

Abstract
Cell culture-based production methods may assist in meeting increasing demand for seasonal influenza vaccines and developing production flexibility required for addressing influenza pandemics. MDCK-33016PF cells are used in propagation of a cell-based seasonal influenza vaccine (Optaflu®); but, like most continuous cell lines, can grow in immunocompromised mice to produce tumors. It is, therefore, essential that no residual cells remain within the vaccine, that cell lysates or DNA are not oncogenic, and that the cell substrate does not contain oncogenic viruses or oncogenic DNA. Multiple, redundant processes ensure the safety of influenza vaccines produced in MDCK-33016PF cells. The probability of a residual cell being present in a dose of vaccine is approximately 1 in 1034. Residual MDCK-DNA is ≤10 ng per dose and the ß-propiolactone used to inactivate influenza virus results in reduction of detectable DNA to less than 200 base pairs (bp). Degenerate PCR and specific PCR confirm exclusion of oncogenic viruses. The manufacturing process has been validated for its capacity to remove and inactivate viruses. We conclude that the theoretical risks arising from manufacturing seasonal influenza vaccine using MDCK-33016PF cells are reduced to levels that are effectively zero by the multiple, orthogonal processes used during production.
Keywords: Influenza vaccine, MDCK cell, Oncogenicity, Tumorigenicity
1. Introduction
Influenza virus infections are the major cause of respiratory illness and an important cause of morbidity and mortality in the elderly, very young, and those with intercurrent disease [1], [2]. Vaccination remains the principal means of control for seasonal infections and is the core strategy in pandemic influenza virus preparedness. The vast majority of influenza vaccines are produced in embryonated hens’ eggs, using decades-old technology, with one egg resulting in approximately one trivalent vaccine dose. While egg produced vaccines have a long history of safety there are issues of bioburden using this production system and a very small proportion of vaccines have an allergic reaction to egg protein [3], [4]. The logistics of egg production require a 6 month lead time before vaccine production can begin. This lengthy lead time results in a manufacturing inflexibility and can result in vaccine shortages as occurred in the 2004–05 influenza season.
To provide for the increasing demand for seasonal influenza vaccines and to develop the production flexibility required for addressing an influenza pandemic, the attention of both industry and government has turned to cell based production methods. These production systems have several potential advantages including being permissive for a wider range of strains than eggs [5], rapid scalability, and a greater antigenic fidelity of the vaccine virus to field strains [6], [7]. Furthermore, the closed nature of cell based manufacturing and the ability to exhaustively test cell banks for contaminating adventitious agents prior to production significantly reduces the risk of microbial contamination.
Several criteria are of importance in selecting a cell line for the production of inactivated influenza vaccines. The cell line must be permissive for a wide range of influenza viruses and permit their replication to high titer. The Madin-Darby canine kidney cell line (MDCK) which has been used in public health surveillance programs, because of its sensitivity and productivity for influenza virus detection, fulfils these requirements [8], [9]. The public health requirement for hundreds of millions of doses of influenza vaccine is facilitated by the use of cells selected for suspension growth in large capacity fermentors [10]. Additional selection of the cells for growth in animal origin free (AOF) media excludes a significant potential source for the introduction of viruses, like those that may be present in bovine serum, during the manufacturing process.
While MDCK cells provide significant advantages in vaccine production, the safety of vaccines derived from these cells is of paramount importance. MDCK cells were derived in the late 1950s from the kidney of a normal Cocker Spaniel. Similar to many continuous cell lines, MDCK cells display a variable capacity to produce tumors when injected into immunocompromised animals [11], [12], [13], [14], [15]. Tumorigenicity is the ability of injected cells to produce tumors consisting of the same cell type in an immunocompromised animal. Oncogenicity is the ability of cellular elements to transform the cells of the injected animal into neoplastic cells that then grow into host animal-derived tumors [16]. The MDCK-33016PF cells used in the manufacture the Novartis cell-based seasonal influenza vaccine Optaflu® are tumorigenic in immunocompromised (Nude) mice. It is, therefore, essential to ensure that no residual cells are present within a vaccine, that cell lysates or DNA are not oncogenic, and that procedures are in place to ensure that the cell substrate does not contain oncogenic viruses. Additional steps in the manufacturing process have been introduced to inactivate viruses and to remove or inactivate functional and potentially oncogenic DNA. We report here on the studies conducted and the multiple and redundant processes used to ensure the safety of an MDCK-33016PF cell-derived influenza vaccine with respect to issues of tumorigenicity and oncogenicity.
2. Methods and results
2.1. Analysis of potential oncogenicity of MDCK cell lysates and DNA
Studies with cell lysates and high molecular weight DNA prepared from end-of-production MDCK-33016PF cells were performed in accordance with EC Directive 86/609/EEC and current international guidelines for the testing of novel cell substrates [16]. These studies evaluated the potential oncogenicity of cell-free MDCK cell components (such as chromatin, host cell proteins, etc.) and purified DNA in neonatal nude mice, rats, and hamsters. 14–16 animals per sex per group (<4 days old) were used in each study. Treatment was by subcutaneous injection of 0.1–0.2 mL per animal. Evaluations included mortality and clinical signs, palpation of injection sites, macroscopic examinations (liver, spleen, kidneys, lungs, brain, lymph nodes and site of inoculation), and microscopic evaluation of selected tissues (muscle from the site of inoculation, lungs, and any lesions from all test article-treated animals and one animal per sex from the control groups).
In the studies with lysates, the control material was ß-propiolactone (BPL)-treated Tris buffer and test materials were an MDCK cell-free lysate prepared by freeze-thawing and a lysate of BPL-treated MDCK cells. The volumes of cell lysate administered to the animals, 0.2 mL to neonatal rats and hamsters and 0.1 mL to neonatal nude mice due to their small size, were the equivalents of 1 × 107 and 5 × 106 cells, respectively. 1 × 107 cells is the average number of cells needed to produce a single clinical dose of trivalent vaccine. The number of treated animals that survived to study termination (∼150 days) and the incidence of tumors are summarized in Table 1 .
Table 1.
Number of lysate-treated animals and tumor incidence.
| Treatment | Neonatal species testeda |
Number of tumors | |||
|---|---|---|---|---|---|
| Nude mice (N) | Rats (N) | Hamsters (N) | Total | ||
| BPL-treated Tris buffer (control) | 14 | 29 | 28 | 71 | 0 |
| MDCK-33016PF | 11 | 30 | 28 | 69 | 0 |
| BPL-treated-MDCK-33016PF | 12 | 28 | 30 | 70 | 0 |
| 210 | 0 | ||||
14–16 animals per sex per group (<4 days old) were used in each study.
Studies were also performed to test the potential oncogenicity of DNA prepared from MDCK-33016PF cells, influenza-infected MDCK-33016PF cells, and influenza-infected BPL-treated MDCK-33016PF cells. Neonatal Nude mice, rats, and hamsters were tested as described above. The control material used in the studies was mouse DNA. The concentration of DNA per 200 μL dose was ∼70 μg and is approximately equivalent to 1 × 107 MDCK-33016PF cells, the average number of cells needed to produce a single clinical dose of trivalent vaccine. The number of treated animals surviving to scheduled necropsy (∼150 days) and the incidence of tumors are summarized in Table 2 .
Table 2.
Number of DNA-treated animals and tumor incidence.
| Treatment | Neonatal species testeda |
Number of tumors | |||
|---|---|---|---|---|---|
| Nude mice (N) | Rats (N) | Hamsters (N) | Total | ||
| Mouse DNA (control) | 11 | 28 | 28 | 67 | 0 |
| MDCK-33016PF | 4 | 29 | 30 | 63 | 0 |
| Influenza-infected MDCK-33016PF | 16 | 28 | 27 | 71 | 0 |
| Influenza-infected, BPL-treated -MDCK-33016PF | 30 | 30 | 30 | 90 | 0 |
| 291 | 0 | ||||
14–16 animals per sex per group (<4 days old) were used in each study.
Mortality in neonatal mice during the first two weeks following dosing across all groups, including controls was ∼50%. The deaths were not treatment-related and were attributable to cannibalism by BALB/c dams provoked by interference with the nests for sexing, sorting, and dosing of pups. No tumors occurred in any tissues in any species. These studies demonstrate that the destruction of intact cells by lysis (freeze-thaw cycles or BPL treatment) abrogates the tumorigenic potential of intact MDCK-33016PF cells, and that DNA or lysates prepared from MDCK-33016PF cells are not oncogenic.
2.2. Clearance of cells in the manufacturing process
Viable MDCK-33016PF cells are removed by multiple processes. An initial centrifugation step effects the removal of approximately 99% of cells. A subsequent ultracentrifugation step also removes any intact cells that might remain that stage of the manufacturing process. The ultracentrifugation step was not, however, validated for its cell removal effect and, as a result, this step is not included in deriving an overall clearance factor.
There are several filtration steps in the manufacturing process, namely, an initial filtration through 0.45 μm filters followed by two filtrations through 0.2 μm filters, and a final sterile filtration through an 0.2 μm filter; these steps have been validated to effect ≥9.2, ≥8.8, ≥8.8, and ≥8.8 log10 reductions of cells, respectively (Fig. 1 ). The cell removal validations were carried out by the filter manufacturers using Saccharomyces cerevisiae for the 0.45 μm filters and Brevundimonas diminuta for the 0.2 μm filters; use of these readily cultured organisms permitted sensitive detection of any organisms that might pass through the filter. The effectiveness of filtration steps in the removal of the larger MDCK-33016PF cells is readily understandable when considering the nominal pore size of the filter (0.2 and 0.45 μm) relative to the diameter of an MDCK-33016PF cell, approximately 15 μm.
Fig. 1.
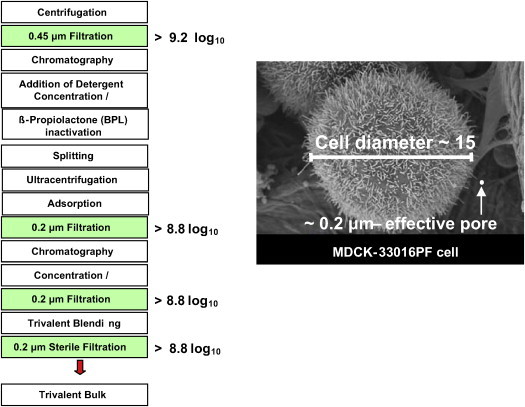
Overview of the manufacturing process. Steps validated for cell removal are shown in filled boxes with the respective log10 reduction values. The picture shows an electron micrograph of a single MDCK-33016PF cell for comparison of the cell diameter to the effective 0.2 μm pore size.
The cell reduction capacities of the chemical compounds that are used to purify, split and inactivate of the influenza virus were investigated. The effects of BPL, cetyltrimethylammonium bromide (CTAB) and polysorbate 80 (Tween 80) were studied to formally document the effects of each chemical on inactivation and depletion of cells. The results of these studies, which mimic the manufacturing process, indicate that in a worst-case challenge, BPL and polysorbate-80 are each capable of reducing the number of viable cells at least 10-fold. The cationic detergent, CTAB, had the strongest effect on cells with a complete destruction of cell cultures within 5 min. The extent of depletion of living cells by CTAB was shown to be ≥4 log10. However, the toxic effect of the reagents on cell viability during processing would likely be much higher than observed in the experimental approach, since the retention period of cells with each reagent would be much longer during the processing than in the experimental approach. Considered from this point of view the reduction capacity of the chemical moieties is unquestionably underestimated and a much higher depletion rate is anticipated. Taken together, the chemical solutions used for the processing of the vaccine have the capability to decrease the number of viable MDCK-33016PF cells by more than 6 log10.
The chromatography steps used in the manufacture of the viral vaccine are also likely to be effective in cell removal. However, as with the ultracentrifugation step, the chromatographic steps have not been specifically validated for cell removal and were therefore not counted in the overall viral clearance factor. The cumulative cell removal potential of the manufacturing process – centrifugations, filtrations, and chemical treatments – when combined, can effect a greater than 41 log10 reduction in cell number. Assuming that approximately 10 million cells are needed to produce each dose of vaccine, intact cells are removed by the manufacturing process to the point that there will be less than one cell in 1034 doses of vaccine. For all practical purposes, the probability of an intact MDCK-33016PF cell being in the influenza vaccine is zero.
2.3. DNA removal and inactivation by the manufacturing process
The specified DNA content of the final trivalent bulk and of the dose-adjusted final drug product is less than 10 ng/vaccine dose. This value is consistent with WHO guidelines and FDA Draft Guidance for the DNA content of vaccines produced from continuous cell lines. The FDA Draft Guidance does, however, note that for vaccines produced from continuous cell lines that are tumorigenic, additional considerations may apply, such as the requirement to degrade the DNA below certain sizes [16].
In the manufacture of Optaflu®, there are several steps that eliminate DNA. These steps include cellulose sulfate ion-exchange chromatography that binds the influenza virus and allows DNA to pass through and a subsequent CTAB precipitation step that, inter alia, precipitates the DNA. The DNA content of the vaccine is monitored throughout the production process by the Threshold™ total DNA assay system (MDS Analytical technologies, CA). The average, residual DNA content, from 18 manufacturing runs, with 3 different vaccine strains was 0.7 ng per dose.
During the Optaflu® manufacture, the viral harvest is inactivated with BPL. BPL is an alkylating agent that reacts, inter alia, with DNA (primarily guanine residues). BPL also reduces DNA size by inducing strand breaks and cross-links DNA with other DNA chains and with proteins. In addition to reducing the size of the DNA, BPL markedly reduces its ability to serve as a template for DNA polymerases [17], [18], [19]. It was noted by these later authors that, under specific conditions, BPL treatment abolishes all traces of DNA activity when evaluated by polymerase chain reaction (PCR).
No DNA greater than 200 base pairs (bps) in length was detected in final monovalent vaccine bulks by capillary gel electrophoresis (CGE). There is, undoubtedly, DNA greater than ca. 200 bps, but the amount is below the level of detection of the CGE method, and there may be some DNA approaching 1000 bps in length, which is sufficient to potentially encode for an oncogenic protein. Therefore, a mathematical (statistical) modeling approach was used to estimate the potential amount of long DNA sequences. Using this approach, the distribution of DNA lengths before BPL treatment was estimated, as was the relative number of breaks induced by the BPL treatment. The distribution of DNA lengths following BPL treatment was then evaluated by a mathematical model similar to that of Bose and Git [20]. The results of this evaluation provide an estimate of the relative amounts of DNA greater than a chosen length, e.g., 500 bps or 1000 bps, that were below the level of detection of the CGE methodology. The model indicated that, for each of the three production runs evaluated, >99.99% of the residual DNA is below 300 bps in length and that the amount of DNA >500 bps approaches zero.
2.4. Induction assays for oncogenic viruses and analysis or the presence of latent DNA viruses
To detect oncogenic DNA viruses a two stage process was implemented (Fig. 2 ). Stage 1: Treatment of the cells with sodium butyrate and 12-O-tetradecanoyl phorbol-13-acetate (TPA) for 5 days to induce viral DNA amplification and lytic replication. Stage 2: Examination of the cell pellet by transmission electron microscopy and by PCR for adenoviruses and canine papillomaviruses. Furthermore, to detect the presence of unknown herpesviruses or polyomaviruses, degenerate PCRs, capable of detecting a wide range of herpesviruses and polyomaviruses, were undertaken.
Fig. 2.
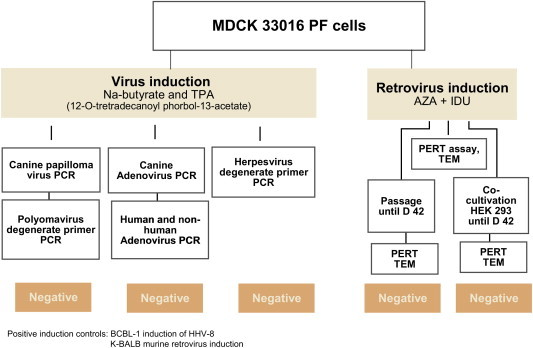
Overview of the virus induction studies.
On day 0 test article MDCK-33016PF cells were seeded at a density of 5 × 104 cells/mL into 60 × 150 cm2 flasks, in 50 mL of culture medium (2.5 × 106 cells/flask). On day 1, once the cells had formed a monolayer, the culture medium was replaced with fresh medium containing 0.3 mM sodium butyrate plus 20 ng/mL of TPA (30 bottles) or with fresh medium alone to act as a control for the induction step (30 bottles). The levels of inducing agents were determined following preliminary cytotoxicity experiments on MDCK-33016PF cells and are in line with levels shown to induce maximum induction of HHV-8 in BCBL-1 cells [21]. Induced and uninduced test article cells and BCBL-1 cells were monitored throughout the induction period for viability. As a further negative control MDCK cells derived directly from the ATCC stock (ATCC CCL-34) were seeded and treated like the untreated test article cells. Supernatant material and cell pellets were prepared from day 2 and day 5 samples. Total DNA from pellets of 1 × 107 cells were extracted using Qiagen DNA extraction kits.
BLCB-1 cells harboring latent HHV-8 were used as a positive control for the induction step. BCBL-1 cells were seeded on day 1 into 24 × 150 cm2 culture flasks in 50 mL of medium (1 × 107 cells flask) in the presence or absence of inducing agents. Preliminary studies had shown that peak induction occurred on day 5 of treatment. Induction was measured by analysis of cells with a specific, validated real-time PCR for HHV-8 genome copy number, normalized against the copy number of a cellular β-actin gene.
To exclude the presence of canine papillomaviruses, a specific, validated, real-time PCR was conducted on the cell pellets of the induced and uninduced cells. Test article and control samples were tested in triplicate. The internal positive control consisted samples of negative nucleic acid spiked with a positive sample 10-fold above the detection limit of the assay. Exogenous internal positive controls were included in all PCR master mixes to establish that all negative reactions were truly negative and not due to failed amplification. Negative controls consisted of the appropriate PCR mix to which negative control, human placental DNA was added. Sentinel extraction controls are designed to detect airborne PCR contamination and consisted of all extraction kit reagents processed alongside the test article. For a valid assay each of the triplicate negative controls were required to be negative and each of the positive controls including the internal positive control had to demonstrate amplification of their target sequences.
Specific PCRs for canine adenovirus had been conducted on the MCB and found to be negative. In this induction experiment, the interest was in excluding the presence of heterologous, potentially transforming, adenoviruses. A conventional PCR using primers targeted to the capsid hexon gene was employed on the induced and uninduced MDCK-33016PF cells. This PCR detects human adenoviruses Ad1, Ad2, Ad5 and Ad6 but in addition has a broader specificity for non-human adenoviruses [22], [23]. Degenerate PCR assays for herpesviruses and polyomaviruses were conducted, as previously described [24], [25], on cells from duplicate bottles of induced and uninduced MDCK-33016PF cells and on control MDCK – ATCC CCL-34 cells. DNA samples were quantified using a NanoDrop® ND-1000 spectrophotometer (Labtech UK) and 1 μg DNA used per reaction. Samples were analyzed for herpesvirus genomes using primers derived from viral polymerase gene sequences. Two outer PCRs were performed and then 1 μl of reaction products from each of these assays was amplified in each of two semi-nested PCRs as shown in Fig. 3 .
Fig. 3.

Conserved motifs in herpesvirus polymerase protein, and assay format.
Reactions included control or test DNA template, PCR primers (4 μM) and hotstart Taq mastermix kit (Qiagen) in a total volume of 50 μl. Thermal cycling was performed on a GenAmp PCR system 2400 (Applied Biosystems) using the following cycling parameters: 95 °C for 15 min followed by 5 cycles of 94 °C for 60 s; 44 °C for 2 min, 72 °C for 3 min, followed by 35 cycles as above but with an annealing temperature of 55 °C and a final extension at 72 °C for 7 min. Reaction products from both the outer and semi-nested reactions were purified using QuickStep™ 2 PCR purification kits (VH Bio Ltd, UK). Purified PCR products were subjected to capillary electrophoresis on an ABI PRISM® 31310 genetic Analyzer and evaluated using GeneScan software (Applied Biosystems). Three positive control samples, HCMV pol plasmid, HHV-6 pol plasmid and EBV positive Raji cell DNA were used at spike levels of 50 000, 5000 and 500 copies. A no template negative control consisting of water was included after every two samples and after the final sample. The MDCK – ATCC CCL-34 was used as a control for non-specific amplification products. Degenerate PCR for polyomaviruses was conducted similarly using primers derived from sequences of the large T antigen as shown in Fig. 4 . Positive control plasmids containing T-antigen sequences of JCV and BPyV were used at 50 000 and 5000 copies. Negative controls were as described for the degenerate herpesvirus assay.
Fig. 4.
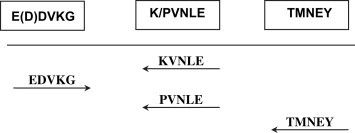
Conserved motifs in polyomavirus large T antigen, and assay format.
Analysis of the induced BCLB-1 cells indicated a 44-fold increase in HHV-8 sequences in the induced cells compared to the uninduced cells after normalization for the amount of β-actin DNA in the samples. Examination of 200 cell profiles of duplicate treatments of uninduced and induced BCBL-1 cells indicated the presence of herpesvirus virions in 3% and 0.5% of the TPA plus sodium butyrate treated cells whereas, no virions were detected in the untreated BCBL-1 cells or in the duplicate samples of induced and uninduced MDCK-33016PF cells. For the PCR reactions all the controls met the appropriate validity criteria. The real time CCR for canine oral papillomavirus and canine papillomaviruses 1 and 2 were negative down to the limit of detection (LOD) of 102 copies per reaction, equivalent to 1 copy in 1 × 105 cells. Similarly the conventional PCR for adenoviruses was negative to the same LOD. In the degenerate PCR for herpesviruses all the positive controls were clearly positive in the semi-nested analysis at 500 copies per reaction. In addition EBV sequences were detectable with primer set 6 and HHV-6 sequences were detectable with primer set 5; emphasizing that some mismatches in the primer sequences are tolerated within these conserved regions of the viral genomes. The induced and uninduced test article MDCK-33016PF cells and the control MDCK cells did not give rise to amplified fragments in the expected size range of herpesvirus products. For the polyomavirus degenerate PCRs the positive controls had the expected specificities and all controls gave positive results at the 5000 copy level. No amplified products of the expected size for a polyomavirus were detected in the induced or uninduced test article cells or the control MDCK cells.
2.5. Analysis for the presence of retroviruses
All vertebrate cells contain defective or non-defective retroviral sequences which may be expressed as mRNA. In the cells of some species, virions are spontaneously produced or may be induced to do so after treatment with agents including mutagens like 5-iodo-2′-deoxyuridine (IdU) or 5-azacytidine (AzaC), an inhibitor of DNA methyltransferases [26]. The capacity of MDCK-33016PF cells to produce retroviruses was evaluated by combined treatment with these agents followed by detection of viruses using transmission electron microscopy (TEM) and a highly sensitive F-PERT assay for reverse transcriptase [27]. In addition, induced cells were co-cultivated with cells permissive for a wide range of retroviruses to amplify potentially low levels of virus.
Duplicate 150 cm2 flasks were seeded with 1 × 106 MDCK-33016PF test article cells on day 0. On day one the medium was replaced with fresh medium containing 2 μg/mL AzaC and 30 μg/mL IDU. Similar MDCK-33016PF cultures were established without inducing agents to evaluate the toxicity of the inducing drugs. Duplicate cultures of human 293 HEK cells (ATCC CRL-1573) infected with feline leukemia virus were used as positive controls and duplicate cultures of murine K-Balb cells (ATCC CCL163.3) acted as a positive control for the induction of retrovirus expression. On day two, 24 h after treatment with the inducing agent, the medium was removed from test and control cultures, and the cells re-fed with medium without inducing agent.
On days 3, 4 and 5 (1, 2 and 3 days after removing the inducing agent), the supernatants were harvested for analysis by PERT, and fresh medium added. The PERT assays were conduced as previously described using a real-time PCR to detect cDNAs reverse transcribed from a plant virus target [27]. On day 5, cells from the induced and uninduced test article MDCK-33016PF cells were prepared for TEM by fixation in glutaraldehyde and paraformaldehyde in a cacodylate buffer. The cells were then post-fixed in 1% osmium tetroxide for 1 h, stained en bloc with uranyl acetate, rinsed, dehydrated in an ethanol series and embedded in Araldite. On day three, 3.3 × 105 cells removed from induced test article MDCK-33016PF cells were co-cultivated in the presence of 2 μg/mL of polybrene with 1 × 106 human HEK 293 cells or with control MDCK cells (ATCC CCL-34). The co-cultivations were maintained for 42 days and cells were passaged at least once a week. Test article cells were also passaged without indicator cells to test for any residual reverse transcriptase activity. As a further control, indicator cells were grown and passaged without test article cells to evaluate background reverse transcriptase activity. As a positive control, HEK 293 cells were infected with 100 infectious units of Feline leukemia virus (FeLV) then cultured and passaged in the same way as the test article co-cultures.
The uninduced K-Balb cultures were positive for reverse transcriptase on each day of analysis. The induced K-Balb cells displayed slightly higher levels of reverse transcriptase on day three compared to the uninduced cells (induced average, cycle threshold (Ct) 31.29, uninduced average Ct 34.14), peaking on day 4 at an average Ct of 24.39 before falling back to an average of 33.27. The F-PERT positive control of 103 retroviral particles, enumerated by electron microscopy, gave an average Ct of 29.74. Neither the uninduced nor induced MDCK cells displayed any reverse transcriptase activity, the Ct values remaining at 40 i.e. negative for all samples. In the co-cultures, the positive controls, 293 cultures infected with 100 infectious units of FeLV, were strongly positive by passage four with supernatant Ct values of 24 or under. After the final, eleventh, passage, all of the test article co-cultures with MDCK or HEK 293 cells were negative for reverse transcriptase in the PERT assay (Ct 40). Similarly, neither the test article cells nor the indicator cells cultured alone were positive for reverse transcriptase.
At least 200 median profiles (i.e. those where a nuclear profile is visible) of the induced and uninduced MDCK cells were examined. Observation of the membrane for budding virions and the cytoplasm for intracellular virions was undertaken but no retroviruses or, other adventitious agents were detected.
2.6. Quantitative virus safety risk assessments
In order to quantitatively assess potential risks represented by adventitious viruses, data were collected about growth properties of various relevant viruses in MDCK-33016PF cells, about the virus inactivating and virus clearance of the vaccine manufacturing process, and about the detection limits of applied PCR methods to detect adventitious viruses.
Virus growth was studied by inoculating representative viruses into MDCK-33016PF suspension cells followed by extended cultivation in the production medium. The DNA viruses studied were Parvovirus (Minute virus of mice), circovirus (porcine circovirus), polyomaviruses (SV-40, BK virus, avian polyomavirus), herpesviruses (herpes simplex virus, human cytomegalovirus, Epstein–Barr virus), and adenoviruses (human adenoviruses type 1, 5 and 6). The RNA viruses studied were reoviruses (mammalian reovirus 3, two avian reovirus strains), enteroviruses (echovirus, Coxsackie A and B virus), rhinoviruses (rhinovirus 1B, 37, Neth.8501841), coronavirus (229E), paramyxoviruses (parainfluenza 3, two human metapneumoviruses, two human respiratory syncytial viruses), Rous sarcoma virus and avian birnavirus.
At least 104 infectious units of virus were inoculated into 100 mL suspension cultures with 106 cells/mL. The cultures were kept in spinner flasks and were incubated at 33 °C for 14 days. Samples of cells and medium were taken directly after inoculation and at intervals thereafter (day 3, 5, 7, 10, 14) to monitor growth of the inoculated agent. By each sampling about 20% of the culture volume and cells were removed and the volume was replaced by fresh medium to maintain a growing culture. The culture samples, including cells, were frozen without separation or removal of cells. At the end of the studies, all samples, including inoculum virus samples were titered together on the same day.
Essentially the same viruses and agents that were used for growth studies were also used to assess the capacity of the BPL inactivation process. Inactivation was studied with maximum impurities present, i.e. with unpurified harvests, applying 0.05% BPL and incubating the inactivation at the shortest specified inactivation time periods. Standard micro titer plate virus titrations were used to measure starting tires and end titers after inactivation. If end titers were expected to be significantly below the detection limit of microtitrations (≤1.5 log10 TCID50/mL), larger volumes (up to 10 mL undiluted sample) were inoculated in culture flasks to test for residual virus at lower detection limits. Starting and end titers were compared and used to calculate the log reduction for each virus.
Selected model viruses were used to study virus inactivation and or removal by selected purification process steps. Infectious virus was spiked into process material before the process step, passed through the respective purification procedure at small scale, and samples after the process step were titered in comparison with the spiked starting material. When necessary, cytotoxic samples were diluted before testing. Residual virus below the detection limit of standard microtitration tests was assayed by plating larger volumes. Two enveloped viruses (herpes simplex virus and murine leukemia virus) and three non-enveloped viruses (reovirus3, simian virus 40, and porcine parvovirus) were used for virus removal studies. Study conditions reflected worst-case conditions such as lowest expected detergent concentrations used to detach surface antigens or shortest processing times during separation steps. These validation studies (altogether 14 studies with 5 model viruses) were conducted in collaboration with Analysis GmbH, now part of Newlab BioQuality AG, Erkrath, Germany.
Combining all data on the virus growth in MDCK-33016PF cells, on the inactivation or process removal of various model viruses, and with consideration of applicable detection limits for virus exclusion tests, a process-specific risk assessment was made using quantitative data relative to infectious doses. The methodology of these quantitative risk assessments has been described elsewhere [28]. It should be noted that further virus removal studies and robustness studies were done after publication, thus results presented here partly vary from those published earlier (Table 3 ).
Table 3.
Quantitative risk assessments for adventitious agents: Worst case residual virus content in a trivalent vaccine dose.
| Virus type/group | log10 human infectious dose/vaccine dosea |
|---|---|
| Influenza vaccine virus | −16.0 |
| Adenovirus | −8.1 |
| Herpes simplex virus | −9.7 |
| Other human Herpesviruses | −10.1 |
| Parainfluenzavirus | −14.0 |
| Respiratory syncytial virus | −14.9 |
| Metapneumovirus | −9.9 |
| Mumps/Measles virus | −9.9 |
| Coronavirus | −14.1 |
| Rhinovirus | −8.8 |
| Enterovirus | −8.1 |
| Polyomavirus (JC/BK virus) | −7.7 |
| Hepatitis B, C, G viruses | −11.0 |
| Human Retroviruses | −11.8 |
| Mammalian Orthoreovirus | −8.3 |
| Chlamydia | −13.4 |
| Mycoplasma | −13.1 |
The values for potential avian and other animal-derived contaminating viruses, which are not known to infect humans, range from −7.1 (avian polyomavirus) to −13.8 (avian retrovirus).
Realistic worst-case assumptions were made to adequately cover variables, such as potential contaminating virus titers, virus passage numbers, dilutions, or inactivation results with different types of the same virus family. Starting with a co-isolated contaminant during isolation of an influenza virus strain from a human throat sample plus further contamination by avian viruses assumed to occur during egg-passage, the virus titers were estimated for all subsequent steps of the vaccine strain selection and passaging to the working seed for vaccine manufacturing. At this level a further contamination by aerosol or droplet infection from the human operator was assumed for all human viruses. The calculation was then continued through the entire manufacturing process, including fermentation, purification, and formulation of a final, trivalent vaccine. Considering also the results obtained during worst-case robustness studies, the end results of the risk assessments in terms of human infectious doses per vaccine dose, were between 10−7.1 (avian polyomavirus) and 10−14.9 (respiratory syncytial virus). The according end result for influenza virus would be 10−16 (Table 3).
3. Discussion
Multiple, independent, tests and processes, implemented under GMP ensure the safety of Optaflu® with regard to tumorigenic and oncogenic risks. The theoretical risk associated with tumorigenicity relates to the possibility of inoculation of a viable MDCK-33016PF cell into a recipient of the vaccine followed by the growth of that cell in the recipient. The risk of this occurrence is, for all practical purposes, zero. MDCK-33016PF cells are partly killed during the replication of influenza virus and multiple specific steps in the manufacturing process remove or inactivate remaining cells. These processes include centrifugation, filtration, chemical inactivation and membrane disruption (viral splitting). The filtration steps, BPL virus inactivating stages, and viral splitting activities have been specifically investigated to determine their ability to remove or inactivate cells and each operates through orthogonal mechanisms. Collectively, the evaluated steps have the capacity to reduce cellular concentration by ≥41 log10 (validated data ≥35.6 log10). Given that up to approximately 10 million cells are required to produce one dose of vaccine, the risk of an MDCK-33016PF cell being in one vial is less than ∼10−34.
In any continuous cell line – indeed, any cell substrate – the possibility that a latent oncogenic virus might be present needs detailed consideration. In the case of DNA viruses, certain members of the herpesvirus, polyomavirus and papillomavirus families can induce tumors in their hosts and other species, while some adenoviruses have been shown to be oncogenic in heterologous hosts. Viruses from these families may also establish latent infections or, become integrated into chromosomal DNA following abortive replication. In addition, viruses might be introduced into the cells from the virus seed, through human contamination or via raw materials. These viral risks were countered by two independent approaches. First a rigorous analysis of the MDCK-33016PF cells was undertaken to exclude the presence of infectious oncogenic viruses or infectious oncogenic viral genomes; secondly the manufacturing process was shown to be capable of inactivating potential virus contaminants at very high levels.
Neonatal animals are highly susceptible to the presence of oncogenic viruses [29] but no tumors were induced in neonatal nude mice, rats or hamsters by cell lysates or high molecular weight DNA extracted from MDCK-33016PF cells, therefore, neither infectious virions nor infectious genomes capable of inducing tumors were present. A more definitive analysis for the presence of oncogenic DNA viruses is provided by degenerate PCR capable of broad detection of herpesviruses and polyomaviruses. These methods have been shown to be capable of detecting new viruses [24] emphasizing their utility in this role. The possibility of detecting oncogenic herpesviruses is enhanced by induction procedures that can result in genome amplification and recrudescence from latency. None of the assays involving, degenerate PCRs for herpesviruses and polyomaviruses, broad PCR for adenoviruses and the specific PCRs for papillomaviruses, revealed the presence of oncogenic DNA virus sequences.
Dogs are not known to be infected by an exogenous retrovirus nor known to possess proviruses encoding an infective endogenous retrovirus. Although claims for canine retroviruses have been made from time to time, none have been independently confirmed. In some species, infectious, endogenous, retroviruses may be expressed after induction with mutagens or inhibitors of DNA methyltransferases. Since these experiments were conducted, Khan and colleagues have formulated a specific algorithmic approach for the exclusion of retroviruses in vaccine cell substrates [30]; the studies conducted here are in accord with that approach. The PERT assay for retroviruses is a highly sensitive method for the detection of this family of viruses and does not rely on specific genome information. No retrovirus production was detected following induction or, after co-culture with cells known to be permissive for a range of retroviruses.
Our growth studies, as well as systematic literature studies, have shown that MDCK cells only support the growth of a very limited spectrum of viruses (herpes simplex virus, parainfluenza virus, and reovirus), which is almost identical to those viruses that grow in embryonated eggs. In contrast to eggs, however, MDCK cells do not support growth of avian viruses. Compared to existing egg-derived vaccines, the MDCK cells used to produce vaccine virus from influenza virus strains isolated in eggs do not introduce new or higher virus-associated risk, as the host spectrum of MDCK cells is similar to that of embryonated eggs. Using a simple risk scoring algorithm to estimate and compare the risks of adventitious agents when different cell substrates are used for virus isolation and vaccine manufacturing, MDCK cells used to produce vaccines from egg-derived isolates gave either the same or lower risk scores than embryonated eggs used for isolation and production [31].
Modern virus production methods as described here have more in common with recombinant protein or, monoclonal antibody, production than with traditional vaccine manufacturing. The downstream manufacturing steps have the capacity to remove and inactivate viruses particularly through the action of BPL. Process-specific and quantitative risk assessments were made for a wide range of potential process contaminants including latent or oncogenic viruses, such as polyomaviruses, adenoviruses, retroviruses, herpesviruses, and parvoviruses. These model calculations showed that even under worst-case conditions and in the unlikely case of a contamination, the process is able to reduce such contaminants to levels which are million-fold below infectious levels. Even if the calculated worst case contaminant levels are considered, the final vaccine product would still be unable to transmit infectious viruses.
Although there are several theoretical ways in which exogenous DNA could contribute to oncogenesis, including insertional inactivation of tumor suppressor genes or transcription of regulatory microRNAs [32], the principal risk relates to dominant oncogenes. For an oncogene to result in transformation it would have to be present as a full-length copy, be transfected into recipient cells and become integrated in a chromosomal region favorable for expression. Burns reported in 1991 that DNA could be directly oncogenic in experiments using high levels of plasmid containing the activated HRAS gene [33]. More recently, scientists at the FDA have made important contributions by demonstrating the quantitative limits to direct DNA oncogenicity [34], [35]. In their initial studies, 12.5 μg of a plasmid capable of expressing both ras and myc were required to induce tumors in mice [35]. More recently, they have lowered this oncogenic level for a dual oncogene plasmid to 1 ng [36]. These recent data have reduced the safety margin estimate for 10 ng of high molecular weight DNA but methods to reduce the size or functional activity of DNA, like BPL treatment, can increase the safety factor (i.e. inverse of risk) by 107 [36].
The conclusions on safety margins are based on intact high molecular weight DNA, but the treatment with BPL radically reduces the functional size of DNA. It should be noted that, even if sequences >1000 bps were present, relatively few 1000-mers will encompass the correct sequence for encoding an oncogene. Our methodology did not enable us to differentiate fragments with still intact structure and BPL-crosslinked and otherwise impaired DNA fragments. However, the residual DNA does not approach that size. Of the maximum amount of 10 ng per dose of DNA present in Optaflu®, less than 0.01% is calculated to be above 300 bps, i.e. less than 1 pg, with an even lower percentage above 500 bps.
The use of cell based vaccines has the potential to transform the strategies for prophylaxis of seasonal and pandemic influenza, but demonstrating the safety of these vaccines is an essential prerequisite. We conclude that the multiple, orthogonal processes used during production that the theoretical risks arising from manufacturing Optaflu® using MDCK-33016PF cells are reduced to levels that are effectively zero.
Acknowledgement
This project has been funded in part with Federal funds from the Office of Public Health Emergency Preparedness, Office of Research and Development Coordination, under Contract No. HHS0100200600012C.
References
- 1.World Health Organization (WHO) Influenza vaccines. Wkly Epidemiol Rec. 2005;80:277–288. [Google Scholar]
- 2.Fiore A.E., Shay D.K., Broder K., Iskander J.K., Uyeki T.M., Mootrey G. Prevention and control of seasonal influenza with vaccines, recommendations of the advisory committee on immunization practices (ACIP) MMWR. 2009;58:1–52. [PubMed] [Google Scholar]
- 3.Coop C.A., Balanon S.K., White K.M., Whisman B.A., Rathkopf M.M. Anaphylaxis from the influenza virus vaccine. Int Arch Allergy Immunol. 2008;146(1):85–88. doi: 10.1159/000112507. [DOI] [PubMed] [Google Scholar]
- 4.Zeiger R.S. Current issues with influenza vaccination in egg allergy. J Allergy Clin Immunol. 2002;110:834–840. doi: 10.1067/mai.2002.129372. [DOI] [PubMed] [Google Scholar]
- 5.Lu B., Zhou H., Chan W., Kemble G., Jin H. Single amino acid substitutions in the hemagglutinin of influenza A/Singapore/21/04 (H3N2) increase virus growth in embryonated chicken eggs. Vaccine. 2006;24:6691–6693. doi: 10.1016/j.vaccine.2006.05.062. [DOI] [PubMed] [Google Scholar]
- 6.Robertson J.S., Cook P., Attwell A.M., Williams S.P. Replicative advantage in tissue culture of egg-adapted influenza virus over tissue-culture derived virus: implications for vaccine manufacture. Vaccine. 1995;13:1583–1588. doi: 10.1016/0264-410x(95)00085-f. [DOI] [PubMed] [Google Scholar]
- 7.Katz J.M., Wang M., Webster R.G. Direct sequencing of the HA gene of influenza (H3N2) virus in original clinical samples reveals sequence identity with mammalian cell-grown virus. J Virol. 1990;64:1808–1811. doi: 10.1128/jvi.64.4.1808-1811.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Noma K., Kiyotani K., Kouchi H., Fujii Y., Egi Y., Tanaka K. Endogenous protease-dependent replication of human influenza viruses in two MDCK cell lines. Arch Virol. 1998;143(10):1893–1909. doi: 10.1007/s007050050428. [DOI] [PubMed] [Google Scholar]
- 9.Oh D.Y., Barr I.G., Mosse J.A., Laurie K.L. MDCK-SIAT1 cells show improved isolation rates for recent human influenza viruses compared to conventional MDCK cells. J Clin Microbiol. 2008 Jul;46(7):2189–2194. doi: 10.1128/JCM.00398-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chu C., Lugotsev V., Golding H., Betenbaugh M., Shiloach J. Conversion of MDCK cell line to suspension culture by transfecting with human siat7e gene and its application for influenza virus production. Proc Natl Acad Sci U S A. 2009;106:14802–14807. doi: 10.1073/pnas.0905912106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stiles C., Desmond W., Chuman L., Sato G., Saier M.H. Relationship of cell growth behavior in vitro to tumorigenicity in athymic nude mice. Cancer Res. 1976;36:3300–3305. [PubMed] [Google Scholar]
- 12.Taub M., Ben U., Chuman L., Rindler M.J., Saier M.H., Jr., Sato G. Alterations in growth requirements of kidney epithelial cells in defined medium associated with malignant transformation. J Supramol Struct Cell Biochem. 1981;15(1):63–72. doi: 10.1002/jsscb.1981.380150107. [DOI] [PubMed] [Google Scholar]
- 13.U Hs, Boerner P., Rindler M.J., Chuman L., Saier M.H. Characterization of chemically and virally transformed variants of Madin-Darby canine kidney (MDCK) epithelial cells. J Cell Physiol. 1985;122(2):299–307. doi: 10.1002/jcp.1041220220. [DOI] [PubMed] [Google Scholar]
- 14.Boerner P., Saier M.H., Jr. Adaptive regulatory control of system A transport activity in a kidney epithelial cell line (MDCK) and in a transformed variant (MDCK-T1) J Cell Physiol. 1985;122(2):308–315. doi: 10.1002/jcp.1041220221. [DOI] [PubMed] [Google Scholar]
- 15.Zhang D.L., Li L.J., Xia G.T., He X.Y., Gao B.X., Bai X.H. Analyses of chromosomal karyotypes and cytogenetic variations of animal cell lines. Yi Chuan Xue Bao. 2001;28(4):327–344. [PubMed] [Google Scholar]
- 16.Derivation and characterisation of cell substrates used for production of biotechnological/biological products, September 97. http://www.ich.org/LOB/media/MEDIA429.pdf FDA Draft Guidance for Industry: characterization and qualification of cell substrates and other biological starting materials used in the production of viral vaccines for the prevention and treatment of infectious diseases, Sep 2006; ICH Q5D. Available from:
- 17.Morgeaux S., Tordo N., Gontier C., Perrin P. Beta-propiolactone treatment impairs tile biological activity of residual DNA from BHK-21 cells infected with rabies virus. Vaccine. 1993;11:82–90. doi: 10.1016/0264-410x(93)90343-v. [DOI] [PubMed] [Google Scholar]
- 18.Perrin P., Morgeaux S. Inactivation of DNA by ß-propiolactone. Biologicals. 1995;23:207–211. doi: 10.1006/biol.1995.0034. [DOI] [PubMed] [Google Scholar]
- 19.Grosseil C., Guerin P., Abramowicz P. Evaluation by polymerase chain reaction on the effect of beta-propiolactone and binary ethyleneimine on DNA. Biologicals. 1995;23:213–220. doi: 10.1006/biol.1995.0035. [DOI] [PubMed] [Google Scholar]
- 20.Bose S.M., Git Y. Mathematical modelling and computer simulation of linear polymer degradation: simple scissions. Macromol Theor Simul. 2004;13:453–473. [Google Scholar]
- 21.Yu Y., Black J.B., Goldsmith C.S., Browning P.J., Bhalla K., Offermann M.K. Induction of human herpesvirus-8 DNA replication and transcription by butyrate and TPA in BCBL-1 cells. J Gen Virol. 1999 Jan;80(Pt 1):83–90. doi: 10.1099/0022-1317-80-1-83. [DOI] [PubMed] [Google Scholar]
- 22.Allard A., Girones R., Juto P., Wadell G. Polymerase chain reaction for detection of adenoviruses in stool samples. J Clin Microbiol. 1991;29(11):2683. doi: 10.1128/jcm.29.11.2683-.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kiss I., Matiz K., Allard A., Wadell G., Benkö M. Detection of homologous DNA sequences in animal adenoviruses by polymerase chain reaction. Acta Vet Hung. 1996;44(2):243–251. [PubMed] [Google Scholar]
- 24.Jarrett R.F., Johnson D., Wilson K.S., Gallagher A. Molecular methods for virus discovery. Dev Biol (Basel) 2006;123:77–88. [PubMed] [Google Scholar]
- 25.Wilson K.S., Gallagher A., Freeland J.M., Shield L.A., Jarrett R.F. Viruses and Hodgkin lymphoma: no evidence of polyomavirus genomes in tumor biopsies. Leuk Lymphoma. 2006;47(7):1315–1321. doi: 10.1080/10428190500525789. [DOI] [PubMed] [Google Scholar]
- 26.Khan A.S., Muller J., Sears J.F. Early detection of endogenous retroviruses in chemically induced mouse cells. Virus Res. 2001;79(1–2):39–45. doi: 10.1016/s0168-1702(01)00280-5. [DOI] [PubMed] [Google Scholar]
- 27.Lovatt A., Black J., Galbraith D., Doherty I., Moran M.W., Shepherd A.J. High throughput detection of retrovirus-associated reverse transcriptase using an improved fluorescent product enhanced reverse transcriptase assay and its comparison to conventional detection methods. J Virol Methods. 1999;82(2):185–200. doi: 10.1016/s0166-0934(99)00111-1. [DOI] [PubMed] [Google Scholar]
- 28.Gregersen J.P. A quantitative risk assessment of exposure to adventitious agents in a cell culture-derived subunit influenza vaccine. Vaccine. 2008;26:3332–3340. doi: 10.1016/j.vaccine.2008.03.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lewis A.M., Jr. Developing an approach to evaluate the use of neoplastic cells as vaccine substrates. Dev Biol (Basel) 2001;106:37–43. [PubMed] [Google Scholar]
- 30.Khan A.S., Ma W., Ma Y., Kumar A., Williams D.K., Muller J. Proposed algorithm to investigate latent and occult viruses in vaccine cell substrates by chemical induction. Biologicals. 2009;37(3):196–201. doi: 10.1016/j.biologicals.2009.02.016. [DOI] [PubMed] [Google Scholar]
- 31.Gregersen J.P. A risk-assessment model to rate the occurrence and relevance of adventitious agents in the production of influenza vaccines. Vaccine. 2008;26:3297–3304. doi: 10.1016/j.vaccine.2008.03.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.He L., Thomson J.M., Hemann M.T., Hernando-Monge E., Mu D., Goodson S. A microRNA polycistron as a potential human oncogene. Nature. 2005;435(7043):828–833. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burns P.A., Jack A., Neilson F., Haddow S., Balmain A. Transformation of mouse skin endothelial cells in vivo by direct application of plasmid DNA encoding the human T24 H-ras oncogene. Oncogene. 1991;6(11):1973–1978. [PubMed] [Google Scholar]
- 34.Sheng-Fowler L., Cai F., Zhu Y., Pal A., Athanasiou M., Orrison B. Oncogenicity of DNA in vivo: tumor induction with expression plasmids for activated H-ras and c-myc. Biologicals. 2008;36(3):184–197. doi: 10.1016/j.biologicals.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 35.Sheng-Fowler L., Lewis A.M., Jr., Peden K. Quantitative determination of the infectivity of the proviral DNA of a retrovirus in vitro: evaluation of methods for DNA inactivation. Biologicals. 2009;37(4):259–269. doi: 10.1016/j.biologicals.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 36.Sheng-Fowler L., Lewis A.M., Jr., Peden K. Issues associated with residual cell-substrate DNA in viral vaccines. Biologicals. 2009;37(3):190–195. doi: 10.1016/j.biologicals.2009.02.015. [DOI] [PubMed] [Google Scholar]