

Abstract
Although nitric oxide (NO) kills or inhibits the replication of a variety of intracellular pathogens, the antimicrobial mechanisms of NO are unknown. Here, we identify a viral protease as a target of NO. The life cycle of many viruses depends upon viral proteases that cleave viral polyproteins into individual polypeptides. NO inactivates the Coxsackievirus protease 3C, an enzyme necessary for the replication of Coxsackievirus. NO S-nitrosylates the cysteine residue in the active site of protease 3C, inhibiting protease activity and interrupting the viral life cycle. Substituting a serine residue for the active site cysteine renders protease 3C resistant to NO inhibition. Since cysteine proteases are critical for virulence or replication of many viruses, bacteria, and parasites, S-nitrosylation of pathogen cysteine proteases may be a general mechanism of antimicrobial host defenses.
Introduction
Nitric oxide (NO) is an antiviral effector of the immune system (45, 14, 44, 56). Viral infection can induce expression of the inducible nitric oxide synthase (iNOS) in cells and in animals. NO inhibits the replication of a variety of viruses (9, 30, 7, 8, 26, 1, 33, 65). We and others have previously shown that NO inhibits the replication of Coxsackievirus and other Picornaviruses (33, 42, 50, 15, 21, 68, 69). Coxsackievirus B3 (CVB3) infection induces iNOS expression in macrophages in mice, and CVB3 replicates more rapidly to higher titers and causes more tissue damage in mice lacking iNOS, compared to infections of wild-type mice. Exogenous NO inhibits replication of Coxsackievirus and poliovirus in vitro, reducing viral RNA and protein synthesis. Thus, iNOS and NO are important components of the immune response to CVB3 infection. However, the viral targets of NO are unknown.
Results
To explore the mechanism by which NO inhibits CVB3 replication, we first determined that NO inhibits CVB3 replication by blocking a step early in the viral life cycle. We infected HeLa cells with CVB3 and then at various times treated the infected cells with NO donors or control compounds; after 10 hr, we measured the amount of CVB3 produced during one viral life cycle. The NO donor S-nitroso-acetyl-penicillamine (SNAP) reduces the amount of CVB3 produced by infected HeLa cells, and the maximum effect of SNAP occurs when the NO donor is added 1–2 hr after infection (Figure 1). In contrast, the control compound acetyl-penicillamine (AP) has no effect. When SNAP is added to HeLa cells, most of the NO is released within 2 hr (data not shown). Since the life cycle of CVB3 lasts approximately 8 hr, these data suggest that NO inhibits an early step in the life cycle of CVB3.
Figure 1.

The NO Donor SNAP Inhibits CVB3 Replication
HeLa cells were infected with CVB3 at a multiplicity of infection of 10. SNAP (200 μM) was added at various times after infection as indicated. The amount of CVB3 after 10 hr of infection was measured by the plaque assay. To one set of infected cells (C), 200 μM AP was added 1 hr after infection. To another set of cells (-1), SNAP was added 1 hr before infection. n = 3 ± SD.
Coxsackievirus, a member of the Picornaviridae, is a nonenveloped virus with a single-stranded RNA genome, whose life cycle includes virion attachment to the host cell, penetration, translation of the viral RNA into a polyprotein, autocleavage of the polyprotein by viral proteases into viral polypeptides, replication of the viral genome, assembly of the virion, and exit from the cell. Since protease activity is a critical step early in the life cycle of Coxsackievirus, we hypothesized that NO inhibits CVB3 proteases.
Exogenous NO Inhibits Protease Processing of an Endogenous Viral Polyprotein
In order to examine the effect of NO upon viral protease processing of viral proteins, we added the NO donor SNAP or its control AP to infected HeLa cells; cell lysates were harvested at various times after infection, fractionated by SDS-PAGE, and analyzed by immunoblotting for viral polypeptides. NO inhibits proteolysis of the viral protein precursor 2ABC, delaying and reducing the amount of the cleavage products 2BC and 2C detected by immunoblotting (Figure 2A). Since 3Cpro is responsible for cleavage of 2ABC, one interpretation of these data is that exogenous NO inhibits the proteolysis of the viral polypeptide 2BC.
Figure 2.

Figure 2. NO Inhibits Proteolysis of CVB3 Polypeptide 2BC in Infected Cells
(A) Exogenous NO donor SNAP inhibits proteolysis of CVB3 polypeptide. HeLa cells were infected with CVB3 at a multiplicity of infection of 10. After 1 hr, 200 μM of SNAP (+) or AP (-) was added, and after 3.5 hr guanidine was added to inhibit new viral RNA synthesis. HeLa cells were harvested at various times indicated after guanidine treatment. Cell lysates were fractionated by SDS-PAGE and immunoblotted with an antibody to CVB3 polypeptide 2C. These experiments were repeated twice with similar results.
(B) Endogenous NO from activated macrophages inhibits proteolysis of CVB3 polypeptide. HeLa cells were infected with CVB3 at a multiplicity of infection of 10. After 1 hr, plastic inserts containing resting (R) or LPS/IFN-stimulated (S) macrophages were added to the wells containing infected HeLa cells. Some cultures were also treated with the NOS inhibitor NAME (0–10 mM). Cells were harvested 5 hr after infection and cell lysates analyzed for CVB3 polypeptide 2C as above. This experiment was repeated five times with similar results.
(C) LPS or IFN do not directly affect CVB3 replication. HeLa cells were infected with CVB3 at a multiplicity of infection of 10. After 1 hr, LPS or IFN or media alone was added to the wells containing infected HeLa cells. Some cultures were also treated with the NOS inhibitor NAME (0–10 mM). Cells were harvested 5 hr after infection and cell lysates analyzed for CVB3 polypeptide 2C as above.
Endogenous NO Inhibits Protease Processing of an Endogenous Viral Polyprotein
To answer the question of whether or not endogenous NO produced by iNOS would also interfere with viral proteolytic processing, we added macrophages that had been induced to express iNOS to infected cells. First, HeLa cells were infected with CVB3, and then cell culture inserts were added that contained RAW 264.7 macrophages, activated or not with lipopolysaccharide and interferon-γ. The macrophages were separated from the infected HeLa cells by a 0.4 μM pore size membrane, which would allow the passage of NO from macrophages to infected cells. The NOS inhibitor nitro-arginine-methyl-ester (NAME) was added to some cocultures of infected HeLa cells and activated macrophages, in order to confirm that any antiviral effect of activated macrophages was a result of NO, and not some other mechanism. Cells were harvested 5 hr after infection, and the cleavage of the viral polyprotein 2ABC was measured by immunoblotting as above.
Activated macrophages block proteolytic processing of the CVB3 polyprotein 2ABC, resulting in a decrease in the amount of 2BC and 2C (Figure 2B). Infected HeLa cells alone (lane 2) contain three prominent polypeptides recognized by the antibody to 2C (as well as another band also found in noninfected cells [lane 1]). These three polypeptides are 2ABC and its two proteolytic products 2BC and 2C. Resting macrophages (lane 3) do not affect the intensity of the three viral polypeptides. However, macrophages activated by lipopolysaccharide (LPS) and interferon-γ (IFNγ) (lane 4) release a soluble mediator that diffuses across the 4 μM pores and inhibits the processing of 2ABC into 2BC and 2C. This soluble mediator may be NO, since increasing concentrations of the NOS inhibitor NAME are associated with decreasing amounts of NO and also with increasing proteolytic processing of the viral polypeptide (lanes 5–7). (LPS or IFN alone have no effect upon viral replication in HeLa cells [Figure 2C].) Thus, NO synthesized by activated macrophages can diffuse into infected cells and inhibit the viral proteases that process the viral polyproteins.
Since exogenous and endogenous NO inhibit proteolytic processing of Coxsackievirus polyproteins, we therefore focused our attention on the effect of NO upon Coxsackievirus proteases.
NO Inhibits CVB3 Protease Activity
Picornaviruses encode two cysteine proteases, 2Apro and 3Cpro (35, 24, 55, 11). The protease 2Apro cleaves the viral polyprotein at the boundary between the structural proteins and the nonstructural proteins, VP1 and 2A, producing two polyproteins. The protease 3Cpro is thought to cleave the resulting polyproteins into ten viral polypeptides, including the cleavage of 2BC into 2B and 2C (27, 22, 51). The protease 3Cpro is a critical component of the Picornavirus life cycle: mutation of any of the three amino acids in the active site of 3Cpro inactivates the protease and abolishes viral replication (27, 22, 51). One of the residues of the catalytic site of 3Cpro is cysteine at amino acid position 147, and since NO can nitrosylate cysteine residues (61, 62, 60), we hypothesized that NO inhibits the CVB3 3Cpro.
We therefore prepared pure 3Cpro and various protease substrates. We subcloned the cDNA encoding 3Cpro into a bacterial expression vector, expressed a fusion protein consisting of 3Cpro tagged with His6 in bacteria, and then purified 3Cpro (Figure 3A) (Ghosh and Lowenstein 1996). To prepare an authentic protease substrate, we subcloned, expressed, and purified a fusion polypeptide, consisting of glutathione-S-transferase, the entire CVB3 3B polypeptide, and the amino acid residues 1–140 of 3C; we refer to this fusion protein as GST-3B-3CT. This fusion protein contains an authentic 3Cpro cleavage site between 3B and 3C, consisting of the 3Cpro recognition motif AXXQG (where Q is the P1 residue) (35, 24, 38, 55). (The fragment of 3C included in the fusion protein consists of the first 140 amino acids of 3C. It lacks the Cys147 of the active site and is thus incapable of protease activity.)
Figure 3.

NO Inhibits Cleavage of a Viral Target by Purified 3Cpro
(A) 3Cpro was expressed in bacteria and purified. Bacterial lysates were subjected to SDS-PAGE: lane 1, noninduced bacteria; lane 2, bacteria induced with IPTG; lane 3, high-speed supernatant of induced bacteria; lane 4, flow-through from a Ni+ column; lanes 5–9, sequential column washes with 10 mM imidazole; and lanes 10–13, fractions from a Ni+ column eluted with 500 mM imidazole.
(B) The purified fusion protein GST-3B-3CT, which contains an authentic 3Cpro cleavage site, was incubated with buffer, purified 3Cpro, or 3Cpro with the control compound AP, or the NO donor SNAP, or SNAP and DTT. The reaction mixture was analyzed by immunoblot with an antibody to GST and quantitated by densitometry. n = 3 ± SD, and * p < 0.05.
(C) The cleavage experiment was then repeated using spermine NONOate (SP-NO) as an NO donor or its control compound spermine (SP). n = 3 ± SD, and * p < 0.05.
To test the activity of recombinant protease, purified 3Cpro was incubated with the protease substrate GST-3B-3CT and the reaction mixture analyzed by immunoblotting with an antibody to GST. Purified 3Cpro is capable of cleaving the polyprotein GST-3B-3CT, as detected by immunoblotting (Figure 3B, immunoblot lanes 1 and 2).
In order to measure the susceptibility of 3Cpro to NO, we added a variety of NO donors or their controls to 3Cpro and a protease substrate. The NO donor SNAP inhibits the proteolytic activity of 3Cpro in a dose-dependent manner, while its control acetyl-penicillamine (AP), which has no nitroso group, has no effect (Figure 3B). DTT, an agent capable of reducing the nitrosylation of cysteine residues, reverses the NO inhibition of 3Cpro. Another NO donor, spermine NONOate, also inhibits 3Cpro cleavage of GST-3B-3CT. DTT also reverses this effect (Figure 3C).
To confirm these results, we used luciferase as another protease substrate (25, 19, 64). Firefly luciferase serves as a 3Cpro substrate because it contains the 3Cpro motif AXXQG. Cleavage of luciferase was measured by monitoring luciferase activity. 3Cpro cleaves luciferase in a dose- and time-dependent manner (Figure 4A and Figure 4B). The NO donors SNAP or spermine NONOate but not their corresponding control drugs inhibit 3Cpro activity, and these inhibitions are reversed by DTT (Figure 4C). It was previously shown that the nonphysiological reagents iodoacetamide and N-ethylmaleimide can inhibit 3Cpro (19, 54). Thus, NO inhibits the activity of 3Cpro.
Figure 4.
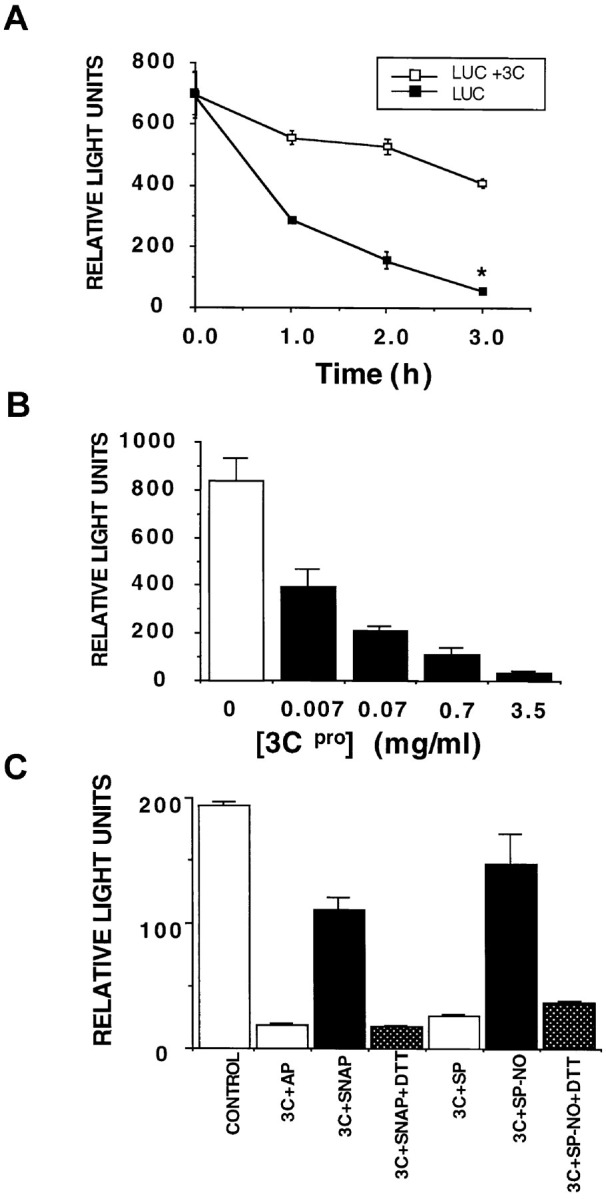
NO Inhibits 3Cpro Cleavage of Luciferase
(A) Purified 3Cpro was added to luciferase, which contains a 3Cpro consensus cleavage motif, and the activity of luciferase remaining at various times was assayed using a luminometer. n = 3 ± SD.
(B) Increasing amounts of 3Cpro were added to luciferase for 3 hr, and the amount of luciferase assayed as above. n = 3 ± SD.
(C) Luciferase was incubated alone, or in the presence of 3Cpro with the following additions: the control compound AP, the NO donor SNAP, SNAP and DTT, the control compound spermine (SP), the NO donor spermine NONOate (SP-NO), or spermine NONOate and DTT. n = 3 ± SD.
NO Nitrosylates the CVB3 Protease Active Site Cysteine
3Cpro has only one cysteine, namely Cys147 in the active site. To determine whether or not NO nitrosylates this cysteine, we first measured the effect of NO donors upon its thiol group with the Ellman assay (13, 70). Purified 3Cpro was incubated with NO donors for 1 hr and then dialyzed. 5,5′- Dithiobis (2-nitrobenzoic acid) was added, the increase of absorbance at 412 nm was measured, and the concentration of thiols was calculated from the molar extinction coefficient of the nitrothiobenzoate ion. NO donors but not their corresponding control compounds eliminate the sulfhydryl group from purified 3Cpro over time (Figure 5A).
Figure 5.
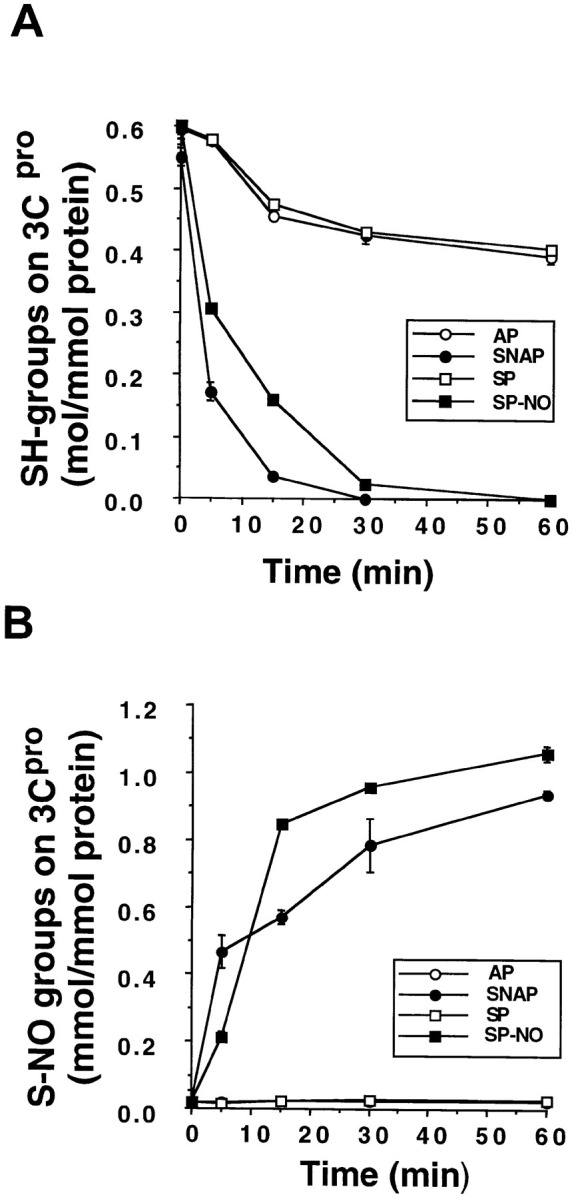
NO Eliminates a Sulfhydryl Group from 3Cpro and Nitrosylates 3Cpro
Purified 3Cpro was incubated with AP, SNAP, spermine, or spermine NONOate. (A) Analysis of the 3Cpro sulfhydryl group. Treated 3Cpro was incubated with an excess of 5,5 dithiobis(2-nitrobenzoate) (DNTB) and its absorption measured at 412 nm. n = 4 ± SD. (B) Nitrosylation of the 3Cpro active site Cys147. 3Cpro was exposed to NO donors or their controls, then incubated with HgCl2, and the amount of nitrite released was measured by the Griess reaction. n = 4 ± SD.
We next measured the effect of NO donors upon the formation of a nitroso-thiol group by the Saville assay (58, 70). Purified 3Cpro was incubated with various NO donors or their controls, and excess NO donors and nitrite were removed from the sample with Sephadex G-25 column chromatography. 3Cpro was then incubated with HgCl2 and the amount of nitrite was measured in the Griess reaction (Stamler et al. 1992a). For every 1 mole of 3Cpro added to the reaction, approximately 1 mole of nitrite is detected (Figure 5B). These data suggest that NO nitrosylates 3Cpro on a cysteine residue. Since the active site Cys147 is the only cysteine residue in 3Cpro, the data imply that NO nitrosylates the active site cysteine residue.
Mutating the CVB3 Protease Active Site Cysteine to Serine Renders the Protease Resistant to NO
To confirm that nitrosylation of the active site Cys147 is responsible for the inhibition of 3Cpro, we created a mutant protease by PCR-directed mutagenesis, substituting a serine for the cysteine at position 147. The resultant mutant protease, C147S3Cpro, still has catalytic activity, although at a level greatly reduced from wild-type protease, as others have shown (27, 22, 51). The effect of NO upon protease activity was determined by adding NO donors and luciferase to either wild-type or mutant protease. NO reduces wild-type 3Cpro activity, and DTT restores wild-type protease activity (Figure 6). However, NO has no effect upon mutant C147S3Cpro activity, nor does DTT change mutant protease activity. Thus, the Cys147 of the catalytic triad is critical in determining the susceptibility of 3Cpro to NO.
Figure 6.

Mutation of the 3Cpro Active Site Cys147 Renders 3Cpro Resistant to NO
A mutant 3Cpro with serine substituted for cysteine at amino acid residue 147 was purified from bacteria. Wild-type 3Cpro (1 μg/ml) and mutated 3Cpro (5 μg/ml) were incubated for 3 hr with luciferase and AP, SNAP, or SNAP and DTT, and the activity of luciferase assayed in a luminometer. n = 3 ± SD, repeated twice with similar results.
Discussion
The major finding of this study is that NO inhibits Coxsackievirus replication at least in part by nitrosylating the cysteine residue in the active site of a viral protease, thereby drastically reducing the activity of this enzyme. This protease is critical to the life cycle of Picornaviruses. Nitrosylation of cysteine residues is one mechanism by which NO can directly modify a polypeptide, and NO in theory can reduce the activity of an enzyme if its catalytic site includes a cysteine residue (62, 63, 60). For example, NO inhibits activity of the cysteine protease caspase-3 (31, 40, 47, 52). Although proteins capable of being nitrosylated at a cysteine residue contain a consensus nitrosylation motif, the 3Cpro lacks this sequence (Stamler et al. 1997). Coxsackievirus may have a selective advantage if its viral protease lacks a nitrosylation consensus sequence, decreasing cysteine reactivity to NO. The other protease of Coxsackievirus, 2Apro, is also a cysteine protease, and may be a target of NO as well.
Previously, we showed that NO inhibits Coxsackievirus replication in vitro and that NO reduces both viral RNA synthesis and viral protein synthesis (Zaragoza et al. 1997). NO inactivation of 3Cpro could explain this reduction in both viral RNA and viral protein levels, given the characteristics of the Coxsackievirus life cycle. After Coxsackievirus enters the cell, viral RNA is translated into a large polyprotein. The 3Cpro excises itself from the polyprotein and cleaves the polyprotein into its components, including 3Dpol. This RNA-dependent RNA polymerase 3Dpol then replicates the viral genome. Thus, 3Cpro processing acts upstream of viral RNA synthesis and is necessary to generate 3Dpol. Therefore, nitrosylation and inactivation of 3Cpro would be expected to block not only viral protein processing but also viral RNA synthesis.
Although our data suggest that NO inhibits activity of Coxsackievirus protease 3Cpro, there are other potential viral targets of NO as well. For example, NO could affect the RNA polymerase activity of 3Dpol. Although 3Dpol lacks the typical molecular targets of NO, cysteine residues or iron in its active site, NO could affect its activity by other mechanisms. It is also possible that NO could inactivate host proteins necessary for viral replication. The lack of effect of NO upon viral replication when NO is added to cells prior to infection argues against this possibility (Figure 1). Nonetheless, NO can affect a variety of host proteins; however, the net effect of NO upon host processes either harmful or beneficial to viral replication is unknown.
The genomes of many viruses encode a polyprotein that after translation is cleaved into smaller polypeptides by a viral protease. These proteases fall into several categories based on their active site residues, including cysteine proteases, serine proteases, and aspartic proteases (35, 24, 4). NO can inhibit the replication of other viruses that encode cysteine proteases, such as members of the Picornavirus family and the Coronavirus family (45, 56). In contrast, NO does not inhibit the replication of some viruses that encode serine proteases, such as alphaviruses. However, NO also inhibits the replication of some viruses that do not encode cysteine proteases, implying that there exist other viral targets of NO. For example, the EBV transcription factor Zta and ribonucleotide reductase may be targets of NO (46, 49). Nonetheless, to our knowledge there are no viruses that have cysteine proteases that are resistant to NO.
Cysteine proteases are also critical to the replication and virulence of a variety of other micro-organisms. Inhibitors of cysteine proteases block the replication of Plasmodium falciparum, Plasmodium berghei, Leishmania major, Schistosoma mansoni, and Trypanosoma cruzi. (5, 12, 23, 36, 39, 48, 67, 59). Furthermore, inhibitors of cysteine proteases also reduce the virulence of Streptococcus pyogenes, Porphyromonas gingivalis, Schistosoma mansoni, Naegleria fowler, and Plasmodium flaciparum. (28, 29, 3, 6, 18, 53, 67, 37, 43, 20). (For example, the malaria parasite uses its cysteine protease falcipain to degrade hemoglobin for use as a source of amino acids, and cysteine protease inhibitors block the ability of the parasite to infect erythrocytes and to replicate [18, 10].) It is striking that NO inhibits the replication of all of these parasites (45, 14, 44, 56). NO production may thus be a general mechanism by which the host defends itself against infection by viruses and other pathogens whose life cycle depends upon cysteine proteases.
Experimental Procedures
Cells and Viruses
Coxsackievirus B3 (CVB3) (Nancy strain, generous gift of C.G. Gauntt) was grown in HeLa cells and measured by the plaque assay (16, 68).
To measure the effect of the NO donor SNAP upon proteolytic processing of CVB3 polypeptides in cells, CVB3 at an MOI of 10 was added to HeLa cell cultures and 200 μM SNAP was added after 1 hr, and cells were harvested at various times afterward and cell lysates fractionated by SDS-PAGE and analyzed by immunoblotting. Guanidine HCl 3 mM, a specific inhibitor of CVB3 RNA replication, was added to all infected cells 3 hr after infection in order to inhibit new viral RNA synthesis.
To measure the effect of endogenously synthesized NO upon proteolytic processing of CVB3 polypeptides in cells, CVB3 at an MOI of 10 was added to HeLa cell cultures in 6-well plates, and then macrophages (that previously had been plated onto cell culture inserts and then stimulated or not for 16 hr with LPS 10 ng/ml plus interferon-μ 50 U/ml) were added to the 6-well plates. In some wells containing infected HeLa cells and activated macrophages, nitro-arginine-methyl-ester (NAME) was added. Cells were harvested 5 hr after infection and cell lysates fractionated by SDS-PAGE and analyzed by immunoblotting.
Immunoblot Analysis
Immunoblotting was performed as described previously, using an antibody to CVB3 2C (generous gift of Christine Hohenadl) or antibody to GST (Pharmacia) (Zaragoza et al. 1997).
Northern Analysis
Total RNA was isolated from infected HeLa cells as described previously and probed with a radiolabeled CVB3 DNA probe (generous gift of R. Kandolf) (32, 68). Blots were rehybridized with a β-actin cDNA as an internal control.
Expression and Purification of Viral Polypeptides
The cDNA encoding CVB3 3Cpro was cloned between the NdeI and BamHI sites of the expression vector pSG04 so that a (His)6 amino terminal tag is fused to 3Cpro (Ghosh and Lowenstein 1996). The 3Cpro mutant C147S3Cpro was constructed using PCR-directed mutagenesis, using as a mutagenic primer GAATGCTTATGTACAACTTCCCCACA AGAGCAGGCCAGTCTGGTGGA. The plasmid encoding the 3Cpro target polypeptide GST-3B-3CT was prepared by inserting the DNA encoding the 6 carboxy terminal amino acids of CVB3 3B and amino acid residues 1–140 of CVB3 3C into plasmid pGEX 4T2 (Pharmacia). The sequence of all constructions was confirmed by sequencing. The 3Cpro and C147S3Cpro were expressed in bacteria as described previously and purified by metal chelate chromatography (Ghosh and Lowenstein 1996). The GST-3B/3CT fusion protein was expressed in bacteria and purified by glutathione-agarose column chromatography according to the manufacturer’s instructions (Pharmacia). Proteins were dialyzed and stored at −70°C.
Measurement of 3Cpro Activity
The activity of the 3Cpro was assayed using two different substrates.
Viral Polypeptide Substrate
The GST-3B/3CT fragment (1 mg/ml) was incubated with 3Cpro (0.7 mg/ml) in a final volume of 500 μl for 16 hr at 30°C in protease buffer (50 mM Tris-HCl [pH 7.5], 0.1 M NaCl, and 1 mM EDTA). The protease reaction mixture was subjected to 15% SDS-PAGE and immunoblotted using an antibody to GST.
Luciferase Substrate
A 1:108 dilution of a 1 mg/ml solution of firefly luciferase (Sigma, St. Louis, MI) was incubated with 3Cpro in a final volume of 200 μl at 30°C in protease buffer. At various times, 20 μl aliquots were harvested, and the activity of the luciferase was measured using luciferin in a luminometer (Turner model 20e).
Determination of Thiol Content of 3Cpro
Purified 3Cpro (0.5 mg) was incubated in protease buffer with various NO donors (5 mM SNAP or spermine NONOate) or their corresponding control drugs (5 mM aminopenicillamine or spermine) and then dialyzed in a buffer containing 0.1 M sodium phosphate, 6 M guanidine HCl, and 1 mM EDTA. 5,5′-dithiobis(2-nitrobenzoate) was added, and the absorbance at 412 nm was measured over time. The concentration of thiols was determined from the molar extinction coefficient.
Determination of S-Nitrosylation of 3Cpro
Purified 3Cpro was incubated with NO donors for 1 hr and then dialyzed. 5,5′-Dithiobis (2-nitrobenzoic acid) was added to remove nitrite from the sample and then a solution of 3% sulfanilamide and 0.25% HgCl2 was added, followed by 0.1% ethylenediamine. The mixture was incubated at 22°C for 10 min and the absorbance measured at 540 nm.
Acknowledgements
Coxsackievirus B3 (Nancy strain) was a generous gift of C. G. Gauntt. The antibody to CVB3 2C is a generous gift of Christine Hohendadl. The CVB3 DNA is a generous gift of R. Kandolf. This work was supported by a grant from the Spanish Ministry of Education and Culture (M. S.), R01 HL53615 and P50 HL52315 from the National Institutes of Health (C. Z. and C. J. L.), a grant from the Cora and John H. Davis Foundation (C. J. L.), and a grant from the Bernard Bernard Foundation (C. J. L.).
Footnotes
To whom correspondence should be addressed (e-mail: clowenst@welchlink.welch.jhu.edu).
References
- 1.Akaike T, Noguchi Y, Ijiri S, Setoguchi K, Suga M, Zheng Y.M, Dietzschold B, Maeda H. Pathogenesis of influenza virus-induced pneumonia: involvement of both nitric oxide and oxygen radicals. Proc. Natl. Acad. Sci. USA. 1996;93:2448–2453. doi: 10.1073/pnas.93.6.2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akarid K, Sinet M, Desforges B, Gougerot-Pocidalo M.A. Inhibitory effect of nitric oxide on the replication of a murine retrovirus in vitro and in vivo. J. Virol. 1995;69:7001–7005. doi: 10.1128/jvi.69.11.7001-7005.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aldape K, Huizinga H, Bouvier J, McKerrow J. Naegleria fowleri: characterization of a secreted histolytic cysteine protease. Exper. Parasitol. 1994;78:230–241. doi: 10.1006/expr.1994.1023. [DOI] [PubMed] [Google Scholar]
- 4.Babe L.M, Craik C.S. Viral proteases: evolution of diverse structural motifs to optimize function. Cell. 1997;91:427–430. doi: 10.1016/s0092-8674(00)80426-2. [DOI] [PubMed] [Google Scholar]
- 5.Bailly E, Jambou R, Savel J, Jaureguiberry G. Plasmodium falciparum: differential sensitivity in vitro to E-64 (cysteine protease inhibitor) and Pepstatin A (aspartyl protease inhibitor) J. Protozool. 1992;39:593–599. doi: 10.1111/j.1550-7408.1992.tb04856.x. [DOI] [PubMed] [Google Scholar]
- 6.Bedi G.S, Williams T. Purification and characterization of a collagen-degrading protease from Porphyromonas gingivalis. J. Biol. Chem. 1994;269:599–606. [PubMed] [Google Scholar]
- 7.Bi Z, Reiss C.S. Inhibition of vesicular stomatitis virus infection by nitric oxide. J. Virol. 1995;69:2208–2213. doi: 10.1128/jvi.69.4.2208-2213.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bukrinsky M.I, Nottet H.S, Schmidtmayerova H, Dubrovsky L, Flanagan C.R, Mullins M.E, Lipton S.A, Gendelman H.E. Regulation of nitric oxide synthase activity in human immunodeficiency virus type 1 (HIV-1)-infected monocytes: implications for HIV-associated neurological disease. J. Exper. Med. 1995;181:735–745. doi: 10.1084/jem.181.2.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Croen K.D. Evidence for antiviral effect of nitric oxide. Inhibition of herpes simplex virus type 1 replication. J. Clin. Invest. 1993;91:2446–2452. doi: 10.1172/JCI116479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dominguez J.N, Lopez S, Charris J, Iarruso L, Lobo G, Semenov A, Olson J.E, Rosenthal P.J. Synthesis and antimalarial effects of phenothiazine inhibitors of a Plasmodium falciparum cysteine protease. J. Med. Chem. 1997;40:2726–2732. doi: 10.1021/jm970266p. [DOI] [PubMed] [Google Scholar]
- 11.Dougherty W.G, Semler B.L. Expression of virus encoded proteinases: functional and structural similarities with cellular enzymes. Microbiol. Rev. 1993;57:781–822. doi: 10.1128/mr.57.4.781-822.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eakin A.E, Mills A.A, Harth G, McKerrow J.H, Craik C.S. The sequence, organization, and expression of the major cysteine protease (cruzain) from Trypanosoma cruzi. J. Biol. Chem. 1992;267:7411–7420. [PubMed] [Google Scholar]
- 13.Ellman, G.L. (1959). Arch. Biochem. Biophys. 82, 70–77. [DOI] [PubMed]
- 14.Fang F.C. Mechanisms of nitric oxide related antimicrobial activity. J. Clin. Invest. 1997;99:2818–2825. doi: 10.1172/JCI119473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flynn D.L, Becker D.P, Dilworth V.M, Highkin M.K, Hippenmeyer P.J, Houseman K.A, Levine L.M, Li M, Moormann A.E, Rankin A, et al. The herpesvirus protease: mechanistic studies and discovery of inhibitors of the human cytomegalovirus protease. Drug. Des. Discov. 1997;15:3–15. [PubMed] [Google Scholar]
- 16.Gauntt C.J, Trousdale M.D, LaBadie D.R, Paque R.E, Nealon T. Properties of coxsackievirus B3 variants which are amyocarditic or myocarditic for mice. J. Med. Virol. 1979;3:207–220. doi: 10.1002/jmv.1890030307. [DOI] [PubMed] [Google Scholar]
- 17.Ghosh S, Lowenstein J.M. A multifunctional vector system for heterologous expression of proteins in E. coli. Gene. 1996;176:249–255. doi: 10.1016/0378-1119(96)00260-0. [DOI] [PubMed] [Google Scholar]
- 18.Gluzman I.Y, Francis S.E, Oksman A, Smith C.E, Duffin K.L, Goldberg D.E. Order and specificity of the Plasmodium falciparum hemoglobin degradation pathway. J. Clin. Invest. 1994;93:1602–1608. doi: 10.1172/JCI117140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gorbalenya A.E, Svitkin Y.V. Protease of encephalomyocarditis virus: purification and role of SH groups in processing of the structural proteins precursor. Biochemistry (USSR) 1983;48:385–395. [Google Scholar]
- 20.Gubba S, Low D.E, Musser J.M. Expression and characterization of group A Streptococcus extracellular cysteine protease recombinant mutant proteins and documentation of seroconversion during human invasive disease episodes. Infect. Immun. 1998;66:765–770. doi: 10.1128/iai.66.2.765-770.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haddad I.Y, Sorscher E.J, Garver R.I, Jr., Hong J, Tzeng E, Matalon S. Modulation of adenovirus-mediated gene transfer by nitric oxide. Am. J. Respir. Cell Mol. Biol. 1997;16:501–509. doi: 10.1165/ajrcmb.16.5.9160832. [DOI] [PubMed] [Google Scholar]
- 22.Hammerle T, Hellen C.U, Wimmer E. Site-directed mutagenesis of the putative catalytic triad of poliovirus 3C proteinase. J. Biol. Chem. 1991;266:5412–5416. [PubMed] [Google Scholar]
- 23.Harth G, Andrews N, Mills A.A, Engel J.C, Smith R, McKerrow J.H. Peptide-fluoromethyl ketones arrest intracellular replication and intercellular transmission of Trypanosoma cruzi. Mol. Biochem. Parasitol. 1993;58:17–24. doi: 10.1016/0166-6851(93)90086-d. [DOI] [PubMed] [Google Scholar]
- 24.Hellen C.U, Kräusslich H.G, Wimmer E. Proteolytic processing of polyproteins in the replication of RNA viruses. Biochemistry. 1989;28:9881–9890. doi: 10.1021/bi00452a001. [DOI] [PubMed] [Google Scholar]
- 25.Holzman T.F, Baldwin T.O. Proteolytic inactivation of luciferases from three species of luminous marine bacteria, Beneckea harveyi, Photobacterium fischeri, and Photobacterium phosphoreum: evidence of a conserved structural feature. Proc. Natl. Acad. Sci. USA. 1980;77:6363–6367. doi: 10.1073/pnas.77.11.6363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hooper D.C, Ohnishi S.T, Kean R, Numagami Y, Dietzschold B, Koprowski H. Local nitric oxide production in viral and autoimmune diseases of the central nervous system. Proc. Natl. Acad. Sci. USA. 1995;92:5312–5316. doi: 10.1073/pnas.92.12.5312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ivanoff L.A, Towatari T, Ray J, Korant B.D, Petteway S.R., Jr. Expression and site-specific mutagenesis of the poliovirus 3C protease in Escherichia coli. Proc. Natl. Acad. Sci. USA. 1986;83:5392–5396. doi: 10.1073/pnas.83.15.5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kapur V, Majesky M.W, Li L.L, Black R.A, Musser J.M. Cleavage of interleukin 1 beta (IL-1 beta) precursor to produce active IL-1 beta by a conserved extracellular cysteine protease from Streptococcus pyogenes. Proc. Natl. Acad. Sci. USA. 1993;90:7676–7680. doi: 10.1073/pnas.90.16.7676. a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kapur V, Topouzis S, Majesky M.W, Li L.L, Hamrick M.R, Hamill R.J, Patti J.M, Musser J.M. A conserved Streptococcus pyogenes extracellular cysteine protease cleaves human fibronectin and degrades vitronectin. Microb. Pathog. 1993;15:327–346. doi: 10.1006/mpat.1993.1083. b. [DOI] [PubMed] [Google Scholar]
- 30.Karupiah G, Xie Q.W, Buller R.M, Nathan C, Duarte C, MacMicking J.D. Inhibition of viral replication by interferon-gamma-induced nitric oxide synthase. Science. 1993;261:1445–1448. doi: 10.1126/science.7690156. [DOI] [PubMed] [Google Scholar]
- 31.Kim Y.M, Talanian R.V, Billiar T.R. Nitric oxide inhibits apoptosis by preventing increases in caspase-3-like activity via two distinct mechanisms. J. Biol. Chem. 1997;272:31138–31148. doi: 10.1074/jbc.272.49.31138. [DOI] [PubMed] [Google Scholar]
- 32.Klump W.M, Bergmann I, Muller B.C, Ameis D, Kandolf R. Complete nucleotide sequence of infectious Coxsackievirus B3 cDNA: two initial 5′ uridine residues are regained during plus-strand RNA synthesis. J. Virol. 1990;64:1573–1583. doi: 10.1128/jvi.64.4.1573-1583.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Komatsu T, Bi Z, Reiss C.S. Interferon-gamma induced type I nitric oxide synthase activity inhibits viral replication in neurons. J. Neuroimmunol. 1996;68:101–108. doi: 10.1016/0165-5728(96)00083-5. [DOI] [PubMed] [Google Scholar]
- 34.Kreil T.R, Eibl M.M. Nitric oxide and viral infection: NO antiviral activity against a flavivirus in vitro, and evidence for contribution to pathogenesis in experimental infection in vivo. Virology. 1996;219:304–306. doi: 10.1006/viro.1996.0252. [DOI] [PubMed] [Google Scholar]
- 35.Kräusslich H.G, Wimmer E. Viral Proteinases. Annu. Rev. Biochem. 1988;57:701–754. doi: 10.1146/annurev.bi.57.070188.003413. [DOI] [PubMed] [Google Scholar]
- 36.Kumar S, van Pelt J.F, O’Dowd C.A, Hollingdale M.R, Sinden R.E. Effects of hormones and cysteine protease modulators on infection of HepG2 cells by Plasmodium berghei sporozoites in vitro determined by ELISA immunoassay. J. Parasitol. 1994;80:414–420. [PubMed] [Google Scholar]
- 37.Kuramitsu H, Tokuda M, Yoneda M, Duncan M, Cho M.I. Multiple colonization defects in a cysteine protease mutant of Porphyromonas gingivalis. J. Period. Res. 1997;32:140–142. doi: 10.1111/j.1600-0765.1997.tb01395.x. [DOI] [PubMed] [Google Scholar]
- 38.Lawson M.A, Semler B.L. Picornavirus protein processing—enzymes, substrates, and genetic regulation. Curr. Top. Microbiol. Immunol. 1990;161:49–80. [PubMed] [Google Scholar]
- 39.Li Z, Chen X.D.E.Z, Mendis C, Ring C.S, Roush W.R, Fegley G, Li R, Rosenthal P.J. Anti-malarial drug development using models of enzyme structure. Chem. Biol. 1994;1:31–37. doi: 10.1016/1074-5521(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 40.Li J, Billiar T.R, Talanian R.V, Kim Y.M. Nitric oxide reversibly inhibits seven members of the caspase family via S-nitrosylation. Biochem. Biophys. Res. Commun. 1997;240:419–424. doi: 10.1006/bbrc.1997.7672. [DOI] [PubMed] [Google Scholar]
- 41.Liu R.H, Jacob J.R, Tennant B.C, Hotchkiss J.H. Nitrite and nitrosamine synthesis by hepatocytes isolated from normal woodchucks (Marmota monax) and woodchucks chronically infected with woodchuck hepatitis virus. Cancer Res. 1992;52:4139–4143. [PubMed] [Google Scholar]
- 42.Lowenstein C.J, Hill S.L, Lafond-Walker A, Wu J, Allen G, Landavere M, Rose N.R, Herskowitz A. Nitric oxide inhibits viral replication in murine myocarditis. J. Clin. Invest. 1996;97:1837–1843. doi: 10.1172/JCI118613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lukomski S, Sreevatsan S, Amberg C, Reichardt W, Woischnik M, Podbielski A, Musser J.M. Inactivation of Streptococcus pyogenes extracellular cysteine protease significantly decreases mouse lethality of serotype M3 and M49 strains. J. Clin. Invest. 1997;99:2574–2580. doi: 10.1172/JCI119445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.MacMicking J.D, Xie Q.W, Nathan C. In: Annual Review of Immunology. Paul W.E, Fathman C.G, Metzger H, editors. Annual Reviews, Inc; Palo Alto, CA: 1997. Nitric oxide and macrophage function. 323–350.pp. [DOI] [PubMed] [Google Scholar]
- 45.Mannick J.B. The antiviral role of nitric oxide. Res. Immunol. 1995;146:693–697. doi: 10.1016/0923-2494(96)84920-0. [DOI] [PubMed] [Google Scholar]
- 46.Mannick J.B, Asano K, Izumi K, Kieff E, Stamler J.S. Nitric oxide produced by human B lymphocytes inhibits apoptosis and Epstein-Barr virus reactivation. Cell. 1994;79:1137–1146. doi: 10.1016/0092-8674(94)90005-1. [DOI] [PubMed] [Google Scholar]
- 47.Mannick J.B, Miao X.Q, Stamler J.S. Nitric oxide inhibits Fas-induced apoptosis. J. Biol. Chem. 1997;272:24125–24128. doi: 10.1074/jbc.272.39.24125. [DOI] [PubMed] [Google Scholar]
- 48.McGrath M.E, Eakin A.E, Engel J.C, McKerrow J.H, Craik C.S, Fletterick R.J. The crystal structure of cruzain: a therapeutic target for Chagas’ disease. J. Mol. Biol. 1995;247:251–259. doi: 10.1006/jmbi.1994.0137. [DOI] [PubMed] [Google Scholar]
- 49.Melkova Z, Esteban M. Inhibition of vaccinia virus DNA replication by inducible expression of nitric oxide synthase. J. Immunol. 1995;155:5711–5718. [PubMed] [Google Scholar]
- 50.Mikami S, Kawashima S, Kanazawa K, Hirata K, Katayama Y, Hotta H, Hayashi Y, Ito H, Yokoyama M. Expression of nitric oxide synthase in a murine model of viral myocarditis induced by coxsackievirus B3. Biochem. Biophys. Res. Comm. 1996;220:983–989. doi: 10.1006/bbrc.1996.0519. [DOI] [PubMed] [Google Scholar]
- 51.Miyashita K, Kusumi M, Utsumi R, Katayama S, Noda M, Komano T, Satoh N. Site-directed mutagenesis of the putative active site residues of 3C proteinase of coxsackievirus B3: evidence of a functional relationship with trypsin-like serine proteinases. Protein. Eng. 1993;6:189–193. doi: 10.1093/protein/6.2.189. [DOI] [PubMed] [Google Scholar]
- 52.Mohr S, Zech B, Lapetina E.G, Brune B. Inhibition of caspase-3 by S-nitrosation and oxidation caused by nitric oxide. Biochem. Biophys. Res. Commun. 1997;238:387–391. doi: 10.1006/bbrc.1997.7304. [DOI] [PubMed] [Google Scholar]
- 53.Musser J.M, Stockbauer K, Kapur V, Rudgers G.W. Substitution of cysteine 192 in a highly conserved Streptococcus pyogenes extracellular cysteine protease (interleukin 1beta convertase) alters proteolytic activity and ablates zymogen processing. Infect. Immun. 1996;64:1913–1917. doi: 10.1128/iai.64.6.1913-1917.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Orr D.C, Long A.C, Kay J, Dunn B.M, Cameron J.M. Hydrolysis of a series of synthetic peptide substrates by the human rhinovirus 14 3C proteinase, cloned and expressed in Escherichia coli. J. Gen. Virol. 1989;70:2931–2942. doi: 10.1099/0022-1317-70-11-2931. [DOI] [PubMed] [Google Scholar]
- 55.Palmberg A. Proteolytic processing of picornaviral polyprotein. Annu. Rev. Microbiol. 1990;44:603–623. doi: 10.1146/annurev.mi.44.100190.003131. [DOI] [PubMed] [Google Scholar]
- 56.Reiss C.S, Komatsu T. Does nitric oxide play a critical role in viral infections? J. Virol. 1998;72:4547–4551. doi: 10.1128/jvi.72.6.4547-4551.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rolph M.S, Ramshaw I.A, Rockett K.A, Ruby J, Cowden W.B. Nitric oxide production is increased during murine vaccinia virus infection, but may not be essential for virus clearance. Virology. 1996;217:470–477. doi: 10.1006/viro.1996.0141. [DOI] [PubMed] [Google Scholar]
- 58.Saville B. A scheme for the colorimetric determination of microgram amounts of thiols. Analyst. 1958;83:670–672. [Google Scholar]
- 59.Selzer P.M, Chen X, Chan V.J, Cheng M, Kenyon G.L, Kuntz I.D, Sakanari J.A, Cohen F.E, McKerrow J.H. Leishmania major: molecular modeling of cysteine proteases and prediction of new nonpeptide inhibitors. Exp. Parasitol. 1997;87:212–221. doi: 10.1006/expr.1997.4220. [DOI] [PubMed] [Google Scholar]
- 60.Stamler J.S. Redox signaling: nitrosylation and related target interactions of nitric oxide. Cell. 1994;78:931–936. doi: 10.1016/0092-8674(94)90269-0. [DOI] [PubMed] [Google Scholar]
- 61.Stamler J.S, Simon D.I, Osborne J.A, Mullins M.E, Jaraki O, Michel T, Singel D.J, Loscalzo J. S-nitrosylation of proteins with nitric oxide: synthesis and characterization of biologically active compounds. Proc. Natl. Acad. Sci. USA. 1992;89:444–448. doi: 10.1073/pnas.89.1.444. a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stamler J.S, Singel D.J, Loscalzo J. Biochemistry of nitric oxide and its redox-activated forms. Science. 1992;258:1898–1902. doi: 10.1126/science.1281928. b. [DOI] [PubMed] [Google Scholar]
- 63.Stamler J.S, Toone E.J, Lipton S.A, Sucher N.J. S)NO Signals: translocation, regulation, and a consensus motif. Neuron. 1997;18:691–696. doi: 10.1016/s0896-6273(00)80310-4. [DOI] [PubMed] [Google Scholar]
- 64.Thompson J.F, Geoghegan K.F, Lloyd D.B, Lanzetti A.J, Magyar R.A, Anderson S.M, Branchini B.R. Mutation of a protease-sensitive region in firefly luciferase alters light emission properties. J. Biol. Chem. 1997;272:18766–18771. doi: 10.1074/jbc.272.30.18766. [DOI] [PubMed] [Google Scholar]
- 65.Tucker P.C, Griffin D.E, Choi S, Bui N, Wesselingh S. Inhibition of nitric oxide synthesis increases mortality in Sindbis virus encephalitis. J. Virol. 1996;70:3972–3977. doi: 10.1128/jvi.70.6.3972-3977.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Van Dam A.M, Bauer J, Man-A-Hing W.K, Marquette C, Tilders F.J, Berkenbosch F. Appearance of inducible nitric oxide synthase in the rat central nervous system after rabies virus infection and during experimental allergic encephalomyelitis but not after peripheral administration of endotoxin. J. Neurosci. Res. 1995;40:251–260. doi: 10.1002/jnr.490400214. [DOI] [PubMed] [Google Scholar]
- 67.Wasilewski M.M, Lim K.C, Phillips J, McKerrow J.H. Cysteine protease inhibitors block schistosome hemoglobin degradation in vitro and decrease worm burden and egg production in vivo. Mol. Biochem. Parasitol. 1996;81:179–189. doi: 10.1016/0166-6851(96)02703-x. [DOI] [PubMed] [Google Scholar]
- 68.Zaragoza C, Ocampo C.J, Saura M, McMillan A, Lowenstein C.J. Nitric oxide inhibition of coxsackievirus replication in vitro. J. Clin. Invest. 1997;100:1760–1767. doi: 10.1172/JCI119702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zaragoza C, Saura M, Liew F.Y, Moncada S, Lowenstein C.J. The role of NOS2 in coxsackievirus myocarditis. Proc. Natl. Acad. Sci. USA. 1998;95:2469–2474. doi: 10.1073/pnas.95.5.2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang Y, Xu A, Nomen M, Walsh M, Keaney J.F, Loscalzo J. Nitrosation of tryptophan residue(s) in serumalbumion and model dipeptides. J. Biol. Chem. 1996;271:14271–14279. [PubMed] [Google Scholar]