

Modern homology modeling techniques applied for the genome-wide prediction of drug target protein structures represent a real chance for an enhancement in the effectiveness of the drug discovery process in the pharmaceutical industry.
Keywords: Homology modeling, structural bioinformatics, structure-based drug design, assessment of target druggability, prediction of animal model suitability, prediction of drug metabolism
Abstract
Advances in bioinformatics and protein modeling algorithms, in addition to the enormous increase in experimental protein structure information, have aided in the generation of databases that comprise homology models of a significant portion of known genomic protein sequences. Currently, 3D structure information can be generated for up to 56% of all known proteins. However, there is considerable controversy concerning the real value of homology models for drug design. This review provides an overview of the latest developments in this area and includes selected examples of successful applications of the homology modeling technique to pharmaceutically relevant questions. In addition, the strengths and limitations of the application of homology models during all phases of the drug discovery process are discussed.
The majority of drugs available today were discovered either from chance observations or from the screening of synthetic or natural product libraries. The chemical modification of lead compounds, on a trial-and-error basis, typically led to compounds with improved potency, selectivity and bioavailability and reduced toxicity. However, this approach is labor- and time-intensive and researchers in the pharmaceutical industry are constantly developing methods with a view to increasing the efficiency of the drug discovery process [1]. Two directions have evolved from these efforts. The ‘random’ approach involves the development of HTS assays and the testing of a large number of compounds. Combinatorial chemistry is used to satisfy the need for extensive compound libraries. The ‘rational’, protein structure-based approach relies on an iterative procedure of the initial determination of the structure of the target protein, followed by the prediction of hypothetical ligands for the target protein from molecular modeling and the subsequent chemical synthesis and biological testing of specific compounds (the structure-based drug design cycle). The rational approach is severely limited to target proteins that are amenable to structure determination. Although the protein data bank (PDB; http://www.rcsb.org/pdb) is growing rapidly (∼13 new entries daily), the 3D structure of only 1-2% of all known proteins has as yet been experimentally characterized. However, advances in sequence comparison, fold recognition and protein-modeling algorithms have enabled the partial closure of the so-called ‘sequence-structure gap’ and the extension of experimental protein structure information to homologous proteins. The quality of these homology models, and thus their applicability to, for example, drug discovery, predominantly depends on the sequence similarity between the protein of known structure (template) and the protein to be modeled (target). Despite the numerous uncertainties that are associated with homology modeling, recent research has shown that this approach can be used to significant advantage in the identification and validation of drug targets, as well as for the identification and optimization of lead compounds. In this review, we will focus on the application of homology models to the drug discovery process.
Homology modeling techniques
Homology, or comparative, modeling uses experimentally determined protein structures to predict the conformation of another protein that has a similar amino acid sequence. The method relies on the observation that in nature the structural conformation of a protein is more highly conserved than its amino acid sequence and that small or medium changes in sequence typically result in only small changes in the 3D structure [2].
Generally, the process of homology modeling involves four steps - fold assignment, sequence alignment, model building and model refinement (Figure 1). The fold assignment process identifies proteins of known 3D structure (template structures) that are related to the polypeptide sequence of unknown structure (the target sequence; this is not to be mistaken with drug target). Next, a sequence database of proteins with known structures (e.g. the PDB-sequence database) is searched with the target sequence using sequence similarity search algorithms or threading techniques [3]. Following identification of a distinct correlation between the target protein and a protein of known 3D structure, the two protein sequences are aligned to identify the optimum correlation between the residues in the template and target sequences. The next stage in the homology modeling process is the model-building phase. Here, a model of the target protein is constructed from the substitution of amino acids in the 3D structure of the template protein and the insertion and/or deletion of amino acids according to the sequence alignment. Finally, the constructed model is checked with regard to conformational aspects and is corrected or energy minimized using force-field approaches.
Figure 1.
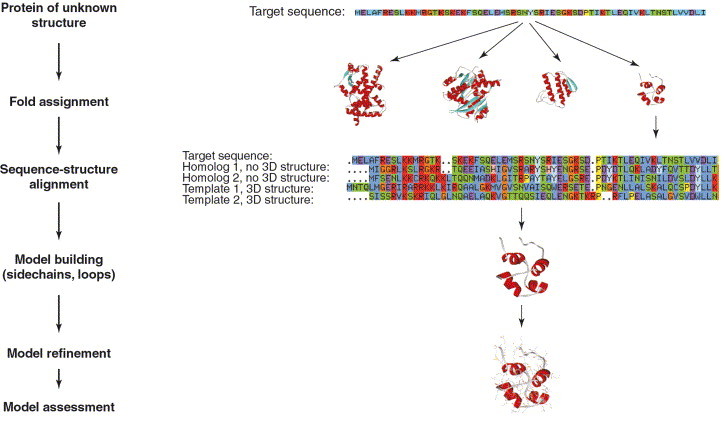
The steps involved in the prediction of protein structure by homology modeling. Structure modeling of the bacterial transcriptional repressor CopR is shown [28]. Although the model is based on a low-sequence identity of only 13.8% between CopR and the P22 c2 repressor, several experimental methods support this homology model. Reproduced, with permission, from Ref. [84]. Abbreviation: CopR, plasmid copy control protein.
Several improvements and modifications of this general homology modeling strategy have been developed and applied to the prediction of protein structures. To subject the available structure prediction methods to a blind test, community-wide experiments on the critical assessment of techniques for protein structure prediction (CASP 1-5) have been performed and their results presented and published. As a result, the current state-of-the-art in protein structure prediction has been established, the progress made has been documented and the areas where future efforts might be most productively concentrated have been highlighted [4, 5].
Experimental protein structure information and the sequence-structure gap
Homology modeling techniques are dependent on the availability of high-resolution experimental protein structure data. The development of effective protein expression systems and major technological advances in the instrumentation used for structure determination (X-ray crystallography and NMR spectroscopy) has contributed to an exponential growth in the number of experimental protein 3D structures. By May 2004, the PDB contained ∼23,000 experimental protein structures for ∼7400 different proteins (proteins with less than 90% sequence identity). A recent analysis of all protein chains in the PDB shows that these proteins can be grouped into 2500 protein families comprising 900 unique protein folds [6] (updates can be found at http://scop.mrc-lmb.cam.ac.uk). The majority of the structures in the PDB (84%) were determined by X-ray crystallography, with 15% of the structures being characterized by NMR spectroscopy. The PDB database encompasses experimental information on an extensive array of ligands (small organic molecules and ions) bound to more than 50,000 different binding sites that can be analyzed using programs including ReliBase (http://relibase.ebi.ac.uk) [7], LigBase (http://alto.compbio.ucsf.edu/ligbase) [8] and PDBsum (http://www.biochem.ucl.ac.uk/bsm/pdbsum) [9].
Although the experimental structure database is growing rapidly, there is still a substantial gap between the number of known annotated sequences [1,182,126 unique sequences in Swiss-Prot-TrEMBL (http://www.expasy.org/sprot) as of 29 August 2003] and known protein 3D structures (23,000). If only significantly different proteins are considered (∼7400), which omits muteins, artificial proteins and multiple structure determinations of the same proteins (e.g. HIV-protease and carbonic anhydrase II), then less than 1% of the 3D structures of known protein sequences have been elucidated. This sequence-structure gap can partly be filled with homology models. For example, the queryable database ModBase (http://alto.compbio.ucsf.edu/modbase-cgi/index.cgi) provides access to an enormous number of annotated comparative protein structure models [10]. The program PSI-BLAST was used to assign protein folds to all 1,182,126 unique sequence entries in Swiss-Prot-TrEMBL. For 56% of these sequences, comparative models with an average model size of 235 amino acids could be built using the program MODELLER [11]. Thus, by August 2003, 659,495 3D structure models of proteins were accessible via the Internet. The models are predicted to have at least 30% of their Cα atoms superimposed within 3.5 Å of their correct positions. Information on binding sites and ligands can be retrieved from this database using LigBase [8]. However, the majority of the models are built on a low sequence identity and it should be realized that this level of accuracy is, in most cases, not sufficient for a detailed structure-based ligand design.
The SWISS-MODEL Repository (http://swissmodel.expasy.org/repository) [12] is also a database of annotated comparative protein 3D structure models, which have been generated using the fully automated homology-modeling pipeline SWISS-MODEL. As of August 2003, this database contained models for 282,096 different protein sequence entries (26%) from the Swiss-Prot-TrEMBL databases (1,073,566 sequences), with an average model size of ∼200 amino acids.
Researchers from Eidogen (http://www.eidogen.com) have created a database system called Target Informatics Platform™ [13] that currently includes homology models for 55,000 proteins. Homology modeling of 26,279 human protein sequences resulted in the construction of 17,442 models for 13,114 different sequences (50%). Thus, putative and known ligand binding pockets can be detected, analyzed and compared and the resulting data used to support target prioritization and lead discovery and/or optimization procedures.
Accelrys (http://www.accelrys.com) produces Discovery Studio (DS) AtlasStore™ as a complete Oracle®-based protein and ligand structural data management solution. Currently, DS AtlasStore™ contains 2,052,000 homology models that have been automatically generated from the sequences of 195,000 proteins from 33 different genomes. In conjunction with homology models, Cengent Therapeutics (http://www.cengent.com) offers dynamic structural information generated from molecular dynamics simulations for 5500 human drug target proteins. This structural information can be used for target prioritization and virtual screening.
Structure information content in homology models
The quality of the homology models is dependent on the level of sequence identity between the protein of known structure and the protein to be modeled [14]. For a sequence identity that is greater than 30%, homology can be assumed; the two proteins probably have a common ancestor and are, therefore, evolutionarily related and are likely to share a common 3D structure. In this case, pairwise and multiple sequence alignment algorithms are reliable and can be used for the generation of homology models (Figure 2).
Figure 2.
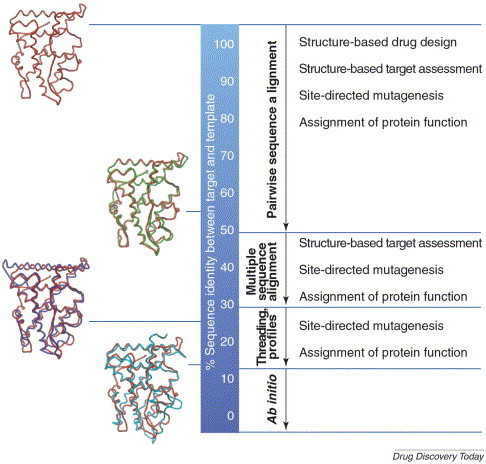
Relationship between target and template sequence identity and the information content of resulting homology models. Arrows indicate the methods that can be used to detect sequence similarity between target and template sequences. Applications of the homology models in drug discovery are listed to the right. The higher the sequence identity, the more accurate the resulting structure information. Homology models that are built on sequence identities above ∼50% can frequently be used for drug design purposes. Superimpositions of X-ray crystal structures of the ligand-binding domains of members of the nuclear receptor family are shown to the left. These X-ray structures illustrate the increase in structure deviation with a decreased sequence identity. The PR is red, the GR is green, the ERα is blue and the TRβ is cyan. Sequence identities: PR:GR, 54%; PR:ERα, 24%; and PR:TRβ, 15%. Abbreviations: ERα, estrogen receptor α GR, glucocorticoid receptor; PR, progesterone receptor; TRβ, thyroid receptor β.
If the sequence identity is below 15%, structure modeling becomes speculative, which could lead to misleading conclusions. When the sequence identity is between 15% and 30%, conventional alignment methods are not sufficiently reliable and only sophisticated, profile-based methods are capable of recognizing homology and predicting fold. For regions of low sequence identity, threading methods [15] are often applied. Protein models that are built on such low sequence identities can be used for the assignment of protein function and for the direction of mutagenesis experiments (Figure 2). Models that have a sequence identity between ∼30% and 50% could facilitate the structure-based prediction of target drugability, the design of mutagenesis experiments and the design of in vitro test assays (Figure 2). If sequence identity is greater than ∼50%, the resulting models are frequently of sufficient quality to be used in the prediction of detailed protein-ligand interactions, such as structure-based drug design and prediction of the preferred sites of metabolism of small molecules (Figure 2).
Application of homology models in the drug discovery process
There are numerous applications for protein structure information and, hence, homology models at various stages of the drug discovery process [16]. The most spectacular successes are clearly those where protein structural information has helped to identify or to optimize compounds that were subsequently progressed to clinical trials or to the drug market [17]. The applications of homology models that had an impact on target identification and/or validation, lead identification and lead optimization are reviewed here (Figure 3).
Figure 3.
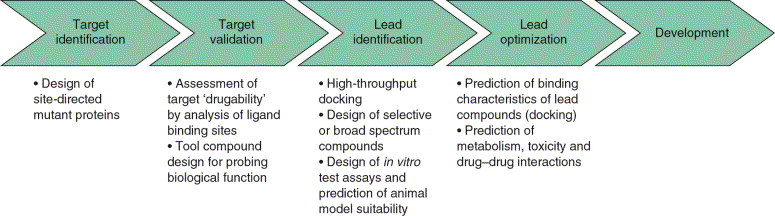
Applications of homology models in the drug discovery process. The enormous amount of protein structure information currently available could not only support lead compound identification and optimization, but could also contribute to target identification and validation. Reproduced, with permission, from [84].
Structure-based assessment of target drugability
It is clear that only a minute fraction of the entire proteome can be affected by drug-like (preferentially orally bioavailable) small molecules. Based on the total numbers of known genes, disease-modifying genes and drugable proteins, the number of drug target proteins, for humans, has been estimated at ∼600-1500 [18]. For small molecules, sets of properties have been established that differentiate drugs from other compounds [19, 20]; these properties can be used to identify compounds with, for example, poor oral absorption properties [21]. Drug molecules and their corresponding target proteins are highly complementary, which suggests that some rules that distinguish good target proteins from others should be deducible [22]. Deep lipophilic pockets that comprise distinct polar interaction sites are clearly superior to shallow highly charged protein surface regions. The inhibition of protein-protein interfaces as a valuable therapeutic principle has recently been shown with inhibitors of the p53-murine double minute clone 2 (MDM2) interaction [23, 24]. The binding site for these inhibitors is a distinct lipophilic pocket that normally interacts with the α-helical surface patches of the p53 tumor suppressor transactivation domain. Advances in the rapid detection, description and analysis of ligand-binding pockets [25, 26, 27], together with the availability of more than 0.5 million homology models, will open new possibilities for the prioritization of proteins with regards to drugability. In the pharmaceutical industry, structural aspects are being increasingly implemented as additional decision criteria on the drugability of potential drug targets. Companies such as Inpharmatica (http://www.inpharmatica.com) have developed an integrated suite of informatics-based discovery technologies that contain software tools for the structure-based assessment of target drugability.
Structure-guided design of mutagenesis experiments
The design of site-directed mutant proteins is one further important option for the application of homology models to target validation. Introducing point mutations and subsequently studying the effects in vitro or in vivo is a common approach in molecular biology. This strategy enables the identification of amino acids that are functionally or structurally important in the protein under investigation, which ultimately contributes to biological knowledge on, for example, potential target proteins. Typically, the amino acids that are to be modified in these studies are selected on the basis of sequence alignments by focusing on conserved residues. However, if at least some structure information is available, the selection of the amino acids that are to be mutated can be much more precise and successful [28]. This approach is even more powerful when applied in conjunction with pharmacologically active compounds. Site-directed mutants of the target protein can be made to render that target sensitive to an existing pharmacological agent. Based on homology models, some members of the mitogen-activated protein (MAP) kinase family were mutated to make them sensitive to a kinase inhibitor from the pyridinyl imidazole class [29]. This enabled the use of the compound for broader target validation studies.
Tool compound design for probing biological function
One of the most attractive ways to validate a target protein is to administer a pharmacologically active compound that selectively acts on that protein and to study the effects in a relevant animal model. Similar strategies have been described under the term ‘chemogenomics’ [30].
It has recently been shown that it is possible to design small molecules based on homology models and then to use these compounds as tools to study the physiological role of the respective target protein of that particular drug [31]. Eight years after the discovery of estrogen receptor β (ERβ), the distinct roles of the two ER isotypes, ERα and ERβ, in mediating the physiological responses to estrogens are not completely understood. Although knockout animal experiments have provided an insight into estrogen signaling, additional information on the function of ERα and ERβ was imparted by the application of isotype selective ER agonists. Based on the crystal structure of the ERα-ligand-binding domain (LBD) and a homology model of the ERβ-LBD (59% sequence identity to ERα), Hillisch et al. [31] designed steroidal ligands that exploit the differences in size and flexibility of the two ligand-binding cavities (Figure 4). Compounds that were predicted to bind preferentially to either ERα or ERβ were synthesized and tested in vitro. This approach led directly to highly ER isotype-selective (200-250-fold) ligands that were also highly potent. To unravel the physiological roles of each of the two receptors, in vivo experiments with rats were conducted using the ERα- and ERβ-selective agonists in parallel with the natural ligand of ER, 17β-estradiol. The ERα agonist was shown to be responsible for most of the known estrogenic effects (e.g. induction of uterine growth and bone-protection), in addition to pituitary (e.g. reduction of luteinizing hormone plasma levels) and liver (e.g. increase in angiotensin I plasma levels) effects [31]. However, the ERβ agonist had distinct effects on the ovary, for example, the stimulation of early folliculogenesis [32], which possibly presents clinicians with a new option for tailoring classical ovarian stimulation protocols. A comparison of the homology model with the X-ray crystal structure of the ERβ-LBD complexed with genistein [33] revealed that the homology model had a root-mean-square deviation (rmsd) of the backbone atoms (not considering helix 12) of 1.4 Å. The X-ray crystal structure confirmed the presence of essential interactions between the ligand and the ERβ and did not reveal, at least in this case, any new aspects for the design of ERβ agonists that were not covered by the homology model. These studies show that it is possible to design highly selective compounds, if structure information on all of the relevant homologs of the target is available, and that the designed tool compounds contribute to the elucidation of the physiological roles of the target protein.
Figure 4.
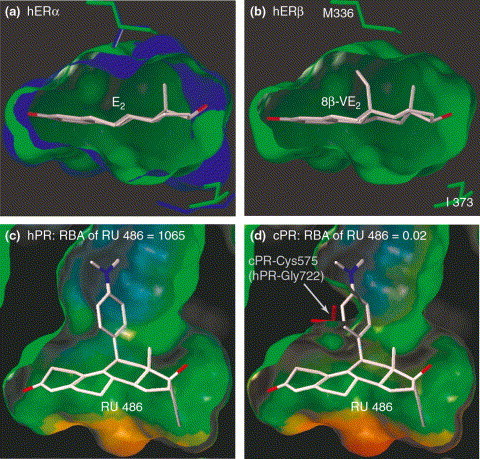
(a,b) Comparison of the two isotypes of the estrogen receptor. In the homology model, ERα (blue) and ERβ (green) ligand-binding pockets are shown in complex with the natural ligand of the ER, 17β-estradiol. The binding of 8β-VE2, a highly potent and selective ERβ agonist, modeled into the ERβ ligand-binding niche is depicted to the right. Reproduced, with permission, from Ref. [31]. (c,d) A model of the antiprogestin RU 486 (Mifepristone) bound to hPR. A single amino acid mutation renders this compound inactive at the cPR and hamster PR. Steric clashes between RU 486 and cPR are shown on the right side. Abbreviations: ER, estrogen receptor; hER, human estrogen receptor; cPR, chicken progesterone receptor; hPR, human progesterone receptor; PR, progesterone receptor; RBA, relative binding affinity; 8β-VE2, 8β-vinylestra-1,3,5(10)-triene-3,17β-diol.
Homology model-based ligand design
There are numerous examples where protein homology models have supported the discovery and the optimization of lead compounds with respect to potency and selectivity.
Currently, the structures of 40 of the 518 known different human protein kinases have been characterized by X-ray crystallography [34]. Homology model-based drug design has been applied to epidermal growth factor-receptor tyrosine kinase protein [35, 36], Bruton's tyrosine kinase [37], Janus kinase 3 [38] and human aurora 1 and 2 kinases [39].
Using the X-ray crystal structure of cyclin-dependent kinase 2 (CDK2), Honma et al. [40] generated a homology model of CDK4. This model guided the design of highly potent and selective CDK4 inhibitors that were targeted towards the ATP binding pocket. The diarylurea class of compounds were subsequently synthesized and tested. In an in vitro inhibition assay, the most potent compound had an IC50 of 42 nM. The predicted binding mode of the lead compound was verified by co-crystallization with CDK2 [40]. Vangrevelinghe et al. [41] identified a CDK2 inhibitor using a homology model of the protein and high-throughput docking.
Siedlecki et al. [42] have demonstrated the utility of homology modeling in the prediction of pharmacologically active compounds. Alterations in DNA methylation patterns play an important role in tumorigenesis; therefore, inhibitors of DNA methyltransferase 1 (DNMT1), which is the protein that represents the major DNA methyltransferase activity in human cells, are desired. Known inhibitors from the 5-azacytidine class were docked into the active site of a DNMT1 homology model, which led to the design of N4-fluoroacetyl-5-azacytidine derivatives that acted as highly potent inhibitors of DNA methylation in vitro.
Thrombin-activatable fibrinolysis inhibitor (TAFI) is an important regulator of fibrinolysis, and inhibitors of this enzyme have potential use in antithrombotic and thrombolytic therapy. Based on a homology model of TAFI (∼50% sequence identity to carboxypeptidases A and B), appropriately substituted imidazole acetic acids were designed and were subsequently found to be potent and selective inhibitors of activated TAFI [43].
Homology models of the voltage-gated K+-channel Kv1.3 and the Ca2+-activated channel IKCa1 were used to design selective IKCa1 inhibitors that were based on the polypeptide toxin charybdotoxin. Comparison of the two models revealed a unique cluster of negatively charged residues in the turret of Kv1.3 that were not present in IKCa1. To exploit this difference, the homology model was used to design novel analogs, which were then synthesized and tested. Research demonstrated that the novel compounds blocked IKCa1 activity with ∼20-fold higher affinity than Kv1.3 [44].
The key proteinase (Mpro, or 3CLpro) of the new coronavirus (CoV) that caused the severe acute respiratory syndrome (SARS) outbreak of 2003 (SARS-CoV) is another example of successful homology model building; in this case, success is defined as the ability to use the model to propose an inhibitor that has significant affinity for the target enzyme. X-ray crystal structures for the Mpros of transmissible gastroenteritis virus (TGEV, a porcine coronavirus) and of human coronavirus 229E [45, 46] have been characterized. These proteinases have 44 and 40% sequence identity, respectively, with the key proteinase of SARS-CoV. Following publication of the genome sequence of the new virus, first on the internet and a few weeks later in print [46, 47], the level of sequence identity between the proteinases enabled Anand et al. [46] to construct a 3D homology model for the Mpro of human CoV. However, the 3D homology model generated was insufficient for the design of inhibitors with reasonable confidence. To establish the structural basis of the interaction with the polypeptide substrate of the Mpro, Anand and co-workers [46] synthesized a substrate-analogous hexapeptidyl chloromethylketone inhibitor that was complexed with TGEV Mpro. The X-ray crystal structure of the complex was then determined, which revealed that, as expected, the chloromethylketone moiety had covalently reacted with the active-site cysteine residue of the proteinase. The P1, P2, and P4 side chains of the inhibitor had bound to, and thereby defined, the specificity binding sites of the target enzyme. The experimentally determined structure of the inhibitor-TGEV Mpro complex was then compared with all inhibitor complexes of cysteine proteinases in the PDB, which revealed a surprisingly similar inhibitor binding mode in the complex of human rhinovirus type 2 (HRV2) 3C proteinase with AG7088 (Figure 5) [48]. At that time, AG7088 was in late Phase II clinical trials as a drug for the treatment of the strain of the common cold that is caused by human rhinovirus. The comparison of the crystal structures of HRV2 in complex with AG7088 and TGEV Mpro in complex with the hexapeptidyl chloromethylketone inhibitor revealed little similarity between the two target enzymes, except in the immediate neighborhood of the catalytic cysteine residue, but an almost perfect match of the inhibitors. To investigate these findings further, AG7088 was docked into the substrate-binding site of the SARS-CoV Mpro model without much difficulty, although it was noted that there could potentially be steric problems with the p-fluorobenzyl group in the S2 pocket, and also with the ethylester moiety in S1′. Therefore, it was proposed that, although AG7088 was not an ideal inhibitor, this compound should be a good starting point for the design of anti-SARS drugs. Indeed, only a few days after the on-line publication of these results in ScienceXpress [46], it was confirmed that AG7088 had anti-SARS activity in vitro. Derivatives of AG7088 with modified P2 residues have since been shown to have K i values in the lower μmolar range (Rao et al., pers. commun.). The crystal structure of the authentic SARS-CoV key proteinase was determined a few months later [49]. Although the dimeric structure showed the expected similarity to the homologous enzymes of TGEV and human CoV 229E, there were interesting differences in detail. In particular, one of the monomers in the dimer was observed to be in an inactive conformation, which was thought to be the result of the low pH of crystallization. The overall rmsd for the entire dimer from the homology model of Anand et al. [46] was >3.0 Å (i.e. no residues excluded from the comparison), which dropped to 2.1 Å when a few outliers at the carboxy terminus were excluded from the comparison, and to <1.8 Å for each of the three individual domains of the enzyme. Other homology models were generated (D. Debe, unpublished and [50]) and virtual screening has been performed using a SARS-CoV Mpro model [51]. Taken together, these findings confirm that homology modeling is often inadequate for the prediction of the mutual orientation of domains in multidomain proteins. However, the homology model generated by Anand et al. [46] also shows that a reasonable model of a substrate-binding site can serve to develop useful ideas for inhibitor design that can inspire medicinal chemists to start a synthesis program long before the 3D structure of the target enzyme is experimentally determined.
Figure 5.
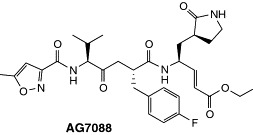
Structure of AG7088. This compound is an inhibitor of HRV2 3C proteinase and, on the basis of a homology model of HRV2 3C proteinase, was suggested as a potential inhibitor of SARS-CoV Mpro. Abbreviations: CoV, coronavirus; HRV2, human rhinovirus type 2; SARS, severe acute respiratory syndrome.
In the case of G-protein-coupled receptors (GPCR), homology-modeling approaches are limited by the lack of experimentally determined structures and the low sequence similarity of those structures that have been characterized with respect to pharmacologically important target proteins. The X-ray crystal structure of only one GPCR, bovine rhodopsin, has been determined [52]. This structure is complemented by bacteriorhodopsin, which is a transmembrane protein that comprises seven helices and is also of relevance for modeling approaches, even though this protein is not a GPCR. Some examples of homology models for GPCRs and their utility have recently been reviewed [53]. High-throughput docking has been applied to verify the ability of homology models to identify agonists (glucocorticoid receptor agonists) [54], antagonists of retinoic acid receptor α [55], D3-dopamine-, M1-muscarinic acetylcholine- and V1a-vasopressin-receptors [56] and inhibitors of thrombin [57]. In the identification of thrombin inhibitors, homology models of thrombin were built retrospectively and were based on homologous serine proteases (28%-40% sequence identity); the best docking solutions were yielded with those models that were derived from proteins of higher sequence identity.
Recently, the performance of docking studies into protein active sites that had been constructed from homology models was assessed using experimental screening datasets of CDK2 and factor VIIa [58]. When the sequence identity between the model and the template near the binding site was greater than ∼50%, there was an approximate fivefold increase in the number of active compounds identified than would have been be detected randomly. This performance is comparable to docking to crystal structures.
Design of in vitro test assays and prediction of animal model suitability
A further application of homology models is the design of test assays for the in vitro pharmacological characterization of compounds or HTS. Based on the structure of the coiled-coil domain of c-Jun, models for α-helical proteins were designed such that they can be used as affinity-tagged proteins that incorporate protease cleavage sites [59]. The resulting 10.5 kDa recombinant proteins were synthesized and used as molecularly defined and uniform substrates for in vitro detection of HIV-1 and IgA endoprotease activity, which enabled the surface plasmon resonance-based screening of inhibitors.
The enormous volume of structure information on entire target protein families that is available might also have an impact on screening cascades. Many drug discovery projects endeavor to identify ligands that are highly selective for particular drug targets. Selective compounds are supposed to be superior because such compounds typically lead to fewer adverse side effects (e.g. COX-2 inhibitors). However, the most important homologs that should not be targeted by the desired drug, with respect to the actual target, are not always clear, particularly within the larger target protein families. The sequence similarity of the full-length proteins or entire domains might not always be representative of the target protein when considering the conservation of the ligand-binding pockets. Comparison of the shape and features of the binding pockets within a protein family could indicate which homologs should be included in the screening cascade for so-called ‘counter screening’. The structure information that is currently available on entire protein families (e.g. proteases, kinases and nuclear receptors) could contribute to the design of selective compounds or better screening cascades, both of which could potentially advance the design of drugs that have fewer side effects.
A detailed structural knowledge of the ligand-binding sites of target proteins was also shown to facilitate the selection of animal models for ex vivo or in vivo experiments. The proposal is that animals having target proteins with significantly different binding sites compared with human orthologs should be excluded as pharmacological models. Many promising compounds showing high-potency in human in vitro assays have not reached clinical trials because efficacy could not be demonstrated in animal models. Single amino acid differences between humans and animals might, in some cases, be sufficient to cause such effects. The ER selectivity of ligands described by Hillisch et al. [31] was shown by homology models and in vitro assays to be crucially dependent on the interaction of ligand substituents with one particular amino acid that differs between ERα and ERβ (Figure 4a) [31]. To ensure that this important interaction is present in estrogen receptors of all animal models that are used to characterize compounds [32], homology models of murine, rat and bovine ERβ were built and compared with the binding pocket of human ERβ (hERβ). A complete conservation of amino acids within the binding pockets of human, murine and rat ERβ was observed. However, bovine ERβ showed one amino acid difference at the exact position that was determined to be crucial for ERβ ligand selectivity. The prediction that the hERβ selective compounds should not bind to bovine ERβ was later verified using transactivation experiments (unpublished results). Thus, the implementation of uninterpretable experiments could be avoided at an early stage and the otherwise attractive bovine tissues (later available in larger amounts) could be excluded from ex vivo investigations. Similarly, information on the structure of progesterone receptors (PR) can be used to explain the abolished binding of the progesterone antagonist mifepristone (RU 486) to chicken PR and hamster PR [60]. A single point mutation (human PR Gly722 to chicken PR Cys575) prevents antiprogestins containing 11β-aryl substituents (e.g. RU 486) from binding to chicken (and hamster) PR (Figure 4c), which therefore excludes hamsters, for example, from pharmacological studies with antiprogestins [61]. In the future, such effects could be predicted and particular species could then be excluded from pharmacological studies at an early stage, which would ultimately reduce attrition rates in the drug discovery process.
Structure-based prediction of drug metabolism and toxicity
One of the challenges in lead optimization is to identify compounds that not only show a high potency at the desired target protein but also have adequate physical properties to reach systemic circulation, to resist metabolic inactivation for a specific time period and to avoid undesired pharmacological effects. Knowledge of the structure of the proteins that are involved in these processes, such as drug-metabolizing enzymes, transcription factors or transporters, could help to design molecules that do not interact with these ‘non-target’ proteins.
The cytochrome P450s (CYP) are an extremely important class of enzymes that are involved in Phase I oxidative metabolism of structurally diverse chemicals. Only ∼10 hepatic CYPs are responsible for the metabolism of 90% of known drugs. Recently, the X-ray crystal structures of three mammalian CYPs, CYP2C5 [62], CYP2C8 [63] and CYP2C9 [64], have been solved and represent a solid basis for the homology modeling of this entire superfamily. Models of CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4 and CYP4A11 have been generated using different structure templates. These models have been used to explain and to predict the probable sites of metabolic attack in a variety of CYP substrates [65, 66, 67, 68, 69, 70, 71, 72]. However, the large lipophilic and highly flexible character of some CYP binding cavities renders pure in silico approaches towards the prediction of the occurrence and site of small molecule metabolism extremely difficult. If protein structure information is combined with pharmacophoric patterns and quantum mechanical calculations, some predictions concerning the preferred sites of metabolism within small molecules are possible [73]. Regarding this aspect of homology modeling, CYP2D6 is a particularly interesting CYP because 5-9% of the Caucasian population does not produce this polymorphic member of the CYP superfamily. The resulting deficiencies in drug oxidation can lead to severe side effects in these individuals. Predictions on whether or not a lead compound could act as a CYP2D6 substrate could help to identify problematic cases early in drug discovery. Combined homology modeling and quantitative SAR approaches are able to predict such CYP inhibitors [74]. Thus, in the future, protein structure information in conjunction with high-throughput docking and pharmacophore-based methods could be used to decide which compounds have the potential to inhibit particular CYPs. This approach could facilitate the detection of potential drug-drug interactions early in the drug discovery process and measures could then be taken to avoid such interactions [75].
CYP substrates and inhibitors are not the only compounds to have been studied using homology models. These approaches have been used recently to investigate CYP inducers. The induction of CYPs is primarily mediated via the activation of ligand-dependent transcription factors, such as the aryl hydrocarbon receptor (AhR) for the CYP1A family, the constitutive androstane receptor (CAR) for the CYP2D family and the pregnane X receptor (PXR), glucocorticoid receptor (GR) and vitamin D receptor (VDR) for the CYP3A family [76]. In principle, the in silico prediction of drug-metabolizing enzyme induction could be reduced to predicting the binding and activation of transcription factors (e.g. AhR and CAR). However, recent X-ray structure analyses of PXR have shown that the LBD of this nuclear receptor contains a large lipophilic and flexible binding pocket [77]. This renders pure in silico structure-based predictions concerning whether or not a small molecule will activate PXR difficult. The homology modeling of CAR [78, 79] and other members of the nuclear receptor family involved in CYP induction [80] have recently been described. These models predict reasonably shaped potential ligand binding pockets. However, further results on the utility of these models are needed.
With respect to the structure-based prediction of adverse health effects, progress has been described with the human ether-a-go-go-related gene (hERG). This tetrameric potassium channel contributes to phase three repolarization of heart muscle cells by opposing the depolarizing Ca2+ influx during the plateau phase. Inhibition of this protein results in cardiovascular toxicity (QT-prolongation) and has caused several drugs to be withdrawn from the market. Therefore, in silico predictions on the probability of the formation of an interaction between a drug and hERG have gained enormous attention and have recently been reviewed [81]. Homology models of hERG, which are based on the X-ray crystal structures of the bacterial KcsA [82] and MthK channels [83], have already shed light on some details of the molecular interactions that initiate hERG inhibition. However, the complexity of this potassium channel signifies that detailed X-ray structure analyses of the protein in the open- and closed-state are required before these molecular interactions can be fully understood and predicted, which has implications for the prediction of cardiotoxicity.
Conclusions and outlook
Numerous examples for the successful application of homology modeling in drug discovery are described here. In the absence of experimental structures of drug target proteins, homology models have supported the design of several potent pharmacological agents. One of the advantages of homology models is that these models can be generated relatively easily and quickly. Furthermore, such models could support the hypotheses of medicinal chemists on how to generate biologically active compounds in the important early conceptual phase of a drug discovery project. The design of compounds that are selectively directed at particular drug target proteins is one of the strengths of this technique. Such selective compounds can even be applied to gain insights into the physiological role of novel drug targets. The in silico protein structure-based prediction of metabolism and toxicity of small molecules, particularly CYP inhibition and induction and hERG inhibition, is currently in its infancy and predictive capabilities could be limited to classification only. However, while complete experimental structures of pharmacologically important proteins are missing, the homology modeling technique provides one approach to bridge the gap until this information becomes available.
Acknowledgements
We gratefully acknowledge fruitful discussions with Mario Lobell (Bayer HealthCare; http://www.bayerhealthcare.com), Derek Debe, Sean Mullen (Eidogen) and Sunil Patel (Accelrys).
References
References
- 1.Giersiefen H. Modern Methods of Drug Discovery: An Introduction. In: Hillisch A., Hilgenfeld R., editors. Modern Methods of Drug Discovery. Birkhäuser Verlag; 2003. pp. 1–18. [DOI] [PubMed] [Google Scholar]
- 2.Lesk A.M., Chothia C. The response of protein structures to amino-acid sequence changes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1986;317:345–356. [Google Scholar]
- 3.Godzik A. Fold Recognition Methods. In: Bourne P., Weissig H., editors. Structural Bioinformatics. Wiley-Liss; 2003. pp. 525–546. [Google Scholar]
- 4.Murzin A.G. Progress in protein structure prediction. Nat. Struct. Biol. 2001;8:110–112. doi: 10.1038/84088. [DOI] [PubMed] [Google Scholar]
- 5.Tramontano A. Assessment of homology-based predictions in CASP5. Proteins. 2003;53:352–368. doi: 10.1002/prot.10543. [DOI] [PubMed] [Google Scholar]
- 6.Murzin A.G. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 7.Hendlich M. Databases for protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 1998;54:1178–1182. doi: 10.1107/s0907444998007124. [DOI] [PubMed] [Google Scholar]
- 8.Stuart A.C. LigBase: a database of families of aligned ligand binding sites in known protein sequences and structures. Bioinformatics. 2002;18:200–201. doi: 10.1093/bioinformatics/18.1.200. [DOI] [PubMed] [Google Scholar]
- 9.Laskowski R.A. PDBsum: summaries and analyses of PDB structures. Nucleic Acids Res. 2001;29:221–222. doi: 10.1093/nar/29.1.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pieper U. MODBASE, a database of annotated comparative protein structure models, and associated resources. Nucleic Acids Res. 2004;32:D217–D222. doi: 10.1093/nar/gkh095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sali A. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 12.Kopp J. The SWISS-MODEL Repository of annotated three-dimensional protein structure homology models. Nucleic Acids Res. 2004;32:D230–D234. doi: 10.1093/nar/gkh008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Debe D.A., Hambly K. Supporting your pipeline with structural knowledge. Curr. Drug Discov. 2004;3:15–18. [Google Scholar]
- 14.Chothia C. The relation between the divergence of sequence and structure in proteins. EMBO J. 1986;5:823–826. doi: 10.1002/j.1460-2075.1986.tb04288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kinch L.N. CASP5 assessment of fold recognition target predictions. Proteins. 2003;53:395–409. doi: 10.1002/prot.10557. [DOI] [PubMed] [Google Scholar]
- 16.Hillisch A., Hilgenfeld R. The role of protein 3D structures in the drug discovery process. In: Hillisch A., Hilgenfeld R., editors. Modern Methods of Drug Discovery. Birkhäuser Verlag; 2003. pp. 157–181. [DOI] [PubMed] [Google Scholar]
- 17.Hardy L.W. The impact of structure-guided drug design on clinical agents. Curr. Drug Discov. 2003;12:15–20. [Google Scholar]
- 18.Hopkins A.L. The druggable genome. Nat. Rev. Drug Discov. 2002;1:727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- 19.Walters W.P. Prediction of ‘drug-likeness’. Adv. Drug Deliv. Rev. 2002;54:255–271. doi: 10.1016/s0169-409x(02)00003-0. [DOI] [PubMed] [Google Scholar]
- 20.Sadowski J. A scoring scheme for discriminating between drugs and nondrugs. J. Med. Chem. 1998;41:3325–3329. doi: 10.1021/jm9706776. [DOI] [PubMed] [Google Scholar]
- 21.Lipinski C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods. 2000;44:235–249. doi: 10.1016/s1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 22.Fauman E.B. Structural bioinformatics in drug discovery. Methods Biochem. Anal. 2003;44:477–497. doi: 10.1002/0471721204.ch23. [DOI] [PubMed] [Google Scholar]
- 23.Chene P. Inhibition of the p53-hdm2 interaction with low molecular weight compounds. Cell Cycle. 2004;3:460–461. doi: 10.4161/cc.3.4.791. [DOI] [PubMed] [Google Scholar]
- 24.Chene P. Inhibition of the p53-MDM2 interaction: targeting a protein-protein interface. Mol. Cancer Res. 2004;2:20–28. [PubMed] [Google Scholar]
- 25.Schmitt S. A new method to detect related function among proteins independent of sequence and fold homology. J. Mol. Biol. 2002;323:387–406. doi: 10.1016/s0022-2836(02)00811-2. [DOI] [PubMed] [Google Scholar]
- 26.Binkowski T.A. Inferring functional relationships of proteins from local sequence and spatial surface patterns. J. Mol. Biol. 2003;332:505–526. doi: 10.1016/s0022-2836(03)00882-9. [DOI] [PubMed] [Google Scholar]
- 27.Naumann T. Structural classification of protein kinases using 3D molecular interaction field analysis of their ligand binding sites: target family landscapes. J. Med. Chem. 2002;45:2366–2378. doi: 10.1021/jm011002c. [DOI] [PubMed] [Google Scholar]
- 28.Steinmetzer K. Transcriptional repressor CopR: structure model-based localization of the deoxyribonucleic acid binding motif. Proteins. 2000;38:393–406. [PubMed] [Google Scholar]
- 29.Eyers P.A. Use of a drug-resistant mutant of stress-activated protein kinase 2a/p38 to validate the in vivo specificity of SB 203580. FEBS Lett. 1999;451:191–196. doi: 10.1016/s0014-5793(99)00552-9. [DOI] [PubMed] [Google Scholar]
- 30.Bredel M. Chemogenomics: an emerging strategy for rapid target and drug discovery. Nat. Rev. Genet. 2004;5:262–275. doi: 10.1038/nrg1317. [DOI] [PubMed] [Google Scholar]
- 31.Hillisch A. Dissecting physiological roles of estrogen receptor alpha and beta with potent selective ligands from structure-based design. Mol. Endocrinol. 2004;18:1599–1609. doi: 10.1210/me.2004-0050. [DOI] [PubMed] [Google Scholar]
- 32.Hegele-Hartung C. Impact of isotype-selective estrogen receptor agonists on ovarian function. Proc. Natl. Acad. Sci. U. S. A. 2004;101:5129–5134. doi: 10.1073/pnas.0306720101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pike A.C. Structure of the ligand-binding domain of oestrogen receptor beta in the presence of a partial agonist and a full antagonist. EMBO J. 1999;18:4608–4618. doi: 10.1093/emboj/18.17.4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Manning G. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 35.Traxler P. Design and synthesis of novel tyrosine kinase inhibitors using a pharmacophore model of the ATP-binding site of the EGF-R. J. Pharm. Belg. 1997;52:88–96. [PubMed] [Google Scholar]
- 36.Ghosh S. Rational design of potent and selective EGFR tyrosine kinase inhibitors as anticancer agents. Curr. Cancer Drug Targets. 2001;1:129–140. doi: 10.2174/1568009013334188. [DOI] [PubMed] [Google Scholar]
- 37.Mahajan S. Rational design and synthesis of a novel anti-leukemic agent targeting Bruton's tyrosine kinase (BTK), LFM-A13. J. Biol. Chem. 1999;274:9587–9599. doi: 10.1074/jbc.274.14.9587. [DOI] [PubMed] [Google Scholar]
- 38.Sudbeck E.A. Structure-based design of specific inhibitors of Janus kinase 3 as apoptosis-inducing antileukemic agents. Clin. Cancer Res. 1999;5:1569–1582. [PubMed] [Google Scholar]
- 39.Vankayalapati H. Targeting aurora2 kinase in oncogenesis: a structural bioinformatics approach to target validation and rational drug design. Mol. Cancer Ther. 2003;2:283–294. [PubMed] [Google Scholar]
- 40.Honma T. Structure-based generation of a new class of potent Cdk4 inhibitors: new de novo design strategy and library design. J. Med. Chem. 2001;44:4615–4627. doi: 10.1021/jm0103256. [DOI] [PubMed] [Google Scholar]
- 41.Vangrevelinghe E. Discovery of a potent and selective protein kinase CK2 inhibitor by high-throughput docking. J. Med. Chem. 2003;46:2656–2662. doi: 10.1021/jm030827e. [DOI] [PubMed] [Google Scholar]
- 42.Siedlecki P. Establishment and functional validation of a structural homology model for human DNA methyltransferase 1. Biochem. Biophys. Res. Commun. 2003;306:558–563. doi: 10.1016/s0006-291x(03)01000-3. [DOI] [PubMed] [Google Scholar]
- 43.Barrow J.C. Synthesis and evaluation of imidazole acetic acid inhibitors of activated thrombin-activatable fibrinolysis inhibitor as novel antithrombotics. J. Med. Chem. 2003;46:5294–5297. doi: 10.1021/jm034141y. [DOI] [PubMed] [Google Scholar]
- 44.Rauer H. Structure-guided transformation of charybdotoxin yields an analog that selectively targets Ca2+-activated over voltage-gated K+ channels. J. Biol. Chem. 2000;275:1201–1208. doi: 10.1074/jbc.275.2.1201. [DOI] [PubMed] [Google Scholar]
- 45.Anand K. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra alpha-helical domain. EMBO J. 2002;21:3213–3224. doi: 10.1093/emboj/cdf327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Anand K. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 47.Rota P.A. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 48.Matthews D.A. Structure-assisted design of mechanism-based irreversible inhibitors of human rhinovirus 3C protease with potent antiviral activity against multiple rhinovirus serotypes. Proc. Natl. Acad. Sci. U. S. A. 1999;96:11000–11007. doi: 10.1073/pnas.96.20.11000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang H. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. U. S. A. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Takeda-Shitaka M. Evaluation of homology modeling of the severe acute respiratory syndrome (SARS) coronavirus main protease for structure-based drug design. Chem. Pharm. Bull. (Tokyo) 2004;52:643–645. doi: 10.1248/cpb.52.643. [DOI] [PubMed] [Google Scholar]
- 51.Xiong B. A 3D model of SARS CoV 3CL proteinase and its inhibitors design by virtual screening. Acta Pharmacol. Sin. 2003;24:497–504. [PubMed] [Google Scholar]
- 52.Palczewski K. Crystal structure of rhodopsin: A G-protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 53.Becker O.M. Modeling the 3D structure of GPCRs: advances and application to drug discovery. Curr. Opin. Drug Discov. Devel. 2003;6:353–361. [PubMed] [Google Scholar]
- 54.Schapira M. Nuclear hormone receptor targeted virtual screening. J. Med. Chem. 2003;46:3045–3059. doi: 10.1021/jm0300173. [DOI] [PubMed] [Google Scholar]
- 55.Schapira M. Rational discovery of novel nuclear hormone receptor antagonists. Proc. Natl. Acad. Sci. U. S. A. 2000;97:1008–1013. doi: 10.1073/pnas.97.3.1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bissantz C. Protein-based virtual screening of chemical databases. II. Are homology models of G-protein-coupled receptors suitable targets? Proteins. 2003;50:5–25. doi: 10.1002/prot.10237. [DOI] [PubMed] [Google Scholar]
- 57.Schafferhans A. Docking ligands onto binding site representations derived from proteins built by homology modelling. J. Mol. Biol. 2001;307:407–427. doi: 10.1006/jmbi.2000.4453. [DOI] [PubMed] [Google Scholar]
- 58.Oshiro C. Performance of 3D database molecular docking studies into homology models. J. Med. Chem. 2004;47:764–767. doi: 10.1021/jm0300781. [DOI] [PubMed] [Google Scholar]
- 59.Steinrücke P. Design of helical proteins for real-time endoprotease assays. Anal. Biochem. 2000;286:26–34. doi: 10.1006/abio.2000.4780. [DOI] [PubMed] [Google Scholar]
- 60.Benhamou B. A single amino acid that determines the sensitivity of progesterone receptors to RU486. Science. 1992;255:206–209. doi: 10.1126/science.1372753. [DOI] [PubMed] [Google Scholar]
- 61.Gray G.O. RU486 is not an antiprogestin in the hamster. J. Steroid Biochem. 1987;28:493–497. doi: 10.1016/0022-4731(87)90507-3. [DOI] [PubMed] [Google Scholar]
- 62.Williams P.A. Mammalian microsomal cytochrome P450 monooxygenase: structural adaptations for membrane binding and functional diversity. Mol. Cell. 2000;5:121–131. doi: 10.1016/s1097-2765(00)80408-6. [DOI] [PubMed] [Google Scholar]
- 63.Schoch G.A. Structure of human microsomal cytochrome P450 2C8. Evidence for a peripheral fatty acid binding site. J. Biol. Chem. 2004;279:9497–9503. doi: 10.1074/jbc.M312516200. [DOI] [PubMed] [Google Scholar]
- 64.Williams P.A. Crystal structure of human cytochrome P450 2C9 with bound warfarin. Nature. 2003;424:464–468. doi: 10.1038/nature01862. [DOI] [PubMed] [Google Scholar]
- 65.Lewis D.F. Molecular modeling of human cytochrome P450-substrate interactions. Drug Metab. Rev. 2002;34:55–67. doi: 10.1081/dmr-120001390. [DOI] [PubMed] [Google Scholar]
- 66.Lewis D.F. Modelling human cytochromes P450 involved in drug metabolism from the CYP2C5 crystallographic template. J. Inorg. Biochem. 2002;91:502–514. doi: 10.1016/s0162-0134(02)00429-4. [DOI] [PubMed] [Google Scholar]
- 67.Lewis D.F. Homology modelling of human CYP1A2 based on the CYP2C5 crystallographic template structure. Xenobiotica. 2003;33:239–254. doi: 10.1080/0049825021000048791. [DOI] [PubMed] [Google Scholar]
- 68.Lewis D.F. Homology modelling of CYP2A6 based on the CYP2C5 crystallographic template: enzyme-substrate interactions and QSARs for binding affinity and inhibition. Toxicol. In Vitro. 2003;17:179–190. doi: 10.1016/s0887-2333(02)00132-7. [DOI] [PubMed] [Google Scholar]
- 69.Lewis D.F. Molecular modelling of CYP2B6 based on homology with the CYP2C5 crystal structure: analysis of enzyme-substrate interactions. Drug Metabol. Drug Interact. 2002;19:115–135. doi: 10.1515/dmdi.2002.19.2.115. [DOI] [PubMed] [Google Scholar]
- 70.Lewis D.F. A molecular model of CYP2D6 constructed by homology with the CYP2C5 crystallographic template: investigation of enzyme-substrate interactions. Drug Metabol. Drug Interact. 2003;19:189–210. doi: 10.1515/dmdi.2003.19.3.189. [DOI] [PubMed] [Google Scholar]
- 71.Lewis D.F. Investigation of enzyme selectivity in the human CYP2C subfamily: homology modelling of CYP2C8, CYP2C9 and CYP2C19 from the CYP2C5 crystallographic template. Drug Metabol. Drug Interact. 2003;19:257–285. doi: 10.1515/dmdi.2003.19.4.257. [DOI] [PubMed] [Google Scholar]
- 72.Vermeulen N.P. Prediction of drug metabolism: the case of cytochrome P450 2D6. Curr. Top. Med. Chem. 2003;3:1227–1239. doi: 10.2174/1568026033451998. [DOI] [PubMed] [Google Scholar]
- 73.De Groot M.J. A novel approach to predicting P450 mediated drug metabolism. CYP2D6 catalyzed N-dealkylation reactions and qualitative metabolite predictions using a combined protein and pharmacophore model for CYP2D6. J. Med. Chem. 1999;42:4062–4070. doi: 10.1021/jm991058v. [DOI] [PubMed] [Google Scholar]
- 74.Afzelius L. Competitive CYP2C9 inhibitors: enzyme inhibition studies, protein homology modeling, and three-dimensional quantitative structure-activity relationship analysis. Mol. Pharmacol. 2001;59:909–919. doi: 10.1124/mol.59.4.909. [DOI] [PubMed] [Google Scholar]
- 75.Szklarz G.D. Molecular basis of P450 inhibition and activation: implications for drug development and drug therapy. Drug Metab. Dispos. 1998;26:1179–1184. [PubMed] [Google Scholar]
- 76.Mankowskia D.C. Prediction of human drug metabolizing enzyme induction. Curr. Drug Metab. 2003;4:381–391. doi: 10.2174/1389200033489352. [DOI] [PubMed] [Google Scholar]
- 77.Watkins R.E. Coactivator binding promotes the specific interaction between ligand and the pregnane X receptor. J. Mol. Biol. 2003;331:815–828. doi: 10.1016/s0022-2836(03)00795-2. [DOI] [PubMed] [Google Scholar]
- 78.Dussault I. A structural model of the constitutive androstane receptor defines novel interactions that mediate ligand-independent activity. Mol. Cell. Biol. 2002;22:5270–5280. doi: 10.1128/MCB.22.15.5270-5280.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xiao L. Insights from a three-dimensional model into ligand binding to constitutive active receptor. Drug Metab. Dispos. 2002;30:951–956. doi: 10.1124/dmd.30.9.951. [DOI] [PubMed] [Google Scholar]
- 80.Lewis D.F. Molecular modelling of the human glucocorticoid receptor (hGR) ligand-binding domain (LBD) by homology with the human estrogen receptor alpha (hERalpha) LBD: quantitative structure-activity relationships within a series of CYP3A4 inducers where induction is mediated via hGR involvement. J. Steroid Biochem. Mol. Biol. 2002;82:195–199. doi: 10.1016/s0960-0760(02)00158-9. [DOI] [PubMed] [Google Scholar]
- 81.Ekins S. Predicting undesirable drug interactions with promiscuous proteins in silico. Drug Discov. Today. 2004;9:276–285. doi: 10.1016/S1359-6446(03)03008-3. [DOI] [PubMed] [Google Scholar]
- 82.Mitcheson J.S. A structural basis for drug-induced long QT syndrome. Proc. Natl. Acad. Sci. U. S. A. 2000;97:12329–12333. doi: 10.1073/pnas.210244497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pearlstein R.A. Characterization of HERG potassium channel inhibition using CoMSiA 3D QSAR and homology modeling approaches. Bioorg. Med. Chem. Lett. 2003;13:1829–1835. doi: 10.1016/s0960-894x(03)00196-3. [DOI] [PubMed] [Google Scholar]
- 84.Hillisch A., Hilgenfeld R. Modern Methods of Drug Discovery. Birkhäuser Verlag AG; 2003. [DOI] [PubMed] [Google Scholar]