH2 decrease Bcr-Abl kinase activity in vitro and in vivo (Roumiantsev et al., 2001) and impair Bcr-Abl transformation and leukemogenesis Pendergast et al. 1991b, Roumiantsev et al. 2001. The precise mechanism of SH2-dependent activation of Bcr-Abl is not known but has been suggested to involve SH2 binding to phosphoserine residues C-terminal to the coiled-coil domain in Bcr (Pendergast et al., 1991b). However, if the similarity between the autoinhibited conformations of Bcr-Abl and c-Abl extends to the SH2 domain, then SH2 should be docked to the C-lobe of the kinase domain when Bcr-Abl is monomeric, and might participate in kinase activation through binding of phosphotyrosine ligands (Hantschel et al., 2003). Testing the effect of mutations in SH2 that are predicted to disrupt H-bonding with the kinase domain C-lobe (Hantschel et al., 2003) on the activity of monomeric Bcr-Abl might clarify the role of SH2. There is also a discrepancy in the oncogenic activity of one of the mutants, p210Y1127F, which was completely defective for transformation of early B-lymphoid progenitors but readily induced myeloproliferative disease when expressed in hematopoietic stem cells. Other Bcr-Abl mutations, for example in the SH2 domain (Roumiantsev et al., 2001), also differentially affect lymphoid and myeloid leukemogenesis. In the case of p210Y1127F, it is possible that phospho-Tyr1127 forms a binding site for an SH2-containing protein that is critical for B-lymphoid transformation but not induction of CML.
Our results have important implications for the mechanism of inhibition of Bcr-Abl by imatinib (STI-571). Structural (Schindler et al., 2000) and enzymatic (Roumiantsev et al., 2002) studies have demonstrated that imatinib preferentially binds to and inhibits the unphosphorylated form of c-Abl. It is very likely that the autoinhibited form of Bcr-Abl we have postulated is also the imatinib-sensitive state of the fusion protein. As equilibrium may exist in vivo between the inactive and active states of Bcr-Abl depicted in Figure 7, imatinib probably acts to trap the enzyme in the autoinhibited, unphosphorylated state (Roumiantsev et al., 2002). Oligomerization of Bcr-Abl and activation of the enzyme through intermolecular autophosphorylation would force the catalytic domain into an open, activated conformation that is inconsistent with binding the phenylaminopyrimidine pharmacophore of the drug. Our studies predict that mutations in Bcr-Abl that impair its ability to assume the autoinhibited conformation, such as those we have defined in the SH3 and SH2-CD linker regions, would tend to drive the enzyme toward the activated state in vivo and could be associated with clinically significant resistance to imatinib. Interestingly, a recent random, unbiased screen for mutations in Bcr-Abl that confer imatinib resistance recovered mutations in the SH3 domain (including P1013Q) that are predicted to abolish ligand binding and in the SH2-CD linker adjacent to Pro1124 (Azam et al., 2003). The authors inferred from these mutants that Bcr-Abl has an autoregulated state that is similar to c-Abl, and our results provide direct proof of this hypothesis. In theory, the same mutations might contribute to the progression of chronic phase CML to blast crisis by increasing in vivo kinase activity of Bcr-Abl. Indeed, some Abl kinase domain mutations found in patients with advanced CML and acquired resistance to imatinib are present at significant frequency in leukemic cells prior to initiation of therapy (Shah et al., 2002), suggesting that these mutations may be selected for during CML disease progression. Last, these results emphasize the need for crystal structures of both full-length c-Abl and Bcr-Abl, both to illuminate the mechanisms of Abl kinase regulation and to guide future rational drug development. Our studies also validate the development of small molecule inhibitors of Bcr-Abl oligomerization as potential therapeutic agents for CML.
Experimental Procedures
Constructs
The retroviral vector pMSCVneop210 has been described previously (Li et al., 1999). The Δcc, F54P, LZA, P1013L, P1124L, and Y1127F mutations were generated by PCR, while the Y1294F mutant was the gift of Dr. Ann Marie Pendergast, Duke University. p210LZA contains the following point mutations: V28A, I31A, L35A, I42A, L45A, V49A, M56A, L59A, L63A. p210ΔBX was generated by digesting the p210 cDNA with BstEII and XbaI, blunting the ends with T4 DNA polymerase, and self-ligation. The resulting mutation truncates p210 Bcr-Abl at amino acid 1677 and generates a protein with a predicted molecular mass of 172 kDa.
Cell Culture, Transfection, and Retroviral Transduction
For coprecipitation studies, 293T cells were transfected with 5 μg of the full-length p210 construct and 3 μg of the corresponding ΔBX construct. Retroviral stocks were prepared by transient transfection of 293T cells as described (Li et al., 1999). All stocks had titers of 6–12 × 106 neomycin-resistant colony-forming units per ml, and gave equivalent proviral copy number as determined by Southern blot analysis of genomic DNA from transduced fibroblasts or primary bone marrow.
Transformation of NIH 3T3 fibroblasts to anchorage-independent growth was as described previously (Van Etten et al., 1995), with results expressed as soft agar colonies/105 cells plated, normalized to one proviral copy/cell as determined by Southern blotting. Assessment of IL-3-independent proliferation of Ba/F3 cells was as described previously (Li et al., 1999). Primary bone marrow cells from Balb/c female mice (Taconic Farms, Germantown, NY) were transduced with retroviruses by co-sedimentation, and serial dilutions plated in 24-well dishes for Whitlock/Witte culture in RPMI 1640 medium supplemented with 20% fetal calf serum, 200 μM L-glutamine, 50 μM 2-mercaptoethanol, and penicillin/streptomycin. Nontransduced bone marrow was added for a total of 106 cells per well.
Immunoprecipitation and Immunoblotting
293T cells were lysed in RIPA buffer, incubated overnight with polyclonal anti-Abl GEX5 or pEX2 antiserum, immune complexes collected with protein A-Sepharose, washed in RIPA buffer, high salt buffer (500 mM NaCl, 20 mM Tris-HCl at pH 7.4, 1% Triton X-100), and low salt buffer (20 mM PIPES at pH 7.2), then subjected to SDS-PAGE and immunoblotting with anti-Abl (8E9, Pharmingen), anti-phospho-Abl (#2861, Cell Signaling Technology), and anti-phosphotyrosine (4G10, Upstate Biotechnology) antibodies.
Bone Marrow Transduction/Transplantation
Transduction of bone marrow and transplantation of Balb/c recipient mice (Taconic Farms, Germantown, MD) was as previously described (Li et al., 1999). The transplanted cell dose was 5 × 105 cells per recipient. Diseased mice were subjected to histopathologic and molecular analysis as described previously (Li et al., 1999).
Acknowledgements
We thank Jennifer Abraham for expert technical assistance. This work was supported by NIH grants HL07623 (K.M.S.) and CA90576 (R.A.V.) and a Leukemia and Lymphoma Society SCOR grant (R.A.V.). R.A.V. is a Scholar of the Leukemia and Lymphoma Society and the Carl and Margaret Walter Scholar in Blood Research at Harvard Medical School.
Published: July 24, 2003
References
- Azam M., Latek R.R., Daley G.Q. Mechanisms of autoinhibition and STI-571/imatinib resistance revealed by mutagenesis of BCR-ABL. Cell. 2003;112:831–843. doi: 10.1016/s0092-8674(03)00190-9. [DOI] [PubMed] [Google Scholar]
- Barila D., Superti-Furga G. An intramolecular SH3domain interaction regulates c-Abl activity. Nat. Genet. 1998;18:280–282. doi: 10.1038/ng0398-280. [DOI] [PubMed] [Google Scholar]
- Brasher B.B., Van Etten R.A. c-Abl has high intrinsic tyrosine kinase activity that is stimulated by mutation of the Src homology 3 domain and by autophosphorylation at two distinct regulatory tyrosines. J. Biol. Chem. 2000;275:35631–35637. doi: 10.1074/jbc.M005401200. [DOI] [PubMed] [Google Scholar]
- Brasher B.B., Roumiantsev S., Van Etten R.A. Mutational analysis of the regulatory function of the c-Abl Src homology 3 domain. Oncogene. 2001;20:7744–7752. doi: 10.1038/sj.onc.1204978. [DOI] [PubMed] [Google Scholar]
- Dorey K., Engen J.R., Kretzschmar J., Wilm M., Neubauer G., Schindler T., Superti-Furga G. Phosphorylation and structure-based functional studies reveal a positive and a negative role for the activation loop of the c-Abl tyrosine kinase. Oncogene. 2001;20:8075–8084. doi: 10.1038/sj.onc.1205017. [DOI] [PubMed] [Google Scholar]
- Echarri A., Pendergast A.M. Activated c-Abl is degraded by the ubiquitin-proteasome pathway. Curr. Biol. 2001;11:1759–1765. doi: 10.1016/s0960-9822(01)00538-3. [DOI] [PubMed] [Google Scholar]
- Franz W.M., Berger P., Wang J.Y.J. Deletion of an N-terminal regulatory domain of the c-abl tyrosine kinase activates its oncogenic potential. EMBO J. 1989;8:137–147. doi: 10.1002/j.1460-2075.1989.tb03358.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hantschel O., Nagar B., Guettler S., Kretzschmar J., Dorey K., Kuriyan J., Superti-Furga G. A myristoyl/phosphotyrosine switch regulates c-Abl. Cell. 2003;112:845–857. doi: 10.1016/s0092-8674(03)00191-0. [DOI] [PubMed] [Google Scholar]
- He Y., Wertheim J.A., Xu L., Miller J.P., Karnell F.G., Choi J.K., Ren R., Pear W.S. The coiled-coil domain and Tyr177 of bcr are required to induce a murine chronic myelogenous leukemia-like disease by bcr/abl. Blood. 2002;99:2957–2968. doi: 10.1182/blood.v99.8.2957. [DOI] [PubMed] [Google Scholar]
- Kantarjian H., Sawyers C.L., Hochhaus A., Guilhot F., Schiffer C., Gambacorti-Passerini C., Niederwieser D., Resta D., Capdeville R., Zoellner U. Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N. Engl. J. Med. 2002;346:645–652. doi: 10.1056/NEJMoa011573. [DOI] [PubMed] [Google Scholar]
- Konopka J.B., Witte O.N. Detection of c-abl tyrosine kinase activity in vitro permits direct comparison of normal and altered abl gene products. Mol. Cell. Biol. 1985;5:3116–3123. doi: 10.1128/mcb.5.11.3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Ilaria R.L., Million R.P., Daley G.Q., Van Etten R.A. The P190, P210, and P230 forms of the BCR/ABL oncogene induce a similar chronic myeloid leukemia-like syndrome in mice but have different lymphoid leukemogenic activity. J. Exp. Med. 1999;189:1399–1412. doi: 10.1084/jem.189.9.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Z., Matthews A.M., Weiss S.R. Amino acid substitutions within the leucine zipper domain of the murine coronavirus spike protein cause defects in oligomerization and the ability to induce cell-to-cell fusion. J. Virol. 1999;73:8152–8159. doi: 10.1128/jvi.73.10.8152-8159.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maru Y., Witte O.N., Shibuya M. Deletion of the ABL SH3 domain reactivates de-oligomerized BCR-ABL for growth factor independence. FEBS Lett. 1996;379:244–246. doi: 10.1016/0014-5793(95)01518-3. [DOI] [PubMed] [Google Scholar]
- McLaughlin J., Chianese E., Witte O.N. In vitro transformation of immature hematopoietic cells by the P210 bcr/abl oncogene product of the Philadelphia chromosome. Proc. Natl. Acad. Sci. USA. 1987;84:6558–6562. doi: 10.1073/pnas.84.18.6558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWhirter J.R., Wang J.Y.J. An actin-binding function contributes to transformation by the Bcr-Abl oncoprotein of the Philadelphia chromosome-positive leukemias. EMBO J. 1993;12:1533–1546. doi: 10.1002/j.1460-2075.1993.tb05797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWhirter J.R., Wang J.Y.J. Effect of Bcr sequences on the cellular function of the Bcr-Abl oncoprotein. Oncogene. 1997;15:1625–1634. doi: 10.1038/sj.onc.1201342. [DOI] [PubMed] [Google Scholar]
- McWhirter J.R., Galasso D.L., Wang J.Y.J. A coiled-coil oligomerization domain of Bcr is essential for the transforming function of Bcr-Abl oncoproteins. Mol. Cell. Biol. 1993;13:7587–7595. doi: 10.1128/mcb.13.12.7587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Million R.P., Van Etten R.A. The Grb2 binding site is required for induction of chronic myeloid leukemia-like disease in mice by the Bcr/Abl tyrosine kinase. Blood. 2000;96:664–670. [PubMed] [Google Scholar]
- Nagar B., Hantschel O., Young M.A., Scheffzek K., Veach D., Bornmann W., Clarkson B., Superti-Furga G., Kuriyan J. Structural basis for the autoinhibition of c-Abl tyrosine kinase. Cell. 2003;112:859–871. doi: 10.1016/s0092-8674(03)00194-6. [DOI] [PubMed] [Google Scholar]
- Pendergast A.M. The Abl family kinases: mechanisms of regulation and signaling. Adv. Cancer Res. 2002;85:51–100. doi: 10.1016/s0065-230x(02)85003-5. [DOI] [PubMed] [Google Scholar]
- Pendergast A.M., Muller A.J., Havlik M.H., Clark R., McCormick F., Witte O.N. Evidence for regulation of the human ABL tyrosine kinase by a cellular inhibitor. Proc. Natl. Acad. Sci. USA. 1991;88:5927–5931. doi: 10.1073/pnas.88.13.5927. a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendergast A.M., Muller A.J., Havlik M.H., Maru Y., Witte O.N. BCR sequences essential for transformation by the BCR-ABL oncogene bind to the ABL SH2 regulatory domain in a non-phosphotyrosine-dependent manner. Cell. 1991;66:161–171. doi: 10.1016/0092-8674(91)90148-r. b. [DOI] [PubMed] [Google Scholar]
- Pendergast A.M., Gishizky M.L., Havlik M.H., Witte O.N. SH1 domain autophosphorylation of P210 BCR/ABL is required for transformation but not growth factor independence. Mol. Cell. Biol. 1993;13:1728–1736. doi: 10.1128/mcb.13.3.1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plattner R., Kadlec L., DeMali K.A., Kazlauskas A., Pendergast A.M. c-Abl is activated by growth factors and Src family kinases and has a role in the cellular response to PDGF. Genes Dev. 1999;13:2400–2411. doi: 10.1101/gad.13.18.2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plattner R., Irvin B.J., Guo S., Blackburn K., Kazlauskas A., Abraham R.T., York Y.D., Pendergast A.M. A new link between the c-Abl tyrosine kinase and phosphoinositide signalling through PLC-gamma1. Nat. Cell Biol. 2003;5:309–319. doi: 10.1038/ncb949. [DOI] [PubMed] [Google Scholar]
- Pluk H., Dorey K., Superti-Furga G. Autoinhibition of c-Abl. Cell. 2002;108:247–260. doi: 10.1016/s0092-8674(02)00623-2. [DOI] [PubMed] [Google Scholar]
- Roumiantsev S., de Aos I., Varticovski L., Ilaria R.L., Van Etten R.A. The Src homology 2 domain of Bcr/Abl is required for efficient induction of chronic myeloid leukemia-like disease in mice but not for lymphoid leukemogenesis or activation of phosphatidylinositol 3-kinase. Blood. 2001;97:4–13. doi: 10.1182/blood.v97.1.4. [DOI] [PubMed] [Google Scholar]
- Roumiantsev S., Shah N.P., Gorre M.E., Nicoll J., Brasher B.B., Sawyers C.L., Van Etten R.A. Clinical resistance to the kinase inhibitor STI-571 in CML by mutation of Tyr253 in the Abl kinase domain P-loop. Proc. Natl. Acad. Sci. USA. 2002;99:10700–10705. doi: 10.1073/pnas.162140299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler T., Bornmann W., Pellicena P., Miller W.T., Clarkson B., Kuriyan J. Structural mechanism for STI-571 inhibition of Abelson tyrosine kinase. Science. 2000;289:1938–1942. doi: 10.1126/science.289.5486.1938. [DOI] [PubMed] [Google Scholar]
- Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;13:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- Shah N.P., Nicoll J.M., Nagar B., Gorre M.E., Paquette R.L., Kuriyan J., Sawyers C.L. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002;2:117–125. doi: 10.1016/s1535-6108(02)00096-x. [DOI] [PubMed] [Google Scholar]
- Sicheri F., Moarefi I., Kuriyan J. Crystal structure of the Src family tyrosine kinase Hck. Nature. 1997;385:602–609. doi: 10.1038/385602a0. [DOI] [PubMed] [Google Scholar]
- Smith K.M., Van Etten R.A. Activation of c-Abl kinase activity and transformation by a chemical inducer of dimerization. J. Biol. Chem. 2001;276:24372–24379. doi: 10.1074/jbc.M100786200. [DOI] [PubMed] [Google Scholar]
- Van Etten R.A. Studying the pathogenesis of BCR-ABL+ leukemia in mice. Oncogene. 2002;21:8643–8651. doi: 10.1038/sj.onc.1206091. [DOI] [PubMed] [Google Scholar]
- Van Etten R.A., Debnath J., Zhou H., Casasnovas J.M. Introduction of a loss-of-function point mutation from the SH3 region of the Caenorhabditis elegans sem-5 gene activates the transforming ability of c-abl in vivo and abolishes binding of proline-rich ligands in vitro. Oncogene. 1995;10:1977–1988. [PubMed] [Google Scholar]
- Wen S.-T., Van Etten R.A. The PAG gene product, a stress-induced protein with antioxidant properties, is an Abl SH3-binding protein and a physiological inhibitor of c-Abl tyrosine kinase activity. Genes Dev. 1997;11:2456–2467. doi: 10.1101/gad.11.19.2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Subrahmanyam R., Wong R., Gross A.W., Ren R. The NH2-terminal coiled-coil domain and tyrosine 177 play important roles in induction of a myeloproliferative disease in mice by Bcr-Abl. Mol. Cell. Biol. 2001;21:840–853. doi: 10.1128/MCB.21.3.840-853.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X., Ghaffari S., Lodish H., Malashkevich V.N., Kim P.S. Structure of the Bcr-Abl oncoprotein oligomerization domain. Nat. Struct. Biol. 2002;9:117–120. doi: 10.1038/nsb747. [DOI] [PubMed] [Google Scholar]