

Abstract
The demands of structural and functional genomics for large quantities of soluble, properly folded proteins in heterologous hosts have been aided by advancements in the field of protein production and purification. Escherichia coli, the preferred host for recombinant protein expression, presents many challenges which must be surmounted in order to over-express heterologous proteins. These challenges include the proteolytic degradation of target proteins, protein misfolding, poor solubility, and the necessity for good purification methodologies. Gene fusion technologies have been able to improve heterologous expression by overcoming many of these challenges. The ability of gene fusions to improve expression, solubility, purification, and decrease proteolytic degradation will be discussed in this review. The main disadvantage, cleaving the protein fusion, will also be addressed. Focus will be given to the newly described SUMO fusion system and the improvements that this technology has advanced over traditional gene fusion systems.
Keywords: Protein expression, Gene fusion, Protein fusion, Ubhiquitin, SUMO, Ubiquitin-like proteins
Efficient recombinant protein expression is a major bottleneck for structural genomics and proteomics. Despite progress in automation, soluble protein expression is frequently the rate-limiting step for many researchers. The preferred host for recombinant protein expression has historically been Escherichia coli (E. coli) due to the simplicity and low costs associated with using this host. While E. coli has proved a successful host for the expression of many heterologous proteins, it is frequently not capable of expressing soluble heterologous proteins. The Southeast Collaboratory for Structural Genomics (SECSG) 1 reports that of the 6386 proteins they have expressed in E. coli only 22.7% (1452) have been soluble (as published on SECSG web page 03/04/2005). Much advancement has been made toward improving recombinant protein expression in E. coli, including the development of strong promoters [1], co-expression with chaperones [2], and through the use of protein fusions. No other technology has been as effective at improving the solubility of recombinant proteins as fusion systems, especially for difficult-to-express proteins. A variety of structures have been used as fusion motifs (Table 1 ). These fusion proteins are frequently employed to enhance protein expression and facilitate purification [3], [4], [5]. There is no particular similarity among these proteins in terms of molecular weight, structure, or function, with the exception of ubiquitin (Ub) and SUMO, which share a common structure [6]. This review will discuss the advantages of fusion technologies. These advantages include the manner in which protein expression is enhanced, proteolytic degradation of the target protein is decreased, protein folding and solubility are increased, and purification and detection are simplified. The main disadvantage of fusion technology, cleaving the protein fusion, is also covered. In addition, focus is given to the newly described SUMO fusion systems. SUMO fusions appear to have all of the advantages of traditional fusion systems, and due to the activity of SUMO proteases do not encounter the same difficulties with cleavage.
Table 1.
Fusion partner proteins, which enhance expression and simplify purification
| Fusion partner | Molecular weight (kDa) | Purification method |
|---|---|---|
| 6× Hisa-SUMO | 12.2 | Ni–NTAa |
| CTHS | 7 | NTHS |
| GST | 27.3 | Glutathione–Sepharose |
| 6× His-NusA | 58.4 | Ni–NTA |
| MBP | 45 | Amylose resin |
| TRX | 14.3 | ThioBond resin |
| 6× HIS-Ub | 8.6 | Ni–NTA |
| Flag peptide | 1 | Anti-Flag antibodies |
Abbreviations used: SUMO, small ubiquitin modifying protein; Ni–NTA, nickel–nitriloacetic acid; CTHS, C-terminal half of SUMO; NTHS, N-terminal half of SUMO; GST, glutathione S-transferase; MBP, maltose binding protein; Trx, thioredoxin; Ub, ubiquitin.
The N-terminus of SUMO is very flexible and can accommodate a variety of affinity tags (e.g., GST). The SUMO system can therefore be tailored to the researcher’s desired affinity tag.
Advantages of recombinant protein fusion technology
Protection from degradation
Proteolysis is highly regulated and plays critical roles in maintaining cellular homeostasis, including removing unwanted or incorrectly folded proteins from the cell [7]. Often, recombinant proteins are viewed as unwanted by cells and are subjected to proteolytic degradation, decreasing the level of recombinant protein expression (reviewed in [8]). Several strategies have been developed to protect recombinant proteins from degradation including the use of protease inhibitors [9], secretion into the periplasm [10] or culture medium [11], and generating protective fusions [12]. Fusions between the N-terminus of target proteins and protein tags (Table 1) have been shown to protect the target protein from degradation [13], [14], [15]. Furthermore, fusions with C-terminus [16], dual C- and N-terminal fusions [17], and tandem fusions of multiple copies of the target gene [18] have been shown to afford protection from proteolytic degradation.
The compartmentalization hypothesis describes the mechanism by which gene fusions protect against proteolytic degradation [19]. Fusions can promote the translocation of their partner proteins to different cellular compartments, thereby decreasing the concentration of the recombinant protein in the protease-rich cytosol. For example, maltose binding protein (MBP) can translocate to the membrane compartment of the cell [20] or SUMO can translocate from the cytosol to the nucleus [21]. The tags can thereby compartmentalize their partner proteins and decrease the susceptibility to proteolytic degradation.
Enhanced recombinant protein expression
Protein expression is a complex process dependent upon mRNA stability and translational efficiency, as well as transcriptional regulation. Enhanced recombinant protein expression is the result of a high mRNA copy number, efficient translational initiation and elongation, stability of the mRNA, and translation enhancers (reviewed in [22]). Many heterologous genes are not translated efficiently due to codon usage bias. Codon bias has been implicated as one of the main reasons for inefficient translation (e.g., malaria parasite genes [23]). Codon bias has been overcome by engineering new strains or cell lines that contain rare tRNAs or by altering the problematic codons to more common prokaryotic codons [24]. Strong and highly regulated promoters from E. coli, yeast, and insect cells are available [14], [25], [26], and as such transcription of the heterologous gene is usually not a rate-limiting factor. It has been observed that many fusion proteins enhance protein expression [14], [15], [27], however, the exact mechanism by which fusion proteins achieve this enhanced expression is unknown. It has been speculated that enhanced expression is the result of the highly conserved structures of these proteins. Attachment of a highly evolved translational frame at the N-terminus of an inefficiently translated protein may help to improve the latter’s efficiency of expression [28].
Improved protein folding
Although E. coli is usually the first choice as a recombinant expression organism, many eukaryotic proteins, especially proteins with disulfide bridges, cannot be expressed as soluble, active, and properly folded proteins in E. coli [29]. When a large quantity of protein is expressed, macromolecular crowding (200–300 mg/ml in the cytoplasm) presents an unfavorable environment for protein folding. Frequently, the result of a high concentration of incorrectly folded recombinant protein is the formation of inclusion bodies [30]. In fact, the level of aggregated protein can increase to the extent that inclusion bodies are observable by light microscopy as round bodies surrounding the cytosol of E. coli [31]. While the formation of inclusion bodies does afford protection from proteolytic degradation, and re-folding can recover active proteins from inactive inclusion bodies, the initial expression of correctly folded, soluble recombinant protein is ideal [32].
Several strategies have been developed to promote the expression of properly folded recombinant protein, including co-expression of molecular chaperones [2] and foldases [33], expression of secreted proteins [34], and expression of protein fusions [35]. Fusion partners (Table 1) have been shown to act as solubility enhancers, although the exact mechanism by which they improve solubility has not been described. It has been hypothesized that these fusions may act as chaperones [36]. The fusion of a stable or conserved structure to an insoluble recombinant protein may serve to stabilize and promote proper folding of the recombinant protein. MBP has been shown to function as a general molecular chaperone in the context of a fusion protein by binding to aggregation-prone folding intermediates of passenger proteins and preventing their self-association [36]. Fusion tags have also been hypothesized to enhance the solubility of the protein target by acting as a nucleus of folding (“molten globule hypothesis”) [37], [38]. This theory suggests that a fusion tag acts as a nucleation site for the folding of the target protein. Ub, which has a highly hydrophobic core, has been shown to be the fastest folding protein known [39]. Ub’s ancient structure is highly conserved in SUMO, as well as in other ubiquitin-like proteins used as fusions [6]. When fused to Ub or SUMO, otherwise insoluble proteins have been observed to fold properly and be soluble [14], [15].
Simplified purification and detection
The use of protein fusion technology offers the opportunity to simplify and facilitate purification and detection of recombinant proteins. Fusing a tag to the target protein provides a one-step purification procedure by passing cell extracts or supernatants over an appropriate affinity matrix. Numerous examples of affinity purification exist for fusion proteins, including nickel–nitriloacetic acid to isolate hexahistidine-fused proteins [40], biotin to isolate streptavidin-fused proteins [41], and amylose to isolate MBP-fused proteins [42]. The fusion tag can also be used to identify the recombinant product through Western blotting by way of anti-tag antibodies [43].
Disadvantages of recombinant protein fusion technology
Cleaving the fusion protein
Cleavage of protein fusions to generate free protein remains the major disadvantage of protein fusion technologies. Cleavage of the fusion is usually necessary because the fusion interferes with the structural or functional properties of the recombinant protein [44]. A variety of chemical and enzymatic methodologies have been developed to cleave fusions [45], [46]. These methods include the use of engineered cleavage sites, which are recognized by the proteases and are positioned between the fusion tag and the protein target. Proteases that have been employed to cleave fusion tags include tobacco etch virus (Tev) protease [47], factor Xa, or thrombin protease (reviewed in [46]). Problems associated with proteolytic cleavage of fusion tags are low yield, precipitation of the protein of interest, labor-intensive optimization of cleavage conditions, expense of proteases, and failure to recover active, structurally intact protein [48].
Another major problem encountered with the cleavage of fusion proteins is the generation of non-native N-terminal amino acids. Many structural and therapeutic proteins require a specific N-terminus, other than methionine, for biological activity (e.g., chemokines). All nascent proteins have methionine as their N-terminal residue; some (e.g., precursor proteins, which undergo proteolytic maturation) then undergo post-translational processing and modification, leading to a variety of N-terminal amino acids. Experiments performed by Varshavsky and co-workers [49] in the 1980s demonstrated that the expression of ubiquitin-β-galactosidase in yeast leads to rapid processing of ubiquitin in the cells. The processing of Ub-X-fusion, where X is any amino acid, was independent of the identity of residue X [19], [49]. It was observed that the in vivo half-life of the resulting protein varied as a function of the N-terminal residue of the protein (residue X). The “N-end rule” proposes that there is a relationship between the identity of a protein’s N-terminal residue and its half-life. For an in depth discussion of the “N-end rule,” please see appropriate publications of Varshavsky and co-workers [19], [49].
Cleavage by the aforementioned proteases results in the retention of several amino acids, which are downstream from the cleavage site and required for protease recognition. For example, thrombin will cleave the sequence LVPRGS at the arginine residue, resulting in an N-terminal extension of the target protein by two amino acids (GS) [46]. Since many proteins require a specific N-terminus for biological activity, half-life, or structural stability, this characteristic of protease cleavage can have serious effects on the ability to recombinantly produce active protein. Notable exceptions include the intein, SUMO, and Ub fusion systems. The intein fusion system utilizes the inducible self-cleavage activity of engineered protein splicing elements (termed inteins). In the presence of thiols such as DTT, β-mercaptoethanol or cysteine, the intein undergoes specific self-cleavage which releases the target protein from the tag. When fused directly to the N-terminus of the target protein cleavage results in the production of a target protein without any extra non-native residues attached to its terminus after cleavage [45].
Unlike intein-mediated cleavage, the Ub and SUMO fusion systems do require the use of proteases for removal of the tag. However, the SUMO proteases and the deubiquitylating enzymes (DUBs) are distinct from other proteases that recognize a peptide sequence, as they recognize the tertiary structure of the SUMO or Ub tag. When the target protein is fused directly to the C-terminus of SUMO or Ub, cleavage will not result in extraneous residues at the N-terminus of the target protein and therefore will yield native-like protein [15], [49]. The DUBs have a major drawback in that complete cleavage of the Ub tag requires a large amount of enzyme (1:10 molar ratio of DUB to target) [50]. The SUMO protease has been reported to be much more robust, requiring only a 1:5000 molar ratio of protease to target [15]. The new generation of SUMO proteases has been shown to be even more catalytically active requiring only 1:100,000 molar ratio of protease to target (Butt et al. unpublished results) (see further discussion below).
SUMO fusion technology
SUMO and SUMO pathways
The SUMO family of proteins has been described extensively in the literature (reviewed in [51]). SUMO and the SUMO pathway are highly conserved in all eukaryotes from yeast to humans, but are absent from prokaryotes (Fig. 1 ) [52], [53], [54]. Saccharomyces cerevisiae has only a single SUMO gene, SMT3 which is essential for viability [55], [56]. In contrast, vertebrates have three SUMO genes, SUMO-1, SUMO-2, and SUMO-3. The three human SUMOs are highly homologous, with human SUMO-1 sharing 50% sequence identity with human SUMO-2 and SUMO-3 [56], and human SUMO-2 and SUMO-3 sharing 87% sequence identity with each other [57]. SMT3 shares 47% sequence identity with human SUMO-1. Although overall sequence identity between Ub and SUMO is only 18%, structure determination by nuclear magnetic resonance (NMR) reveals that the two proteins possess a common three-dimensional structure that is characterized by a tightly packed globular fold with β-sheets wrapped around one α-helix [6].
Fig. 1.

The SUMO cycle. SUMO is synthesized as a precursor and cleaved by SUMO proteases (yeast Ulp1 or Ulp2). Activating enzyme (E1), conjugating enzyme (E2), and ligase (E3) are named according to yeast where most were discovered.
The family of SUMO proteins function, like Ub, as covalent modifiers of other proteins, and sumoylation occurs in a similar fashion to the ubiquitination of proteins [57], [58], [59] (Fig. 1). SUMO is activated in an ATP-dependent step by the formation of a thioester bond with the SUMO activating enzyme E1. Following activation, SUMO is transferred to SUMO E2, or SUMO-conjugating enzyme. Following conjugation to E2, SUMO is transferred to its target protein through the activity of SUMO E3. These modifications can have a variety of cellular consequences. Sumoylation can antagonize the action of ubiquitination by preventing ubiquitin-meditated proteolysis [60]. SUMO conjugation has also been observed to impact higher-order chromatin structure [61], transcriptional regulation [62], DNA repair pathways [63], nuclear transport [64], and signal transduction pathways [65].
SUMO conjugation to target proteins is a dynamic process, which changes in response to a variety of stimuli [66]. SUMO can be removed from target proteins enzymatically by SUMO C-terminal hydrolases-isopeptidases, several of which are now known in many species including yeast (Ulp1 and Ulp2) [67], [68], [69], Arabidopsis [70], and humans [59], [71], [72], [73]. SUMO proteases share a common C-terminal domain (Ulp domain), and have no sequence homology to the DUBs [67], [68], [69].
SUMO fusion enhances protein expression, solubility, and purification in prokaryotes
Recently, SUMO has been fused to the N-terminus of several proteins, including matrix metalloprotease (MMP13), green fluorescent protein (GFP), and SARS-CoV 3CL protease, and used as a recombinant expression system [15], [74] (www.lifesensors.com). SUMO fusion leads to enhanced expression and solubility (Fig. 2 A and unpublished results). For example, when MMP13 is expressed without SUMO fusion in E. coli, it is contained primarily in inclusion bodies. However, when MMP13 is expressed as a SUMO fusion, MMP13 was observed primarily in the soluble fraction [15]. The effect that SUMO has on enhancing protein solubility has been explained in part by the structure of SUMO. SUMO has an external hydrophilic surface and inner hydrophobic core, which may exert a detergent-like effect on otherwise insoluble proteins [15]. A hexahistidine SUMO fusion construct has been shown to enhance expression and facilitate purification with Ni–NTA chromatography. Ni–NTA chromatography has been used to purify the fusion from the cellular lysate (Fig. 2B) [15].
Fig. 2.

Enhanced expression and purification using the SUMO fusion system. (A) Enhanced expression of SARS-CoV 3CL protease (3CL) by SUMO fusion in E. coli. Cells grown in Luria–Bertani (LB) media were induced at the temperatures and for the lengths of time indicated. Just before expression was induced and after induction was completed the cells from a 1.5 ml aliquot of culture were lysed. Samples of whole cell lysates (∼7.5 μl) from the various expression conditions were resolved in 12% SDS-gels and stained with Coomassie blue. Molecular weights (in kDa) were as indicated, and arrowheads highlight expected/observed positions of respective expressed protein bands [74]. (B) An SDS gel depicting the purification of SARS-CoV 3CL protease. Total cell lysate was passed over a Ni–NTA column, washed with 40 mM imidazole, and eluted with 300 mM imidazole (affinity purified). Cleavage with the SUMO protease was conducted under standard conditions, and the sample was passed over another Ni–NTA column to remove the SUMO protease and tag (subtracted). Aliquots of the samples (each containing ∼5 μg protein) were separated on a 12% SDS-gel and stained with Coomassie blue. The migration positions of the SUMO fusion and the proteins resulting from the cleavage are as indicated [74].
SUMO proteases
As described above, the major disadvantage of fusion protein technology is cleavage of the tag. Commonly used proteases do not cleave all fusions efficiently, accurately and, moreover, can generate extraneous amino acids at the N-terminus of the target protein [75]. SUMO proteases, which are members of the cysteine protease superfamily, are able to overcome these difficulties. SUMO proteases (LifeSensors) are accurate and efficient in cleavage of the SUMO tag and allow for retention of the desired N-terminus. To date, we have cleaved approximately 100 SUMO fusions and never observed erroneous cleavage within the partner protein. Unlike other proteases that recognize a peptide sequence, SUMO proteases recognize the tertiary structure of the SUMO tag and cleavage does not result in an extended the N-terminus in the partner protein. Owing to its unique structure, SUMO protease can accommodate a variety of SUMO fusion partner proteins (Fig. 4). SUMO proteases have been observed to completely cleave a broad range (6–110 kDa) of partner proteins fused to SUMO [15], [74]. In addition, SUMO protease is able to cleave efficiently under a wide range of conditions, including pH, temperature, and ionic strength (Fig. 3 ). Unfortunately, SUMO protease is not able to cleave target proteins which contain an N-terminal proline. SUMO proteases have a constrictive hydrophobic tunnel within the active site, and substrates must pass through this tunnel during cleavage (Fig. 4 ) [76]. It has been hypothesized that the constrictive tunnel is unable to accommodate the structural changes induced by prolines near the cleavage site.
Fig. 4.

The X-ray crystal structure of human SUMO protease (Senp2, grey) in complex with human SUMO-1 (black) [76]. SUMO-1 must pass through a constrictive hydrophobic tunnel (arrow) within the active site in order to be cleaved by Senp2.
Fig. 3.

Effect of various conditions on the activity of SUMO protease. SUMO-green fluorescent protein fusion (SUMO-GFP) (2.5 μg) was incubated with SUMO protease (1:5000 molar ratio of SUMO-GFP to protease) for 1 h under conditions described in the figure: temperatures of 4 or 37 °C, and concentration ranges of imidazole (at 25 °C), sodium dodecyl sulfate (SDS) (at 25 °C), Triton X-100 (at 25 °C), urea (at 25 °C), or guanidine hydrochloride (at 25 °C). The data show that the enzyme is active over a broad temperature range and tolerates highly adverse biochemical conditions [15].
SUMO fusion in eukaryotic hosts, the split SUMO solution
This review is primarily focused on the role of SUMO fusion expression systems in prokaryotic cells. However, it is relevant to mention that fusion of Ub to under-expressed genes in eukaryotes enhances the level of protein production, even though the Ub tag is cleaved by endogenous DUBs [77]. We have also observed dramatic enhancement of under-expressed proteins in eukaryotes after fusion with SUMO. Similar to Ub, the SUMO tag is cleaved soon after translation (Edavettal and Butt, unpublished). These studies suggest that protein expression enhancing properties of Ub and like proteins (SUMO) are preserved in eukaryotes.
The lack of an endogenous SUMO protease in E. coli facilitates the use of SUMO as a purification tag in this host organism. However, in eukaryotic systems, endogenous SUMO proteases immediately cleave SUMO fusions, making purification using the SUMO tag impossible. A novel fusion system, split SUMO, has been developed to overcome this problem. In the split SUMO system, SUMO is bifurcated into N- and C-terminal halves (NTHS and CTHS) (Fig. 5 A). Fusion of the CTHS to the N-terminus of a target protein allows enhancement of expression in eukaryotes and, most importantly, the fusion is not recognized by endogenous SUMO proteases; therefore it is not cleaved in vivo and can facilitate purification. CTHS has strong hydrophobic interactions with NTHS, and mixture of NTHS with the CTHS fusion allows for reconstitution of SUMO in vitro. Reconstitution of intact SUMO by non-covalent interactions between the two halves in vitro generates a substrate for SUMO protease, permitting cleavage and purification of the partner protein (Fig. 5B). We have also observed that fusion of a secretory signal to the N-terminus of the SUMO fusion permits enhanced expression and prevents cleavage of the SUMO tag (Edavettal et al., unpublished). We believe that the secretory signal affords protection from the endogenous SUMO proteases because the nascent protein is captured by the endoplasmic reticulum, and secreted into the media, therefore bypassing the protease-rich cytosol.
Fig. 5.
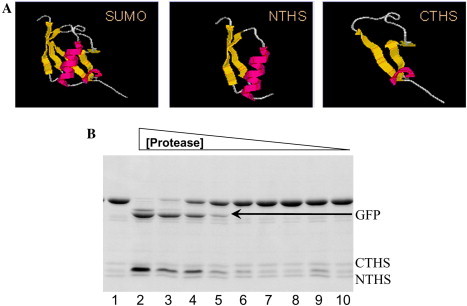
The split SUMO expression system. (A) The structure of SUMO, and the N- and C-terminal halves (NTHS and CTHS). The target protein is fused to the CTHS, and the full SUMO structure is reconstituted after purification by incubating with NTHS. (B) The reconstitution of cleavable structure on the CTHS fusion in vitro and cleavage by Ulp1. An SDS PAGE of 6× His-CTHS-GFP (8 μg) fusion protein purified from E. coli that was incubated for 30 min at 30 °C with purified 6× His-NTHS (2 μg) and increasing concentrations of SUMO protease (lane 1, CTHS-GFP + NTHS and lanes 2–9, CTHS-GFP + NTHS with decreasing concentrations of SUMO protease (1000, 500, 250, 125, 62.5, 31.3, 15.6, 7.8, and 3.9 ng)). Note the release of free GFP indicating the reconstitution of the full SUMO structure at high protease concentrations.
Conclusions and future directions
Rapid, efficient, and cost-effective protein expression and purification strategies are required for high throughput structural genomics and the production of therapeutic proteins. Fusion protein technology represents one strategy to achieve these goals. Fusion protein technology allows for the enhanced expression of recombinant, proteins, which are protected from degradation and have improved solubility and simplified purification and detection. The major drawback to most fusion systems is cleavage of the fusion tag, which can result in the generation of non-native N-termini and erroneous cleavage. The SUMO fusion expression system affords the advantages of other fusion technologies, but also the SUMO protease is efficient, accurate, and does not result in the extraneous residues at the N-terminus of the target protein. We are currently developing the second generation of SUMO tags, which have demonstrated added enhanced expression of under-expressed proteins, and the second generation of SUMO proteases, which are even more robust than Ulp1.
The SUMO and split SUMO fusion technologies allow for efficient recombinant expression in both prokaryotic and eukaryotic hosts. This technology has been used to efficiently express and purify a variety of proteins, including membrane proteins [78]. Another innovative application of the SUMO fusion technology is its use as an immobilization tool for protein arrays. Applications for protein arrays include expression profiling, protein isolation and purification, protein–protein interaction studies, and small molecular drug discovery [79], [80], [81]. SUMO has both flexible N- and C-termini, allowing dynamic processes to occur with relative ease, and the complex can be released from the solid support by the action of SUMO protease, which facilitates the identification and characterization of the partner protein(s). SUMO and other fusion technologies will allow for efficient recombinant expression of proteins, aiding in numerous future discoveries.
Acknowledgments
In 2004, the Nobel Prize in chemistry was awarded to Drs. Aaron Ciechanover, Avram Hershko, and Irwin Rose for their contributions towards the discovery of the ubiquitin pathway. We thank them and others, including Drs. Alex Varshavsky, Keith Wilkinson, and Arthur Haas, for their seminal work in this fascinating field. The SUMO fusion technology described here is based on their life’s work. Tauseef Butt is grateful to these colleagues for the support of his ubiquitin and UBL work. This work is supported by grants R43 AI 51752, R43 HL 69744, R43 GM 067271, R43 GM 068404, and 1R43-AI058584-01A1 to T.R.B.
Footnotes
Abbreviations used: SECSG, Southeast Collaboratory for Structural Genomics; SUMO, small ubiquitin modifying protein; Ni–NTA, nickel–nitriloacetic acid; CTHS, C-terminal half of SUMO; NTHS, N-terminal half of SUMO; GST, glutathione S-transferase; MBP, maltose binding protein; Trx, thioredoxin; Ub, ubiquitin; Tev, tobacco etch virus; DUBs, deubiquitylating enzymes.
References
- 1.Studier F.W., Rosenberg A.H., Dunn J.J., Dubendorff J.W. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 1990;185:60–89. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- 2.Ikura K., Kokubu T., Natsuka S., Ichikawa A., Adachi M., Nishihara K., Yanagi H., Utsumi S. Co-overexpression of folding modulators improves the solubility of the recombinant guinea pig liver transglutaminase in Escherichia coli. Prep. Biochem. Biotechnol. 2002;32:189–205. doi: 10.1081/PB-120004130. [DOI] [PubMed] [Google Scholar]
- 3.De Marco V., Stier G., Blandin S., de Marco A. The solubility and stability of recombinant proteins are increased by their fusion to NusA. Biochem. Biophys. Res. Commun. 2004;322:766–771. doi: 10.1016/j.bbrc.2004.07.189. [DOI] [PubMed] [Google Scholar]
- 4.Wang C., Castro A.F., Wilkes D.M., Altenberg G.A. Expression and purification of the first nucleotide-binding domain and linker region of human multidrug resistance gene product: comparison of fusions to glutathione S-transferase, thioredoxin and maltose-binding protein. Biochem. J. 1999;338:77–81. [PMC free article] [PubMed] [Google Scholar]
- 5.Pryor K.D., Leiting B. High-level expression of soluble protein in Escherichia coli using a His6-tag and maltose-binding-protein double-affinity fusion system. Protein Express. Purif. 1997;10:309–319. doi: 10.1006/prep.1997.0759. [DOI] [PubMed] [Google Scholar]
- 6.Bayer P., Arndt A., Metzger S., Mahajan R., Melchior F., Jaenicke R., Becker J. Structure determination of the small ubiquitin-related modifier SUMO-1. J. Mol. Biol. 1998;280:275–286. doi: 10.1006/jmbi.1998.1839. [DOI] [PubMed] [Google Scholar]
- 7.Goldberg A.L, Dice J.F. Intracellular protein degradation in mammalian and bacterial cells. Annu. Rev. Biochem. 1974;43:835–869. doi: 10.1146/annurev.bi.43.070174.004155. [DOI] [PubMed] [Google Scholar]
- 8.Rozkov A., Enfors S.-O. Analysis and control of proteolysis of recombinant proteins in Escherichia coli. Adv. Biochem. Eng./Biotechnol. 2004;89:163–195. doi: 10.1007/b95567. [DOI] [PubMed] [Google Scholar]
- 9.Prouty W., Goldberg A. Efffects of protease inhibitors on protein breakdown in Escherichia coli. J. Biol. Chem. 1972;247:3341–3352. [PubMed] [Google Scholar]
- 10.Talmadge K., Gilbert W. Cellular location affects protein stability in Escherichia coli. Proc. Natl. Acad. Sci. USA. 1982;79:1830–1833. doi: 10.1073/pnas.79.6.1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hogset A., Blingsom O.R., Saether O., Gautvik V.T., Holmgren E., Hartmanis M., Josephson S., Gabrielsen O.S., Gordeladze J.O., Alestrom P. Expression and characterization of a recombinant human parathyroid hormone secreted by Escherichia coli employing the staphylococcal protein A promoter and signal sequence. J. Biol. Chem. 1990;265:7338–7344. [PubMed] [Google Scholar]
- 12.Murby M., Uhlen M., Stahl S. Upstream strategies to minimize proteolytic degradation upon recombinant production in Escherichia coli. Protein Express. Purif. 1996;7:129–136. doi: 10.1006/prep.1996.0018. [DOI] [PubMed] [Google Scholar]
- 13.Martinez A., Knappskog P.M., Olafsdottir S., Doskeland A.P., Eiken H.G., Svebak R.M., Bozzini M., Apold J., Flatmark T. Expression of recombinant human phenylalanine hydroxylase as fusion protein in Escherichia coli circumvents proteolytic degradation by host cell proteases. Isolation and characterization of the wild-type enzyme. Biochem. J. 1995;306:589–597. doi: 10.1042/bj3060589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Butt T.R., Jonnalagadda S., Monia B.P., Sternberg E.J., Marsh J.A., Stadel J.M., Ecker D.J., Crooke S.T. Ubiquitin fusion augments the yield of cloned gene products in Escherichia coli. Proc. Natl. Acad. Sci. USA. 1989;86:2540–2544. doi: 10.1073/pnas.86.8.2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malakhov M.P., Malakhova O.A., Drinker M., Weeks S., Butt T.R. SUMO fusion and SUMO-specific proteases for efficient expression and purification of proteins. J. Struct. Funct. Genom. 2004;5:75–86. doi: 10.1023/B:JSFG.0000029237.70316.52. [DOI] [PubMed] [Google Scholar]
- 16.Koken M.H., Odijk H.H., van Duin M., Fornerod M., Hoeijmakers J.H. Augmentation of protein production by a combination of the T7 RNA polymerase system and ubiquitin fusion: overproduction of the human DNA repair protein, ERCC1, as an ubiquitin fusion protein in Escherichia coli. Biochem. Biophys. Res. Commun. 1993;195:643–653. doi: 10.1006/bbrc.1993.2094. [DOI] [PubMed] [Google Scholar]
- 17.Murby M., Cedergren L., Nilsson J., Nygren P.A., Hammarberg B., Nilsson B., Enfors S.O., Uhlen M. Stabilization of recombinant proteins from proteolytic degradation in Escherichia coli using a dual affinity fusion strategy. Biotechnol. Appl. Biochem. 1991;14:336–346. [PubMed] [Google Scholar]
- 18.Shen S.H. Multiple joined genes prevent product degradation in Escherichia coli. Proc. Natl. Acad. Sci. USA. 1984;81:4627–4631. doi: 10.1073/pnas.81.15.4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Varshavsky A. The N-end rule: functions, mysteries, uses. Proc. Natl. Acad. Sci. USA. 1996;93:12142–12149. doi: 10.1073/pnas.93.22.12142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nikaido H. Maltose transport system of Escherichia coli: an ABC-type transporter. FEBS Lett. 1994;346:55–58. doi: 10.1016/0014-5793(94)00315-7. [DOI] [PubMed] [Google Scholar]
- 21.Kishi A., Nakamura T., Nishio Y., Maegawa H., Kashiwagi A. Sumoylation of Pdx1 is associated with its nuclear localization and insulin gene activation. Am. J. Physiol. Endocrinol. Metab. 2003;284:E830–E840. doi: 10.1152/ajpendo.00390.2002. [DOI] [PubMed] [Google Scholar]
- 22.Makrides A.C. Strategies for achieving high-level expression of genes in Escherichia coli. Microbiol. Rev. 1996;60:512–538. doi: 10.1128/mr.60.3.512-538.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Narum D.L., Kumar S., Rogers W.O., Fuhrmann S.R., Liang H., Oakley M., Taye A., Sim B.K., Hoffman S.L. Codon optimization of gene fragments encoding Plasmodium falciparum merzoite proteins enhances DNA vaccine protein expression and immunogenicity in mice. Infect. Immun. 2001;69:7250–7253. doi: 10.1128/IAI.69.12.7250-7253.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brinkmann U., Mattes R.E., Buckel P. High-level expression of recombinant genes in Escherichia coli is dependent on the availability of the dnaY gene product. Gene. 1989;85:109–114. doi: 10.1016/0378-1119(89)90470-8. [DOI] [PubMed] [Google Scholar]
- 25.Dubendorff J.W., Studier F.W. Controlling basal expression in an inducible T7 expression system by blocking the target T7 promoter with the lac repressor. J. Mol. Biol. 1991;219:45–59. doi: 10.1016/0022-2836(91)90856-2. [DOI] [PubMed] [Google Scholar]
- 26.Thiem S.M., Miller L.K. Differential gene expression mediated by late, very late and hybrid baculovirus promoters. Gene. 1990;91:87–94. doi: 10.1016/0378-1119(90)90166-o. [DOI] [PubMed] [Google Scholar]
- 27.Sachdev D., Chirgwin J.M. Fusions to maltose-binding protein: control of folding and solubility in protein purification. Methods Enzymol. 2000;326:312–321. doi: 10.1016/s0076-6879(00)26062-x. [DOI] [PubMed] [Google Scholar]
- 28.Arechaga I., Miroux B., Runswick M.J., Walker J.E. Over-expression of Escherichia coli F1F(o)-ATPase subunit α is inhibited by instability of the uncB gene transcript. FEBS Lett. 2003;547:97–100. doi: 10.1016/s0014-5793(03)00677-x. [DOI] [PubMed] [Google Scholar]
- 29.Prinz W.A., Aslund F., Holmgren A, Beckwith J. The role of the thioredoxin and glutaredoxin pathways in reducing protein disulfide bonds in the Escherichia coli cytoplasm. J. Biol. Chem. 1997;272:15661–15667. doi: 10.1074/jbc.272.25.15661. [DOI] [PubMed] [Google Scholar]
- 30.Chen J., Acton T.B., Basu S.K., Montelione G.T., Inouye M. Enhancement of the solubility of proteins overexpressed in Escherichia coli by heat shock. J. Mol. Microbiol. Biotechnol. 2002;4:519–524. [PubMed] [Google Scholar]
- 31.Carrio M.M., Cubarsi R., Villaverde A. Fine architecture of bacterial inclusion bodies. FEBS Lett. 2000;471:7–11. doi: 10.1016/s0014-5793(00)01357-0. [DOI] [PubMed] [Google Scholar]
- 32.Georgiou G., Valax P. Expression of correctly folded proteins in Escherichia coli. Curr. Opin. Biotechnol. 1996;7:190–197. doi: 10.1016/s0958-1669(96)80012-7. [DOI] [PubMed] [Google Scholar]
- 33.Wulfing C., Pluckthun A. Correctly folded T-cell receptor fragments in the periplasm of Escherichia coli. Influence of folding catalysts. J. Mol. Biol. 1994;242:655–669. doi: 10.1006/jmbi.1994.1615. [DOI] [PubMed] [Google Scholar]
- 34.Sugamata Y., Shiba T. Improved secretory production of recombinant proteins by random mutagenesis of hlyB, an α-hemolysin transporter from Escherichia coli. Appl. Environ. Microbiol. 2005;71:656–662. doi: 10.1128/AEM.71.2.656-662.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chopra A.K., Brasier A.R., Das M., Xu X.J., Peterson J.W. Improved synthesis of Salmonella typhimurium enterotoxin using gene fusion expression systems. Gene. 1994;144:81–85. doi: 10.1016/0378-1119(94)90207-0. [DOI] [PubMed] [Google Scholar]
- 36.Fox J.D., Kapust R.B., Waugh D.S. Single amino acid substitutions on the surface of Escherichia coli maltose-binding protein can have a profound impact on the solubility of fusion proteins. Protein Sci. 2001;10:622–630. doi: 10.1110/ps.45201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Englander S.W. Protein folding intermediates and pathways studied by hydrogen exchange. Annu. Rev. Biophys. Biomol. Struct. 2000;29:213–238. doi: 10.1146/annurev.biophys.29.1.213. [DOI] [PubMed] [Google Scholar]
- 38.Creighton T.E. How important is the molten globule for correct protein folding? Trends Biochem. Sci. 1997;22:6–10. doi: 10.1016/s0968-0004(96)20030-1. [DOI] [PubMed] [Google Scholar]
- 39.Khorasanizadeh S., Peters I.D., Roder H. Evidence for a three-state model of protein folding from kinetic analysis of ubiquitin variants with altered core residues. Nat. Struct. Biol. 1996;3:193–205. doi: 10.1038/nsb0296-193. [DOI] [PubMed] [Google Scholar]
- 40.Sharma S.K., Evans D.B., Vosters A.F., McQuade T.J., Tarpley W.G. Metal affinity chromatography of recombinant HIV-1 reverse transcriptase containing a human renin cleavable metal binding domain. Biotechnol. Appl. Biochem. 1991;14:69–81. [PubMed] [Google Scholar]
- 41.Ford C.F., Suominen I., Glatz C.E. Fusion tails for the recovery and purification of recombinant proteins. Protein Express. Purif. 1991;2:95–107. doi: 10.1016/1046-5928(91)90057-p. [DOI] [PubMed] [Google Scholar]
- 42.diGuan C., Li P., Riggs P.D., Inouye H. Vectors that facilitate the expression and purification of foreign peptides in Escherichia coli by fusion to maltose-binding protein. Gene. 1988;67:21–30. doi: 10.1016/0378-1119(88)90004-2. [DOI] [PubMed] [Google Scholar]
- 43.Sun Q.M., Cao L., Fang L., Chen C., Dai J., Chen L.L., Hua Z.C. Expression, purification of human vasostatin120–180 in Escherichia coli and its anti-angiogenic characterization. Protein Express. Purif. 2005;39:288–295. doi: 10.1016/j.pep.2004.10.024. [DOI] [PubMed] [Google Scholar]
- 44.Balbas P. Understanding the art of producing protein and nonprotein molecules in Escherichia coli. Mol. Biotechnol. 2001;19:251–267. doi: 10.1385/MB:19:3:251. [DOI] [PubMed] [Google Scholar]
- 45.Chong S., Mersha F.B., Comb D.G., Scott M.E., Landry D., Vence L.M., Perler F.B., Benner J., Kucera R.B., Hirvonen C.A., Pelletier J.J., Paulus H., Xu M.Q. Single-column purification of free recombinant proteins using a self-cleavable affinity tag derived from a protein splicing element. Gene. 1997;192:277–281. doi: 10.1016/s0378-1119(97)00105-4. [DOI] [PubMed] [Google Scholar]
- 46.Jenny R.J., Mann K.G., Lundblad R.L. A critical review of the methods for cleavage of fusion proteins with thrombin and factor Xa. Protein Express. Purif. 2003;31:1–11. doi: 10.1016/s1046-5928(03)00168-2. [DOI] [PubMed] [Google Scholar]
- 47.Carrington J.C., Cary S.M., Parks T.D., Dougherty W.G. A second proteinase encoded by a plant polyvirus genome. Embo J. 1989;8:365–370. doi: 10.1002/j.1460-2075.1989.tb03386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baneyx F. Recombinant protein expression in Escherichia coli. Curr. Opin. Biotechnol. 1999;10:411–421. doi: 10.1016/s0958-1669(99)00003-8. [DOI] [PubMed] [Google Scholar]
- 49.Bachmair A., Finley D., Varshavsky A. In vivo half-life of a protein is a function of its amino-terminal residue. Science. 1986;234:179–186. doi: 10.1126/science.3018930. [DOI] [PubMed] [Google Scholar]
- 50.Catanzariti A., Soboleva T.A., Jans D.A., Board P.G., Baker R.T. An efficient system for high-level expression and easy purification of authentic recombinant proteins. Protein Sci. 2004;13:1331–1339. doi: 10.1110/ps.04618904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Johnson E.S. Protein modification by SUMO. Annu. Rev. Biochem. 2004;73:355–382. doi: 10.1146/annurev.biochem.73.011303.074118. [DOI] [PubMed] [Google Scholar]
- 52.Hochstrasser M. Evolution and function of ubiquitin-like protein-conjugation systems. Nat. Cell Biol. 2000;2:E153–E157. doi: 10.1038/35019643. [DOI] [PubMed] [Google Scholar]
- 53.Jentsch S., Pyrowolakis G. Ubiquitin and its kin: how close are the family ties? Trends Cell Biol. 2000;10:335–342. doi: 10.1016/s0962-8924(00)01785-2. [DOI] [PubMed] [Google Scholar]
- 54.Muller S., Hoege C., Pyrowolakis G., Jentsch S. SUMO, ubiquitin’s mysterious cousin. Nat. Rev. Mol. Cell Biol. 2001;2:202–210. doi: 10.1038/35056591. [DOI] [PubMed] [Google Scholar]
- 55.Johnson E.S., Schwienhorst I., Dohmen R.J., Blobel G. The ubiquitin-like protein Smt3p is activated for conjugation to other proteins by an Aos1p/Uba2p heterodimer. Embo J. 1997;16:5509–5519. doi: 10.1093/emboj/16.18.5509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Muller S., Matunis M.J., Dejean A. Conjugation with the ubiquitin-related modifier SUMO-1 regulates the partitioning of PML within the nucleus. Embo J. 1998;17:61–70. doi: 10.1093/emboj/17.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Melchior F. SUMO—nonclassical ubiquitin. Annu. Rev. Cell Dev. Biol. 2000;16:591–626. doi: 10.1146/annurev.cellbio.16.1.591. [DOI] [PubMed] [Google Scholar]
- 58.Hochstrasser M. SP-RING for SUMO: new functions bloom for a ubiquitin-like protein. Cell. 2001;107:5–8. doi: 10.1016/s0092-8674(01)00519-0. [DOI] [PubMed] [Google Scholar]
- 59.Yeh E.T., Gong L., Kamitani T. Ubiquitin-like proteins: new wines in new bottles. Gene. 2000;248:1–14. doi: 10.1016/s0378-1119(00)00139-6. [DOI] [PubMed] [Google Scholar]
- 60.Steffan J.S., Agrawal N., Pallos J., Rockabrand E., Trotman L.C., Slepko N., Illes K., Lukacsovich T., Zhu Y.Z., Cattaneo E., Pandolfi P.P., Thompson L.M., Marsh J.L. SUMO modification of Huntingtin and Huntington’s disease pathology. Science. 2004;304:100–104. doi: 10.1126/science.1092194. [DOI] [PubMed] [Google Scholar]
- 61.Shiio Y., Eisenman R.N. Histone sumoylation is associated with transcriptional repression. Proc. Natl. Acad. Sci. USA. 2003;100:13225–13230. doi: 10.1073/pnas.1735528100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zheng G., Yang Y.C. ZNF76, a novel transcriptional repressor targeting TATA-binding protein, is modulated by sumoylation. J. Biol. Chem. 2004;41:42410–42421. doi: 10.1074/jbc.M407287200. [DOI] [PubMed] [Google Scholar]
- 63.Matunis M.J. On the road to repair: PCNA encounters SUMO and ubiquitin modifications. Mol. Cell. 2002;10:441–442. doi: 10.1016/s1097-2765(02)00653-6. [DOI] [PubMed] [Google Scholar]
- 64.Wood L.D., Irvin B.J., Nucifora G., Luce K.S., Hiebert S.W. Small ubiquitin-like modifier conjugation regulates nuclear export of TEL, a putative tumor suppressor. Proc. Natl. Acad. Sci. USA. 2003;100:3257–3262. doi: 10.1073/pnas.0637114100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huang T.T., Wuerzberger-Davis S.M., Wu Z.H., Miyamoto S. Sequential modification of NEMO/IKKgamma by SUMO-1 and ubiquitin mediates NF-κB activation by genotoxic stress. Cell. 2003;115:565–576. doi: 10.1016/s0092-8674(03)00895-x. [DOI] [PubMed] [Google Scholar]
- 66.Manza L.L., Codreanu S.G., Stamer S.L., Smith D.L., Wells K.S., Roberts R.L., Liebler D.C. Global shifts in protein sumoylation in response to electrophile and oxidative stress. Chem. Res. Toxicol. 2004;17:1706–1715. doi: 10.1021/tx049767l. [DOI] [PubMed] [Google Scholar]
- 67.Li S.J., Hochstrasser M. A new protease required for cell-cycle progression in yeast. Nature. 1999;398:246–251. doi: 10.1038/18457. [DOI] [PubMed] [Google Scholar]
- 68.Li S.J., Hochstrasser M. The yeast ULP2 (SMT4) gene encodes a novel protease specific for the ubiquitin-like Smt3 protein. Mol. Cell. Biol. 2000;20:2367–2377. doi: 10.1128/mcb.20.7.2367-2377.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li S.J., Hochstrasser M. The Ulp1 SUMO isopeptidase: distinct domains required for viability, nuclear envelope localization, and substrate specificity. J. Cell Biol. 2003;160:1069–1081. doi: 10.1083/jcb.200212052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Murtas G., Reeves P.H., Fu Y.F., Bancroft I., Dean C., Coupland G. A nuclear protease required for flowering-time regulation in Arabidopsis reduces the abundance of small ubiquitin-related modifier conjugates. Plant Cell. 2003;15:2308–2319. doi: 10.1105/tpc.015487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cheng J., Wang D., Wang Z., Yeh E.T. SENP1 enhances androgen receptor-dependent transcription through desumoylation of histone deacetylase 1. Mol. Cell. Biol. 2004;24:6021–6028. doi: 10.1128/MCB.24.13.6021-6028.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kadoya T., Yamamoto H., Suzuki T., Yukita A., Fukui A., Michiue T., Asahara T., Tanaka K., Asashima M., Kikuchi A. Desumoylation activity of Axam, a novel Axin-binding protein, is involved in downregulation of beta-catenin. Mol. Cell. Biol. 2002;22:3803–3819. doi: 10.1128/MCB.22.11.3803-3819.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kurepa J., Walker J.M., Smalle J., Gosink M.M., Davis S.J., Durham T.L., Sung D.-Y., Vierstra R.D. The small ubiquitin-like modifier (SUMO) protein modification system in Arabidopsis: accumulation of SUMO-1 and -2 conjugates is increased by stress. J. Biol. Chem. 2003;278:6862–6872. doi: 10.1074/jbc.M209694200. [DOI] [PubMed] [Google Scholar]
- 74.X. Zuo, M.R. Mattern, R. Tan, S. Li, J. Hall, D.E. Sterner, J. Shoo, H. Tran, P. Lim, S.G. Sarafianos, L. Kazi, S. Navas-Martin, S.R. Weiss, T.R. Butt, Expression and purification of SARS Coronavirus proteins using SUMO fusions, Protein Expess. Purif. (2005) in press, doi:10.1016/j.pep.2005.02.004. [DOI] [PMC free article] [PubMed]
- 75.Schaefer F., Schaefer A., Steinert K. A highly specific system for efficient removal of tags from recombinant proteins. J. Biomol. Techn. 2002;13:158–171. [PMC free article] [PubMed] [Google Scholar]
- 76.Reverter D., Lima C.D. A basis for SUMO protease specificity provided by analysis of human Senp2 and a Senp2–SUMO complex. Structure. 2004;12:1519–1531. doi: 10.1016/j.str.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 77.Ecker D.J., Butt T.R., Marsh J., Sternberg E., Shatzman A., Dixon J.S., Weber P.L., Crooke S.T. Ubiquitin function studied by disulfide engineering. J. Biol. Chem. 1989;264:1887–1893. [PubMed] [Google Scholar]
- 78.X. Zuo, S. Li, J. Hall, M.R. Mattern, H. Tran, J. Shoo, R. Tan, S.R. Weiss, T.R. Butt, Enhanced expression and purification of membrane proteins by SUMO fusion in Escherichia coli, J. Struct. Funct. Genom. (2005) in press. [DOI] [PMC free article] [PubMed]
- 79.MacBeath G., Schreiber S.L. Printing proteins as microarrays for high-throughput function determination. Science. 2000;289:1760–1763. doi: 10.1126/science.289.5485.1760. [DOI] [PubMed] [Google Scholar]
- 80.Mitchell P. A perspective on protein microarrays. Nat. Biotechnol. 2002;20:225–229. doi: 10.1038/nbt0302-225. [DOI] [PubMed] [Google Scholar]
- 81.Zhu H., Bilgin M., Bangham R, Hall D., Casamayor A., Bertone P., Lan N., Jansen R, Bidlingmaier S., Houfek T., Mitchell T., Miller P., Dean R.A., Gerstein M., Snyder M. Global analysis of protein activities using proteome chips. Science. 2001;293:2101–2105. doi: 10.1126/science.1062191. [DOI] [PubMed] [Google Scholar]