

Highlights
-
•
Type I interferon represents the first line of innate host defense against a number of invading pathogens that also instructs adaptive immunity.
-
•
Type I interferon induction downstream germ-line-encoded pattern recognition receptors triggers a signaling pathway that results in the expression of hundreds of IFN-stimulated genes with both direct anti-pathogen as well as immunomodulatory activities.
-
•
An inverse interference to escape at every step the IFN system is used by pathogens to eventually promote disease progression or establish a chronic infection.
-
•
Learning from bacterial and viral escape mechanisms may help in understanding how to cheat the pathogen and fit out the immune system to shift the balance in favor of the host.
Abbreviations: AIM2, absent in melanoma 2; APOBEC, apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like; ASC, apoptosis-associated speck-like protein containing a CARD; CARD, caspase recruitment domain; CDNs, cyclic dinucleotides; CLRs, C-type lectin receptors; cGAS, cyclic GMP-AMP synthase; cIAP, cellular inhibitors of apoptosis; CoV, Coronavirus; CypA, cyclophilin A; CPSF6, cleavage and polyadenylation specificity factor subunit 6; DDX, DEAD box protein; DHX, DEAH box protein; dsRNA, double-strand RNA; DAI, DNA-dependent activator of IFN-regulatory factors; DCs, dendritic cells; DENV, dengue virus; DNA, deoxyribonucleic acid; dNTPase, deoxynucleotide triphosphate triphosphohydrolase; ER, endoplasmic reticulum; HCV, hepatitis C virus; HIV-1, human immunodeficiency virus 1; IFN, interferon; IFI16, interferon gamma-interferon-inducible protein 16; IFIT, IFN-induced tetratricopeptide repeat proteins; IFITM, IFN-induced transmembrane protein; IKK, inhibitor of kB kinase; IPS1, IFN-β-promoter stimulator 1; IRAK, IL-1R-associated kinases; IRF, IFN regulatory factor; ISGs, IFN stimulated genes; ISGF3, interferon-stimulated gene factor 3; ISG15, IFN-stimulated gene 15; ISRE, IFN-stimulated response element; ITIM, immunoreceptor tyrosine-based inhibition motif; JEV, Japanese encephalitis virus; JAK, Janus kinase; LPS, lipopolysaccharide; LRRFIP1, extrachromosomal histone; H2B, leucine rich repeat (in FLII) interacting protein; MAM, mitochondrial associated membranes; MAPKs, mitogen-activated protein kinases; MAVS, mitochondrial antiviral signaling protein; MDA5, melanoma differentiation-associated gene-5; MERS, Middle East respiratory syndrome; mRNA, messenger RNA; miR, microRNA; MyD88, myeloid differentiation primary response gene 88; NADPH, nicotinamide adenine dinucleotide phosphate; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NLRs, nucleotide-binding oligomerization domain-like receptors; NLRP3, NACHT LRR and PYD domains-containing protein 3; NSP, nonstructural protein; NO, nitric oxide; iNOS, inducible nitric oxide synthase 1; NOD, nucleotide-binding oligomerization domain; PACT, PKR-associated activator; PAMP, pathogen-associated molecular pattern; pDCs, plasmacytoid dendritic cells; PKR, double-stranded RNA-activated protein kinase; PLP, papain-like protease; PRR, pattern-recognition receptors; RIG-I, retinoic acid-inducible gene 1; RIP, receptor-interacting serine-threonine kinase; RNA, ribonucleic acid; RLRs, (RIG)-I-like receptors; RT, reverse transcription; SAMHD1, SAM domain and HD domain-containing protein 1; SARS, severe acute respiratory syndrome; SIV, simian immunodeficiency virus; SLFN11, schlafen family member 11; ssDNA, single-stranded DNA; ssRNA, single-stranded RNA; SOCS, suppressor of cytokine signaling; STAT, signal transducer and activator of transcription; STING, stimulator of IFN genes protein; TBK1, TANK-binding kinase 1; TIR, Toll-Interleukin-1 receptor; TRAF, TNF receptor associated factor; TREX1, three prime repair exonuclease 1; TRIF, TIR domain-containing adaptor inducing IFN-β; TRIM, tripartite motif; tRNA, transfer RNA; TLRs, Toll-like receptors; IFN-α/β, IFN-I; Type II IFN, IFN-II; WNV, West Nile virus
Keywords: Interferon, Signaling, IFN-stimulated genes, Evasion mechanism, Virus, Bacteria
Abstract
Type I interferon (IFN) comprises a class of cytokines first discovered more than 50 years ago and initially characterized for their ability to interfere with viral replication and restrict locally viral propagation. As such, their induction downstream of germ-line encoded pattern recognition receptors (PRRs) upon recognition of pathogen-associated molecular patterns (PAMPs) is a hallmark of the host antiviral response. The acknowledgment that several PAMPs, not just of viral origin, may induce IFN, pinpoints at these molecules as a first line of host defense against a number of invading pathogens. Acting in both autocrine and paracrine manner, IFN interferes with viral replication by inducing hundreds of different IFN-stimulated genes with both direct anti-pathogenic as well as immunomodulatory activities, therefore functioning as a bridge between innate and adaptive immunity. On the other hand an inverse interference to escape the IFN system is largely exploited by pathogens through a number of tactics and tricks aimed at evading, inhibiting or manipulating the IFN pathway, that result in progression of infection or establishment of chronic disease.
In this review we discuss the interplay between the IFN system and some selected clinically important and challenging viruses and bacteria, highlighting the wide array of pathogen-triggered molecular mechanisms involved in evasion strategies.
1. Introduction
The ability of the host to respond to invading pathogens relies on the activation of the innate immune system that orchestrates adaptive immune responses for pathogen clearance. In recent years, our understanding of the mechanisms involved in the activation of the innate response has evolved significantly with the identification and characterization of the mammalian system of pathogen recognition.
The innate immune system detects the presence of a pathogen through a set of germline-encoded membrane-associated or cytoplasmic receptors, termed pattern recognition receptors (PRRs) that are engaged by microbial-derived products named pathogen-associated molecular patterns (PAMPs). Major classes of PRR include Toll-like receptors (TLRs), nucleotide-binding oligomerization domain (NOD)-like receptors (NLR), C-type lectin receptors (CLRs), retinoic-acid inducible gene (RIG)-I-like receptors (RLRs) and a growing list of cytosolic DNA-sensing receptors [1], [2], [3], [4], [5], [6], [7]. Upon engagement, these receptors recruit a number of adaptor proteins to signal downstream and activate three major pathways: the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), the mitogen-activated protein kinases (MAPKs) and the IFN regulatory factor (IRF) pathway [8], [9]. Downstream TLRs and RLRs, type I IFN and pro-inflammatory cytokines are mainly produced, while NLRs predominantly activate inflammasomes, resulting in the release of IL-1 and multiple inflammatory cytokines (Fig. 1 ).
Fig. 1.
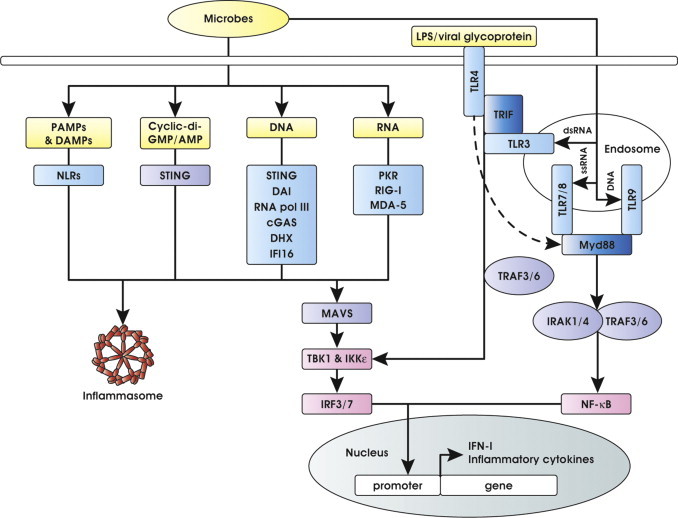
Schematic representation of IFN-I and inflammatory cytokine induction downstream pattern recognition receptors and adaptors signaling. Viral and bacterial components are detected by sensors present on host membranes or in the cytoplasm. These sensors signal through a limited number of shared adaptors initiating a cascade of events that results in transcription factor activation and induction of IFN-I and inflammatory cytokines. See the text for more details and references.
Based on the structure of their receptors, interferons are broadly classified into three groups, type I, II and III IFNs. Type I IFN comprises the largest IFN class that includes IFN-α, constituted by several partially homologous genes and IFN-β, IFN-ɛ, IFN-κ, IFN-ω represented by a single gene. The IFN-α and IFN-β are the best characterized as antiviral and the most broadly expressed [10], [11], [12] and will be referred as IFN-I from now on. The IFN-γ, the only Type II IFN, is released by activated T and NK cells; type III IFNs, which include IFN-λ 1–4, similarly to IFN-I are believed to regulate the antiviral response [13]. According to the most common view, IFN-I exerts primarily an anti-viral action while IFN-II acts predominantly on macrophages to induce a microbicidal state against ingested intracellular, non-viral pathogens.
Anti-microbial IFN-I activity is not intrinsic, but mediated, in both autocrine and paracrine manner, by a unique set of induced genes named IFN-stimulated genes (ISGs) [10], [11], [14]. Secreted IFN-I, indeed, binds to a common heterodimeric ubiquitously expressed receptor composed of IFNAR1 and IFNAR2 chains, and initiates a signaling cascade that has been characterized in detail and reviewed elsewhere [10], [11], [12]. Briefly, canonical IFN-I pathway results in activation of the Janus kinase (JAK) family (cytoplasmic tyrosine kinases) that, in turn, activate by phosphorylation the signal transducers and activators of transcription (STATs) [15]. Activated STATs complex with IRF9 to form the heterocomplex IFN-stimulated gene factor 3 (ISGF3), that translocates in the nucleus, binds to upstream sequence elements named IFN-stimulated response elements (ISRE), and activates the transcription of ISGs (Fig. 2 ). These genes act to promote viral clearance and establish an antiviral state in uninfected bystander cells, or to induce apoptosis and several anti-microbial mechanisms in infected cells. They also stimulate cells at the interface of innate and adaptive immunity, such as macrophages and dendritic cells (DCs) that trigger the adaptive response [16], [17].
Fig. 2.
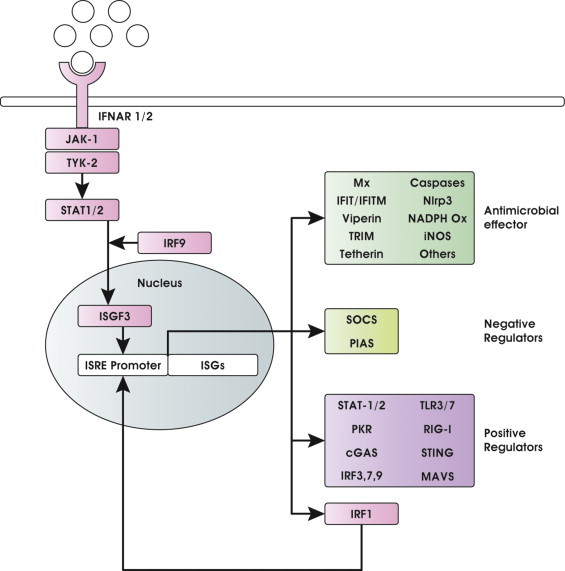
IFN-I signaling pathway and ISG antimicrobial effectors. IFN-I produced downstream PRRs is secreted by infected cells and acts in autocrine and paracrine manner through the JAK-STAT pathway, leading to widespread ISG induction that promotes an antimicrobial state also in bystander uninfected cells. ISGs can be classified in antimicrobial effectors and IFN signaling negative and positive regulators. IRF1 represents a unique example of positive regulator that upon expression directly translocates into the nucleus to enhance the expression of a subset of ISGs.
The ability to break in innate immunity before the onset of the adaptive response is, thus, crucial for the survival of virtually all mammalian pathogens. On the other side of the coin, an essential part of the early response to pathogens is aimed at limiting their ability to hijack host cellular machinery and evade the IFN-mediated antimicrobial mechanisms.
Nowadays, while it is largely accepted that also bacteria can induce the production of IFN-I, the role of the IFN system in the pathogenesis of bacterial infections can be either detrimental (mainly in intracellular bacterial infections) or protective (mainly in extracellular bacterial infections). The route and tropism of bacterial infection, viral co-infections and last, but not least, the balance between IFN-I and IFN-II effects are all determinants of the different outcome [18]. The ratio between IFN-I and II species produced in response to infection, might differ as consequence of the capacity of the pathogen to stimulate specific cell types in the infected tissues. Moreover, taking into consideration that several antibacterial IFN-II-induced genes are stimulated to a much lesser degree by IFN-I, it is quite difficult in vivo to separate the activities strictly dependent on one of the two IFN families. A more reliable view consists of a crosstalk between the two pathways: if the bacterium is able to stimulate IFN-I production, generally it occurs early after the infection as an immediate innate immune response, while IFN-II intervenes later on when immune cells (such as T cell subsets and NK cells) are activated. Also due to this complex picture, while a number of viral evasion strategies have been mechanistically defined so far, studies aimed at characterizing the bacterial components that inhibit the IFN system are only recently starting to be elucidated in molecular details [19], [20], [21].
Main strategies that pathogens have evolved to disarm the IFN-I response include: (i) blocking IFN-I production by modifying, curtailing or limiting production of their PAMPS to make them inaccessible to PRRs and/or hitting the components of PRRs signaling pathways; (ii) interfering with the IFN-I signaling by inhibiting signal transducers; (iii) blocking or disturbing the action of ISGs; (iv) hijacking host proteins or components of the IFN system. Each of these strategies involves a number of different molecular mechanisms and the combination of more strategies may be necessary to overcome the IFN-I response by a single pathogen. Depending on the nature of the pathogen, these countermeasures that tip the balance toward the pathogen, may result in increased replication or in the establishment of a persistent infection.
Leaving out strategies that envision IFN-I repression due to a general inhibition of cellular gene expression, reviewed elsewhere [22], [23], here we summarize recent findings on evasion tricks utilized by pathogens to specifically subvert the IFN system. A special focus is put on few highly pathogenic bacteria and emerging or re-emerging viruses, which represent a major threat to human health due to the lack of effective vaccines and/or therapeutics. For an in-depth coverage of other pathogens and strategies to escape the host immune response, the reader is referred to more comprehensive and specific reviews [19], [20], [21], [23], [24], [25], [26].
2. Hiding detection and/or interfering with sensor pathways
Recent years have seen major advances in our understanding of the innate response to infectious pathogens and specifically of how pathogen recognition promotes IFN-I release [8], [27], [28].
Cytoplasmic and membrane-associated PRRs recognize a variety of pathogen components. Viral PAMPs mainly consist of nucleic acids originating from the uncoating of infecting virions, the transcription of viral genomes and the replication of genomic intermediates, in the form of single-stranded (ss) and double-stranded (ds) DNA, and ss and ds RNA, as well as viral glycoproteins. Bacterial PAMPs include various molecules, ranging from lipoproteins, lipopolysaccharide (LPS), flagellin and peptidoglycan to unique bacterial nucleic acid structures, such as cyclic dinucleotides (CDNs).
Multiple endosome-associated TLRs (TLR3, 7, 8 and 9) are specialized in the detection of viral and bacterial nucleic acids. TLR3 recognizes dsRNA, TLR7/8 are bound by ssRNA and TLR9 by CpG-containing DNA. TLR2, 4 and 13, previously considered sensors only for bacterial components, are now been involved also in recognition of viral ligands and in the induction of IFN-I [29]. All TLRs contain an intracellular domain, the Toll-Interleukin-1 receptor (TIR), which recruits one or more TIR-containing adaptor proteins to transmit signals downstream. TLR3 signals through the adaptor TIR domain-containing adaptor inducing IFN-β (TRIF) to activate the two related kinases, inhibitor of kB kinase (IKK)-ɛ and TANK-binding kinase 1 (TBK1) that mediates activation by phosphorylation of IRF3 and IRF7. TLR7/8 and TLR9 use, as adaptor, myeloid differentiation primary-response protein 88 (MyD88) that then initiates signaling cascades involving IL-1R-associated kinases (IRAK1-4) and TNFR-associated factor (TRAF) 3/6 proteins, which finally converge at the activation of the IκB kinase (IKK) family members IKK-α, IKK-β, IKK-ɛ and TBK1 responsible for activation of NF-κB and IRF3/7 [4].
In the cytoplasm, two closely related helicases, retinoic acid-inducible gene 1 (RIG-I) and melanoma differentiation-associated gene-5 (MDA5), recognize dsRNA of many replicating viruses in a TLR-independent manner. RIG-I preferentially senses short dsRNA and ssRNA with a 5′-triphosphate (5′-ppp RNA), while MDA5 recognizes long dsRNA and poly I:C [30], [31], [32]. Like viral RNAs, bacterial RNAs can possess 5′-ppp termini and secondary structures that make them RIG-I agonists. Consistent with this notion, RIG-I can act as a sensor of bacterial RNA and may help maintain homeostasis to gut microbiota [33], [34]. Upon recognition of non-self RNA, RIG-I and MDA5 are recruited to the mitochondrial antiviral signaling protein (MAVS; also known as CARDIF, VISA or IPS1), which triggers a signaling cascade that leads to the activation of IKK-ɛ and TBK1 and, in turn, IRF3/7 phosphorylation [35]. Stimulator of IFN genes (STING), initially identified as a cytosolic DNA sensor, also participates in the RIG-I signalling [36]. STING interacts with the adaptor MAVS at the mitochondrial associated membranes (MAM) facilitating the recruitment of TBK1 and the activation of IRF3 [37]. A more general role of STING in innate immune responses, not only limited to its function as adaptor of RNA and DNA sensors, has been now established [38].
In addition to these RNA sensors, viral RNAs may also be recognized by effector molecules that are themselves IFN-induced proteins with antiviral functions. These molecules include dsRNA-activated protein kinase (PKR) that binds and is activated by dsRNA from viruses and bacteria [39], IFN-induced tetratricopeptide repeat proteins 1/2/3 (IFIT1, IFIT2, and IFIT3) that, as RIG-I, bind 5′-ppp RNA and may recognize viral mRNAs that lack 2′-O-methylation [1], [40].
A growing list of cytosolic DNA sensors then recognizes DNA from different sources including viruses, bacteria and apoptotic cells [3], [6], [7]. More than ten DNA cytosolic receptors have been proposed so far that include DAI (DNA-dependent activator of IRF), RNA polymerase-III, IFN-inducible Interferon gamma-interferon-inducible protein 16 (IFI16), STING, extrachromosomal histone H2B, leucine rich repeat (in FLII) interacting protein (LRRFIP1), Ku70, DEAH box protein (DHX) 9 and DHX36, cyclic GMP-AMP synthase (c-GAS). As other sensors, DNA sensor activation results in the production of IFN-I and proinflammatory cytokines and chemokines via the STING-TBK1-IRF3 axis. In addition to the activation of IRF3- and NF-κB-dependent signaling cascades, cytosolic DNA can also promote an apoptosis-associated speck-like protein containing a CARD (ASC)-dependent inflammasome-mediated response resulting in the secretion of proinflammatory cytokines [3], [7], [25], [27], [28].
The intracellular NLRs scaffold large signaling complexes to mediate innate immunity and inflammatory responses. They may trigger the assembly of inflammasomes and modulate the NF-κB, MAPK and IRF signaling pathways. In particular, NOD1 and NOD2 are important for immune detection of intracellular bacterial pathogens and are also involved in a variety of immune homeostatic functions [41]. Upon the recognition of bacterial peptidoglycans by NOD1 and NOD2, receptor-interacting serine-threonine kinase 2 (RIP2) is activated via cellular inhibitors of apoptosis 1 and 2 (cIAP1 and 2), subsequently leading to ubiquitination of NF-κB essential modulator (NEMO) and the activation of the proinflammatory NF-κB pathway. In parallel, the recognition of muramyl dipeptide present in all peptidoglycans, can also lead to the activation of MAPK pathway via RIP2, which contributes to cytokine production. For the induction of IFN-I, NOD2 activated by viral RNA, signals through the mitochondrial MAVS independently of RIP2.
Then, some sensors can aggregate with adaptors as apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) and caspase 1 to form multimeric structures named inflammasome [42] (Fig. 1).
As first line strategy to face these sensing pathways, pathogens hide detection by modifying their PAMPs. They may also degrade/inactivate target key signal transduction hubs by counteracting host-induced post-translational modifications required for signaling molecule activity and use concomitant different strategies as better described below and also discussed by Thomas Kufer and Igor Brodsky in this issue. We focus here on some members of Coronaviruses, Flaviviruses, Hepaciviruses, Filoviruses and Retroviruses as well as bacteria whose PAMPs could in principle activate the IFN system, but that efficiently betray host sensing and effector pathways (Fig. 3 ).
Fig. 3.
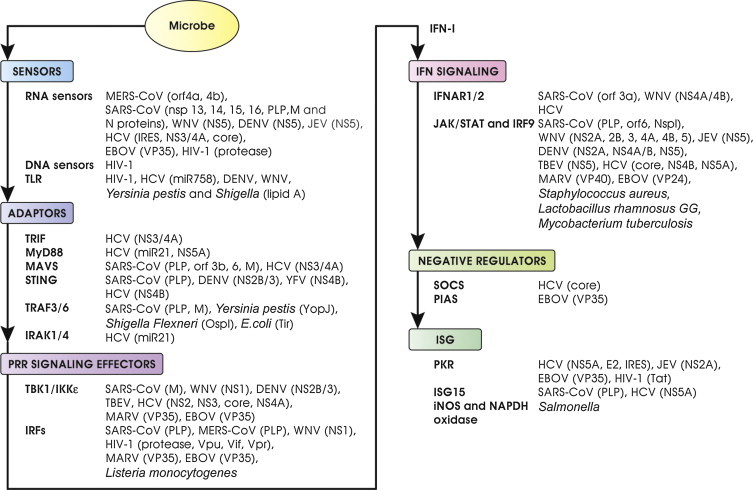
Examples of viral and bacterial antagonists that block subvert or exploit the IFN system. Pathogens affect at every step the IFN system by multiple mechanisms. Sites of intervention by several antagonists are indicated. IFN antagonists may prevent PRR recognition by hiding or modifying PAMPs, may inhibit PRR signaling by directly targeting adaptors and signaling effectors, may interfere with the IFN signaling by impairing signaling transducers and may block or disturb the action of ISGs. Some antagonists have more than one cellular target while others target common signaling molecules, effectively blocking IFN induction from a variety of PAMPs. See the text for more details and references.
Coronaviruses (CoV) are large enveloped RNA viruses, in the family Coronaviridae, of both veterinary and clinical importance. Two newly emerging viruses in the family, the severe acute respiratory syndrome CoV (SARS-CoV) and the Middle East respiratory syndrome CoV (MERS-CoV), have been recently responsible for severe disease in humans (reviewed in [43], [44], [45], [46], [47]). A common trait in their pathogenesis is the lack of induction of a robust IFN-I response in infected cells [48], [49]. To do this, CoV have developed multiple strategies (recently reviewed in [50], [51]).
RIG-I, MDA5 and the host ISGs IFIT1 and 2 are critically involved in sensing of CoV infection. However, as first line of hiding from recognition, CoV encode several highly conserved nonstructural proteins (nsps) implicated in viral RNA capping activity in order to mimick N7- and 2′-O-methylated 5′ cap structure of cellular mRNAs [52]. These viral proteins include a RNA-triphosphatase, a guanine-N7-methyltransferase, and a 2′-O-methyltransferase encoded by nsp13, 14 and 16, respectively [50], [53]. Consistently, human and mouse CoV mutants lacking 2′-O-methyltransferase activity induce higher expression of IFN-I and are highly sensitive to IFN-I effects [54], [55]. SARS-CoV nsp14 also encode a 3′,5′ exoribonuclease that is involved in RNA-proofreading, but probably it also functions in degrading viral PAMPs, further hiding immune detection [50], [56]. Similarly, nsp11 encodes a ribonuclease that may degrade viral PAMPs [57].
Another strategy used to avoid detection is the replication in protected sites as the double membrane vesicles (DMVs) that the virus induces in the host cytoplasm. In the case of SARS-CoV these membranes contain the replicase complex and the viral genomic RNA suggesting that replication occurs in these sites and the generated nucleic acids are soon after shielded [58], [59]. In addition, the highly basic nucleocapsid (N) protein of SARS-CoV has been reported to directly inhibit IFN-I production induced by both poly(I:C) or Sendai virus, by a mechanism that involves steps upstream RIG-I. Thus, it is conceivable that by binding dsRNA, N protein prevents RIG-I/MDA5 activation [60]. Others SARS-CoV-encoded proteins, as ORF-3b and ORF6, may also interfere with the RLR recruitment of the adaptor MAVS based on their preferential localization at the mitochondrial membrane even if their mechanism of action is not yet elucidated [61], [62]. The SARS-CoV Membrane (M) protein blocks transcription of IFN-I when stimulated by dsRNA or members of the RIG-I signaling pathway including RIG-I, MAVS, IKK-ɛ, and TBK1, but it does not influence the transcriptional activity of the IFN promoter when IRF3 or IRF7 are overexpressed. The physical association of SARS-CoV M with RIG-I, TBK1, IKK-ɛ, and TRAF3 suggests that M protein may prevent the formation of the functional complex with TBK1 thereby inhibiting activation of IRF3/IRF7 and IFN-I transcription [63]. Similarly, the papain-like protease (PLP) domain contained in the SARS-CoV nsp3 protein, an essential component of the viral replicase complex, interacts with the adaptor STING blocking the binding to MAVS and the recruitment of the TBK1/IRF3 complex [64], [65], [66]. Finally, through its deubiquitinating activity, the PLP protein removes Ubiquitin from several components of the RLR pathway blocking their activation (reviewed in [50], [51], [67], [68]).
Recently, two MERS-CoV accessory proteins, the ORF-4a and ORF-4b products, have also been identified as immunosuppressive factors. MERS-CoV 4a is a RNA-binding protein that interacts in a mRNA-dependent manner with PKR-associated activator (PACT), a cellular dsRNA-binding protein, which potently stimulates RIG-I-induced IFN production by binding to the C-terminal repression domain of RIG-I [69]. So, ORF-4a inhibits RIG-I/MDA5 pathway without a direct binding to the sensor, but by perturbing the function of a stimulator of RIG-I signaling as PACT [70]. The ORF-4b-encoded accessory protein is also able to inhibit IFN-I induction in vitro, however, the mechanism involved is not yet elucidated [71].
The genus Flavivirus, of the Flaviridae family, includes West Nile virus (WNV), dengue virus (DENV), Japanese encephalitis virus (JEV), yellow fever virus (YFV), Tick-borne encephalitis virus (TBEV) and several other viruses all causing serious medical problems in humans [72], [73], [74], [75]. Currently, vaccines for humans are available only for YFV, JEV, and TBEV and no clinically approved antiviral therapy is available for the treatment of flavivirus infections [75]. In particular, DENV and WNV are re-emerging as global life-threatening human pathogens [75], [76]. Both viruses are sensitive to IFN antiviral effects and the severity of the disease is mostly dependent on the ability to avoid and/or attenuate induction of IFN-I and its effector responses through several viral encoded IFN-antagonists [73], [74]. Interestingly, some of these strategies are shared with CoV.
The most relevant PRRs for the detection of WNV and DENV products, described so far, are the TLR3, 7, 8 and the RLRs, RIG-I/MDA-5. Suppression of both TLR- and RLR-mediated IFN induction has been shown to be important for viral replication [77], [78]. As CoV, Flaviviruses contain a cap structure that is generated by a methyltransferase mapped to the N-terminal region of the NS5 protein [74], [79]. Through 2′-O-methylation of the viral mRNA cap WNV, DENV and JEV evade IFIT1-dependent and -independent mechanisms of host restriction in vitro and in vivo [54], [80], [81]. Moreover, to hide their nucleic acids during the replicative cycle, both DENV and WNV induce the formation of convoluted membranes in the endoplasmic reticulum (ER) and Golgi apparatus that envelop the virus replication complex [82], [83] protecting viral nucleic acids from both TLR and RLR recognition, as it occurs along the infection with CoV. So far, an antagonism at the level of sensing signaling has been clearly defined only for DENV that fails to induce an IFN response in myeloid cells where it replicates, despite other pro-inflammatory cytokines and chemokines are produced [84], [85]. The ability to inhibit IFN-I production is due to the viral NS2B/3 protease that binds and cleaves the adaptor/sensor STING [85], [86], [87]. Interestingly, first evidences indicate that the NS2B/3 proteases of other Flaviviruses, as JEV or YFV, are not able to cleave STING, while the NS4B of YFV can do it [68].
To the same Flaviviridae family belongs the Hepacivirus genus, distantly related to Flaviviruses, that includes Hepatitis C virus (HCV) a major cause of chronic liver disease [88], [89]. HCV uses several strategies to efficiently evade innate immunity and this escape is considered the main determinant of viral persistence that leads to a chronic infection in 70–80% of infected people [88], [90]. HCV RNA can be sensed by different PRRs, namely RLRs, TLRs, NLRs, and, as recently reported, by protein kinase R (PKR). RIG-I is the best described sensor for the poly U/UC region located within the 3′-untranslated region (UTR) of the viral RNA, along with a 5′-ppp. This region is essential for viral replication and, thus, highly conserved among HCV genotypes. The key viral protein involved in the evasion strategies is the NS3/4A protease, which consists of NS3 and NS4A. The complex is essential for several steps in the viral cycle including viral RNA replication, polyprotein processing and viral assembly [91]. Taking advantage of its serine protease activity, NS3/4A cleaves the adaptor MAVS, preventing its dimerization and downstream signaling [92], [93]. After cleavage, MAVS dissociates from the mitochondrial associated endoplasmic reticulum membranes (MAM) where upon HCV-induced RIG-I activation it is recruited and colocalizes with IKK-ɛ [94], thus impairing IFN-I expression [95]. Importantly, the cleavage of MAVS by the viral protease has been confirmed in patients [96]. To antagonize IFN-I production, NS4B protein instead targets STING. HCV NS4B, indeed, contains a STING homology domain and interacts with STING in the ER blocking STING interaction with MAVS and TBK1 [97], [98]. Even if the exact molecular mechanism involved in NS4B inhibition of STING signaling has not yet been defined in the context of viral infection, NS4B likely cooperates with NS3/4A in targeting the RIG-I signaling pathway. Interestingly, NS2B/3 and NS4B of other members of the Flaviviridae family including DENV and YFV as mentioned above also block STING signaling and possess the same STING homology domain, indicating a conserved mechanism of STING antagonisms between flaviviruses and hepaciviruses.
Intracellularly, both TLR3 and TLR7 have been shown to sense HCV RNA, depending on the infected cell type considered [88]. Sensing by TLR3 may occur in liver cells, as hepatocytes, and liver resident macrophages Kupffer cells, while TLR7 sensing occurs predominantly in plasmacytoid DCs (pDCs) and macrophages. As reported above for the MAVS adaptor, the serine protease NS3/4A also cleaves the key adaptor of TLR3, TRIF [99], thus preventing an IFN response in productively-infected hepatocytes. In these cells, the HCV-induced miR-21 has been recently reported to be involved in evasion of IFN-I production and stimulation of HCV replication, upon suppression of MyD88 and IRAK1 expression, that is required for the TLR7-mediated sensing of the virus [100]. In macrophages, MyD88 signaling is instead targeted by the NS5A protein that, upon the direct binding to the adaptor, impairs the recruitment of IRAK-1 and cytokine production in response to TLR ligands [101]. Interestingly, TLR3 and TLR7 levels are decreased in patients chronically infected with HCV [102], [103] and this has been recently correlated with increased levels of miR-758 [104].
HCV-infected cells, however, can trigger IFN-I production in non-productively-infected pDCs in a cell–cell contact- and TLR7-dependent manner depending on the intracellular HCV RNA level of cocultured infected cells [105]. This production of IFN-I by pDCs may thus account for the strong IFN-I response observed in the liver of infected people [106]. Interestingly, a robust expression of ISGs correlated with a decreased response to IFN therapy [107] supporting a pathogenetic role of a high IFN signature in chronically infected individuals where the virus has established a persistent infection.
In contrast, in monocytes and macrophages upon clathrin-mediated endocytosis and recognition of the virus by TLR7, HCV activates the inflammasome and not IFN-I production in an infection-independent process [108]. An association between TLR-polymorphisms and cytokine production in response to TLR7 agonist in vitro has also been reported, supporting a pathogenic role of TLR7-mediated sensing in immune cells [109]. Interestingly, this cell-type dependent stimulation of IFN-I and inflammasome in response to HCV infection is also observed in human immunodeficiency virus type 1 (HIV-1) infection that as HCV can establish a persistent infection (see discussion below).
PKR has been shown to sense HCV RNA very early in infection even prior to RIG-I sensing [110]. PKR is a dsRNA binding protein that upon activation phosphorylates the α subunit of the eukaryotic translation initiation factor 2 (eIF2 α) to inhibit translation of host capped mRNA but not of non-capped mRNA, as that of HCV. PKR, upon binding HCV RNA and independently of its kinase-activity, interacts with MAVS to induce the transcription of a number of early ISGs including IFN stimulated gene 15 (ISG15), but not IFN-I. ISG15, in turn, deubiquitinates RIG-I inhibiting its functions [110]. By doing so, HCV blocks sensing by PKR and reinforces HCV evasion from RIG-1 signaling.
Amongst RNA viruses that, as HCV, can establish a persistent infection, HIV-1, a lentivirus from the Retroviridae family, represents a paradigm for its ability to prevent or circumvent the innate immune response mediated by IFN-I. In spite of evidence that a sustained IFN-I response occurs in HIV-infected patients, it fails to clear the infection in the first place and to prevent the early establishment of long-lived HIV-1 reservoirs [111], [112], [113]. HIV-1 has, indeed, developed a number of strategies to block the IFN signaling and the activity of IFN-induced host restriction factors. Here, we only briefly summarize these strategies some of which have been only recently discovered, leading to the identification of immune pathways, thus far, unrecognized (as recently reviewed elsewhere [114], [115], [116], [117], [118], [119], [120]).
In the past few years, the knowledge on innate immunity against HIV-1 has evolved enormously with the recent identification and the characterization of the molecular basis of retroviral recognition by PRRs [116], [118], [121]. Retroviral replication generates several structural and intermediate molecules as ssRNA, hybrids RNA/DNA, ss and ds DNA produced upon reverse transcriptase, that are potentially available for recognition by cellular PRRs as “non-self”.
With the exception of pDCs, which produce high levels of IFN-I upon detection of HIV-1 ssRNA by TLR7, in all other immune cells IFN-I production is prevented or barely detectable unless viral countermeasures are disabled. In conventional DCs (cDCs), monocytes and resting CD4+ T cells, indeed, HIV-1 sensing is prevented by the restriction factor Sterile Alpha Motif (SAM) and the hystidine/aspartic acid (HD) domain-containing protein1 (SAMHD1) that blocks reverse transcription or directly degrades genomic RNA, thus preventing PRR recognition [122], [123]. This SAMHD1-mediated restriction is overcome by the viral protein Vpx [124], [125] that is a HIV-2 and SIV accessory protein absent in HIV-1. This apparent disadvantage is, however, effectively exploited by the virus that maintains a reward by not replicating in myeloid cells and by reducing the impact of IFN-I production in these cells. The block of productive infection in non-cycling cells where SAMDH1 is active, particularly in cDCs, results in lack of maturation and thus in impairment in priming of naive, HIV-1-specific T cells for optimal anti-HIV-1 immunity [126]. Furthermore, in the small fraction of cDCs that become infected, the recognition of HIV-1 genomic RNA by TLR8 paradoxically licenses HIV-1 transcription [127]. In this case, productive DC infection allows an increased transmission to T cells while inhibiting IFN-I production. In monocytes instead, recognition of viral RNA by TLR8, does not trigger IFN-I production but, as in the case of HCV, leads to the formation of the NLRP3 inflammasome with activation of caspase-1 and IL-1β production, favoring the establishment of an inflammatory milieu that fuels HIV-1 replication [108], [126]. In contrast, in cells that are target of a productive infection, such as macrophages and T lymphocytes, IFN-I production is prevented by an escape mechanism mediated by the HIV-1 protease that drives RIG-I to the lysosomes [128].
The ssDNA derived from proviral DNA upon RT can, instead, be sensed by the newly identified DNA sensor Interferon gamma-interferon-inducible protein 16 (IFI16) [129], however, HIV exploits the host cytosolic nuclease 3′ repair exonuclease1 (TREX1) to digest HIV-1 DNA generated during infection that, thus, does not accumulate at levels sufficient to be detected by IFI16, unless TREX1 activity is blocked [130]. Finally, resting CD4+ T cells are not permissive to virus replication due to the expression of an active SAMHD1 that, as mentioned before, degrades genomic RNA and prevents efficient RT and recognition of ssDNA. However, this prevention of sensing may not be complete and partial recognition of RT intermediates by the IFI16 sensor not only leads to the initiation of IFN-I production, but also to the activation of the inflammasome, triggering cell death mechanisms including pyroptosis and apoptosis [131]. Finally, the viral capsid exploits two cellular proteins, cyclophilin A and CPSF6, and binds just a right amount of both to allow opening of the capsid and RT process, while preventing sensing of the viral cDNA before integration with the following production of IFN-I [132], [133].
Overall, viruses as HCV and HIV-1 have evolved nifty strategies to dampen the host innate response in cells where a productive infection may take place, while they induce infection-independent mechanisms in non-permissive cells to facilitate the viral life cycle and promote a chronic inflammation.
The genus Filoviruses from the Filoviridae family are among the most virulent known human pathogens and comprise one species of Marburg virus and five species of Ebola viruses, including Zaire ebolavirus (EBOV) that is the most lethal and responsible of the recent severe outbreak of hemorrhagic fevers causing up to 90% mortality in untreated humans [134]. Several immunoevasion strategies that result in total impairment of the innate immune system are responsible for most of EBOV virulence [135]. A major target of these strategies that are exerted by few viral proteins, is the IFN-I response that controls in vivo filoviruses infection [136]. At the level of viral sensing, the EBOV VP35 inhibits RIG-I signaling [137], [138]. VP35 binds with high affinity to dsRNA and 5′-ppp dsRNA in a sequence-independent manner. Four crystallographic structures of EBOV VP35 RBD/IID from Zaire and Reston viruses and of MARV VP35 RBD/IID have elucidated how VP35 RBD/IID dimers bind to RNA strands and how the dimers mimic the RLR shape, hiding the RNA recognition site [139], [140]. Interestingly, the residues involved in both protein–protein interactions at the RBD/IID dimer interface and involved in dsRNA binding are highly conserved among all known EBOV and MARV species. Moreover, all residues that are important for dsRNA binding are also crucial for IFN-I inhibition.
As the knowledge on the importance of IFN-I in controlling the immunity against bacteria increases, studies aimed at characterizing the evasion mechanisms that these pathogens employ to evade, inhibit, or otherwise manipulate the innate immune response are arising.
The mechanisms induced by bacteria for IFN-I expression in different cell types are various and reflect the heterogeneity of host–pathogen interactions established along bacterial infections. While the extracellular bacteria activate IFN signaling mainly through the interaction with molecules present on the cell surface, the intracellular bacteria are recognized as they enter a cell by cell-surface or endosome/phagosome bound receptors or by cytoplasmic pathogen sensors once they escape from these compartments [141]. Here, we report only few examples of bacterial evasion mostly related to signaling pathways converging in IFN-I stimulation. A more exhaustive discussion of bacterial evasion strategies from PRR-signaling pathways and inflammasome is provided by the contribution of Thomas Kufer and Igor Brodsky in this issue.
To elude PRR recognition many bacterial pathogens, like viruses, have modified the molecular structure of their PAMPs. LPS, an ubiquitous component of Gram-negative bacterial cell wall, is a PAMP that is recognized by the TLR4/MD-2 complex present on the cell surface. Some bacterial species have evolved an alternative form of LPS resulting in a weak antagonist of TLR4/MD-2 signaling. This strategy is utilized by Yersinia pestis, the causative agent of pestis infection. The acylation status of lipid A from hexa- to tetra-acylated is reduced when the temperature increases from 21 °C (flea temperature) to 37 °C (human temperature). This alteration renders lipid A less recognizable LPS by TLR4 and, following transmission from fleas to humans, contributes predominantly to the virulence of the bacterium [142]. Another example is Shigella that, after internalization and proliferation within epithelial cells, hypoacylates lipid A to become less visible to the immune system once leave the infected epithelial cells [143].
In addition to LPS, other bacterial components that may induce IFN-I, include bacterial nucleic acids and peptidoglycans. These PAMPs can be recognized outside or inside the host cells, leading to the activation of distinct signaling pathways [141]. Intracellularly bacterial RNA and DNA nucleic acids are recognized by the several intracellular receptors that also sense viral PAMPs, while bacterial peptidoglycans are detected by NOD1 and NOD2 system. Most of these pathways then converge in the activation of the STING/TBK1/IRF3 axis [6].
As viruses, bacteria also encode proteins with enzymatic activity that interfere with the activation of adaptor molecules involved in PRR signaling. This is the case of the Yersinia pestis virulence factor YopJ that, besides being a potent inhibitor of the NF-κB and MAPK signaling pathways, also inhibits TLR-mediated IFN response. As a deubiquitinating protease, YopJ prevents or removes the K63-polymerized ubiquitin conjugates, which are required for TRAF3 and TRAF6 activation in the signal transduction pathway leading to IRF3 activation [144]. Similarly, the type III effector OspI of Shigella Flexneri inactivates by deamination the E2 ubiquitin ligase UBC13, a factor important for TRAF6 auto-polyubiquitinylation and activation [145].
Another interesting example is represented by the translocated intimin receptor (Tir), which is one of the first type III effector proteins discovered in A/E pathogens including the enteropathogenic E. coli (EPEC), enterohemorrhagic E. coli O157:H7 (EHEC), and Citrobacter rodentium. In addition to the role played in the attachment to the host membrane, this factor shares sequence similarities with conserved regions present in the cytoplasmic tails of inhibitory receptors of the host immune system, such as the immunoreceptor tyrosine-based inhibition motifs (ITIMs). Tir utilizes these ITIM-like motifs to mimic an endogenous innate immunoregulatory mechanism. In particular, Tir recruits the host Src-homology-region-2-domain-containing phosphatase 1 to the adaptor TRAF6 and thus prevents polyubiquitinylation and activation of TRAF6 in the IFN-stimulated pathway [146].
3. Evasion/subversion of downstream PRR signaling: IKKs and IRFs
Adaptor molecules downstream sensors transmit signals to classical IκB kinase complex, including NEMO/IKK-γ, TANK and to the atypical IKK-related kinases IKK-ɛ and TBK1 that trigger activation of NF-κB and IRFs, respectively [147], [148]. IFN-I production downstream of these signaling pathways depends essentially on the presence and activation of IRFs and their contribution changes depending on the cell type considered [9]. The IRF family is presently composed of nine mammalian members namely IRF1 to 9, coded by distinct but related genes that exert a number of functions in the regulation of innate and adaptive immune responses. The IRFs with intrinsic antiviral function include IRF3 and IRF7 that are essential for the PRR-mediated IFN gene transcription, but also induce some IFN effectors in an IFN-independent manner. IRF9, as mentioned before, is part of the heterocomplex ISGF3 that drives the expression of most ISGs including IRF1 (Fig. 2). Although IRF1 is itself an ISGs, which affects different aspects of the immune response even independently from IFN-I production [149], [150], it also represents a positive regulator of IFN-I gene expression in response to specific stimuli in a cell type specific manner [151]. Moreover, IRF1 plays a crucial role in regulating MAVS-dependent signaling from peroxisomes [152]. In this respect, IRF1 regulates the transcriptional profile of antiviral genes unique to that induced by IFN-I and cooperatively promotes an effective antiviral program against a broad spectrum of viruses [152], [153]. IRF5 is instead specifically involved in inflammatory cytokines induction [9]. Given the unique functions exerted by IRFs, viruses have evolved strategies aimed at the specific destruction of these transcription factors. With regard to the viruses covered here, most of them inhibit IRF activation either indirectly by acting on sensors and elements of the signaling pathway that activate them, as described above, or directly by impairing/hijacking IRF activity (Fig. 3).
As described above, the SARS-CoV PLP affects the activation of both IRF3 and NF-κB not directly, as initially suggested [66], [154], but by targeting RIG-I, MAVS, TRAF3 [63] and, as more recently reported, STING [64], [65]. Interestingly, this activity is independent from PLP protease activity. Recently, it has been reported that the PLP of MERS-CoV also suppresses IFN-I transcription by interfering with IRF3 phosphorylation and nuclear translocation [155]. As SARS-CoV, MERS-CoV PLP is a viral deubiquitinating enzyme that acts on both K48- and K63-linked ubiquitination and ISG15-linked ISGylation, two posttranslational modifications that play important roles in regulating the RIG-I and STING/IRF3 and NF-κB activation [156]. Whether the deubiquitination and deISGylation activity of MERS-CoV PLP are directly responsible for inactivation of IRF3/NF-κB or upstream signaling pathway, it remains unclear.
The WNV NS1 protein inhibits the TLR3-induced activation of IFN-I and IL-6 transcription through inhibition of nuclear translocation of IRF3 and NF-κB [157]. Recent studies indicate that this effect seems to be dependent on NS1 domains that control viral replication [158]. Interestingly, in the draining lymph nodes the protein released predominantly from macrophages and DCs can inhibit the innate immune signaling pathways in uninfected cells and impairs cytokine production in response to infection [159], thus suggesting that NS1 could also influence the development of the adaptive immune response directed to WNV.
The NS2B/3 serine protease of DENV, instead, blocks the serine 386 phosphorylation and nuclear translocation of IRF3 by directly interacting with IKK-ɛ and masking the kinase domain [160].
Two tick-borne flaviviruses, LGTV and TBEV, have been recently reported to inhibit IRF1 independently of their ability to antagonize IFN signaling. In particular, a weak expression of IRF1 protein and nuclear localization, without reduction in IRF1 mRNA expression, was observed in DCs, an early cellular target of infection [161].
Several HCV proteins interfere with IRF activity. The HCV NS3 protease impairs IRF3 activation by blocking the interaction with TBK1 and IRF3 [162]. In addition, the NS2 protein inhibits, in a dose-dependent manner, IKK-ɛ- and especially TBK1-induced IRF3 phosphorylation [163]. The basic amino acid region 1 (BR1) in the N-terminal region of the core protein is also crucial in inhibiting IRF3 dimerization as well as phosphorylation induced by NDV infection and poly (I:C) [164]. Interestingly, this domain has been identified as the binding region for a DEAD box protein the DDX3, which has been recently found to enhance the TBK1/IKK-ɛ-induced IFN-β promoter activity upon binding to the adaptor MAVS [165]. The HCV core also decreased the expression levels of DDX3 suggesting that the IRF3 inhibition may be mediated by the core effect on DDX3. Moreover, through binding to DDX3, the HCV core protein also promotes HCV replication. Thus, the core protein appears to switch DDX3 from an IFN-inducing mode to an HCV-replication mode [166]. IRF1 expression is, instead, suppressed by the HCV core at the transcriptional level. This event blocks the expression of several antiviral and immunomodulatory genes of both innate and adaptive immunity and, in doing so, facilitates the establishment of HCV persistent infection [167]. In line with this hypothesis, accumulating evidence suggests that HCV also targets DCs to control the host antiviral response and trigger persistence. As WNV NS1, HCV core is, indeed, a secreted protein found in the peripheral blood of patients with chronic infection that may thus affect directly DC functions. In this context, it has been recently reported that the core protein suppresses IFN-I production in response to TLR agonists and to RIG-I stimulated by HCV PAMP in a cell culture model of pDCs, through the reduced levels of IRF7 and of phosphorylated STAT1 protein [168]. The effect on IRF7 is, however, not direct but probably mediated by the reduced levels of IFN-I production by core-stimulated pDCs. Whether or not this also occurs along the natural infection and contributes to HCV persistency remains to be determined.
To increase virus replication and establish viral persistence and latency, HIV-1, besides to dismantle or exploit almost all cell intrinsic innate recognition pathways, as discussed above, also directly hits IRFs [113], [119], [169].
In T cells, the virion-associated accessory proteins, Vif, Vpr and Vpu, directly target IRF3 for ubiquitin-associated proteasome degradation [170], [171], [172]. Recently, this Vpu effect on IRF3 degradation has been, however, challenged and it has been reported that Vpu, instead, mediates a partial cleavage of IRF3 in a caspase-dependent manner. Interestingly, this cleavage produces a C-terminal fragment that can act as a negative regulator of IRF3-dependent gene activation [173]. Thus, HIV-1, as already reported for several viruses, can also exploit the apoptotic machinery to interfere with IRF3 function [174].
In myeloid cells, instead, HIV-1 does not inhibit but, rather, stimulates both IRF1 and IRF7 expression. IRF1 activity is, however, exploited by the virus to induce a distinct subset of ISGs that despite displays intrinsic and unique antiviral actions, does not restrict viral infection [175]. IRF1 is also induced in HIV-1-productively infected T cells where it may regulate viral promoter activity even in the absence of the viral transactivator Tat driving initial transcription of the viral genome [176], [177]. Later on, however, when viral replication is mostly accomplished by the viral transactivator, IRF1 is sequestered by Tat to accelerate proteasomal-mediated IRF1 degradation (Remoli AL and Battistini A, unpublished) and to quench IRF1 transcriptional activity on target genes [178]. By so doing, Tat disarms the unique antiviral response against viral infections that IRF1 could exert [153]. A block of IFN transcription in primary CD4+ T cells may also depend on CD3/CD28-mediated activation of IKK-ɛ. We, indeed, recently reported that IKK-ɛ activation results in a peculiar pattern of IRF phosphorylation in T cells, including a splicing isoform of IRF3, which may function as an inhibitor of IFN-β expression, and phosphorylation of IRF1 that blocks its activity on IFN-I promoter [179].
The EBOV VP35, in addition to prevent RLR recognition, inhibits IFN-I promoter activation mediated by TBK-1/IKK-ɛ overexpression but not by a constitutively active IRF3, strongly suggesting a specific inhibition of the kinase activity of TBK-1/IKK-ɛ. Indeed, VP35 interacts with the TBK-1/IKK-ɛ kinase domain and functions as a substitute substrate, thus inhibiting both the kinase activity and the binding of the physiological IRF3/7 substrate [180], [181]. Notably, mutations of VP35 residues, involved in this IFN antagonism, do not alter the function of VP35 in viral replication and transcription [182]. In DCs, VP35 targets IRF7. By interacting with the small ubiquitin-like modifier SUMO E2 enzyme Ubc9 and the E3 ligase PIAS1, VP35 promotes IRF7 sumoylation, a post-translational modification that prevents IRF translocation into the nucleus and, in turn, IFN-I gene transcription. A similar effect of VP35 was also reported for IRF3 [183]. This VP35 activity is independent of its ability to recognize dsRNA and maps to the N-terminus, which is essential for interactions with both IRF7 and PIAS1. Interestingly, SUMO modification of IRF3/7 is a part of the negative feedback loop of normal IFN-I signaling [184] that is exploited by EBOV to weaken host innate immunity.
Downstream PRRs, bacteria mainly target the MAPK pathway and the NEMO-IKK-NF-κB signaling axis, which primarily induces inflammatory cytokines (reviewed in [19] and Thomas Kuper and Igor Brodsky in this issue). As an example of bacteria that target the IRF pathway. Listeria monocytogenes suppresses IFN-I gene induction downstream of TLR-triggered MyD88 signaling pathway, acting on IRF3. Indeed, a MAPK phosphatase renders IRF3 hypophosphorylated by enhancing the formation of a MAPK phosphatase-IRF3-TBK-1 ternary complex in response to infection [185].
4. Interfering with the IFN signaling pathway
In Fig. 2 is illustrated the IFN-I signaling pathway downstream the IFN-I receptor (IFNAR) (a heterodimer of IFNAR1 and IFNAR2) that is activated upon binding of virus-infected cell-secreted IFN-I, and some ISGs, including positive and negative feed-back regulators. Most of the viruses here covered and some bacteria also target these pathways (Fig. 3).
Upstream the JAK/STAT pathway, the SARS-CoV 3a promotes serine phosphorylation within the IFNAR1 degradation motif and increases IFNAR1 ubiquitination [186]. The PLP from SARS-CoV has a complex mechanism of interference with the JAK/STAT pathway. Through its de-ubiquitinase activity upregulates the expression of the ubiquitinating enzyme E2-25k, leading to degradation of the ERK kinase that, in turn, interferes with STAT1 phosphorylation [187]. The ORF6-encoded protein, instead, antagonizes STAT1 function by interacting and sequestering in the ER components of the nuclear import complex, as karyopherin alpha 2 and karyopherin beta 1. By doing so, ORF-6 competes for the binding of the nuclear import complex to STAT1, thus inhibiting STAT1 nuclear import [188]. However, the majority of these evidences have been obtained by overexpression or stably expression of individual viral components in cell culture, which represents an experimental setting that may not accurately reflect the innate immune signaling occurring during SARS-CoV infection in vivo.
WNV interferes with IFNAR complex by promoting phosphorylation-dependent ubiquitination and degradation of IFNAR1. This effect is mediated by the hydrophobic NS4A and NS4B proteins that potently induce the unfolded protein response (UPR). This pathway is physiologically induced by different stimuli, including the accumulation of misfolded proteins in the ER [189], [190] that, as mentioned before, represents the site of Flaviviruses replication. Activation of the UPR pathway inhibits IFN activation and induces a general ER stress response, thus facilitating viral replication. The methyltransferase domain of NS5 from Langat virus and JEV, instead, binds directly to IFNAR through its methyltransferase domain and inhibits the activation of kinases associated to the receptor [191], [192]. Several WNV non structural proteins, as NS2A, NS2B, NS3, NS4A, NS4B and NS5, have been reported to prevent the phoshorylation of JAK1, TYK2 and, as a consequence, the activation of STAT1/2 (recently reviewed in [73]). Likewise, expression of DENV nonstructural protein NS2A, NS4A, or NS4B proteins impairs the JAK/STAT signaling pathway by reducing the phosphorylation and nuclear translocation of STAT1 [193]. Phosphorylation of STAT2 is also blocked by DENV NS5 through inhibition of Jak1 and Tyk2 activity [194]. NS5 also binds to the coiled-coil region in the first half of the human STAT2 protein and acts as a bridge between UBR-4, a member of the N-recognin family, and STAT2 leading to STAT2 ubiquitination and proteasomal-mediated degradation [195], [196]. Interestingly, only proteolytically-processed NS5 can efficiently mediate STAT2 degradation, though both unprocessed and processed NS5 proteins are able to bind STAT2.
The JEV NS5 protein greatly reduces TYK2 and, partially, STAT1 phosphorylation, probably through its phosphatase activity [197]. The TBEV NS5, instead, blocks STAT1 phosphorylation by promoting the association with the PDZ membrane protein scribble [198].
By activating the Ras/Raf/MEK pathway, HCV replication has been shown to increase the phosphorylation of a motif contained in the cytoplasmic tail of IFNAR1, which is involved in controlling the receptor ubiquitin-dependent endocytosis and attenuation of STAT1/2 phosphorylation [199]. Several HCV proteins have also been implicated in the regulation of the IFN response pathway interfering directly with the JAK/STAT signaling. However, contrasting results have been reported that probably stem from the use of different cell lines or different HCV expression/replication systems [200], [201], [202], [203], [204]. The core protein has been reported to upregulate the expression of SOCS3, thereby inhibiting tyrosine phosphorylation of STAT1 [201], although the decreased STAT1 phoshorylation has not been detected in other studies [200], [205]. The HCV core protein, expressed alone, has been reported to directly bind to STAT1 and to prevent its phosphorylation and subsequent expression of downstream ISGs [206]. The NS5A, similarly, binds and prevents STAT1 phosphorylation specifically in hepatocyte-derived cell lines [207].
Two strategies are used by EBOV VP24 to limit the JAK1/TYK2-mediated activation of STAT1/2 and their subsequent nuclear localization. VP24 binds within the tyrosine-phosphorylated-STAT1 binding region located in the C terminus of members of the NPI-1 subfamily of karyopherin alpha nuclear localization signal receptors preventing their binding and shuttling to the nucleus of tyrosine-phosphorylated-STAT1 [208], [209]. The crystal structure of human KPNalpha5 C terminus in complex with VP24 has been recently resolved and a unique nonclassical nuclear localization signal binding site on KPNA5 has been identified. This motif is necessary for binding and efficient nuclear import of tyrosine-phosphorylated-STAT1 [210]. EBOV VP24 can also directly bind STAT1; whether the binding occurs with the unphosphorylated or phosphorylated STAT1 or both isoforms it is not yet clear [211]. However, also unphosphorylated STAT1 enters the nucleus to activate and sustain the expression of a number of IFN-induced immune regulatory genes, which are distinct from those activated by the phosphorylated STAT1. Thus, EBOV VP24 binding and inhibition of either forms of STAT1 may be important in the suppression of an antiviral state.
Notably, in spite of the high similarity of EBOV and MARV genome organization and high sequence homology between many of the EBOV and MARV encoded proteins, MARV VP24 unlike EBOV VP24, does not inhibit JAK/STAT signaling [212]. This is consistent with the observation that regions important for karyopherin alpha binding are different between these two VP24s [210]. In contrast, tyrosine phosphorylation of JAK1 and STAT is inhibited by the MARV matrix protein VP40 that also inhibits this IFN-II signaling. Interestingly, also JAK1-dependent and IL-6-induced tyrosine phosphorylation of STAT1 and STAT3 are targeted by MARV VP40 suggesting that MARV may globally inhibit JAK1-dependent cytokine signaling with mechanisms different from that employed by EBOV [212].
Among the escape mechanisms used by bacteria to evade immune responses, some have been recently reported to target IFN-I signaling molecules. Having found that influenza viruses replicate to a higher efficiency in cells co-infected with Staphylococcus aureus, Warnking et al. [213] demonstrated that an impaired STAT1/STAT2 dimerization is responsible for a poor induction of ISG transcription in spite of an abundant secretion of IFN-I driven by the FLU virus infection. Similarly, the inhibition of the response to IFN-I by Mycobacterium tuberculosis was observed in human macrophages and correlated with mycobacterial pathogenicity [214]. In primary cells and THP-1 cells, indeed, Mycobacterium tuberculosis specifically inhibits IFN-I signal transduction pathway by impairing the activation of STAT1, while the avirulent Mycobacterium bovis BCG fails to do so. Alteration in ISGF-3 complex formation was instead observed in human macrophages infected with nonpathogenic Lactobacillus rhamnosus GG where only STAT1 homodimers were found. In contrast, the pathogenic Streptococcus pyogenes led to formation of not only STAT1 homodimers but also of ISGF-3 [215]. Based on these finding the authors speculated that the efficient induction of IFN-I production and related transcription factor activation by streptococci would lead to fast and effective immune responses that, however, could play a role in the pathogenesis.
5. Interfering with ISGs
Although the first antiviral ISGs were discovered decades ago, until recently the mechanisms of action was defined for only a limited number of ISG-encoded proteins. The renewed interest in the innate immune response to retroviruses with the identification of how several host restriction factors may limit retroviral infection [118], [119], [120], as well as large-scale functional screening of ISGs, have identified genes that coordinately control the infection of a range of RNA and DNA viruses and have begun to dissect their mechanism of action [153], [216]. Interestingly, some of the most potent antiviral effectors reinforce the system by further inducing IFN or ISGs. Thus, by directly disarming and/or making the use of individual IFN-induced effector proteins, the antiviral effect of host cells may still be attenuated even though IFN-I is induced, and viruses can ensure protection from both autocrine and paracrine effects of secreted IFN-I.
Here, we will primarily focus on ISGs for which viral countermeasures have been identified in the context of viruses covered in the present review. Recent reviews report more extensively on ISG viral antagonists and specifically on IFN-induced HIV restriction factors that are not covered here [117], [118], [119], [120], [217], [218], [219].
5.1. PKR
PKR is one of the major effectors of the IFN-I-induced antiviral state. Upon activation from binding dsRNA molecules via a dsRNA binding domain, PKR phosphorylates the translation initiation factor, eIF2α, thus blocking cellular and viral protein synthesis in infected cells. Due to its ability to bind dsRNA molecules PKR is also a nucleic acid sensor, as mentioned above [220].
Viruses have evolved specific mechanisms to inhibit PKR activity or escape its action downstream. These include: the production of small and highly structured RNA molecules that prevent the dsRNA-induced dimerization and activation of PKR; expression of proteins that bind directly to and inhibit the activity of PKR or PKR activators; proteins that behave as pseudosubstrate and competitive inhibitors of PKR [221], [222], [223].
Amongst viruses covered in this review, both antiviral and proviral roles of PKR have been reported for HCV. The HCV NS5A and E2 proteins directly interfere with the antiviral action of PKR. NS5A binds directly to PKR, while the glyco-protein E2 acts as competitive substrate with eIF2 α for PKR binding, resulting in inhibition of PKR kinase activity and in increased HCV replication. The full length IRES of HCV RNA, which is recognized by PKR, may mediate either activation or inhibition of PKR [224]. Moreover, NS5A, by binding to various domains of the IRES, can alter the activation of PKR [225]. Another indirect inhibitory strategy mediated by the HCV IRES has been recently reported. Upon HCV RNA sensing, PKR activates the MAVS/TRAF3/IRF3 pathway that, however, does not induce IFN but a set of ISGs including ISG15. As mentioned, ISG15 deubiquitinates RIG-I to negatively control the RIG-I/MAVS pathway and prevent uncontrolled detrimental IFN-I expression in physiological conditions. Thus, HCV hijacks a protective cellular pathway to curtail host innate response [110]. Through a specific IRES-mediated inhibition of eIF2α-dependent translation, the HCV IRES also regulates the translational activity of PKR [224]. eIF2 α is, indeed, essential to the translation of capped mRNA, as those of ISGs, while non-capped mRNAs, as those of HCV, are translated independently from this factor. A general attenuation of ISGs expression by HCV IRES can be achieved also through a direct activation of PKR [226]. Interestingly, this attenuation of ISG expression is observed in acute but not persistent infection, where, instead, a sustained ISG expression occurs [206], [227].
Other proteins from Flaviviruses have been shown to inhibit PKR including the NS2A protein of JEV that physically interacts with PKR and blocks its activation in response to several stimuli [228].
EBOV VP35 also interferes with the pathway regulated by PKR by blocking and also reversing PKR activation, thereby preventing translational arrest of viral mRNAs. This PKR antagonism seems to be functionally different from dsRNA binding and IRF3 inhibition [229], [230], [231]. EBOV VP 35 also associates with PACT (PKR-associated activator). This complex abolishes PACT interaction with the C-terminal domain of RIG-I, which is required for full activation of RIG-I [232].
As for retroviruses, HIV-1 infection does not activate PKR due to both viral and cellular controls (reviewed in [233]). In vitro, PKR is activated by low amounts of TAR RNA, whereas high concentrations inhibit the kinase function. The viral Tat protein also counteracts PKR activation by several other mechanisms: it sequesters the activating dsRNA; it can act as a substrate homologue of eIF2α preventing the PKR mediated inhibition of protein synthesis; it prevents PKR auto-phosphorylation and exploits PKR activity to get phosphorylated and increase its binding to TAR RNA. Moreover, HIV-1 replicates only in cells that have high levels of the TAR RNA binding protein (TRBP), a strong inhibitor of PKR activation. Interestingly, during HIV-1 infection of lymphocytes, when HIV-1 replicates at high levels, increased amounts of ADAR1, an IFN-induced RNA editing enzyme that binds to PKR to inhibit its activation, have been observed. Moreover, PACT contributes to PKR dephosphorylation during HIV-1 replication probably due to its binding to ADAR1 [234]. Thus, HIV-1 has evolved to replicate in cells with high levels of TRBP, to induce the expression of ADAR1 and to change the function of PACT for PKR inhibition [235].
5.2. ISG15
ISG15 is an ubiquitin-like modifier that is induced rapidly by IFN-I and possesses antiviral activity against a number of viruses. ISG15 antiviral functions include inhibition of virus release, ISGylation of both viral and host proteins and immunomodulatory cytokine-like properties in its unconjugated and secreted form, as recently reviewed [236], [237].
More than 160 host proteins that are ISGylated have been identified including IRF3, PKR and RIG-I. ISGylation preferentially targets newly translated proteins and, as a consequence of ISGylation, degradation of the target protein is reduced by competition with ubiquitin conjugation [238], [239].
To date, only few viral proteins have been shown to be ISGylated and functional characterizations of ISG15 conjugation has not been always verified under conditions of endogenous protein expression [237]. Moreover, often, ISG15-mediated protection might not be a result of direct antagonism of virus replication. As an example, ISG15 has been shown to inhibit the release of HIV-1 and EBOV. This effect is mediated by an ubiquitin antagonism. ISG15 disrupts the ubiquitin-mediated regulation of EBOV VP40, necessary to produce budding and release of VP40 VLPs [240], as well as the ubiquitination of the HIV-1 Gag protein, which is required for the interaction with the cellular protein Tsg101 to mediate HIV-1 budding and release [241].
Several viruses have, thus, developed countermeasures against ISG15 and/or its conjugation. Strategies, identified so far, include viral proteins that bind ISG15 or that remove ISG15 from target proteins (reviewed in [237]).
SARS-CoV PLP has both deubiquitinating and deISGylating activities [242], [243]. Recently, the structural basis of recognition and processing of deubiquitin and ISG15 by PLP has been reported [244]. Interestingly, despite MERS-CoV encodes a single PLP similar to SARS-CoV, there is little to no sequence conservation among residues important for the deubiquitinating and deISGylating activity, suggesting that MERS-CoV PLP is likely to recognize and process ubiquitin and ISG15 substrates differently than SARS-CoV PLP [155], [244]. However, by affecting this post-translational modification, both viruses may modify cellular protein localization, protein activity and stability as well as signal transduction in order to increase viral replication and severity of infection. Nevertheless, although these results stem from in vitro overexpression or mutant studies, the direct evidence for ISG15 antagonism by these proteins remains to be demonstrated during viral infection.
Despite the well-characterized role in restricting replication of several viruses, ISG15 may actually also promote the replication of specific viruses, as HCV. In HCV infections both anti-viral or pro-viral effects exerted by ISG15 could be related to the net effect of ISGylation on the various viral and host proteins targeted by ISG15. An antiviral effect has been reported when ISG15 could conjugate to HCV NS5A, thereby enhancing the inhibitory effect of IFN-I on HCV replication [245]. In contrast, several groups have reported that ISG15 and ISGylation promote HCV production in a cell culture model independently of upstream IFN signaling [246], [247]. Although counter-intuitive, this finding may be explained by the observed inhibition of IFN-I induction by HCV upon ISG15 overexpression that negatively controls the RIG-I/MAVS pathway at the level of RIG-I ubiquitination [110]. A negative regulation of IFN-I expression may also be mediated by USP18, an ISG displaying a potent inhibitory effect on the IFN-I pathway, to prevent autoinflammatory consequences of uncontrolled IFN-I production. Similarly, USP18 may dampen the detrimental role that the hyperactivation of IFN-I signaling plays in the pathogenesis of some viral and bacterial infections, including HIV-1, HCV and Mycobacterium tuberculosis. USP18 is, indeed, stabilized by ISG15 in an unconjugated free form [248]. In this respect, the suggested potential role of the ISG15/USP18 pathway in HCV persistence is consistent with the observation that increased expression of hepatic ISGs before IFN treatment is associated with an absent or poor response in patients chronically infected with HCV [249]. Thus, ISG15/USP18 pathway might explain the paradox that the preactivation of the endogenous IFN system, while fails to clear the infection, instead, may stimulate HCV production and blunt the effect of exogenous IFN-I.
USP18 is similarly induced during some bacterial infections, including Salmonella and Mycobacterium tuberculosis. The decreased survival of mice that carry a point mutation in USP18 results from higher Salmonella load in the spleen and liver, an increased inflammatory response and increased IFN-I signaling. Similarly, these USP18 mutant mice are more susceptible to Mycobacterium tuberculosis infection and have increased bacterial load in the lung and spleen, elevated inflammatory cytokine production and more severe lung pathology [250]. In line with these findings, the results of Dorhoi et al. [251] have shown that IFNAR1 deficient mice were protected from death upon aerogenic infection with Mycobacterium tuberculosis. Moreover, a rather detrimental effect of IFN-I was also found in whole blood of patients with tuberculosis, where a neutrophil-driven, IFN-inducible transcriptional signature was associated with clinical severity [252], [253], [254]. These results thus reveal that some viruses and bacteria, to push replication and persistence, utilize an opposite, but as much as effectual strategy, consisting in enhancing/perpetuating an IFN-I response by targeting negative regulators of IFN-I expression.
5.3. iNOS and NAPDH
Anti-microbial molecules, such as nitric oxide (NO) radicals and reactive oxygen species (ROS) mediate the antibacterial properties of IFN-I. The key producers of NO is inducible nitric oxide synthase (iNOS), an enzyme that can be induced by both IFN-I and -II, although the latter is the conventional inducer that plays a crucial role in fighting the infection of intracellular bacteria [255]. Similarly, IFN-I and -II induce the subunits of the phagocyte nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, which generates ROS for killing organisms [256]. Thus, pathogens have evolved several ways of avoiding ROS- and NO-mediated killing. In spite of the fact that no data are available, so far, on the strategies exploited by bacteria to contrast the IFN-mediated transcriptional regulation of iNOS and NADPH oxidase, a common theme for successful intracellular pathogens is the ability to avoid the colocalization with these harmful host enzymes. Intracellular Salmonella, which resides within a specialized membrane compartment called the Salmonella-containing vacuole (SCV) in macrophages, uses a T3SS called Salmonella Pathogenicity Island 2 (Spi2) to mediate protection from NO and ROS intermediates [257]. Intracellular organisms have also developed mechanisms to detoxify and repair NO-mediated damage [255], as well as to avoid the induction of iNOS activity [258].
Given the crucial role played by these antimicrobial enzymes in contrasting bacterial infection, it is likely that strategies pinpointed by pathogens to inhibit the IFN-driven expression of these molecules will be discovered in the nearest future.
6. Conclusions
To effectively resist the continual microbial threat from the environment, vertebrates possess several defense mechanisms including innate and adaptive immunity. As an essential component of the innate immunity, the IFN system constitutes the first line of defense against a number of pathogens to clear an incoming infection and instructing an ensuing adaptive response.
Successful pathogens have, thus, evolved sophisticated strategies to subvert and/or exploit the host immune system where blocking the IFN response, in the first place, is required to replicate and survive. The elucidation of some of these strategies has led to the identification of several, thus far, poorly recognized features of the innate immune response. In parallel, with the enormous recent advances in the comprehension of the molecular mechanisms of innate immune responses to pathogens, specific processes by which pathogenic microorganisms subvert these innate immune pathways, including the IFN system, is becoming progressively appreciated and it is reasonable to assume that many more will be discovered in the near future. By learning from the anti-immune strategies of pathogens we can, thus, not only identify key pathogen regulators as useful target to exploit to the host advantage, but we can also unveil weaknesses of host defenses and intervene to more precisely tune the immune response.
The number and diversity of pathogen strategies for counteracting at each step the IFN system is stupefying. Although beyond the scope of this review to discuss all antagonisms in detail, the ones that we have here reported, represent common and recurrent strategies used by a number of pathogens. This is illustrated by the existence of both viral and bacterial examples of PAMP modifications as well as of viral and bacterial proteins that share cellular-like domain or cellular-like enzymatic properties that can compete with the host counterparts to dampen their physiological activities. In this respect, common hubs in the signaling pathways downstream pathogen sensors that trigger IFN-I, as few common adaptors or cofactors and transcription factors, are attractive targets of pathogen antagonism.
The recognition of the mechanisms involved in microbial countermeasures may have several translation implications. The definition of microbial ability to elude the detection, as the methylation of their RNA caps, can suggest strategies to utilize Methyltransferase mutants as successful vaccine candidates against a number of different viruses as already suggested for DENV [259].
Many of the pathogen proteins responsible for IFN-I antagonism are also determinants of virulence and pathogenesis and, as such, they are highly conserved and may, thus, constitute attractive targets for the development of promising therapeutics against various clinically relevant pathogens reducing the bias of resistance mutations. In turn, some of the cellular identified targets of pathogen proteins as well as protein interacting partners might turn out to be new drug targets for treating a range of different diseases that disarme common components of the IFN pathway. In this respect, the resolution of the crystallographic structures of viral antagonists in complex with their different viral and cellular ligands, is then crucial for the rational design of new drugs.
The system biology approach and the ability to simultaneously investigate diverse pathways has led to appreciate the interconnection between these pathways also in terms of shared components and stimulation by different pathogens. Thus, a single therapeutic strategy could modulate multiple pathways to the host benefit. Nevertheless, each approach needs to be complemented with effective treatments that also overcome other concurrent strategies that often the same pathogen put in place.
On the other side of the coin, recovery of a full innate response must be finely tuned. IFN-I is, indeed, not always protective but can instead play a pathogenic role as reported for some bacterial and viral infections, where an uncontrolled IFN production is a determinant of disease progression. Even in these cases, however, insights in the mechanisms involved in turning an IFN protective response into a pathogenetic one, may be as well relevant for non-pathogen-induced diseases, as autoimmunity and inflammatory diseases. In this context, cell death and inflammasome activation have been described as crucial IFN-I-regulated events exploited by both pathogen and host to get their own advantage. Despite inflammasome facilitates pathogen clearance and is beneficial to the host, in some instances, IFN-induced non-canonical NLRP3 inflammasome activation and pyroptosis appear to be detrimental due to excessive cell death, inflammation, and collateral tissue damage in vital organs [260]. So pathogen proteins themselves or modified versions of them could be used as therapeutics working in suppressing inappropriate immune activation. This would be a bright way of hijacking molecules evolved during pathogen adaptation and associated fitness, to shift the balance to the host advantage. Similarly, some identified targets of viral proteins in PRR signaling pathways might be turned out to be new targets for treating a range of diseases.
To find the way to generally induce an IFN response that is protective against a number of different infectious diseases, may be particularly relevant during an outbreak of unknown etiology or during the arising of newly emerging and re-emerging strains. Likewise, finding the key to unlock the detrimental outcome of excessive IFN-I production during an infectious disease can open the way to cure autoimmune and inflammatory diseases.
Disclosure statement
The authors declare no competing financial interests.
Acknowledgments
We apologize to the many colleagues whose data and influence have been overlooked due to space or our knowledge limitations. A special thank to members of the E.M. Coccia's and A. Battistini's laboratory for helpful discussion and critical reading of the manuscript and Eugenio Morassi for preparing drawings. Our work is supported in part by grant RF-2010235199 from Italian Ministry of Health (to EMC) and from Istituto Superiore di Sanità (to AB).
References
- 1.Wu J., Chen Z.J. Innate immune sensing and signaling of cytosolic nucleic acids. Annu. Rev. Immunol. 2014;32:461–488. doi: 10.1146/annurev-immunol-032713-120156. [DOI] [PubMed] [Google Scholar]
- 2.Sancho D., Reis e Sousa C. Signaling by myeloid C-type lectin receptors in immunity and homeostasis. Annu. Rev. Immunol. 2012;30:491–529. doi: 10.1146/annurev-immunol-031210-101352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paludan S.R., Bowie A.G. Immune sensing of DNA. Immunity. 2013;38:870–880. doi: 10.1016/j.immuni.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blasius A.L., Beutler B. Intracellular toll-like receptors. Immunity. 2010;32:305–315. doi: 10.1016/j.immuni.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 5.Barbe F., Douglas T., Saleh M. Advances in Nod-like receptors (NLR) biology. Cytokine Growth Factor Rev. 2014 doi: 10.1016/j.cytogfr.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 6.Unterholzner L. The interferon response to intracellular DNA: why so many receptors? Immunobiology. 2013;218:1312–1321. doi: 10.1016/j.imbio.2013.07.007. [DOI] [PubMed] [Google Scholar]
- 7.Mansur D.S., Smith G.L., Ferguson B.J. Intracellular sensing of viral DNA by the innate immune system. Microbes Infect. 2014 doi: 10.1016/j.micinf.2014.09.010. [DOI] [PubMed] [Google Scholar]
- 8.Akira S., Uematsu S., Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 9.Honda K., Taniguchi T. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 2006;6:644–658. doi: 10.1038/nri1900. [DOI] [PubMed] [Google Scholar]
- 10.Ivashkiv L.B., Donlin L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014;14:36–49. doi: 10.1038/nri3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schneider W.M., Chevillotte M.D., Rice C.M. Interferon-stimulated genes: a complex web of host defenses. Annu. Rev. Immunol. 2014;32:513–545. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Platanias L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005;5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- 13.Reid E., Charleston B. Type I and III interferon production in response to RNA viruses. J. Interferon Cytokine Res. 2014;34:649–658. doi: 10.1089/jir.2014.0066. [DOI] [PubMed] [Google Scholar]
- 14.Samarajiwa S.A., Forster S., Auchettl K., Hertzog P.J. INTERFEROME: the database of interferon regulated genes. Nucleic Acids Res. 2009;37:D852–D857. doi: 10.1093/nar/gkn732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stark G.R., Darnell J.E., Jr. The JAK-STAT pathway at twenty. Immunity. 2012;36:503–514. doi: 10.1016/j.immuni.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stetson D.B., Medzhitov R. Type I interferons in host defense. Immunity. 2006;25:373–381. doi: 10.1016/j.immuni.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 17.Coccia E.M. IFN regulation and functions in myeloid dendritic cells. Cytokine Growth Factor Rev. 2008;19:21–32. doi: 10.1016/j.cytogfr.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 18.Carrero J.A. Confounding roles for type I interferons during bacterial and viral pathogenesis. Int. Immunol. 2013;25:663–669. doi: 10.1093/intimm/dxt050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reddick L.E., Alto N.M. Bacteria fighting back: how pathogens target and subvert the host innate immune system. Mol. Cell. 2014;54:321–328. doi: 10.1016/j.molcel.2014.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Finlay B.B., McFadden G. Anti-immunology: evasion of the host immune system by bacterial and viral pathogens. Cell. 2006;124:767–782. doi: 10.1016/j.cell.2006.01.034. [DOI] [PubMed] [Google Scholar]
- 21.Baxt L.A., Garza-Mayers A.C., Goldberg M.B. Bacterial subversion of host innate immune pathways. Science. 2013;340:697–701. doi: 10.1126/science.1235771. [DOI] [PubMed] [Google Scholar]
- 22.Weber F., Kochs G., Haller O. Inverse interference: how viruses fight the interferon system. Viral Immunol. 2004;17:498–515. doi: 10.1089/vim.2004.17.498. [DOI] [PubMed] [Google Scholar]
- 23.Versteeg G.A., Garcia-Sastre A. Viral tricks to grid-lock the type I interferon system. Curr. Opin. Microbiol. 2010;13:508–516. doi: 10.1016/j.mib.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bowie A.G., Unterholzner L. Viral evasion and subversion of pattern-recognition receptor signalling. Nat. Rev. Immunol. 2008;8:911–922. doi: 10.1038/nri2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Orzalli M.H., Knipe D.M. Cellular sensing of viral DNA and viral evasion mechanisms. Annu. Rev. Microbiol. 2014;68:477–492. doi: 10.1146/annurev-micro-091313-103409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rosadini C.V., Kagan J.C. Microbial strategies for antagonizing Toll-like-receptor signal transduction. Curr. Opin. Immunol. 2015;32C:61–70. doi: 10.1016/j.coi.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kawai T., Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 28.Iwasaki A. A virological view of innate immune recognition. Annu. Rev. Microbiol. 2012;66:177–196. doi: 10.1146/annurev-micro-092611-150203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pandey S., Kawai T., Akira S. Microbial sensing by toll-like receptors and intracellular nucleic acid sensors. Cold Spring Harb. Perspect. Biol. 2015:7a016246. doi: 10.1101/cshperspect.a016246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kato H., Takeuchi O., Mikamo-Satoh E., Hirai R., Kawai T., Matsushita K. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 2008;205:1601–1610. doi: 10.1084/jem.20080091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pichlmair A., Schulz O., Tan C.P., Naslund T.I., Liljestrom P., Weber F. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science. 2006;314:997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- 32.Hornung V., Ellegast J., Kim S., Brzozka K., Jung A., Kato H. 5′-Triphosphate RNA is the ligand for RIG-I. Science. 2006;314:994–997. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- 33.Abdullah Z., Schlee M., Roth S., Mraheil M.A., Barchet W., Bottcher J. RIG-I detects infection with live Listeria by sensing secreted bacterial nucleic acids. EMBO J. 2012;31:4153–4164. doi: 10.1038/emboj.2012.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li X.D., Chiu Y.H., Ismail A.S., Behrendt C.L., Wight-Carter M., Hooper L.V. Mitochondrial antiviral signaling protein (MAVS) monitors commensal bacteria and induces an immune response that prevents experimental colitis. Proc. Natl. Acad. Sci. U.S.A. 2011;108:17390–17395. doi: 10.1073/pnas.1107114108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seth R.B., Sun L., Ea C.K., Chen Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 36.Ishikawa H., Barber G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhong B., Yang Y., Li S., Wang Y.Y., Li Y., Diao F. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity. 2008;29:538–550. doi: 10.1016/j.immuni.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 38.Ran Y., Shu H.B., Wang Y.Y. MITA/STING: a central and multifaceted mediator in innate immune response. Cytokine Growth Factor Rev. 2014;25:631–639. doi: 10.1016/j.cytogfr.2014.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dabo S., Meurs E.F. dsRNA-dependent protein kinase PKR and its role in stress, signaling and HCV infection. Viruses. 2012;4:2598–2635. doi: 10.3390/v4112598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Diamond M.S. IFIT1: a dual sensor and effector molecule that detects non-2′-O methylated viral RNA and inhibits its translation. Cytokine Growth Factor Rev. 2014 doi: 10.1016/j.cytogfr.2014.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Philpott D.J., Sorbara M.T., Robertson S.J., Croitoru K., Girardin S.E. NOD proteins: regulators of inflammation in health and disease. Nat. Rev. Immunol. 2014;14:9–23. doi: 10.1038/nri3565. [DOI] [PubMed] [Google Scholar]
- 42.Vanaja S.K., Rathinam V.A., Fitzgerald K.A. Mechanisms of inflammasome activation: recent advances and novel insights. Trends Cell Biol. 2015 doi: 10.1016/j.tcb.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hui D.S., Wong G.W. Advancements in the battle against severe acute respiratory syndrome. Expert Opin. Pharmacother. 2004;5:1687–1693. doi: 10.1517/14656566.5.8.1687. [DOI] [PubMed] [Google Scholar]
- 44.de Groot R.J., Baker S.C., Baric R.S., Brown C.S., Drosten C., Enjuanes L. Middle East respiratory syndrome coronavirus (MERS-CoV): announcement of the Coronavirus Study Group. J. Virol. 2013;87:7790–7792. doi: 10.1128/JVI.01244-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Coleman C.M., Frieman M.B. Coronaviruses: important emerging human pathogens. J. Virol. 2014;88:5209–5212. doi: 10.1128/JVI.03488-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hilgenfeld R., Peiris M. From SARS to MERS: 10 years of research on highly pathogenic human coronaviruses. Antiviral Res. 2013;100:286–295. doi: 10.1016/j.antiviral.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hui D.S., Memish Z.A., Zumla A. Severe acute respiratory syndrome vs. the Middle East respiratory syndrome. Curr. Opin. Pulm. Med. 2014;20:233–241. doi: 10.1097/MCP.0000000000000046. [DOI] [PubMed] [Google Scholar]
- 48.Zielecki F., Weber M., Eickmann M., Spiegelberg L., Zaki A.M., Matrosovich M. Human cell tropism and innate immune system interactions of human respiratory coronavirus EMC compared to those of severe acute respiratory syndrome coronavirus. J. Virol. 2013;87:5300–5304. doi: 10.1128/JVI.03496-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kindler E., Jonsdottir H.R., Muth D., Hamming O.J., Hartmann R., Rodriguez R. Efficient replication of the novel human betacoronavirus EMC on primary human epithelium highlights its zoonotic potential. MBio. 2013;4:e00611–e00612. doi: 10.1128/mBio.00611-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kindler E., Thiel V. To sense or not to sense viral RNA – essentials of coronavirus innate immune evasion. Curr. Opin. Microbiol. 2014;20:69–75. doi: 10.1016/j.mib.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Totura A.L., Baric R.S. SARS coronavirus pathogenesis: host innate immune responses and viral antagonism of interferon. Curr. Opin. Virol. 2012;2:264–275. doi: 10.1016/j.coviro.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zust R., Cervantes-Barragan L., Habjan M., Maier R., Neuman B.W., Ziebuhr J. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat. Immunol. 2011;12:137–143. doi: 10.1038/ni.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Menachery V.D., Debbink K., Baric R.S. Coronavirus non-structural protein 16: evasion, attenuation, and possible treatments. Virus Res. 2014 doi: 10.1016/j.virusres.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Daffis S., Szretter K.J., Schriewer J., Li J., Youn S., Errett J. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature. 2010;468:452–456. doi: 10.1038/nature09489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Menachery V.D., Yount B.L., Jr., Josset L., Gralinski L.E., Scobey T., Agnihothram S. Attenuation and restoration of severe acute respiratory syndrome coronavirus mutant lacking 2′-O-methyltransferase activity. J. Virol. 2014;88:4251–4264. doi: 10.1128/JVI.03571-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bouvet M., Imbert I., Subissi L., Gluais L., Canard B., Decroly E. RNA 3′-end mismatch excision by the severe acute respiratory syndrome coronavirus nonstructural protein nsp10/nsp14 exoribonuclease complex. Proc. Natl. Acad. Sci. U.S.A. 2012;109:9372–9377. doi: 10.1073/pnas.1201130109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shi X., Wang L., Li X., Zhang G., Guo J., Zhao D. Endoribonuclease activities of porcine reproductive and respiratory syndrome virus nsp11 was essential for nsp11 to inhibit IFN-beta induction. Mol. Immunol. 2011;48:1568–1572. doi: 10.1016/j.molimm.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Knoops K., Kikkert M., Worm S.H., Zevenhoven-Dobbe J.C., van der Meer Y., Koster A.J. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 2008;6:e226. doi: 10.1371/journal.pbio.0060226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.de Wilde A.H., Raj V.S., Oudshoorn D., Bestebroer T.M., van Nieuwkoop S., Limpens R.W. MERS-coronavirus replication induces severe in vitro cytopathology and is strongly inhibited by cyclosporin A or interferon-alpha treatment. J. Gen. Virol. 2013;94:1749–1760. doi: 10.1099/vir.0.052910-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lu X., Pan J., Tao J., Guo D. SARS-CoV nucleocapsid protein antagonizes IFN-beta response by targeting initial step of IFN-beta induction pathway, and its C-terminal region is critical for the antagonism. Virus Genes. 2011;42:37–45. doi: 10.1007/s11262-010-0544-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kopecky-Bromberg S.A., Martinez-Sobrido L., Frieman M., Baric R.A., Palese P. Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J. Virol. 2007;81:548–557. doi: 10.1128/JVI.01782-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Freundt E.C., Yu L., Park E., Lenardo M.J., Xu X.N. Molecular determinants for subcellular localization of the severe acute respiratory syndrome coronavirus open reading frame 3b protein. J. Virol. 2009;83:6631–6640. doi: 10.1128/JVI.00367-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Siu K.L., Kok K.H., Ng M.H., Poon V.K., Yuen K.Y., Zheng B.J. Severe acute respiratory syndrome coronavirus M protein inhibits type I interferon production by impeding the formation of TRAF3.TANK.TBK1/IKKepsilon complex. J. Biol. Chem. 2009;284:16202–16209. doi: 10.1074/jbc.M109.008227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen X., Yang X., Zheng Y., Yang Y., Xing Y., Chen Z. SARS coronavirus papain-like protease inhibits the type I interferon signaling pathway through interaction with the STING-TRAF3-TBK1 complex. Protein Cell. 2014;5:369–381. doi: 10.1007/s13238-014-0026-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sun L., Xing Y., Chen X., Zheng Y., Yang Y., Nichols D.B. Coronavirus papain-like proteases negatively regulate antiviral innate immune response through disruption of STING-mediated signaling. PLoS One. 2012;7:e30802. doi: 10.1371/journal.pone.0030802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Frieman M., Ratia K., Johnston R.E., Mesecar A.D., Baric R.S. Severe acute respiratory syndrome coronavirus papain-like protease ubiquitin-like domain and catalytic domain regulate antagonism of IRF3 and NF-kappaB signaling. J. Virol. 2009;83:6689–6705. doi: 10.1128/JVI.02220-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zinzula L., Tramontano E. Strategies of highly pathogenic RNA viruses to block dsRNA detection by RIG-I-like receptors: hide, mask, hit. Antiviral Res. 2013;100:615–635. doi: 10.1016/j.antiviral.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Maringer K., Fernandez-Sesma A. Message in a bottle: lessons learned from antagonism of STING signalling during RNA virus infection. Cytokine Growth Factor Rev. 2014 doi: 10.1016/j.cytogfr.2014.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kok K.H., Lui P.Y., Ng M.H., Siu K.L., Au S.W., Jin D.Y. The double-stranded RNA-binding protein PACT functions as a cellular activator of RIG-I to facilitate innate antiviral response. Cell Host Microbe. 2011;9:299–309. doi: 10.1016/j.chom.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 70.Siu K.L., Yeung M.L., Kok K.H., Yuen K.S., Kew C., Lui P.Y. Middle east respiratory syndrome coronavirus 4a protein is a double-stranded RNA-binding protein that suppresses PACT-induced activation of RIG-I and MDA5 in the innate antiviral response. J. Virol. 2014;88:4866–4876. doi: 10.1128/JVI.03649-13. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 71.Matthews K.L., Coleman C.M., van der Meer Y., Snijder E.J., Frieman M.B. The ORF4b-encoded accessory proteins of Middle East respiratory syndrome coronavirus and two related bat coronaviruses localize to the nucleus and inhibit innate immune signalling. J. Gen. Virol. 2014;95:874–882. doi: 10.1099/vir.0.062059-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bidet K., Garcia-Blanco M.A. Flaviviral RNAs: weapons and targets in the war between virus and host. Biochem. J. 2014;462:215–230. doi: 10.1042/BJ20140456. [DOI] [PubMed] [Google Scholar]
- 73.Ye J., Zhu B., Fu Z.F., Chen H., Cao S. Immune evasion strategies of flaviviruses. Vaccine. 2013;31:461–471. doi: 10.1016/j.vaccine.2012.11.015. [DOI] [PubMed] [Google Scholar]
- 74.Liu L., Dong H., Chen H., Zhang J., Ling H., Li Z. Flavivirus RNA cap methyltransferase: structure, function, and inhibition. Front. Biol. (Beijing) 2010;5:286–303. doi: 10.1007/s11515-010-0660-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Heinz F.X., Stiasny K. Flaviviruses and flavivirus vaccines. Vaccine. 2012;30:4301–4306. doi: 10.1016/j.vaccine.2011.09.114. [DOI] [PubMed] [Google Scholar]
- 76.Marston H.D., Folkers G.K., Morens D.M., Fauci A.S. Emerging viral diseases: confronting threats with new technologies. Sci. Transl. Med. 2014;6:253ps210. doi: 10.1126/scitranslmed.3009872. [DOI] [PubMed] [Google Scholar]
- 77.Morrison J., Aguirre S., Fernandez-Sesma A. Innate immunity evasion by dengue virus. Viruses. 2012;4:397–413. doi: 10.3390/v4030397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Suthar M.S., Diamond M.S., Gale M., Jr. West Nile virus infection and immunity. Nat. Rev. Microbiol. 2013;11:115–128. doi: 10.1038/nrmicro2950. [DOI] [PubMed] [Google Scholar]
- 79.Dong H., Zhang B., Shi P.Y. Flavivirus methyltransferase: a novel antiviral target. Antiviral Res. 2008;80:1–10. doi: 10.1016/j.antiviral.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Szretter K.J., Daniels B.P., Cho H., Gainey M.D., Yokoyama W.M., Gale M., Jr. 2′-O methylation of the viral mRNA cap by West Nile virus evades ifit1-dependent and -independent mechanisms of host restriction in vivo. PLoS Pathog. 2012;8:e1002698. doi: 10.1371/journal.ppat.1002698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kimura T., Katoh H., Kayama H., Saiga H., Okuyama M., Okamoto T. Ifit1 inhibits Japanese encephalitis virus replication through binding to 5′ capped 2′-O unmethylated RNA. J. Virol. 2013;87:9997–10003. doi: 10.1128/JVI.00883-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gillespie L.K., Hoenen A., Morgan G., Mackenzie J.M. The endoplasmic reticulum provides the membrane platform for biogenesis of the flavivirus replication complex. J. Virol. 2010;84:10438–10447. doi: 10.1128/JVI.00986-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Welsch S., Miller S., Romero-Brey I., Merz A., Bleck C.K., Walther P. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe. 2009;5:365–375. doi: 10.1016/j.chom.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rodriguez-Madoz J.R., Bernal-Rubio D., Kaminski D., Boyd K., Fernandez-Sesma A. Dengue virus inhibits the production of type I interferon in primary human dendritic cells. J. Virol. 2010;84:4845–4850. doi: 10.1128/JVI.02514-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rodriguez-Madoz J.R., Belicha-Villanueva A., Bernal-Rubio D., Ashour J., Ayllon J., Fernandez-Sesma A. Inhibition of the type I interferon response in human dendritic cells by dengue virus infection requires a catalytically active NS2B3 complex. J. Virol. 2010;84:9760–9774. doi: 10.1128/JVI.01051-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Aguirre S., Maestre A.M., Pagni S., Patel J.R., Savage T., Gutman D. DENV inhibits type I IFN production in infected cells by cleaving human STING. PLoS Pathog. 2012;8:e1002934. doi: 10.1371/journal.ppat.1002934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yu C.Y., Chang T.H., Liang J.J., Chiang R.L., Lee Y.L., Liao C.L. Dengue virus targets the adaptor protein MITA to subvert host innate immunity. PLoS Pathog. 2012;8:e1002780. doi: 10.1371/journal.ppat.1002780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Horner S.M. Activation and evasion of antiviral innate immunity by hepatitis C virus. J. Mol. Biol. 2014;426:1198–1209. doi: 10.1016/j.jmb.2013.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lavanchy D. The global burden of hepatitis C. Liver Int. 2009;29(Suppl. 1):74–81. doi: 10.1111/j.1478-3231.2008.01934.x. [DOI] [PubMed] [Google Scholar]
- 90.Metz P., Reuter A., Bender S., Bartenschlager R. Interferon-stimulated genes and their role in controlling hepatitis C virus. J. Hepatol. 2013;59:1331–1341. doi: 10.1016/j.jhep.2013.07.033. [DOI] [PubMed] [Google Scholar]
- 91.Morikawa K., Lange C.M., Gouttenoire J., Meylan E., Brass V., Penin F. Nonstructural protein 3-4A: the Swiss army knife of hepatitis C virus. J. Viral Hepat. 2011;18:305–315. doi: 10.1111/j.1365-2893.2011.01451.x. [DOI] [PubMed] [Google Scholar]
- 92.Meylan E., Curran J., Hofmann K., Moradpour D., Binder M., Bartenschlager R. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 93.Li X.D., Sun L., Seth R.B., Pineda G., Chen Z.J. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. U.S.A. 2005;102:17717–17722. doi: 10.1073/pnas.0508531102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Horner S.M., Liu H.M., Park H.S., Briley J., Gale M., Jr. Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc. Natl. Acad. Sci. U.S.A. 2011;108:14590–14595. doi: 10.1073/pnas.1110133108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lin R., Lacoste J., Nakhaei P., Sun Q., Yang L., Paz S. Dissociation of a MAVS/IPS-1/VISA/Cardif-IKKepsilon molecular complex from the mitochondrial outer membrane by hepatitis C virus NS3-4A proteolytic cleavage. J. Virol. 2006;80:6072–6083. doi: 10.1128/JVI.02495-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bellecave P., Sarasin-Filipowicz M., Donze O., Kennel A., Gouttenoire J., Meylan E. Cleavage of mitochondrial antiviral signaling protein in the liver of patients with chronic hepatitis C correlates with a reduced activation of the endogenous interferon system. Hepatology. 2010;51:1127–1136. doi: 10.1002/hep.23426. [DOI] [PubMed] [Google Scholar]
- 97.Nitta S., Sakamoto N., Nakagawa M., Kakinuma S., Mishima K., Kusano-Kitazume A. Hepatitis C virus NS4B protein targets STING and abrogates RIG-I-mediated type I interferon-dependent innate immunity. Hepatology. 2013;57:46–58. doi: 10.1002/hep.26017. [DOI] [PubMed] [Google Scholar]
- 98.Ding Q., Cao X., Lu J., Huang B., Liu Y.J., Kato N. Hepatitis C virus NS4B blocks the interaction of STING and TBK1 to evade host innate immunity. J. Hepatol. 2013;59:52–58. doi: 10.1016/j.jhep.2013.03.019. [DOI] [PubMed] [Google Scholar]
- 99.Li K., Foy E., Ferreon J.C., Nakamura M., Ferreon A.C., Ikeda M. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc. Natl. Acad. Sci. U.S.A. 2005;102:2992–2997. doi: 10.1073/pnas.0408824102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chen Y., Chen J., Wang H., Shi J., Wu K., Liu S. HCV-induced miR-21 contributes to evasion of host immune system by targeting MyD88 and IRAK1. PLoS Pathog. 2013;9:e1003248. doi: 10.1371/journal.ppat.1003248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Abe T., Kaname Y., Hamamoto I., Tsuda Y., Wen X., Taguwa S. Hepatitis C virus nonstructural protein 5A modulates the toll-like receptor-MyD88-dependent signaling pathway in macrophage cell lines. J. Virol. 2007;81:8953–8966. doi: 10.1128/JVI.00649-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sato K., Ishikawa T., Okumura A., Yamauchi T., Sato S., Ayada M. Expression of Toll-like receptors in chronic hepatitis C virus infection. J. Gastroenterol. Hepatol. 2007;22:1627–1632. doi: 10.1111/j.1440-1746.2006.04783.x. [DOI] [PubMed] [Google Scholar]
- 103.Motavaf M., Noorbakhsh F., Alavian S.M., Sharifi Z. Distinct toll-like receptor 3 and 7 expression in peripheral blood mononuclear cells from patients with chronic hepatitis C infection. Hepat. Mon. 2014;14:e16421. doi: 10.5812/hepatmon.16421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yang Q., Fu S., Wang J. Hepatitis C virus infection decreases the expression of Toll-like receptors 3 and 7 via upregulation of miR-758. Arch. Virol. 2014;159:2997–3003. doi: 10.1007/s00705-014-2167-3. [DOI] [PubMed] [Google Scholar]
- 105.Takahashi K., Asabe S., Wieland S., Garaigorta U., Gastaminza P., Isogawa M. Plasmacytoid dendritic cells sense hepatitis C virus-infected cells, produce interferon, and inhibit infection. Proc. Natl. Acad. Sci. U.S.A. 2010;107:7431–7436. doi: 10.1073/pnas.1002301107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Alter H.J., Seeff L.B. Recovery, persistence, and sequelae in hepatitis C virus infection: a perspective on long-term outcome. Semin. Liver Dis. 2000;20:17–35. doi: 10.1055/s-2000-9505. [DOI] [PubMed] [Google Scholar]
- 107.Dill M.T., Duong F.H., Vogt J.E., Bibert S., Bochud P.Y., Terracciano L. Interferon-induced gene expression is a stronger predictor of treatment response than IL28B genotype in patients with hepatitis C. Gastroenterology. 2011;140:1021–1031. doi: 10.1053/j.gastro.2010.11.039. [DOI] [PubMed] [Google Scholar]
- 108.Chattergoon M.A., Latanich R., Quinn J., Winter M.E., Buckheit R.W., 3rd, Blankson J.N. HIV and HCV activate the inflammasome in monocytes and macrophages via endosomal Toll-like receptors without induction of type 1 interferon. PLoS Pathog. 2014;10:e1004082. doi: 10.1371/journal.ppat.1004082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yue M., Feng L., Tang S.D., Wang J.J., Xue X.X., Ding W.L. Sex-specific association between X-linked Toll-like receptor 7 with the outcomes of hepatitis C virus infection. Gene. 2014;548:244–250. doi: 10.1016/j.gene.2014.07.040. [DOI] [PubMed] [Google Scholar]
- 110.Arnaud N., Dabo S., Akazawa D., Fukasawa M., Shinkai-Ouchi F., Hugon J. Hepatitis C virus reveals a novel early control in acute immune response. PLoS Pathog. 2011;7:e1002289. doi: 10.1371/journal.ppat.1002289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Eisele E., Siliciano R.F. Redefining the viral reservoirs that prevent HIV-1 eradication. Immunity. 2012;37:377–388. doi: 10.1016/j.immuni.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sgarbanti M., Battistini A. Therapeutics for HIV-1 reactivation from latency. Curr. Opin. Virol. 2013;3:394–401. doi: 10.1016/j.coviro.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 113.Battistini A., Sgarbanti M. HIV-1 latency: an update of molecular mechanisms and therapeutic strategies. Viruses. 2014;6:1715–1758. doi: 10.3390/v6041715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Marsili G., Remoli A.L., Sgarbanti M., Perrotti E., Fragale A., Battistini A. HIV-1, interferon and the interferon regulatory factor system: an interplay between induction, antiviral responses and viral evasion. Cytokine Growth Factor Rev. 2012;23:255–270. doi: 10.1016/j.cytogfr.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 115.Rustagi A., Gale M., Jr. Innate antiviral immune signaling, viral evasion and modulation by HIV-1. J. Mol. Biol. 2014;426:1161–1177. doi: 10.1016/j.jmb.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Iwasaki A. Innate immune recognition of HIV-1. Immunity. 2012;37:389–398. doi: 10.1016/j.immuni.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Douville R.N., Hiscott J. The interface between the innate interferon response and expression of host retroviral restriction factors. Cytokine. 2010;52:108–115. doi: 10.1016/j.cyto.2010.04.010. [DOI] [PubMed] [Google Scholar]
- 118.van Montfoort N., Olagnier D., Hiscott J. Unmasking immune sensing of retroviruses: interplay between innate sensors and host effectors. Cytokine Growth Factor Rev. 2014 doi: 10.1016/j.cytogfr.2014.08.006. [DOI] [PubMed] [Google Scholar]
- 119.Acchioni C., Marsili G., Perrotti E., Remoli A.L., Sgarbanti M., Battistini A. Type I IFN – a blunt spear in fighting HIV-1 infection. Cytokine Growth Factor Rev. 2015;26:143–158. doi: 10.1016/j.cytogfr.2014.10.004. [DOI] [PubMed] [Google Scholar]
- 120.Towers G.J., Noursadeghi M. Interactions between HIV-1 and the cell-autonomous innate immune system. Cell Host Microbe. 2014;16:10–18. doi: 10.1016/j.chom.2014.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Luban J. Innate immune sensing of HIV-1 by dendritic cells. Cell Host Microbe. 2012;12:408–418. doi: 10.1016/j.chom.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ryoo J., Choi J., Oh C., Kim S., Seo M., Kim S.Y. The ribonuclease activity of SAMHD1 is required for HIV-1 restriction. Nat. Med. 2014;20:936–941. doi: 10.1038/nm.3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sze A., Olagnier D., Lin R., van Grevenynghe J., Hiscott J. SAMHD1 host restriction factor: a link with innate immune sensing of retrovirus infection. J. Mol. Biol. 2013;425:4981–4994. doi: 10.1016/j.jmb.2013.10.022. [DOI] [PubMed] [Google Scholar]
- 124.Laguette N., Sobhian B., Casartelli N., Ringeard M., Chable-Bessia C., Segeral E. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature. 2011;474:654–657. doi: 10.1038/nature10117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hrecka K., Hao C., Gierszewska M., Swanson S.K., Kesik-Brodacka M., Srivastava S. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature. 2011;474:658–661. doi: 10.1038/nature10195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Manches O., Frleta D., Bhardwaj N. Dendritic cells in progression and pathology of HIV infection. Trends Immunol. 2014;35:114–122. doi: 10.1016/j.it.2013.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Gringhuis S.I., van der Vlist M., van den Berg L.M., den Dunnen J., Litjens M., Geijtenbeek T.B. HIV-1 exploits innate signaling by TLR8 and DC-SIGN for productive infection of dendritic cells. Nat. Immunol. 2010;11:419–426. doi: 10.1038/ni.1858. [DOI] [PubMed] [Google Scholar]
- 128.Solis M., Nakhaei P., Jalalirad M., Lacoste J., Douville R., Arguello M. RIG-I-mediated antiviral signaling is inhibited in HIV-1 infection by a protease-mediated sequestration of RIG-I. J. Virol. 2011;85:1224–1236. doi: 10.1128/JVI.01635-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Jakobsen M.R., Bak R.O., Andersen A., Berg R.K., Jensen S.B., Tengchuan J. IFI16 senses DNA forms of the lentiviral replication cycle and controls HIV-1 replication. Proc. Natl. Acad. Sci. U.S.A. 2013;110:E4571–E4580. doi: 10.1073/pnas.1311669110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yan N., Regalado-Magdos A.D., Stiggelbout B., Lee-Kirsch M.A., Lieberman J. The cytosolic exonuclease TREX1 inhibits the innate immune response to human immunodeficiency virus type 1. Nat. Immunol. 2010;11:1005–1013. doi: 10.1038/ni.1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Doitsh G., Galloway N.L., Geng X., Yang Z., Monroe K.M., Zepeda O. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature. 2014;505:509–514. doi: 10.1038/nature12940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lahaye X., Satoh T., Gentili M., Cerboni S., Conrad C., Hurbain I. The capsids of HIV-1 and HIV-2 determine immune detection of the viral cDNA by the innate sensor cGAS in dendritic cells. Immunity. 2013;39:1132–1142. doi: 10.1016/j.immuni.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 133.Rasaiyaah J., Tan C.P., Fletcher A.J., Price A.J., Blondeau C., Hilditch L. HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature. 2013;503:402–405. doi: 10.1038/nature12769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Chowell G., Nishiura H. Transmission dynamics and control of Ebola virus disease (EVD): a review. BMC Med. 2014;12:196. doi: 10.1186/s12916-014-0196-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wong G., Kobinger G.P., Qiu X. Characterization of host immune responses in Ebola virus infections. Expert Rev. Clin. Immunol. 2014;10:781–790. doi: 10.1586/1744666X.2014.908705. [DOI] [PubMed] [Google Scholar]
- 136.Bray M. The role of the Type I interferon response in the resistance of mice to filovirus infection. J. Gen. Virol. 2001;82:1365–1373. doi: 10.1099/0022-1317-82-6-1365. [DOI] [PubMed] [Google Scholar]
- 137.Basler C.F., Amarasinghe G.K. Evasion of interferon responses by Ebola and Marburg viruses. J. Interferon Cytokine Res. 2009;29:511–520. doi: 10.1089/jir.2009.0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ramanan P., Shabman R.S., Brown C.S., Amarasinghe G.K., Basler C.F., Leung D.W. Filoviral immune evasion mechanisms. Viruses. 2011;3:1634–1649. doi: 10.3390/v3091634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bale S., Julien J.P., Bornholdt Z.A., Kimberlin C.R., Halfmann P., Zandonatti M.A. Marburg virus VP35 can both fully coat the backbone and cap the ends of dsRNA for interferon antagonism. PLoS Pathog. 2012;8:e1002916. doi: 10.1371/journal.ppat.1002916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ramanan P., Edwards M.R., Shabman R.S., Leung D.W., Endlich-Frazier A.C., Borek D.M. Structural basis for Marburg virus VP35-mediated immune evasion mechanisms. Proc. Natl. Acad. Sci. U.S.A. 2012;109:20661–20666. doi: 10.1073/pnas.1213559109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Decker T., Muller M., Stockinger S. The yin and yang of type I interferon activity in bacterial infection. Nat. Rev. Immunol. 2005;5:675–687. doi: 10.1038/nri1684. [DOI] [PubMed] [Google Scholar]
- 142.Matsuura M. Structural modifications of bacterial lipopolysaccharide that facilitate gram-negative bacteria evasion of host innate immunity. Front. Immunol. 2013;4:109. doi: 10.3389/fimmu.2013.00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Paciello I., Silipo A., Lembo-Fazio L., Curcuru L., Zumsteg A., Noel G. Intracellular Shigella remodels its LPS to dampen the innate immune recognition and evade inflammasome activation. Proc. Natl. Acad. Sci. U.S.A. 2013;110:E4345–E4354. doi: 10.1073/pnas.1303641110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Sweet C.R., Conlon J., Golenbock D.T., Goguen J., Silverman N. YopJ targets TRAF proteins to inhibit TLR-mediated NF-kappaB, MAPK and IRF3 signal transduction. Cell. Microbiol. 2007;9:2700–2715. doi: 10.1111/j.1462-5822.2007.00990.x. [DOI] [PubMed] [Google Scholar]
- 145.Sanada T., Kim M., Mimuro H., Suzuki M., Ogawa M., Oyama A. The Shigella flexneri effector OspI deamidates UBC13 to dampen the inflammatory response. Nature. 2012;483:623–626. doi: 10.1038/nature10894. [DOI] [PubMed] [Google Scholar]
- 146.Yan D., Wang X., Luo L., Cao X., Ge B. Inhibition of TLR signaling by a bacterial protein containing immunoreceptor tyrosine-based inhibitory motifs. Nat. Immunol. 2012;13:1063–1071. doi: 10.1038/ni.2417. [DOI] [PubMed] [Google Scholar]
- 147.Sharma S., tenOever B.R., Grandvaux N., Zhou G.P., Lin R., Hiscott J. Triggering the interferon antiviral response through an IKK-related pathway. Science. 2003;300:1148–1151. doi: 10.1126/science.1081315. [DOI] [PubMed] [Google Scholar]
- 148.Fitzgerald K.A., McWhirter S.M., Faia K.L., Rowe D.C., Latz E., Golenbock D.T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003;4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- 149.Savitsky D., Tamura T., Yanai H., Taniguchi T. Regulation of immunity and oncogenesis by the IRF transcription factor family. Cancer Immunol. Immunother. 2010;59:489–510. doi: 10.1007/s00262-009-0804-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Battistini A. Nova Science Publishers, Inc.; 2012. Interferon Regulatory Factors in Immune Cell Development and Host Response to Infection. [Google Scholar]
- 151.Negishi H., Fujita Y., Yanai H., Sakaguchi S., Ouyang X., Shinohara M. Evidence for licensing of IFN-gamma-induced IFN regulatory factor 1 transcription factor by MyD88 in Toll-like receptor-dependent gene induction program. Proc. Natl. Acad. Sci. U.S.A. 2006;103:15136–15141. doi: 10.1073/pnas.0607181103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Dixit E., Boulant S., Zhang Y., Lee A.S., Odendall C., Shum B. Peroxisomes are signaling platforms for antiviral innate immunity. Cell. 2010;141:668–681. doi: 10.1016/j.cell.2010.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Schoggins J.W., Wilson S.J., Panis M., Murphy M.Y., Jones C.T., Bieniasz P. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature. 2011;472:481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Devaraj S.G., Wang N., Chen Z., Chen Z., Tseng M., Barretto N. Regulation of IRF-3-dependent innate immunity by the papain-like protease domain of the severe acute respiratory syndrome coronavirus. J. Biol. Chem. 2007;282:32208–32221. doi: 10.1074/jbc.M704870200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Yang X., Chen X., Bian G., Tu J., Xing Y., Wang Y. Proteolytic processing, deubiquitinase and interferon antagonist activities of Middle East respiratory syndrome coronavirus papain-like protease. J. Gen. Virol. 2014;95:614–626. doi: 10.1099/vir.0.059014-0. [DOI] [PubMed] [Google Scholar]
- 156.Liu Y.C., Penninger J., Karin M. Immunity by ubiquitylation: a reversible process of modification. Nat. Rev. Immunol. 2005;5:941–952. doi: 10.1038/nri1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Wilson J.R., de Sessions P.F., Leon M.A., Scholle F. West Nile virus nonstructural protein 1 inhibits TLR3 signal transduction. J. Virol. 2008;82:8262–8271. doi: 10.1128/JVI.00226-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Morrison C.R., Scholle F. Abrogation of TLR3 inhibition by discrete amino acid changes in the C-terminal half of the West Nile virus NS1 protein. Virology. 2014;456–457:96–107. doi: 10.1016/j.virol.2014.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Crook K.R., Miller-Kittrell M., Morrison C.R., Scholle F. Modulation of innate immune signaling by the secreted form of the West Nile virus NS1 glycoprotein. Virology. 2014;458–459:172–182. doi: 10.1016/j.virol.2014.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Anglero-Rodriguez Y.I., Pantoja P., Sariol C.A. Dengue virus subverts the interferon induction pathway via NS2B/3 protease-IkappaB kinase epsilon interaction. Clin. Vaccine Immunol. 2014;21:29–38. doi: 10.1128/CVI.00500-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Robertson S.J., Lubick K.J., Freedman B.A., Carmody A.B., Best S.M. Tick-borne flaviviruses antagonize both IRF-1 and type I IFN signaling to inhibit dendritic cell function. J. Immunol. 2014;192:2744–2755. doi: 10.4049/jimmunol.1302110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Otsuka M., Kato N., Moriyama M., Taniguchi H., Wang Y., Dharel N. Interaction between the HCV NS3 protein and the host TBK1 protein leads to inhibition of cellular antiviral responses. Hepatology. 2005;41:1004–1012. doi: 10.1002/hep.20666. [DOI] [PubMed] [Google Scholar]
- 163.Kaukinen P., Sillanpaa M., Nousiainen L., Melen K., Julkunen I. Hepatitis C virus NS2 protease inhibits host cell antiviral response by inhibiting IKKepsilon and TBK1 functions. J. Med. Virol. 2013;85:71–82. doi: 10.1002/jmv.23442. [DOI] [PubMed] [Google Scholar]
- 164.Inoue K., Tsukiyama-Kohara K., Matsuda C., Yoneyama M., Fujita T., Kuge S. Impairment of interferon regulatory factor-3 activation by hepatitis C virus core protein basic amino acid region 1. Biochem. Biophys. Res. Commun. 2012;428:494–499. doi: 10.1016/j.bbrc.2012.10.079. [DOI] [PubMed] [Google Scholar]
- 165.Oshiumi H., Sakai K., Matsumoto M., Seya T. DEAD/H BOX 3 (DDX3) helicase binds the RIG-I adaptor IPS-1 to up-regulate IFN-beta-inducing potential. Eur. J. Immunol. 2010;40:940–948. doi: 10.1002/eji.200940203. [DOI] [PubMed] [Google Scholar]
- 166.Oshiumi H., Ikeda M., Matsumoto M., Watanabe A., Takeuchi O., Akira S. Hepatitis C virus core protein abrogates the DDX3 function that enhances IPS-1-mediated IFN-beta induction. PLoS One. 2010;5:e14258. doi: 10.1371/journal.pone.0014258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Ciccaglione A.R., Stellacci E., Marcantonio C., Muto V., Equestre M., Marsili G. Repression of interferon regulatory factor 1 by hepatitis C virus core protein results in inhibition of antiviral and immunomodulatory genes. J. Virol. 2007;81:202–214. doi: 10.1128/JVI.01011-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Stone A.E., Mitchell A., Brownell J., Miklin D.J., Golden-Mason L., Polyak S.J. Hepatitis C virus core protein inhibits interferon production by a human plasmacytoid dendritic cell line and dysregulates interferon regulatory factor-7 and signal transducer and activator of transcription (STAT) 1 protein expression. PLoS One. 2014;9:e95627. doi: 10.1371/journal.pone.0095627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Remoli A.L., Marsili G., Sgarbanti M., Perrotti E., Fragale A., Orsatti R., Battistini A. HIV-1 targeting of IFN regulatory factors. Fut. Virol. 2011;6:1397–1405. [Google Scholar]
- 170.Doehle B.P., Hladik F., McNevin J.P., McElrath M.J., Gale M., Jr. Human immunodeficiency virus type 1 mediates global disruption of innate antiviral signaling and immune defenses within infected cells. J. Virol. 2009;83:10395–10405. doi: 10.1128/JVI.00849-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Okumura A., Alce T., Lubyova B., Ezelle H., Strebel K., Pitha P.M. HIV-1 accessory proteins VPR and Vif modulate antiviral response by targeting IRF-3 for degradation. Virology. 2008;373:85–97. doi: 10.1016/j.virol.2007.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Doehle B.P., Chang K., Fleming L., McNevin J., Hladik F., McElrath M.J. Vpu-deficient HIV strains stimulate innate immune signaling responses in target cells. J. Virol. 2012;86:8499–8506. doi: 10.1128/JVI.00424-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Park S.Y., Waheed A.A., Zhang Z.R., Freed E.O., Bonifacino J.S. HIV-1 Vpu induces caspase-mediated cleavage of IRF3. J. Biol. Chem. 2014 doi: 10.1074/jbc.M114.597062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Gougeon M.L. Apoptosis as an HIV strategy to escape immune attack. Nat. Rev. Immunol. 2003;3:392–404. doi: 10.1038/nri1087. [DOI] [PubMed] [Google Scholar]
- 175.Harman A.N., Lai J., Turville S., Samarajiwa S., Gray L., Marsden V. HIV infection of dendritic cells subverts the IFN induction pathway via IRF-1 and inhibits type 1 IFN production. Blood. 2011;118:298–308. doi: 10.1182/blood-2010-07-297721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Sgarbanti M., Remoli A.L., Marsili G., Ridolfi B., Borsetti A., Perrotti E. IRF-1 is required for full NF-kappaB transcriptional activity at the human immunodeficiency virus type 1 long terminal repeat enhancer. J. Virol. 2008;82:3632–3641. doi: 10.1128/JVI.00599-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Sgarbanti M., Borsetti A., Moscufo N., Bellocchi M.C., Ridolfi B., Nappi F. Modulation of human immunodeficiency virus 1 replication by interferon regulatory factors. J. Exp. Med. 2002;195:1359–1370. doi: 10.1084/jem.20010753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Remoli A.L., Marsili G., Perrotti E., Gallerani E., Ilari R., Nappi F. Intracellular HIV-1 Tat protein represses constitutive LMP2 transcription increasing proteasome activity by interfering with the binding of IRF-1 to STAT1. Biochem. J. 2006;396:371–380. doi: 10.1042/BJ20051570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Sgarbanti M., Marsili G., Remoli A.L., Stellacci E., Mai A., Rotili D. IkappaB kinase epsilon targets interferon regulatory factor 1 in activated T lymphocytes. Mol. Cell. Biol. 2014;34:1054–1065. doi: 10.1128/MCB.01161-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Basler C.F., Mikulasova A., Martinez-Sobrido L., Paragas J., Muhlberger E., Bray M. The Ebola virus VP35 protein inhibits activation of interferon regulatory factor 3. J. Virol. 2003;77:7945–7956. doi: 10.1128/JVI.77.14.7945-7956.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Prins K.C., Cardenas W.B., Basler C.F. Ebola virus protein VP35 impairs the function of interferon regulatory factor-activating kinases IKKepsilon and TBK-1. J. Virol. 2009;83:3069–3077. doi: 10.1128/JVI.01875-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Prins K.C., Binning J.M., Shabman R.S., Leung D.W., Amarasinghe G.K., Basler C.F. Basic residues within the ebolavirus VP35 protein are required for its viral polymerase cofactor function. J. Virol. 2010;84:10581–10591. doi: 10.1128/JVI.00925-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Chang T.H., Kubota T., Matsuoka M., Jones S., Bradfute S.B., Bray M. Ebola Zaire virus blocks type I interferon production by exploiting the host SUMO modification machinery. PLoS Pathog. 2009;5:e1000493. doi: 10.1371/journal.ppat.1000493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Kubota T., Matsuoka M., Chang T.H., Tailor P., Sasaki T., Tashiro M. Virus infection triggers SUMOylation of IRF3 and IRF7, leading to the negative regulation of type I interferon gene expression. J. Biol. Chem. 2008;283:25660–25670. doi: 10.1074/jbc.M804479200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Negishi H., Matsuki K., Endo N., Sarashina H., Miki S., Matsuda A. Beneficial innate signaling interference for antibacterial responses by a Toll-like receptor-mediated enhancement of the MKP-IRF3 axis. Proc. Natl. Acad. Sci. U.S.A. 2013;110:19884–19889. doi: 10.1073/pnas.1320145110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Minakshi R., Padhan K., Rani M., Khan N., Ahmad F., Jameel S. The SARS Coronavirus 3a protein causes endoplasmic reticulum stress and induces ligand-independent downregulation of the type 1 interferon receptor. PLoS One. 2009;4:e8342. doi: 10.1371/journal.pone.0008342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Li S.W., Lai C.C., Ping J.F., Tsai F.J., Wan L., Lin Y.J. Severe acute respiratory syndrome coronavirus papain-like protease suppressed alpha interferon-induced responses through downregulation of extracellular signal-regulated kinase 1-mediated signalling pathways. J. Gen. Virol. 2011;92:1127–1140. doi: 10.1099/vir.0.028936-0. [DOI] [PubMed] [Google Scholar]
- 188.Frieman M., Yount B., Heise M., Kopecky-Bromberg S.A., Palese P., Baric R.S. Severe acute respiratory syndrome coronavirus ORF6 antagonizes STAT1 function by sequestering nuclear import factors on the rough endoplasmic reticulum/Golgi membrane. J. Virol. 2007;81:9812–9824. doi: 10.1128/JVI.01012-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Liu J., HuangFu W.C., Kumar K.G., Qian J., Casey J.P., Hamanaka R.B. Virus-induced unfolded protein response attenuates antiviral defenses via phosphorylation-dependent degradation of the type I interferon receptor. Cell Host Microbe. 2009;5:72–83. doi: 10.1016/j.chom.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Ambrose R.L., Mackenzie J.M. A conserved peptide in West Nile virus NS4A protein contributes to proteolytic processing and is essential for replication. J. Virol. 2011;85:11274–11282. doi: 10.1128/JVI.05864-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Best S.M., Morris K.L., Shannon J.G., Robertson S.J., Mitzel D.N., Park G.S. Inhibition of interferon-stimulated JAK-STAT signaling by a tick-borne flavivirus and identification of NS5 as an interferon antagonist. J. Virol. 2005;79:12828–12839. doi: 10.1128/JVI.79.20.12828-12839.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Park G.S., Morris K.L., Hallett R.G., Bloom M.E., Best S.M. Identification of residues critical for the interferon antagonist function of Langat virus NS5 reveals a role for the RNA-dependent RNA polymerase domain. J. Virol. 2007;81:6936–6946. doi: 10.1128/JVI.02830-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Munoz-Jordan J.L., Laurent-Rolle M., Ashour J., Martinez-Sobrido L., Ashok M., Lipkin W.I. Inhibition of alpha/beta interferon signaling by the NS4B protein of flaviviruses. J. Virol. 2005;79:8004–8013. doi: 10.1128/JVI.79.13.8004-8013.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Mazzon M., Jones M., Davidson A., Chain B., Jacobs M. Dengue virus NS5 inhibits interferon-alpha signaling by blocking signal transducer and activator of transcription 2 phosphorylation. J. Infect. Dis. 2009;200:1261–1270. doi: 10.1086/605847. [DOI] [PubMed] [Google Scholar]
- 195.Ashour J., Laurent-Rolle M., Shi P.Y., Garcia-Sastre A. NS5 of dengue virus mediates STAT2 binding and degradation. J. Virol. 2009;83:5408–5418. doi: 10.1128/JVI.02188-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Morrison J., Laurent-Rolle M., Maestre A.M., Rajsbaum R., Pisanelli G., Simon V. Dengue virus co-opts UBR4 to degrade STAT2 and antagonize type I interferon signaling. PLoS Pathog. 2013;9:e1003265. doi: 10.1371/journal.ppat.1003265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Lin R.J., Chang B.L., Yu H.P., Liao C.L., Lin Y.L. Blocking of interferon-induced Jak-Stat signaling by Japanese encephalitis virus NS5 through a protein tyrosine phosphatase-mediated mechanism. J. Virol. 2006;80:5908–5918. doi: 10.1128/JVI.02714-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Werme K., Wigerius M., Johansson M. Tick-borne encephalitis virus NS5 associates with membrane protein scribble and impairs interferon-stimulated JAK-STAT signalling. Cell. Microbiol. 2008;10:696–712. doi: 10.1111/j.1462-5822.2007.01076.x. [DOI] [PubMed] [Google Scholar]
- 199.Zhang Q., Gong R., Qu J., Zhou Y., Liu W., Chen M. Activation of the Ras/Raf/MEK pathway facilitates hepatitis C virus replication via attenuation of the interferon-JAK-STAT pathway. J. Virol. 2012;86:1544–1554. doi: 10.1128/JVI.00688-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Heim M.H., Moradpour D., Blum H.E. Expression of hepatitis C virus proteins inhibits signal transduction through the Jak-STAT pathway. J. Virol. 1999;73:8469–8475. doi: 10.1128/jvi.73.10.8469-8475.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Bode J.G., Ludwig S., Ehrhardt C., Albrecht U., Erhardt A., Schaper F. IFN-alpha antagonistic activity of HCV core protein involves induction of suppressor of cytokine signaling-3. FASEB J. 2003;17:488–490. doi: 10.1096/fj.02-0664fje. [DOI] [PubMed] [Google Scholar]
- 202.Luquin E., Larrea E., Civeira M.P., Prieto J., Aldabe R. HCV structural proteins interfere with interferon-alpha Jak/STAT signalling pathway. Antiviral Res. 2007;76:194–197. doi: 10.1016/j.antiviral.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 203.Keskinen P., Melen K., Julkunen I. Expression of HCV structural proteins impairs IFN-mediated antiviral response. Virology. 2002;299:164–171. doi: 10.1006/viro.2002.1527. [DOI] [PubMed] [Google Scholar]
- 204.Xu J., Liu S., Xu Y., Tien P., Gao G. Identification of the nonstructural protein 4B of hepatitis C virus as a factor that inhibits the antiviral activity of interferon-alpha. Virus Res. 2009;141:55–62. doi: 10.1016/j.virusres.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 205.Duong F.H., Filipowicz M., Tripodi M., La Monica N., Heim M.H. Hepatitis C virus inhibits interferon signaling through up-regulation of protein phosphatase 2A. Gastroenterology. 2004;126:263–277. doi: 10.1053/j.gastro.2003.10.076. [DOI] [PubMed] [Google Scholar]
- 206.Horner S.M., Gale M., Jr. Intracellular innate immune cascades and interferon defenses that control hepatitis C virus. J. Interferon Cytokine Res. 2009;29:489–498. doi: 10.1089/jir.2009.0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Lan K.H., Lan K.L., Lee W.P., Sheu M.L., Chen M.Y., Lee Y.L. HCV NS5A inhibits interferon-alpha signaling through suppression of STAT1 phosphorylation in hepatocyte-derived cell lines. J. Hepatol. 2007;46:759–767. doi: 10.1016/j.jhep.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 208.Reid S.P., Valmas C., Martinez O., Sanchez F.M., Basler C.F. Ebola virus VP24 proteins inhibit the interaction of NPI-1 subfamily karyopherin alpha proteins with activated STAT1. J. Virol. 2007;81:13469–13477. doi: 10.1128/JVI.01097-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Mateo M., Reid S.P., Leung L.W., Basler C.F., Volchkov V.E. Ebolavirus VP24 binding to karyopherins is required for inhibition of interferon signaling. J. Virol. 2010;84:1169–1175. doi: 10.1128/JVI.01372-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Xu W., Edwards M.R., Borek D.M., Feagins A.R., Mittal A., Alinger J.B. Ebola virus VP24 targets a unique NLS binding site on karyopherin alpha 5 to selectively compete with nuclear import of phosphorylated STAT1. Cell Host Microbe. 2014;16:187–200. doi: 10.1016/j.chom.2014.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Zhang A.P., Bornholdt Z.A., Liu T., Abelson D.M., Lee D.E., Li S. The ebola virus interferon antagonist VP24 directly binds STAT1 and has a novel, pyramidal fold. PLoS Pathog. 2012;8:e1002550. doi: 10.1371/journal.ppat.1002550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Valmas C., Grosch M.N., Schumann M., Olejnik J., Martinez O., Best S.M. Marburg virus evades interferon responses by a mechanism distinct from ebola virus. PLoS Pathog. 2010;6:e1000721. doi: 10.1371/journal.ppat.1000721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Warnking K., Klemm C., Loffler B., Niemann S., van Kruchten A., Peters G. Super-infection with Staphylococcus aureus inhibits influenza virus-induced type I IFN signaling through impaired STAT1-STAT2 dimerization. Cell. Microbiol. 2014 doi: 10.1111/cmi.12375. [DOI] [PubMed] [Google Scholar]
- 214.Prabhakar S., Qiao Y., Hoshino Y., Weiden M., Canova A., Giacomini E. Inhibition of response to alpha interferon by Mycobacterium tuberculosis. Infect. Immun. 2003;71:2487–2497. doi: 10.1128/IAI.71.5.2487-2497.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Miettinen M., Lehtonen A., Julkunen I., Matikainen S. Lactobacilli and Streptococci activate NF-kappa B and STAT signaling pathways in human macrophages. J. Immunol. 2000;164:3733–3740. doi: 10.4049/jimmunol.164.7.3733. [DOI] [PubMed] [Google Scholar]
- 216.Schoggins J.W. Interferon-stimulated genes: roles in viral pathogenesis. Curr. Opin. Virol. 2014;6:40–46. doi: 10.1016/j.coviro.2014.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Zheng Y.H., Jeang K.T., Tokunaga K. Host restriction factors in retroviral infection: promises in virus–host interaction. Retrovirology. 2012;9:112. doi: 10.1186/1742-4690-9-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Yan N., Chen Z.J. Intrinsic antiviral immunity. Nat. Immunol. 2012;13:214–222. doi: 10.1038/ni.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Duggal N.K., Emerman M. Evolutionary conflicts between viruses and restriction factors shape immunity. Nat. Rev. Immunol. 2012;12:687–695. doi: 10.1038/nri3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Pfaller C.K., Li Z., George C.X., Samuel C.E. Protein kinase PKR and RNA adenosine deaminase ADAR1: new roles for old players as modulators of the interferon response. Curr. Opin. Immunol. 2011;23:573–582. doi: 10.1016/j.coi.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Garcia M.A., Meurs E.F., Esteban M. The dsRNA protein kinase PKR: virus and cell control. Biochimie. 2007;89:799–811. doi: 10.1016/j.biochi.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 222.Langland J.O., Cameron J.M., Heck M.C., Jancovich J.K., Jacobs B.L. Inhibition of PKR by RNA and DNA viruses. Virus Res. 2006;119:100–110. doi: 10.1016/j.virusres.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 223.Dauber B., Wolff T. Activation of the antiviral kinase PKR and viral countermeasures. Viruses. 2009;1:523–544. doi: 10.3390/v1030523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Arnaud N., Dabo S., Maillard P., Budkowska A., Kalliampakou K.I., Mavromara P. Hepatitis C virus controls interferon production through PKR activation. PLoS One. 2010;5:e10575. doi: 10.1371/journal.pone.0010575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Toroney R., Nallagatla S.R., Boyer J.A., Cameron C.E., Bevilacqua P.C. Regulation of PKR by HCV IRES RNA: importance of domain II and NS5A. J. Mol. Biol. 2010;400:393–412. doi: 10.1016/j.jmb.2010.04.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Garaigorta U., Chisari F.V. Hepatitis C virus blocks interferon effector function by inducing protein kinase R phosphorylation. Cell Host Microbe. 2009;6:513–522. doi: 10.1016/j.chom.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Thimme R., Binder M., Bartenschlager R. Failure of innate and adaptive immune responses in controlling hepatitis C virus infection. FEMS Microbiol. Rev. 2012;36:663–683. doi: 10.1111/j.1574-6976.2011.00319.x. [DOI] [PubMed] [Google Scholar]
- 228.Tu Y.C., Yu C.Y., Liang J.J., Lin E., Liao C.L., Lin Y.L. Blocking double-stranded RNA-activated protein kinase PKR by Japanese encephalitis virus nonstructural protein 2A. J. Virol. 2012;86:10347–10358. doi: 10.1128/JVI.00525-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Schumann M., Gantke T., Muhlberger E. Ebola virus VP35 antagonizes PKR activity through its C-terminal interferon inhibitory domain. J. Virol. 2009;83:8993–8997. doi: 10.1128/JVI.00523-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Feng Z., Cerveny M., Yan Z., He B. The VP35 protein of Ebola virus inhibits the antiviral effect mediated by double-stranded RNA-dependent protein kinase PKR. J. Virol. 2007;81:182–192. doi: 10.1128/JVI.01006-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Cardenas W.B., Loo Y.M., Gale M., Jr., Hartman A.L., Kimberlin C.R., Martinez-Sobrido L. Ebola virus VP35 protein binds double-stranded RNA and inhibits alpha/beta interferon production induced by RIG-I signaling. J. Virol. 2006;80:5168–5178. doi: 10.1128/JVI.02199-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Luthra P., Ramanan P., Mire C.E., Weisend C., Tsuda Y., Yen B. Mutual antagonism between the Ebola virus VP35 protein and the RIG-I activator PACT determines infection outcome. Cell Host Microbe. 2013;14:74–84. doi: 10.1016/j.chom.2013.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Clerzius G., Gelinas J.F., Gatignol A. Multiple levels of PKR inhibition during HIV-1 replication. Rev. Med. Virol. 2011;21:42–53. doi: 10.1002/rmv.674. [DOI] [PubMed] [Google Scholar]
- 234.Clerzius G., Shaw E., Daher A., Burugu S., Gelinas J.F., Ear T. The PKR activator, PACT, becomes a PKR inhibitor during HIV-1 replication. Retrovirology. 2013;10:96. doi: 10.1186/1742-4690-10-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Burugu S., Daher A., Meurs E.F., Gatignol A. HIV-1 translation and its regulation by cellular factors PKR and PACT. Virus Res. 2014 doi: 10.1016/j.virusres.2014.07.014. [DOI] [PubMed] [Google Scholar]
- 236.Morales D.J., Lenschow D.J. The antiviral activities of ISG15. J. Mol. Biol. 2013;425:4995–5008. doi: 10.1016/j.jmb.2013.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Zhao C., Collins M.N., Hsiang T.Y., Krug R.M. Interferon-induced ISG15 pathway: an ongoing virus-host battle. Trends Microbiol. 2013;21:181–186. doi: 10.1016/j.tim.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Shi H.X., Yang K., Liu X., Liu X.Y., Wei B., Shan Y.F. Positive regulation of interferon regulatory factor 3 activation by Herc5 via ISG15 modification. Mol. Cell. Biol. 2010;30:2424–2436. doi: 10.1128/MCB.01466-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Durfee L.A., Lyon N., Seo K., Huibregtse J.M. The ISG15 conjugation system broadly targets newly synthesized proteins: implications for the antiviral function of ISG15. Mol. Cell. 2010;38:722–732. doi: 10.1016/j.molcel.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Okumura A., Pitha P.M., Harty R.N. ISG15 inhibits Ebola VP40 VLP budding in an L-domain-dependent manner by blocking Nedd4 ligase activity. Proc. Natl. Acad. Sci. U.S.A. 2008;105:3974–3979. doi: 10.1073/pnas.0710629105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Okumura A., Lu G., Pitha-Rowe I., Pitha P.M. Innate antiviral response targets HIV-1 release by the induction of ubiquitin-like protein ISG15. Proc. Natl. Acad. Sci. U.S.A. 2006;103:1440–1445. doi: 10.1073/pnas.0510518103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Lindner H.A., Fotouhi-Ardakani N., Lytvyn V., Lachance P., Sulea T., Menard R. The papain-like protease from the severe acute respiratory syndrome coronavirus is a deubiquitinating enzyme. J. Virol. 2005;79:15199–15208. doi: 10.1128/JVI.79.24.15199-15208.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Clementz M.A., Chen Z., Banach B.S., Wang Y., Sun L., Ratia K. Deubiquitinating and interferon antagonism activities of coronavirus papain-like proteases. J. Virol. 2010;84:4619–4629. doi: 10.1128/JVI.02406-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Ratia K., Kilianski A., Baez-Santos Y.M., Baker S.C., Mesecar A. Structural basis for the ubiquitin-linkage specificity and deISGylating activity of SARS-CoV papain-like protease. PLoS Pathog. 2014;10:e1004113. doi: 10.1371/journal.ppat.1004113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Kim M.J., Yoo J.Y. Inhibition of hepatitis C virus replication by IFN-mediated ISGylation of HCV-NS5A. J. Immunol. 2010;185:4311–4318. doi: 10.4049/jimmunol.1000098. [DOI] [PubMed] [Google Scholar]
- 246.Broering R., Zhang X., Kottilil S., Trippler M., Jiang M., Lu M. The interferon stimulated gene 15 functions as a proviral factor for the hepatitis C virus and as a regulator of the IFN response. Gut. 2010;59:1111–1119. doi: 10.1136/gut.2009.195545. [DOI] [PubMed] [Google Scholar]
- 247.Chen L., Sun J., Meng L., Heathcote J., Edwards A.M., McGilvray I.D. ISG15, a ubiquitin-like interferon-stimulated gene, promotes hepatitis C virus production in vitro: implications for chronic infection and response to treatment. J. Gen. Virol. 2010;91:382–388. doi: 10.1099/vir.0.015388-0. [DOI] [PubMed] [Google Scholar]
- 248.Zhang X., Bogunovic D., Payelle-Brogard B., Francois-Newton V., Speer S.D., Yuan C. Human intracellular ISG15 prevents interferon-alpha/beta over-amplification and auto-inflammation. Nature. 2014 doi: 10.1038/nature13801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Li Y., Li S., Duan X., Liu B., Yang C., Zeng P. Activation of endogenous type I IFN signaling contributes to persistent HCV infection. Rev. Med. Virol. 2014;24:332–342. doi: 10.1002/rmv.1795. [DOI] [PubMed] [Google Scholar]
- 250.Dauphinee S.M., Richer E., Eva M.M., McIntosh F., Paquet M., Dangoor D. Contribution of increased ISG15, ISGylation and deregulated type I IFN signaling in Usp18 mutant mice during the course of bacterial infections. Genes Immun. 2014;15:282–292. doi: 10.1038/gene.2014.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Dorhoi A., Yeremeev V., Nouailles G., Weiner J., 3rd, Jorg S., Heinemann E. Type I IFN signaling triggers immunopathology in tuberculosis-susceptible mice by modulating lung phagocyte dynamics. Eur. J. Immunol. 2014;44:2380–2393. doi: 10.1002/eji.201344219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Ottenhoff T.H., Dass R.H., Yang N., Zhang M.M., Wong H.E., Sahiratmadja E. Genome-wide expression profiling identifies type 1 interferon response pathways in active tuberculosis. PloS One. 2012;7:e45839. doi: 10.1371/journal.pone.0045839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Berry M.P., Graham C.M., McNab F.W., Xu Z., Bloch S.A., Oni T. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature. 2010;466:973–977. doi: 10.1038/nature09247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Maertzdorf J., Weiner J., 3rd, Mollenkopf H.J., Bauer T., Prasse A., Muller-Quernheim J. Common patterns and disease-related signatures in tuberculosis and sarcoidosis. Proc. Natl. Acad. Sci. U.S.A. 2012;109:7853–7858. doi: 10.1073/pnas.1121072109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Chakravortty D., Hensel M. Inducible nitric oxide synthase and control of intracellular bacterial pathogens. Microbes Infect. 2003;5:621–627. doi: 10.1016/s1286-4579(03)00096-0. [DOI] [PubMed] [Google Scholar]
- 256.Panday A., Sahoo M.K., Osorio D., Batra S. NADPH oxidases: an overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2014 doi: 10.1038/cmi.2014.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Vazquez-Torres A., Xu Y., Jones-Carson J., Holden D.W., Lucia S.M., Dinauer M.C. Salmonella pathogenicity island 2-dependent evasion of the phagocyte NADPH oxidase. Science. 2000;287:1655–1658. doi: 10.1126/science.287.5458.1655. [DOI] [PubMed] [Google Scholar]
- 258.Vallance B.A., Deng W., De Grado M., Chan C., Jacobson K., Finlay B.B. Modulation of inducible nitric oxide synthase expression by the attaching and effacing bacterial pathogen Citrobacter rodentium in infected mice. Infect. Immun. 2002;70:6424–6435. doi: 10.1128/IAI.70.11.6424-6435.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Zust R., Dong H., Li X.F., Chang D.C., Zhang B., Balakrishnan T. Rational design of a live attenuated dengue vaccine: 2′-O-methyltransferase mutants are highly attenuated and immunogenic in mice and macaques. PLoS Pathog. 2013;9:e1003521. doi: 10.1371/journal.ppat.1003521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Malireddi R.K., Kanneganti T.D. Role of type I interferons in inflammasome activation, cell death, and disease during microbial infection. Front. Cell. Infect. Microbiol. 2013;3:77. doi: 10.3389/fcimb.2013.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]