

Highlights
-
•
Thromboinflammation is a major obstacle in modern therapeutic medicine.
-
•
It is triggered both by biomaterials and cell surfaces in contact with blood.
-
•
Thromboinflammation can be lowered by specific inhibitors bound to surfaces.
-
•
This is a promising emerging strategy to improve clinical outcome.
Keywords: Biomaterials, Coagulation, Complement, Inflammation, Platelets, Therapeutic medicine
Abstract
Therapeutic medicine today includes a vast number of procedures involving the use of biomaterials, transplantation of therapeutic cells or cell clusters, as well as of solid organs. These treatment modalities are obviously of great benefit to the patient, but also present a great challenge to the innate immune system, since they involve direct exposure of non-biological materials, cells of non-hematological origin as well as endothelial cells, damaged by ischemia-perfusion in solid organs to proteins and cells in the blood. The result of such an exposure may be an inappropriate activation of the complement and contact/kallikrein systems, which produce mediators capable of triggering the platelets and PMNs and monocytes, which can ultimately result in thrombotic and inflammatory (i.e., a thrombo-inflammatory) response to the treatment modality. In this concept review, we give an overview of the mechanisms of recognition within the innate immunity system, with the aim to identify suitable points for intervention. Finally, we discuss emerging and promising techniques for surface modification of biomaterials and cells with specific inhibitors in order to diminish thromboinflammation and improve clinical outcome.
1. Introduction
Inappropriate and uncontrolled activation of the cascade systems in the blood is a driving force in adverse inflammatory and thrombotic (thromboinflammatory) reactions in therapeutic medicine, such as procedures involving biomaterials, drug delivery devices, or the transplantation of cells, cell clusters, or solid organs. Such therapies are often associated with adverse reactions involving the innate immune system, which under physiological conditions is an important participant in our immediate defense against alien/foreign substances such as microorganisms. In a similar manner, the reactive surfaces of medical equipment as well as the surfaces of transplanted cells or solid organs will trigger an innate immune response, orchestrating a thrombotic/inflammatory reaction that ultimately results in a poor outcome of the therapy and/or damage to the host.
In this conceptual review, we describe the basic mechanisms by which the cascade systems are activated under various therapeutic conditions, and we highlight strategies to dampen these adverse reactions. We focus on a number of emerging techniques to link specific inhibitors directed against key components of the various cascade systems directly to the surface of a biomaterial or cellular surface, thereby providing local inhibition. In addition, we briefly mention the two clinically available inhibitors for systemic administration.
2. Concept and constituents of thromboinflammation
2.1. Concept of thromboinflammation
Thromboinflammation is a complex process that involves the activation of the humoral innate immunity system, which consists of the cascade systems in the blood, i.e, the complement, contact activation/coagulation, and fibrinolytic systems. These initial triggers induce activation of the cellular systems of the blood: platelets, subpopulations of leukocytes within the innate branch of the immune system, and the endothelial cell lining of the blood vessels. There are multiple points of interaction or cross-talk between the humoral and cellular portions of the innate immune system, meaning that a moderate initial activating signal can be rapidly and dramatically amplified [1]. When these processes are triggered by medical treatment procedures, such as treatment with biomaterials or drug delivery devices or the transplantation of cells, cell clusters, or solid organs, the end result may be thrombotic/inflammatory reactions that can diminish the efficacy of the treatment or (in the case of transplantations) induce graft impairment or rejection, processes that are detrimental to the patient undergoing treatment (reviewed in [2]).
2.2. The complement system
Complement plays important roles in the body, not only in identifying and removing intruders such as pathogens but also in clearing apoptotic and necrotic autologous cells or antigen-antibody complexes. In order to carry out these functions, it uses its capacity to detect differences between healthy autologous cells (“self”) and damaged or apoptotic autologous cells (“altered self”), as well as pathogens and other microorganisms (“non-self”). The identified non-self and altered-self structures trigger cascade reactions that ultimately lead to destruction and removal of the unwanted cells or pathogens, either by phagocytosis or by direct lysis.
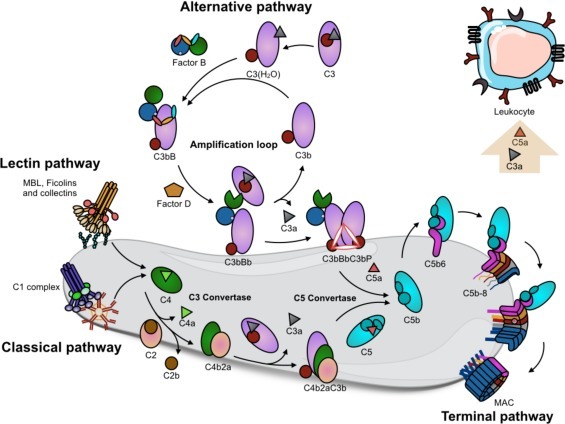
The complement system comprises ≈50 proteins present in the fluid phase (plasma) or bound to autologous cells, where they function as receptors or regulators. Activation of complement occurs via three different pathways: the classical pathway (CP), the lectin pathway (LP), and the alternative pathway (AP). All three pathways lead to the assembly of the CP/LP C3-convertase C4bC2a and the AP C3-convertase C3bBb, both of which cleave C3 into the anaphylatoxin C3a and the opsonin C3b. Every generated C3b moiety is a potential nucleus for a new AP C3-convertase complex (C3bBb). Hence, the AP provides a potent amplification loop of complement activation, regardless of the nature of the initial trigger.
The CP is initiated when the C1q moiety of the C1 complex (which also consists of the proenzymes C1r and C1s) binds to target-bound antibodies, pentraxins, DNA, or other negatively charged substances. The LP is initiated when the recognition molecules mannan-binding lectin (MBL) or ficolins-1, 2, and 3, which are in complex with the MBL-associated serine proteases (MASPs)-1 and −2, bind to carbohydrates. In each case, binding to target surfaces induces the activation of enzymatic compounds that mediate the proteolytic activation of C4 and C2, leading to the formation of the CP/LP C3 convertase, C4bC2a. Activation of the AP can be facilitated by properdin binding and also via the so-called “tick-over” mechanism, whereby the internal thiol ester of C3 is disrupted and the C3(H2O) or iC3 formed acquires C3b-like properties and can form an initial AP C3 convertase, (C3(H2O)Bb), in the fluid phase.
Additional C3b-molecules in association with either of the C3 convertases alter their substrate specificity from C3 to C5. The result of this switch is the cleavage of C5 into the anaphylatoxin C5a and C5b; this cleavage is the first step in the terminal pathway, which ultimately forms the membrane attack complex (MAC), consisting of C5b, C6, C7, C8 and multiple copies of C9. The MAC promotes cell lysis via its insertion into the lipid bilayer of cell membranes. The terminal complex is also present complexed with vitronectin in a soluble, aborted form, termed sC5b-9. Other important effector functions of complement, in addition to direct cell lysis by the MAC, include the recruitment and activation of PMNs and monocytes by the anaphylotoxins C3a and C5a, with C5a being the more potent. Phagocytosis of the target particles is facilitated by the interaction between the target-bound opsonin C3b, and its fragments iC3b and C3dg with complement receptors (CR1, CR3, CR4, and CRIg), which are upregulated on phagocytic cells in response to anaphylatoxins.
In vivo, the complement system is controlled by several soluble and membrane-bound regulators that protect cells against damage caused by autologous complement. Most of the regulators are members of the regulators of complement activation (RCA) superfamily, which predominately regulate the convertases. The plasma proteins factor H (a regulator of the AP) and C4b-binding protein (C4BP, a regulator of the CP and the LP), and membrane-bound proteins; membrane cofactor protein (MCP), decay acceleration factor (DAF), and CR1, all belong to this family. These fluid-phase regulators are recruited to host cells by binding to cell-surface glyocosaminoglycans [3], [4]. In addition, cell-bound CD59 as well as fluid-phase vitronectin and clusterin prevent the formation of the lytic MAC (C5b-9 complex) on host cells. Regulation of complement activation on autologous cells is shown in Fig. 1 .
Fig. 1.
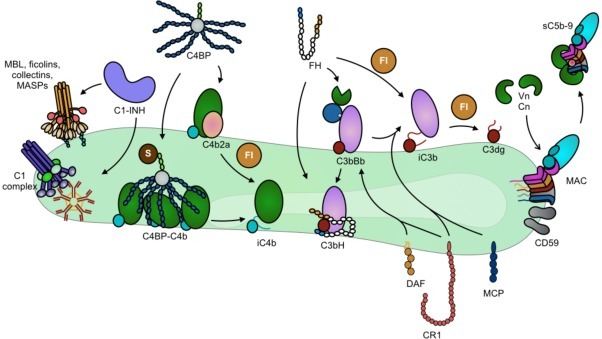
Complement regulation on a host cell surface. C1-INH acts by both preventing initiation complex formation and disassociating formed C1-complex/MBL-MASP, and by inhibiting activated C1r/s and MASP-1/2. C4BP, factor H (FH), MCP, CR1, and DAF act as cofactors for FI to inactivate C4b/C3b and/or accelerate the decay of the C3 convertases. (For the sake of clarity, interaction between DAF, CR1 and MCP is indicated for the AP C3 convertase only.) Vitronectin (Vn) and clusterin (Cn) in the fluid phase, together with cell-bound CD59, act to prevent MAC formation and insertion into the cell membrane.
2.3. The contact/kallikrein system
The proteins of the contact system are factor (F) XII, FXI, and prekallikrein (PK), all of which circulate as zymogens, and high molecular weight kininogen (HK), which functions as a co-factor in the cleavage of both FXI and PK. Binding of FXII to a material surface induces a conformational change in the FXII protein, inducing a non-proteolytic autoactivation of the molecule into an active enzyme form, α-FXIIa, which can cleave soluble FXII to enzymatically active β-FXIIa. β − FXIIa can then initiate two different pathways: One of its substrates is PK, which becomes cleaved to kallikrein (KK), with HK as a co-factor. The generated KK activates soluble FXII to β-FXIIa, which in turn generates more KK, providing a very effective amplification loop. In addition, KK also cleaves HK, releasing bradykinin, which is a nonapeptide with potent pro-inflammatory and vasoactive properties [5].
The other substrate for β-FXIIa is FXI, which is cleaved to FXIa, again with HK as a co-factor, thereby initiating the intrinsic pathway of coagulation and ultimately ending in the activation of thrombin from pro-thrombin and subsequent fibrin formation. Fibrin clots are able to promote further activation of FXII, establishing another amplification loop related to contact system activation.
The generated contact activation proteases FXIIa, FXIa, and KK are subsequently regulated by the serine protease inhibitors (serpins) C1 inhibitor (C1-INH) and antithrombin (AT), which are ubiquitously present in plasma. C1-INH is the predominant inhibitor of the contact system proteases when activation has been initiated by negatively charged surfaces such as glass or kaolin [6], [7]. In contrast, in situations in which the contact system is activated following platelet activation or triggered by preformed fibrin clots, protease-AT complexes, but almost no protease-C1-INH complexes, are detected [7], [8], [9]. In addition to its activation by fibrin clots or platelets, contact system activation has also been demonstrated to occur upon interaction with activated endothelial cells [10], [11]. Our current understanding of the activation and regulation of the contact activation pathway is presented in Fig. 2 .
Fig. 2.
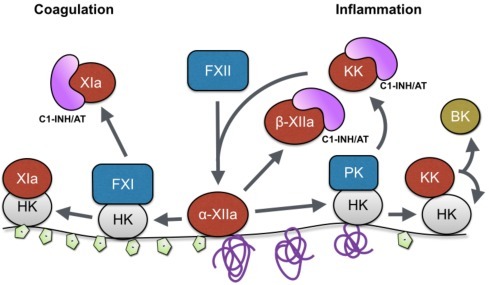
Contact activation on a negatively charged surface or misfolded protein surface. FXII adsorbed on a negatively charged surface or misfolded protein surface undergoes a conformational change that generates surface-adsorbed α-FXIIa and subsequently liberates β-FXIIa. Two pathways are initiated. To the left: activation of FXI that subsequently leads to coagulation activation; to the right: activation of kallikrein pathway that induces an inflammatory response. Fluid phase-activated components (β-FXIIa, FXIa, KK) will be inhibited by either C1-INH or AT.
2.4. Interaction between platelets and cascade system proteins
Apart from the traditional role of platelets as mediators of hemostasis, evidence is emerging to indicate that platelet activation during thrombotic events is closely associated with activation of the complement and contact/kallikrein systems, leading to inflammation; that is, the platelet acts as an innate immune cell.
Chondroitin sulfate A (CS-A) that is released from α-granules during platelet activation is a potent mediator of cross-talk between platelets and the complement system. CS-A activates complement in the fluid phase, generating anaphylatoxins that mediate leukocyte activation [12]. Initially, no complement activation seems to occur on the platelet surface, but C3 in the form of C3(H2O) is bound to the surfaces of activated platelets. In addition, several other complement components (C1q, C4, C9, ficolins-1, -2, and -3, and MASP-1 and MASP-2) are all detected on the surface of activated platelets [13], [14]. The lack of complement activation despite the abundance of bound complement proteins is consistent with the strong expression of membrane-bound complement regulators and the high density of plasma-derived complement regulators (factor H, C4BP, vitronectin), partially bound via CS-A to the platelet surface [15]. Platelet-bound C3(H2O) acts as a ligand for leukocyte CR1(CD35) and CR3 (CD11b/CD18), thereby serving as an important ligand in the tethering of activated platelets to PMNs [16].
MASP-1 and MASP-2 become activated by their interaction with activated platelets and during clotting and are regulated by C1-INH [14]. The MASPs have previously been reported to be involved in coagulation; MASP-2 cleaves prothrombin to thrombin [17], and MASP-1 has a number of thrombin-like effects, in that it can cleave fibrinogen, FXIII, HK, and thrombin-activatable fibrinolysis inhibitor (TAFI) and is thus able to mediate the formation of insoluble fibrin [18], [19], [20]. We have recently reported that both MASP-1 and MASP-2 become activated by interaction with preformed fibrin, produced using purified fibrinogen and thrombin, without the involvement of platelets [14]. Hence, the interaction between fibrin and the MASPs may represent a crossroad between the complement and coagulation systems as well as another amplification mechanism, analogous to that between FXII and fibrin mentioned above (Section 2.3). When activated on fibrin, MASP-1 and MASP-2 are inactivated exclusively by AT, without the presence of heparin as a co-factor [14]. The physiological relevance of these findings is underscored by the observation that MASP-1/2-serpin complexes are detected in plasma samples from patients with different thromboinflammation-related diagnoses [14].
As mentioned above (Section 2.3), contact system activation has been linked to platelet activation, and the generated proteases FXIIa, FXIa, and KK are in this case preferentially regulated by AT, and not by C1-INH, both in vitro and in vivo [7], [8], [9]. Activated platelets bind high amounts of all four contact activation proteins (FXII, FXI, PK, and HK), and the enzymatic activity of all three proteases is found on the surface of activated, but not on non-activated platelets [9].
2.5. Activation of platelets, leukocytes, and endothelial cells by cascade system activation products
The anaphylatoxins C3a and C5a cause smooth muscle contraction, histamine release from mast cells, and enhanced vascular permeability. In addition, they recruit PMNs and monocytes by chemotaxis and activate them, increasing the expression and activation of CR3 (CD11b/CD18), as reviewed in [21]. C5a has also been demonstrated to up-regulate tissue factor (TF) on monocytes and PMNs [22], [23], and both C5a [24] and the cytolytically inactive sC5b-9 [25] can induce TF expression on human endothelial cells. Furthermore, sC5b-9 is capable of activating platelets indirectly via immune complexes (as in [26]). Leukocytes and endothelial cells are activated by bradykinin, produced by the contact system. Thrombin, which is the major end product of the contact activation/coagulation system, is a potent platelet activator [27].
3. Examples of medical treatment modalities associated with thromboinflammation
Upon contact with blood, foreign structures on the surfaces of non-biological (bio)materials, liposomes for drug delivery, non-hematological cells, and altered-self structures on the endothelial lining of solid organs, are all be identified as alien by the recognition molecules of the intravascular innate immune system, triggering response mechanisms that are directed against the initiating agent. The mechanisms for activation and subsequent adverse advents differ, and thus the need for intervention is diverse. It should be pointed out that the aim of this review article is to describe general processes leading to thromboinflammation and that the subsequent events that may ultimately lead to treatment failure and/or host damage are multifactoral, depending, for example, on the composition and structure of the biomaterial, the purity of cells used for cell treatment, the quality of the organ transplanted (determining factors may be the age or condition of the donor, the time of ischemia, and others), and most importantly, the underlying disease and condition of the patient receiving treatment.
3.1. Model of interaction of plasma proteins with non-biological surfaces
When an artificial (bio)material comes into contact with blood, the first event to occur is an instantaneous protein adsorption onto the material surface, which can be observed within the first second [28], [29]. The plasma proteins form a monolayer on the material surface, and the composition of the protein layer and the amount of the individual protein deposited is mainly affected by the material’s inherent chemical and physical properties [30], [31]. Among the plasma proteins, IgG [32] and C3 [33] preferentially bind to material surfaces in their altered conformations, thereby triggering CP and AP activation, respectively. Once the complement system is activated, C3b covalently binds to the protein film surface and activates the amplification loop, generating more C3b fragments. These fragments will ultimately conceal the initial protein layer, enhancing the release of C3a and C5a as well as sC5b-9 into the fluid phase [29]. These anaphylatoxins are potent chemo-attractants that bind to PMNs and monocytes and recruit them to the biomaterial site. Active leukocytes recognize surface-bound C3b fragments via ligands such as CD11b/CD18 and initiate opsonization and cytokine release [34]. Thrombi on biomaterial surfaces are formed by platelet-mediated reactions and coagulation activation [35]. Plasma proteins such as fibrinogen, fibronectin, and vitronectin are potent mediators of platelet adhesion, and they are prone to bind to material surfaces. A minute amount of fibrinogen is sufficient to induce primary platelet activation via integrin αIIbβ3 interactions [36]. In addition, FXII adsorbed onto material surfaces automatically activates the contact system pathway of the coagulation cascade, generating thrombin that can, in turn, intensively activate platelets. Furthermore, IgG, IgM, and human serum albumin (HSA) have been demonstrated to potentiate activation of FXII, leading to contact pathway activation [37], [38]. Generally, biomaterial surfaces lack complement or coagulation regulators. Therefore, once the cascade systems are activated, they can spin out of control, resulting in the accumulation of anaphylatoxins and the activation of immune cells and generation of thrombi. This sequence of events is summarized in Fig. 3 .
Fig. 3.
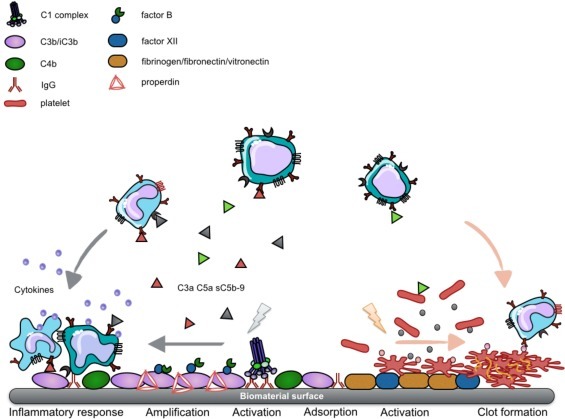
Cascade system activation on a biomaterial surface. Plasma proteins will quickly adsorb to a material surface when it makes contact with blood. On top of the plasma protein film, complement (left) and coagulation (right) will be triggered and amplified, generating either anaphylatoxins that recruit leukocytes and induce an inflammatory response or thrombin that recruits platelets and catalyzes clot formation.
We have recently studied the protein adsorption from plasma as well as complement and contact system activation and cytokine generation in whole blood, which can be induced by model biomaterials: six novel well-characterized artificial polymers and three reference polymers [39]. In brief, the polymers were ground to particles, which were incubated in: (1) EDTA-plasma to monitor the adsorption of 20 selected proteins; (2) plasma containing lepirudin (a specific thrombin inhibitor) to evaluate contact system activation, monitored by the formation of complexes between the generated proteases FXIIa, FXIa, and KK and the serpins C1-INH and AT; (3) lepirudin-anticoagulated whole blood to determine complement activation, monitored as the generation of C3a, C5a, and sC5b-9 and cytokine generation and release.
Our main findings were that: (1) the C4/C4BP ratio (representing regulation of the complement system) and the FXII/C1INH ratio (representing regulation of the contact activation system) adsorbed from EDTA-plasma were negatively correlated; (2) negative correlations were found between C3a, C5a, and sC5b-9 and the contact activation-serpin complexes, implying that these surfaces preferentially activate one of the systems; (3) strong correlations were found between (a) the C4/C4BP ratio (positive) and the FXII/C1INH ratio (negative) and (b) the generation of 10 cytokines (mainly pro-inflammatory), including IL-17, IFN-γ, and IL-6; (4) the correlations between the generation of these cytokines and complement activation products were negligible, corroborating their poor predictive values as biocompatibility markers. The general conclusion from this work was that artificial surfaces in contact with blood preferentially activate the complement system or the contact system. The ratios of C4/C4BP and/or FXII/C1INH adsorbed from EDTA-plasma were shown to be useful surrogate markers for cytokine release and inflammatory response to materials intended for blood contact [40], [41].
3.2. Thromboinflammation on the surface of materials, cells, and organs
In several extracorporeal circulation devices designed for uses such as cardiopulmonary bypass (CPB), extracorporeal membrane oxygenation (ECMO), hemodialysis, hemofiltration, and plasmapheresis, the artificial materials involved are often exposed to fresh blood and other tissue fluids. Since the surface free energy of these materials is high, protein adsorption usually occurs onto the material surfaces once the surfaces come in contact with blood and other tissue fluids. This protein adsorption evokes a sequence of events involving inflammatory reactions in which activation of the complement and coagulation systems is induced. The immunocompetent cells recognized these materials as foreign bodies and adhere to the surfaces of the artificial material.
These sequential events result in serious thromboinflammatory incompatibility reactions when materials are implanted [34], [42]. For example, life expectancy can be significantly decreased in uremic patients as a result of arteriosclerosis, partly due to that chronic whole-body inflammation is triggered by hemodialysis [43]. There is a higher risk of myocardial infarction for hemodialysis patients than for healthy individuals [44]. In addition, CPB and ECMO procedures have increased over the past decade as patients who have had vascular bypass surgery have begun to suffer from long-lasting infections affecting the lungs, e.g., swine influenza, severe acute respiratory syndrome (SARS) [45]. The CPB and ECMO procedures are usually associated with systemic inflammatory response syndrome (SIRS), in which contact between the blood and the material surfaces evokes cellular and humoral defense reactions [46]. Even vascular stents, which are small implants, induce fibrosis, restenosis, and thrombosis when they are exposed to the bloodstream [47]. Cardiac aids and pumps can trigger thrombotic reactions, leading to emboli [48]. Thus, the thromboinflammatory reactions induced at the implantation site lead to deleterious effects.
3.3. Thromboinflammation on the surface of transplanted cells and organs
We also see similar inflammatory reactions, with simultaneous activation of innate immunity and the thrombotic cascade, when therapeutic cells such as islets of Langerhans [49], [50], [51], mesenchymal stem/stromal cells (MSC) [52], [53], and hepatocytes [54] are infused into the human body. The administration of these cells is usually associated with a large loss of transplanted cells as the result of thromboinflammation [49], [50], [51], [54], which is also termed the instant blood-mediated inflammatory reaction (IBMIR). Once the cell surface is exposed to host blood, the innate immune system is triggered and thromboinflammation occurs on the cell surface due to expression of TF and rapid binding of platelets and infiltration of leukocytes into the clot, resulting in early loss of the transplanted cells. This results in the loss of transplanted cells. In addition to TF, collagen is also involved in the activation of the thromboinflammation [53]; for example, when MSCs were systemically infused into 44 patients, a reduction in the platelet count and a significant increase in thrombin-antithrombin (TAT; i.e., a marker of thrombin activation) and elevated levels of C3a were detected within 48 h [53]. Also, we have reported a case of clinical hepatocyte transplantation in which an increase in TAT and C3a levels and a reduction in platelet concentration were found within an hour after transplantation [54]. In conclusion, transplanted cells may be expected to be destroyed by thromboinflammatory reactions immediately after infusion.
The corresponding reactions in whole-organ transplantation are ischemia-reperfusion injury (IRI) and xenogenic/allogeneic antibody-mediated rejection, the major mediator of cell damage during transplantation [42], which is also triggered by complement activation and thrombotic reaction-induced thromboinflammation. IRI occurs upon re-establishment of the blood supply to cells, tissues, or organs that have been exposed to prolonged imbalance between oxygen/nutrient supply and demand [55]. This critical reaction is an inevitable event during organ transplantation, but IRI also occurs in a wide range of other clinically relevant situations such as brain death, sepsis, and cardiac surgery, as well as revascularization after myocardial infarction or stroke. IRI is a complex pathophysiological process involving a cascade of adverse events including hypoxia, ATP exhaustion, mitochondrial damage, increased production of reactive oxygen species, and ionic imbalance, which ultimately leads to structural lesions of the endothelial cells and initial activation of innate immunity and, subsequently, of adaptive immunity. The endothelial cell surface is normally covered by a negatively charged glycocalyx, which consists of a complex network of proteoglycans, glycosaminoglycans, and glycoproteins as well as a number of anti-inflammatory and anti-coagulant proteins that ensure microvascular homeostasis and prevent thromboinflammation. Moreover, this thick shield engulfs adhesion molecules such as ICAM-1, preventing the recruitment of leukocytes and platelets on the endothelial surface. In IRI, vascular protective barriers such as the vascular glycocalyx are lost, resulting in a decrease in adenylate cyclase activity and an increase in vascular permeability [56]. Under hypoxic conditions, TF, together with proinflammatory cytokines and chemokines, is expressed on endothelial and stromal cell surfaces. Also, there is a deposition of complement components onto the cell surface. These reactions lead to the triggering of local inflammation, with the binding of platelets and infiltration of leukocytes, particularly PMNs. This response leads to a further loss of integrity of the endothelial cells, ultimately causing vascular damage. After exposure to circulating fresh blood, the hypoxic cells are also attacked by the innate immune system, which recognizes the cells as “altered self” structures. This attack further aggravates the condition and finally leads to cell death and apoptosis. Recognition molecules in this process are FXII, MBL, and the ficolins; also, CRP and natural antibodies of the IgM isotype (both of which most likely also involve C1q) have also been implicated in these reactions (Fig. 4 ).
Fig. 4.
Complement dysregulation on a damaged or ischemic allogeneic cell surface. The innate immunity recognition molecules MBL ficolins, and collectins recognize “altered self” structures on the cell, and natural IgM may also bind and initiate complement activation. If complement regulators are not present in sufficient density at the cell surface, complement mediated attack may lead to cell death and apoptosis.
4. Therapeutic regulation of thromboinflammation
4.1. Systemic complement inhibition
Development of fluid-phase complement inhibitors is a rapidly growing field with a number of promising candidate molecules in the pipeline. A recent survey of the field is found in [57]. At present, only two complement inhibitors are available in the clinic, C1-INH (Behrinet®, plasma-derived or Ruconest®, recombinant) and the humanized anti-C5 monoclonal antibody eculizumab (Soliris®). C1-INH is not a specific complement inhibitor, in that it also regulates proteases generated by the contact system (Section 2.3). This property is most likely the reason for its success in the treatment of hereditary angioedema, which is its major indication. However, C1-INH has also showed promising results in several disease models, including transplantation [58].
In contrast, eculizumab is complement-specific and inhibits the cleavage of C5 by both C5 convertases, thereby preventing the generation of both C5a and the sC5b-9 and MAC complexes. Its main indication is in paroxysmal nocturnal hemoglobinuria (PNH) and atypical hemolytic uremic syndrome (aHUS), but it has also been shown to produce a prompt reversal of severe complement activation in a patient undergoing intentional ABO-incompatible pancreas and kidney transplantation [59].
4.2. Local regulation of thromboinflammation
Immobilization of ADP-degrading enzymes on the surface of materials and cells is effective in regulating the coagulation system. When platelets are activated, a multi-step process begins that involves adhesion, aggregation, contraction, and secretion. Released ADP is essential for recruiting and aggregating platelets; therefore, it is an obvious target for achieving platelet inhibition [60]. Nilsson et al., have reported that the ADP-degrading enzyme apyrase, immobilized onto substrate surfaces, can inhibit both platelet activation and platelet-dependent activation of the coagulation system [61], [62]. The idea comes from CD39 (NTPDase-1, EC 3.6.1.5), an enzyme expressed on human endothelial cells, which has an ADP-degrading activity similar to that of apyrase [63]. In order to regulate platelet activation, vascular ADP is continuously degraded under homeostatic conditions by CD39. In fact, the immobilization of apyrase can attenuate the activation of platelet and the coagulation system.
An ideal approach would be to mimic the properties of the endothelial cells lining the native vascular wall. Heparan sulfate, a heparin-like molecule expressed on endothelial cell surfaces, plays an important role in regulating coagulation as well as complement and platelet activation. Therefore, the immobilization of heparin is a rational approach for regulating thromboinflammation. The immobilization of heparin onto stents prevents not only platelet activation but also coagulation activation [64].
It is also possible to immobilize apyrase or heparin on the cell surface with an amphiphilic polymer, a poly(ethylene glycol)-conjugated phospholipid (PEG-lipid) derivative (Fig. 5 ). The PEG-lipid interacts with the lipid bilayer of the cell membrane through hydrophobic interactions without causing either cytotoxicity or a volume increase [65], [66]. The other end of the PEG derivative facing the fluid phase can be functionalized to allow binding of peptides, proteins, or oligonucleotides [66]. Apyrase immobilized on the cell surfaces of porcine aortic endothelial cells (PAECs) has been shown to prevent coagulation activation as well as platelet activation in human whole blood [62]. In addition, heparin conjugates can be immobilized onto the cell surface using peptides with high-affinity towards heparin. On cell surfaces modified with the heparin-binding peptide-conjugated PEG-lipid, heparin conjugates can bind the AT, thereby demonstrating its native conformation. When human MSC and hepatocytes are modified with heparin conjugates, thromboinflammation can be regulated in human whole blood [67].
Fig. 5.
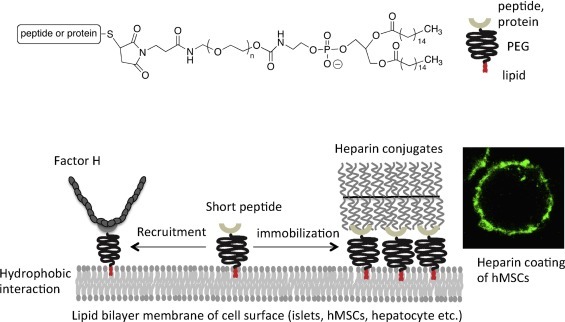
Chemical structure of PEG-lipid derivative. Maleimide group at the PEG end is used to conjugate a peptide or protein via thiol-maleimide reaction. When a specific peptide is conjugated to the PEG-lipid, heparin conjugates and factor H are specifically immobilized onto the cell surface. The confocal picture is human MSC (hMSC), which is modified with heparin conjugates where bound Alexa488-AT is detected.
Regarding the regulation of complement system, utilization of the fluid-phase RCAs factor H and C4BP is effective for local regulation on the surfaces of materials and cells. Several studies have indicated that complement activation on material surfaces is initiated through the CP [29], [68]. The concept of exploiting those complement-regulatory effects on foreign surfaces has been reported. Engberg et al. have used streptococcal M protein-derived peptides to specifically recruit human C4BP to substrate surfaces, thereby reducing complement activation via the CP and suggesting that RCA-binding peptides may be promising regulators of complement activation on the surface [69]. The AP C3 convertase is of major importance because of its ability to amplify the reaction on the surface [33], [70], [71]. Therefore, it is critical to regulate the AP C3 convertase in order to avoid complement attack. The mobilization of factor H to the surface of materials and cells offers a possible approach to regulating complement systems through the AP that has been explored using purified, covalently immobilized factor H to regulate complement activation [72], [73]. Another approach has been the use of factor H-binding peptides, which can specifically recruit factor H from the plasma of the host. Various peptides with high affinity for various domains of human factor H were created, and one of these peptides (5C6) recruited factor H without interfering with its regulatory function, since it bound to a region of this regulator that does not interact with the C3 convertase [74]. By immobilizing 5C6 onto materials, factor H can be specifically recruited from the host blood onto the surface, thereby mimicking the host surface and protecting the host cells from attack by complement [75], [76]. Moreover, it is also possible to co-immobilize 5C6 with apyrase, which is autoregulatory against thromboinflammation. This surface modification with 5C6 and apyrase can be applied to both substrates (artificial materials) and cells [62].
Platelets are intimately involved in the incompatibility processes that occur on foreign surfaces and are directly activated by surfaces [1]. Not only are platelets closely connected to the coagulation cascade, but they can also interact with leukocytes and trigger the activation of complement, thereby acting as an important hub that mediates cross-talk between these components [1], [12]. Therefore, it is critical to regulate both coagulation/platelet activation and complement activation. Using cell-surface modification techniques employing PEG-lipid derivatives, it is possible to co-immobilize apyrase and 5C6 onto the cell surfaces of PAECs. When exposed to human whole blood, factor H is specifically recruited to the modified surfaces and inhibits complement attack. In addition, activation of platelets and coagulation is efficiently attenuated as a result of ADP degradation. Thus, by inhibiting thromboinflammation with a multicomponent approach, we can create a hybrid surface with the potential to greatly reduce incompatibility reactions involving biomaterials and transplants.
4.3. Coating of materials and cell surfaces with synthetic polymers
The coating of material surfaces and cells with synthetic polymers such as PEG and poly(2-methacryloyloxyethyl phosphoryl choline) (PMPC) is also effective in improving blood compatibility and biocompatibility (Fig. 6 ). Coating with PEG and PMPC can prevent protein adsorption of plasma proteins when the materials and cells surfaces are exposed to blood and other tissue fluids; thus, the coated surfaces reduce the risk of incompatibility reactions [77], [78], [79], [80], [81]. Since the MPC polymer is effective to suppress protein adsorption, platelet adhesion, and platelet activation, material coating with MPC polymers have been used in various biomedical applications, such as blood-contacting and cardiovascular devices.
Fig. 6.
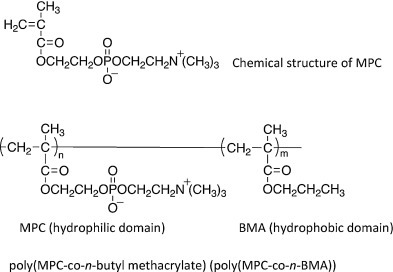
Chemical structure of MPC monomer and MPC polymer. MPC is a methacrylate monomer with a phosphorylcholine (PC) group. The most common MPC polymer for a coating on the material is PMB with 30 mol% of the MPC unit in the polymer. The BMA unit is used for coating onto the material surface.
5. Conclusions
Thromboinflammation is a major obstacle in modern therapeutic medicine. The term “thromboinflammation” refers to adverse inflammatory and thrombotic crosstalk- reactions, which are initiated and propagated by the different constituents of the innante immune system, which ultimately may result in thrombosis and/or chronic inflammation. Thromboinflammation is triggered by biomaterials, which lack the physiological inhibitors present on autologous cells, as well as by therapeutic cells in contact with blood. If uncontrolled, these adverse reactions may cause damage to an implant, a graft, and/or the recipient. Therefore, control of thromboinflammation is a major hurdle to overcome in order to improve results in a number of treatments using biomaterials and transplantation of cells and/or whole organs. Targeting the surface of biomaterials and the surface for therapeutic cells and solid organs with specific inhibitors is a promising approach to decrease thromboinflammation and thereby improve clinical outcome.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Acknowledgements
We thank Dr. Deborah McClellan for excellent editorial assistance. This work was supported by grant 2013-65X-05647-34-4 from the Swedish Research Council (VR), the European Community’s Seventh Framework Programme under the grant agreement n°602699 (DIREKT), the Swedish Foundation for International Cooperation in Research and Higher Education (STINT), the Bilateral Joint Research Projects (Japan-Sweden) of the Japan Society for the Promotion of Science (JSPS), and faculty grants from the Linnæus University.
References
- 1.Markiewski M.M., Nilsson B., Ekdahl K.N., Mollnes T.E., Lambris J.D. Complement and coagulation: strangers or partners in crime. Trends Immunol. 2007;28:184–192. doi: 10.1016/j.it.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 2.Nilsson B., Korsgren O., Lambris J.D., Ekdahl K.N. Can cells and biomaterials in therapeutic medicine be shielded from innate immune recognition. Trends Immunol. 2010;31:32–38. doi: 10.1016/j.it.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hessing M., Vlooswijk R.A., Hackeng T.M., Kanters D., Bouma B.N. The localization of heparin-binding fragments on human C4b-binding protein. J. Immunol. 1990;144:204–208. [PubMed] [Google Scholar]
- 4.Schmidt C.Q., Herbert A.P., Hocking H.G., Uhrin D., Barlow P.N. Translational mini-review series on complement factor H: structural and functional correlations for factor H. Clin. Exp. Immunol. 2008;151:14–24. doi: 10.1111/j.1365-2249.2007.03553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Colman R. Lippincott Williams & Wilkins; Philadelphia: 2006. Contact Activation (kallikrein-kinin) Pathway: Multiple Physiologic and Pathophysiologic Activities. [Google Scholar]
- 6.de Agostini A., Lijnen H., Pixley R., Colman R., Schapira M. Inactivation of factor XII active fragment in normal plasma: predominant role of C-1-inhibitor. J. Clin. Invest. 1984;73:1542–1549. doi: 10.1172/JCI111360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bäck J., Huber-Lang M., Elgue G., Kalbitz M., Sanchez J., Ekdahl K.N. Distinctive regulation of contact activation by antithrombin and C1-inhibitor on activated platelets and material surfaces. Biomaterials. 2009;30:6573–6580. doi: 10.1016/j.biomaterials.2009.07.052. [DOI] [PubMed] [Google Scholar]
- 8.Bäck J., Lood C., Bengtsson A.A., Ekdahl K.N., Nilsson B. Contact activation products are new potential biomarkers to evaluate the risk of thrombotic events in systemic lupus erythematosus. Arthritis Res. Ther. 2013;15:R206. doi: 10.1186/ar4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bäck J., Sanchez J., Elgue G., Ekdahl K.N., Nilsson B. Activated human platelets induce factor XIIa-mediated contact activation. Biochem. Biophys. Res. Commun. 2010;391:11–17. doi: 10.1016/j.bbrc.2009.10.123. [DOI] [PubMed] [Google Scholar]
- 10.Joseph K., Shibayama Y., Ghebrehiwet B., Kaplan A. Factor XII-dependent contact activation on endothelial cells and binding proteins gC1qR and cytokeratin 1. Thromb. Haemost. 2001;85:119–124. [PubMed] [Google Scholar]
- 11.Mahdi F., Madar Z., Figueroa C., Schmaier A. Factor XII interacts with the multiprotein assembly of urokinase plasminogen activator receptor, gC1qR, and cytokeratin 1 on endothelial cell membranes. Blood. 2002;99:3585–3596. doi: 10.1182/blood.v99.10.3585. [DOI] [PubMed] [Google Scholar]
- 12.Hamad O.A., Ekdahl K.N., Nilsson P.H., Andersson J., Magotti P., Lambris J.D. Complement activation triggered by chondroitin sulfate released by thrombin receptor-activated platelets. J. Thromb. Haemost. 2008;6:1413–1421. doi: 10.1111/j.1538-7836.2008.03034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hamad O.A., Nilsson P.H., Wouters D., Lambris J.D., Ekdahl K.N., Nilsson B. Complement component C3 binds to activated normal platelets without preceding proteolytic activation and promotes binding to complement receptor 1. J. Immunol. 2010;184:2686–2692. doi: 10.4049/jimmunol.0902810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kozarcanin H., Lood C., Munthe-Fog L., Sandholm K., Hamad O.A., Bengtsson A.A. The lectin complement pathway serine proteases (MASPs) represent a possible crossroad between the coagulation and complement systems in thromboinflammation. J. Thromb. Haemost. 2015 doi: 10.1111/jth.13208. [DOI] [PubMed] [Google Scholar]
- 15.Hamad O.A., Nilsson P.H., Lasaosa M., Ricklin D., Lambris J.D., Nilsson B. Contribution of chondroitin sulfate A to the binding of complement proteins to activated platelets. PLoS One. 2010;5:e12889. doi: 10.1371/journal.pone.0012889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hamad O.A., Mitroulis I., Fromell K., Kozarcanin H., Chavakis T., Ricklin D. Non-proteolytically activated C3 promotes binding of activated platelets and platelet derived microparticles to leukocytes via CD11b/CD18. Thromb. Haemost. 2015:2015. doi: 10.1160/TH15-02-0162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krarup A., Wallis R., Presanis J.S., Gal P., Sim R.B. Simultaneous activation of complement and coagulation by MBL-associated serine protease 2. PLoS One. 2007;2:e623. doi: 10.1371/journal.pone.0000623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krarup A., Gulla K.C., Gal P., Hajela K., Sim R.B. The action of MBL-associated serine protease 1 (MASP1) on factor XIII and fibrinogen. Biochim. Biophys. Acta. 2008;1784:1294–1300. doi: 10.1016/j.bbapap.2008.03.020. [DOI] [PubMed] [Google Scholar]
- 19.Hess K., Ajjan R., Phoenix F., Dobo J., Gal P., Schroeder V. Effects of MASP-1 of the complement system on activation of coagulation factors and plasma clot formation. PLoS One. 2012;7:e35690. doi: 10.1371/journal.pone.0035690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dobo J., Major B., Kekesi K.A., Szabo I., Megyeri M., Hajela K. Cleavage of kininogen and subsequent bradykinin release by the complement component: mannose-binding lectin-associated serine protease (MASP)-1. PLoS One. 2011;6:e20036. doi: 10.1371/journal.pone.0020036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ricklin D., Hajishengallis G., Yang K., Lambris J.D. Complement: a key system for immune surveillance and homeostasis. Nat. Immunol. 2010;11:785–797. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Osterud B., Bjorklid E. Tissue factor in blood cells and endothelial cells. Front. Biosci. 2012;1:289–299. doi: 10.2741/e376. [DOI] [PubMed] [Google Scholar]
- 23.Ritis K., Doumas M., Mastellos D., Micheli A., Giaglis S., Magotti P. A novel C5a receptor-tissue factor cross-talk in neutrophils links innate immunity to coagulation pathways. J. Immunol. 2006;177:4794–4802. doi: 10.4049/jimmunol.177.7.4794. [DOI] [PubMed] [Google Scholar]
- 24.Ikeda K., Nagasawa K., Horiuchi T., Tsuru T., Nishizaka H.Y.N. C5a induces tissue factor activity on endothelial cells. Thromb. Haemost. 1997;77:394–398. [PubMed] [Google Scholar]
- 25.Tedesco F., Pausa M., Nardon E., Introna M., Mantovani A., Dobrina A. The cytolytically inactive terminal complement complex activates endothelial cells to express adhesion molecules and tissue factor procoagulant activity. J. Exp. Med. 1997;185:1619–1627. doi: 10.1084/jem.185.9.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sims P.J., Wiedmer T. The response of human platelets to activated components of the complement system. Immunol. Today. 1991;12:338–342. doi: 10.1016/0167-5699(91)90012-I. [DOI] [PubMed] [Google Scholar]
- 27.Brass L.F. Thrombin and platelet activation. Chest. 2003;124:18S–25S. doi: 10.1378/chest.124.3_suppl.18s. [DOI] [PubMed] [Google Scholar]
- 28.Arnebrant T., Wahlgren M.C. Protein surfactant interactions at solid surfaces. Acs Sym. Ser. 1995;602:239–254. [Google Scholar]
- 29.Andersson J., Ekdahl K.N., Lambris J.D., Nilsson B. Binding of C3 fragments on top of adsorbed plasma proteins during complement activation on a model biomaterial surface. Biomaterials. 2005;26:1477–1485. doi: 10.1016/j.biomaterials.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 30.Horbett T.A. Principles underlying the role of adsorbed plasma proteins in blood interactions with foreign materials. Cardiovasc. Pathol. 1993;2:137–148. Chapter 13. [Google Scholar]
- 31.Nakanishi K., Sakiyama T., Imamura K. On the adsorption of proteins on solid surfaces, a common but very complicated phenomenon. J. Biosci. Bioeng. 2001;91:233–244. doi: 10.1263/jbb.91.233. [DOI] [PubMed] [Google Scholar]
- 32.Wettero J., Bengtsson T., Tengvall P. C1q-independent activation of neutrophils by immunoglobulin M-coated surfaces. J. Biomed. Mater. Res. 2001;57:550–558. doi: 10.1002/1097-4636(20011215)57:4<550::aid-jbm1201>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 33.Andersson J., Ekdahl K.N., Larsson R., Nilsson U.R., Nilsson B. C3 adsorbed to a polymer surface can form an initiating alternative pathway convertase. J. Immunol. 2002;168:5786–5791. doi: 10.4049/jimmunol.168.11.5786. [DOI] [PubMed] [Google Scholar]
- 34.Ekdahl K.N., Lambris J.D., Elwing H., Ricklin D., Nilsson P.H., Teramura Y. Innate immunity activation on biomaterial surfaces: a mechanistic model and coping strategies. Adv. Drug Deliv. Rev. 2011;63:1042–1050. doi: 10.1016/j.addr.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vogler E.A., Siedlecki C.A. Contact activation of blood-plasma coagulation. Biomaterials. 2009;30:1857–1869. doi: 10.1016/j.biomaterials.2008.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsai W.B., Grunkemeier J.M., Horbett T.A. Human plasma fibrinogen adsorption and platelet adhesion to polystyrene. J. Biomed. Mater. Res. 1999;44:130–139. doi: 10.1002/(sici)1097-4636(199902)44:2<130::aid-jbm2>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 37.Zhuo R., Siedlecki C.A., Vogler E.A. Competitive-protein adsorption in contact activation of blood factor XII. Biomaterials. 2007;28:4355–4369. doi: 10.1016/j.biomaterials.2007.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Golias C., Charalabopoulos A., Stagikas D., Charalabopoulos K., Batistatou A. The kinin system–bradykinin: biological effects and clinical implications: multiple role of the kinin system–bradykinin. Hippokratia. 2007;11:124–128. [PMC free article] [PubMed] [Google Scholar]
- 39.Engberg A.E., Rosengren-Holmberg J.P., Chen H., Nilsson B., Lambris J.D., Nicholls I.A. Blood protein-polymer adsorption: implications for understanding complement-mediated hemoincompatibility. J. Biomed. Mater. Res. A. 2011;97a:74–84. doi: 10.1002/jbm.a.33030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Engberg A.E., Nilsson P.H., Huang S., Fromell K., Hamad O.A., Mollnes T.E. Prediction of inflammatory responses induced by biomaterials in contact with human blood using protein fingerprint from plasma. Biomaterials. 2015;36:55–65. doi: 10.1016/j.biomaterials.2014.09.011. [DOI] [PubMed] [Google Scholar]
- 41.Huang S., Engberg A.E., Jonsson N., Sandholm K., Hong J., Nicholls I.A. Reciprocal relationship between the contact and the complement system activation on artificial polymers exposed to whole human blood. Biomaterials. 2016;77:111–119. doi: 10.1016/j.biomaterials.2015.10.067. [DOI] [PubMed] [Google Scholar]
- 42.Nilsson B., Ekdahl K.N., Mollnes T.E., Lambris J.D. The role of complement in biomaterial-induced inflammation. Mol. Immunol. 2007;44:82–94. doi: 10.1016/j.molimm.2006.06.020. [DOI] [PubMed] [Google Scholar]
- 43.Cavalli A., Del Vecchio L., Manzoni C., Locatelli F. Hemodialysis: yesterday, today and tomorrow. Minerva Urol. Nefrol. 2010;62:1–11. [PubMed] [Google Scholar]
- 44.Miyamoto T., Carrero J.J., Stenvinkel P. Inflammation as a risk factor and target for therapy in chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2011;20:662–668. doi: 10.1097/MNH.0b013e32834ad504. [DOI] [PubMed] [Google Scholar]
- 45.Murphy D.A., Hockings L.E., Andrews R.K., Aubron C., Gardiner E.E., Pellegrino V.A. Extracorporeal membrane oxygenation-hemostatic complications. Transfus. Med. Rev. 2015;29:90–101. doi: 10.1016/j.tmrv.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 46.Punjabi P.P., Taylor K.M. The science and practice of cardiopulmonary bypass: from cross circulation to ECMO and SIRS. Glob. Cardiol. Sci. Pract. 2013;2013:249–260. doi: 10.5339/gcsp.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Simard T., Hibbert B., Ramirez F.D., Froeschl M., Chen Y.X., O'Brien E.R. The evolution of coronary stents: a brief review. Can. J. Cardiol. 2014;30:35–45. doi: 10.1016/j.cjca.2013.09.012. [DOI] [PubMed] [Google Scholar]
- 48.Blitz A. Pump thrombosis-A riddle wrapped in a mystery inside an enigma. Ann. Cardiothorac. Surg. 2014;3:450–471. doi: 10.3978/j.issn.2225-319X.2014.09.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bennet W., Sundberg B., Groth C.G., Brendel M.D., Brandhorst D., Brandhorst H. Incompatibility between human blood and isolated islets of Langerhans: a finding with implications for clinical intraportal islet transplantation. Diabetes. 1999;48:1907–1914. doi: 10.2337/diabetes.48.10.1907. [DOI] [PubMed] [Google Scholar]
- 50.Bennet W., Sundberg B., Lundgren T., Tibell A., Groth C.G., Richards A. Damage to porcine islets of Langerhans after exposure to human blood in vitro, or after intraportal transplantation to cynomologus monkeys: protective effects of sCR1 and heparin. Transplantation. 2000;69:711–719. doi: 10.1097/00007890-200003150-00007. [DOI] [PubMed] [Google Scholar]
- 51.Moberg L., Johansson H., Lukinius A., Berne C., Foss A., Kallen R. Production of tissue factor by pancreatic islet cells as a trigger of detrimental thrombotic reactions in clinical islet transplantation. Lancet. 2002;360:2039–2045. doi: 10.1016/s0140-6736(02)12020-4. [DOI] [PubMed] [Google Scholar]
- 52.Moll G., Jitschin R., von Bahr L., Rasmusson-Duprez I., Sundberg B., Lonnies L. Mesenchymal stromal cells engage complement and complement receptor bearing innate effector cells to modulate immune responses. PLoS One. 2011;6:e21703. doi: 10.1371/journal.pone.0021703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moll G., Rasmusson-Duprez I., von Bahr L., Connolly-Andersen A.M., Elgue G., Funke L. Are therapeutic human mesenchymal stromal cells compatible with human blood. Stem Cells. 2012;30:1565–1574. doi: 10.1002/stem.1111. [DOI] [PubMed] [Google Scholar]
- 54.Gustafson E.K., Elgue G., Hughes R.D., Mitry R.R., Sanchez J., Haglund U. The instant blood-mediated inflammatory reaction characterized in hepatocyte transplantation. Transplantation. 2011;91:632–638. doi: 10.1097/TP.0b013e31820ae459. [DOI] [PubMed] [Google Scholar]
- 55.Eltzschig H.K., Eckle T. Ischemia and reperfusion–from mechanism to translation. Nat. Med. 2011;17:1391–1401. doi: 10.1038/nm.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Singh A., Ramnath R.D., Foster R.R., Wylie E.C., Friden V., Dasgupta I. Reactive oxygen species modulate the barrier function of the human glomerular endothelial glycocalyx. PLoS One. 2013;8:e55852. doi: 10.1371/journal.pone.0055852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ricklin D., Lambris J.D. Complement in immune and inflammatory disorders: therapeutic interventions. J. Immunol. 2013;190:3839–3847. doi: 10.4049/jimmunol.1203200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tillou X., Poirier N. Le bas-Bernardet S, hervouet J, minault D, renaudin K, et al: recombinant human C1-inhibitor prevents acute antibody-mediated rejection in alloimmunized baboons. Kidney Int. 2010;78:152–159. doi: 10.1038/ki.2010.75. [DOI] [PubMed] [Google Scholar]
- 59.Biglarnia A.R., Nilsson B. Nilsson T, von Zur-Muhlen B, Wagner M, Berne C, et al: prompt reversal of a severe complement activation by eculizumab in a patient undergoing intentional ABO-incompatible pancreas and kidney transplantation. Transpl. Int. 2011;24:e61–6. doi: 10.1111/j.1432-2277.2011.01290.x. [DOI] [PubMed] [Google Scholar]
- 60.Cattaneo M., Gachet C. ADP receptors and clinical bleeding disorders. Arterioscler. Thromb. Vasc. Biol. 1999;19:2281–2285. doi: 10.1161/01.atv.19.10.2281. [DOI] [PubMed] [Google Scholar]
- 61.Nilsson P.H., Engberg A.E., Back J., Faxalv L., Lindahl T.L., Nilsson B. The creation of an antithrombotic surface by apyrase immobilization. Biomaterials. 2010;31:4484–4491. doi: 10.1016/j.biomaterials.2010.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nilsson P.H., Ekdahl K.N., Magnusson P.U., Qu H., Iwata H., Ricklin D. Autoregulation of thromboinflammation on biomaterial surfaces by a multicomponent therapeutic coating. Biomaterials. 2013;34:985–994. doi: 10.1016/j.biomaterials.2012.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Marcus A.J., Broekman M.J., Drosopoulos J.H., Islam N., Alyonycheva T.N., Safier L.B. The endothelial cell ecto-ADPase responsible for inhibition of platelet function is CD39. J. Clin. Invest. 1997;99:1351–1360. doi: 10.1172/JCI119294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Christensen K., Larsson R., Emanuelsson H., Elgue G., Larsson A. Coagulation and complement activation. Biomaterials. 2001;22:349–355. doi: 10.1016/s0142-9612(00)00190-3. [DOI] [PubMed] [Google Scholar]
- 65.Teramura Y., Iwata H. Cell surface modification with polymers for biomedical studies. Soft Matter. 2010;6:1081–1091. [Google Scholar]
- 66.Teramura Y., Kaneda Y., Iwata H. Islet-encapsulation in ultra-thin layer-by-layer membranes of poly(vinyl alcohol) anchored to poly(ethylene glycol)-lipids in the cell membrane. Biomaterials. 2007;28:4818–4825. doi: 10.1016/j.biomaterials.2007.07.050. [DOI] [PubMed] [Google Scholar]
- 67.Asif S., Ekdahl K.N., Fromell K., Gustafson E., Barbu A., Le Blanc K. Heparinization of cell surfaces with short peptide-conjugated PEG-lipid regulates thromboinflammation in transplantation of human MSCs and hepatocytes. Acta Biomater. 2016 doi: 10.1016/j.actbio.2016.02.018. Feb 12 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 68.Nilsson U.R., Storm K.E., Elwing H., Nilsson B. Conformational epitopes of C3 reflecting its mode of binding to an artificial polymer surface. Mol. Immunol. 1993;30:211–219. doi: 10.1016/0161-5890(93)90050-l. [DOI] [PubMed] [Google Scholar]
- 69.Engberg A.E., Sandholm K., Bexborn F., Persson J., Nilsson B., Lindahl G. Inhibition of complement activation on a model biomaterial surface by streptococcal M protein-derived peptides. Biomaterials. 2009;30:2653–2659. doi: 10.1016/j.biomaterials.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Murakami Y., Iwata H., Kitano E., Kitamura H., Ikada Y. Interaction of poly(styrene sulfonic acid) with the alternative pathway of the serum complement system. J. Biomater. Sci. Polym. Ed. 2005;16:381–395. doi: 10.1163/1568562053654095. [DOI] [PubMed] [Google Scholar]
- 71.Tengvall P., Askendal A., Lundstrom I. Temporal studies on the deposition of complement on human colostrum IgA and serum IgG immobilized on methylated silicon. J. Biomed. Mater. Res. 1997;35:81–92. doi: 10.1002/(sici)1097-4636(199704)35:1<81::aid-jbm8>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 72.Andersson J., Larsson R., Richter R., Ekdahl K.N., Nilsson B. Binding of a model regulator of complement activation (RCA) to a biomaterial surface: surface-bound factor H inhibits complement activation. Biomaterials. 2001;22:2435–2443. doi: 10.1016/s0142-9612(00)00431-2. [DOI] [PubMed] [Google Scholar]
- 73.Andersson J., Bexborn F., Klinth J., Nilsson B., Ekdahl K.N. Surface-attached PEO in the form of activated Pluronic with immobilized factor H reduces both coagulation and complement activation in a whole-blood model. J. Biomed. Mater. Res. A. 2006;76:25–34. doi: 10.1002/jbm.a.30377. [DOI] [PubMed] [Google Scholar]
- 74.Wu Y.Q., Qu H., Sfyroera G., Tzekou A., Kay B.K., Nilsson B. Protection of nonself surfaces from complement attack by factor h-binding peptides: implications for therapeutic medicine. J. Immunol. 2011;186:4269–4277. doi: 10.4049/jimmunol.1003802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wu J., Wu Y.Q., Ricklin D., Janssen B.J., Lambris J.D., Gros P. Structure of complement fragment C3b-factor H and implications for host protection by complement regulators. Nat. Immunol. 2009;10:728–733. doi: 10.1038/ni.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Serruto D., Rappuoli R., Scarselli M. Gros P, van Strijp JA. Molecular mechanisms of complement evasion: learning from staphylococci and meningococci. Nat. Rev. Microbiol. 2010;8:393–399. doi: 10.1038/nrmicro2366. [DOI] [PubMed] [Google Scholar]
- 77.Ishihara K., Fukumoto K., Iwasaki Y., Nakabayashi N. Modification of polysulfone with phospholipid polymer for improvement of the blood compatibility: part 2. Protein adsorption and platelet adhesion. Biomaterials. 1999;20:1553–1559. doi: 10.1016/s0142-9612(98)00206-3. [DOI] [PubMed] [Google Scholar]
- 78.Ishihara K., Oshida H., Endo Y., Ueda T., Watanabe A., Nakabayashi N. Hemocompatibility of human whole-Blood on polymers with a phospholipid polar group and its mechanism. J. Biomed. Mater. Res. 1992;26:1543–1552. doi: 10.1002/jbm.820261202. [DOI] [PubMed] [Google Scholar]
- 79.Iwasaki Y., Ishihara K. Cell membrane-inspired phospholipid polymers for developing medical devices with excellent biointerfaces. Sci. Technol. Adv. Mat. 2012;13 doi: 10.1088/1468-6996/13/6/064101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nagahashi K., Teramura Y., Takai M. Stable surface coating of silicone elastomer with phosphorylcholine and organosilane copolymer with cross-linking for repelling proteins. Colloids Surf. B: Biointerfaces. 2015;134:384–391. doi: 10.1016/j.colsurfb.2015.07.040. [DOI] [PubMed] [Google Scholar]
- 81.Snyder T.A., Tsukui H., Kihara S., Akimoto T., Litwak K.N., Kameneva M.V. Preclinical biocompatibility assessment of the EVAHEART ventricular assist device: coating comparison and platelet activation. J. Biomed. Mater. Res. A. 2007;81:85–92. doi: 10.1002/jbm.a.31006. [DOI] [PubMed] [Google Scholar]