

Abstract
During the last decade, the application of both qualitative and quantitative nucleic acid detection techniques has had a major impact on diagnostics in clinical virology. Both signal and target amplification‐based systems are currently used routinely in most if not all virology laboratories. However, commercial assays are only available for a very limited number of targets, and this has resulted in the development and introduction of assays developed in‐house for most viral targets. With improved and automated nucleic acid sample isolation techniques, as well as real‐time detection methods, a new generation of assays for most clinically important viruses is being developed. These technological improvements also make it possible to generate results with a very short turnaround time. As an example of a more individual‐patient disease‐management concept, we have introduced in our clinical setting the quantitative detection of Epstein–Barr virus (EBV) in T‐cell‐depleted allogeneic stem cell transplant patients. This has enabled us to develop models for pre‐emptive anti‐B‐cell immunotherapy for EBV reactivation, and for reducing not only the incidence of EBV lymphoproliferative disease (EBV‐LPD), but the virus‐related mortality. It is now also feasible to introduce molecular testing for those viruses that can easily be detected using classical virological methods, such as culture techniques or antigen detection. Prospective studies are needed to evaluate the clinical importance of the additional positive samples detected. It should, however, be clear that a complete exchange of technology is unlikely to occur, and that complementary methods should stay operational, making possible the discovery of new viruses. Furthermore, the ability to characterise viruses more easily by sequencing opens new possibilities for epidemiological studies. There is also an urgent need, with regard to molecular diagnostic methods, for the introduction and use of standardised materials and participation in international quality control programmes. Finally, with the introduction of a universal internal control throughout the whole procedure, the accuracy of the results generated is warranted.
Keywords: Disease management, quantitative DNA assays, real‐time detection, standardisation
Introduction
Advances have been made during the last decade making possible the sensitive detection and characterisation of viral nucleic acids. Amplification methods, such as PCR and Nucleic Acid Sequence‐Based Amplification (NASBA), in principal make possible the amplification of any target of interest. Technological improvements in sequence detection systems make it possible to characterise a virus fully, and to determine, for instance, the subtype, genotype, variant, mutant, and genotypic resistance patterns. The introduction of nucleic acid methods into routine diagnostics represents a great step forward, with the introduction of real‐time methods to detect amplified products easily. For an overview, see Mackay et al.[1] and Niesters [2]. This method is able to quantify target nucleic acids in a single sample over a larger dynamic range than most other quantitative methods, although qualitative detection is, of course, also possible. Since both amplification and detection can currently be performed automatically, the most labour‐intensive and critical step remaining is the efficient extraction of nucleic acids from different clinical samples.
A second advance in clinical virology is the increasing number of antiviral strategies available, not only for the commercially interesting viruses HIV, hepatitis B virus (HBV) and hepatitis C virus (HCV), but also for more ‘general’ viruses such as Epstein–Barr virus (EBV) and enteroviruses. These new antivirals can, however, only be used successfully in combination with an optimal diagnostic strategy and employed in a disease‐management context with an integrated approach combining diagnostics and treatment.
Bottlenecks in molecular diagnostics
There are still several problems that must be solved before molecular diagnostics can be routinely implemented (Table 1). First, it is recognised that commercially standardised assays for the clinical laboratory are available for only a limited number of targets (mainly HIV type 1, HBV and HCV, and, to a limited extent, cytomegalovirus (CMV) and human papilloma virus), and it is unlikely that the number of target viruses will increase rapidly in the near future. However, it should be emphasised that there is a large and growing list of viruses for which the implementation and use of nucleic acid methods is advantageous. In Table 2, a tentative list of ‘commercially interesting’ viruses vs. viral targets with limited commercial interest is given.
Table 1.
Bottlenecks in molecular diagnostics
| Problem identified | Possible solution |
|---|---|
| 1. Quantitative techniques | Real‐time amplification techniques |
| 2. Internal and external control | Well defined universal internal controls (PhHV, PDV) |
| Quality control programmes | |
| 3. Sample preparation still matrix dependent | New extraction techniques |
| 4. Hands‐on time and turnaround time results | Automation |
| 5. Commercially interesting viruses vs. viruses of no commercial interest | Well characterised home‐brew assays |
Table 2.
Commercially available assays vs. assays not commercially available for clinical virology: targets of interest
| Commercially interesting targets | Important in‐house viral targets to be developed |
|---|---|
| HIV type 1 | HIV type 2 |
| Hepatitis B virus | Epstein–Barr virus |
| Hepatitis C Virus | Herpes simplex virus types 1 and 2 |
| Cytomegalovirus | Cytomegalovirus |
| Human papilloma virus | Herpesvirus type 6 |
| Varicella zoster virus | |
| Enterovirus | |
| Rhinovirus | |
| Influenza virus | |
| Coronavirus | |
| Respiratory syncitial virus | |
| Human metapneumovirus | |
| Adenovirus | |
| Parvovirus B19 | |
| Astrovirus | |
| Calicivirus |
The second bottleneck is the easy and reproducible isolation of all nucleic acids (both RNA and DNA) from several clinical specimens. A fully automated system with high reliability is essential for successful implementation of nucleic acid detection in a day‐to‐day clinical virological setting. This will enable the laboratory to generate results with a short turnaround time, which is essential for disease management.
The third bottleneck concerns standardisation and quality control programmes. It has been known since the early 1990s that one of the greatest problems to overcome with molecular methods is false positivity caused by contamination, and false negativity associated with the great differences in sensitivity of, in most cases, home‐brew assays [3, 4, 5]. Furthermore, difficulties in detecting all genotypes of, for example, HIV‐1 or HCV, equally, with the same amplification efficiency, have also been reported [6, 7]. Programmes within Europe have been initiated, and have, indeed, shown that there is a need to obtain more standardised material and a need to participate in these programmes [8, 9]. A successor of one of these initiatives is called QCMD (Quality Control for Molecular Diagnostics: http://www.qcmd.org), endorsed by the European Society for Clinical Virology (ESCV) and the European Society of Clinical Microbiology and Infectious Diseases (ESCMID), which is starting to provide these quality control programmes for an increasing number of viral targets, whether commercially interesting or not. Besides the need for a well‐defined quality control programme, there is still a lack of standardisation of quantitative assays, mainly because standardisation is limited to a select number of blood‐borne viruses, such as HIV‐1, HBV and HCV, for which internationally accepted (WHO) standards are available [10, 11]. These standards are unfortunately only for single viral genotypes or subtypes, and in most cases are focused more specifically on blood bank screening and not on routine virological diagnostics. This is one of the problems that must be solved if we are to develop assays for the ‘non‐commercially interesting’ viral targets.
Universal internal controls
We have introduced the MagnaPure LC system (Roche Applied Science, Penzberg, Germany) in our laboratory as an open extraction system, initially for the extraction of RNA and DNA from serum and plasma. Combined with real‐time detection, whether TaqMan technology or NASBA amplification, this has enabled us to extract automatically and subsequently detect both RNA and DNA over a large dynamic range from a single clinical sample. To monitor this extraction process for loss or inhibition, we introduced the use of a universal internal viral control, which consists of a complete non‐human seal herpesvirus (a DNA virus, PhHV type 1). A real‐time and quantitative TaqMan assay was developed for this seal herpesvirus, which can be grown relatively easily in cell culture. The assumption is that a virus used as a universal control behaves more similarly in the extraction procedure to the target virus of interest than would, for example, a plasmid used as internal control. A low and fixed amount of this virus (equal to an amount giving a cycle threshold value in the real‐time assay of c. 30–33, depending on the lot number used) is added to each clinical sample (in our case, mostly serum or plasma) before the extraction procedure starts. This virus is coextracted and subsequently amplified in a quantitative manner, while still in a separate tube. This method monitors the combined effect of extraction (loss of sample) and amplification (inhibition), and enables us to be more confident about the quantitative results generated for the viral target of interest. In addition, negative results from clinical samples (or better, no target detected) can therefore be generated with high confidence. The data from the internal control are routinely imported into a spreadsheet program, with defined thresholds for approval or rejection of the final results. A typical example is shown in Fig. 1. Currently, multiplexing of the clinical sample with the internal control is under development. This strategy, furthermore, enables us to monitor every new approach, equipment or technique for extraction very rapidly, and enables us to check whether different lot numbers of reagents behave identically (or not).
Figure 1.
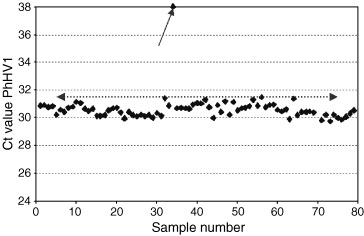
Data with the C t value of the amplification plot of seal herpes virus type 1 (PhHV‐1) added at a fixed concentration of approximately 5000–8000 copies/mL to each clinical sample. The results of a single experiment are shown, imported into a spreadsheet. The virus was coextracted using the MagnaPure LC isolation station, and quantified on an ABI7700 sequence detection system, using TaqMan universal reaction mixture (Applied Biosystems, Nieuwerkerk aan de Ijssel, The Netherlands). Primers and probe sequence were: forward primer 5′‐GGGCGAATCACAGATTGAATC, reverse primer 5′‐GCGGTTCCAAACGTACCAA, probe (labelled with TET) 5′‐TTTTTATGTGTCCGCCACCATCTGGATC. An amplification product of 89 bp within the gB gene is generated. The average C t value was 30.68 (average from 2359 samples analysed with this lot number), with an SD of 0.9. Samples in which the C t value for the internal control was > 32.5 (average plus 2 SD) had to be repeated (one in this run, see arrow).
External standards should also be more or less identical to the samples of interest, with the same or similar amplification efficiency as the clinical samples. The matrix in which the external control is extracted (if it is extracted at all) should be similar to the samples of interest. This implies that cloned material as an external control might be problematic.
Problems with standards and international standards have already been addressed above, but the conclusion is that absolute quantification is very hard to achieve, especially for those viral targets that would classify as not at present commercially interesting. Only international collaboration, in which diagnostic laboratories should take the lead, will enable us to tackle these problems. This does not, of course, exclude using real‐time methods to quantify viral targets, since this has been shown to give invaluable information on how viruses and their hosts interact.
Quantification of viral targets
With the recent development of real‐time methods and the detection of amplification products generated by PCR or NASBA methods, the quantification of both RNA and DNA in clinical samples has been made much easier. Also, the availability of ready‐to‐go reagents for both (RT)‐PCR and NASBA is a step forward. Numerous examples of detection and quantification of viral targets have been published already (for an overview, see Niesters [2]), and the list is growing rapidly. The whole process, from isolation to detection, can now be automated, but, for each target, the optimal strategy still needs to be established. It has to be determined which threshold values have clinical importance, or which detection levels need to be reached with each assay.
We have been involved in the development of a pre‐emptive strategy to reduce the incidence of EBV reactivation and the development of EBV‐related lymphomas (known as post‐transplant lymphoproliferative disease (PTLD)) in allogeneic T‐cell‐depleted stem cell transplant patients. A TaqMan‐based assay for the gene encoding the p143 BNRF protein has been developed and used to investigate the incidence of EBV reactivation and the development of EBV‐related PTLDs [12, 13, 14, 15]. Moreover, the assay is performed very frequently (currently five times a week), enabling us to determine the kinetics of EBV replication in our patient population. Several conclusions could be drawn, one of which was that each 10‐fold stepwise increase of EBV DNA measured in plasma yielded a hazard ratio of 2.9 (95% confidence interval 1.7–4.8) for developing PTLD for those patients receiving a T‐cell‐depleted graft (p < 0.001). Furthermore, it was shown that those patients receiving antithymocyte globulin (ATG) as part of their conditioning regimen belonged to the population with the highest risk, and that the T‐cell depletion itself was not the determinant. The problem now was to find at what viral load one should intervene with a pre‐emptive antiviral strategy, i.e., to identify precisely those patients at risk. A threshold of 1000 copies/mL in plasma was chosen as an indication to start treatment with rituximab, a monoclonal antibody directed against CD20, which is the receptor binding site for EBV. Although this resulted in overtreatment of patients who experienced EBV reactivation and most likely would not develop EBV PTLD, a reduction of EBV PTLD incidence and a complete abrogation of PTLD mortality at 6 months was observed [12]. An example of the kinetic pattern observed with EBV DNA measured in plasma is shown in Fig. 2. It can be seen that the EBV DNA viral load can increase very rapidly, making it necessary to perform the real‐time testing on a regular basis. Monitoring patients at risk, and follow‐up after the initiation of treatment, is thus frequently necessary.
Figure 2.
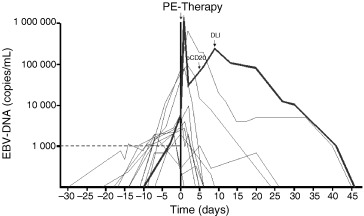
Pre‐emptive (PE) anti‐B‐cell immunotherapy for EBV. To monitor patients at risk for EBV PTLD, EBV DNA load was measured on a regular basis. At a level of 1000 copies/mL, the patients were recalled to the hospital, and PE therapy with rituximab (anti‐CD20 therapy) was initiated. Monitoring was performed five times a week, until the EBV DNA level was twice below the limit of detection of the assay (50 copies/mL). If the signal did not decrease rapidly (or an increase was observed), a second infusion of rituximab or an infusion of donor lymphocytes (DLI) was given (see data represented by a thick line). The initiation of PE therapy is represented as day 0. Each line represents an individual patient.
This strategy of monitoring patients at risk has also been used successfully for cytomegalovirus (CMV) [16, 17, 18]. Viral load kinetic patterns are used to identify patients who are more likely to have recurrence of CMV diseases after the initiation of antiviral therapy, as well as to identify patients who should be treated.
The precise description of the virus
Once we are able to detect and quantify in principal any virus of interest, the question remains whether a more precise characterisation of the isolated or detected virus is necessary for patient management. For the blood‐borne viruses, characterisation of the virus is becoming more or less a routine practice in clinical virology. For HIV, the genotypic resistance pattern, usually obtained by sequencing both the reverse transcriptase and protease genes, generates much information for the determination of optimal antiretroviral therapy. This is used mostly in combination with quantitative RNA data. For HCV, the genotype is important, combined again with the viral load, for the determination of the duration of therapy using interferon and ribavirin [19]. Also, for HBV, there are accumulating data indicating a relationship between genotype and the success of antiviral treatment [20].
Sequencing is, in principle, the ultimate method of characterisation. A disadvantage, however, is that, during antiviral treatment, a mixture of both wild‐type and variant virus exists. It has been observed that sequencing is not very suitable for the detection of minor populations of variant viruses [21]. Other methods, such as reverse hybridisation [22, 23, 24] or restriction fragment length polymorphism analysis [25, 26], might be more suitable, but it should be clear that they can only be successful if the sequence changes in a specific gene are known. Again, viral load measurements can be very helpful when searching for new variants during antiviral treatment, as has been recently described for the discovery of a new HBV variant resistant to lamivudine [26].
As sequencing technology is implemented in more laboratory settings, the question is whether it should be used for the characterisation of viral targets for which serotyping is currently used. It has already been shown that sequencing can be successful for the characterisation of enteroviruses and rhinoviruses, with results comparable to those obtained with serotyping [27, 28].
Is there still a place for virus culture?
The implementation of molecular methods has resulted not only in an improvement in diagnostics for a number of viruses, using the quantitative detection of nucleic acids and the monitoring of antiviral therapy, especially for HIV‐1, HBV and HCV, but also in the development of an amplification assay for almost every human virus, including those viruses that can be cultured relatively easily, e.g., the herpesviruses HSV types 1 and 2 [29]. These new molecular methods have several advantages, especially in those settings in which routine virus culture is not available. However, it should also be noted that improved sensitivity over virus culture is observed in almost all instances. The introduction of molecular diagnostics into routine clinical diagnostic virology is proceeding rapidly, and it will ultimately replace or reduce the use of virus culture techniques. The implementation of automated extraction and detection methods, combined with an extensive quality control programme, should ultimately convince both clinicians and virologists that molecular diagnostics is important in clinical virology. A critical remark often made is that not all positive results will have clinical significance, but this will be countered by good clinical and epidemiological information, combined with quantitative information on the virus(es) present. However, the ability to exclude viral infections has the advantage that unnecessary therapeutic options are not implemented, such as the wrong antivirals or antibiotics, which inevitably will reduce the overall costs of patient care. However, financial figures are needed to prove this. Thus, with improved accuracy and sensitivity, combined with a more rapid turnaround time, these molecular techniques are important for patient care, providing information to guide the best therapeutic options.
There is, of course, still a need for virus culture, as otherwise we will be unable to identify new viruses circulating, such as the recently described human metapneumovirus [30], but it is clear that a major transition from virus culture to molecular diagnostics is at hand.
Conclusions
The availability of open technologies to isolate and amplify nucleic acids makes possible not only the routine isolation of nucleic acids from an increasing range of clinical materials, but also, because of the use of real‐time detection systems, the whole process of semi‐automatic, quantitative detection of viral RNA and DNA targets. Furthermore, the risk of contamination is minimised, and the turnaround time for the generation of results, with minimal handling by personnel, whether positive or negative, is becoming shorter. Results can be generated easily within a working day, and it is expected that, in the near future, this will be reduced to hours.
Molecular techniques are becoming part of routine clinical virology, and the home‐brew assays are maturing, since standardised reagents and improved quality control programmes are enhancing laboratory performance. Most importantly, these standardised reagents are getting cheaper, making the discussion concerning high costs obsolete.
One of the greatest advantages, however, is the ability to detect more positive target sequences in clinical samples. This will not only give us insights into the relationship between virus and host, but, importantly, will also allow patients at risk to be treated more specifically. Molecular testing has been successful in those areas in which conventional virological techniques did not exist or could be improved. Currently, molecular testing is also available for those targets for which conventional technology is available. Logistics within the laboratory, price of assays, and clinical benefit will be the new criteria that will make these technological improvements a fundamental part of the new era in a diagnostic setting.
References
- 1. Mackay IM, Arden KE, Nitsche A. Real‐time PCR in virology. Nucleic Acids Res 2002; 30: 1292–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Niesters HGM. Quantitation of viral load using real‐time amplification techniques. Methods 2001; 25: 419–429. [DOI] [PubMed] [Google Scholar]
- 3. Quint WGV, Heijtink RA, Schirm J, Gerlich WH, Niesters HGM. Reliability of methods for hepatitis B virus DNA detection. J Clin Microbiol 1995; 33: 225–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schuurman R, Descamps D, Weverling GJ et al. Multicenter comparison of three commercial methods for quantification of human immunodeficiency virus type 1 RNA in plasma. J Clin Microbiol 1996; 34: 3016–3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zaaijer HL, Cuypers HT, Reesink HW, Winkel IN, Gerken G, Lelie PN. Reliability of polymerase chain reaction for detection of hepatitis C virus. Lancet 1993; 341: 722–724. [DOI] [PubMed] [Google Scholar]
- 6. Damen M, Cuypers HT, Zaaijer HL et al. International collaborative study on the second EUROHEP HCV‐RNA reference panel. J Virol Methods 1996; 58: 175–185. [DOI] [PubMed] [Google Scholar]
- 7. Zaaijer HL, Ter Borg F, Cuypers HT, Hermus MC, Lelie PN. Comparison of methods for detection of hepatitis B virus DNA. J Clin Microbiol 1994; 32: 2088–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Valentine‐Thon E, Van Loon AM, Schirm J, Reid J, Klapper PE, Cleator GM. European Proficiency Testing Program for molecular detection and quantitation of hepatitis B virus DNA. J Clin Microbiol 2001; 39: 4407–4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Van Vliet KE, Muir P, Echevarria JM, Klapper PE, Cleator GM, Van Loon AM. Multicenter proficiency testing of nucleic acid amplification methods for the detection of enteroviruses. J Clin Microbiol 2001; 39: 3390–3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Saldanha J, Lelie N, Heath A. Establishment of the first international standard for nucleic acid amplification technology (NAT) assays for HCV RNA. WHO Collaborative Study Group. Vox Sang 1999; 76: 149–158. [DOI] [PubMed] [Google Scholar]
- 11. Saldanha J, Gerlich W, Lelie N, Dawson P, Heermann K, Heath A. An international collaborative study to establish a World Health Organization international standard for hepatitis B virus DNA nucleic acid amplification techniques. Vox Sang 2001; 80: 63–71. [DOI] [PubMed] [Google Scholar]
- 12. Van Esser JW, Niesters HGM, Van Der Holt B et al. Prevention of Epstein–Barr virus‐lymphoproliferative disease by molecular monitoring and preemptive rituximab in high‐risk patients after allogeneic stem cell transplantation. Blood 2002; 99: 4364–4369. [DOI] [PubMed] [Google Scholar]
- 13. Van Esser JW, Van Der Holt B, Meijer E et al. Epstein–Barr virus (EBV) reactivation is a frequent event after allogeneic stem cell transplantation (SCT) and quantitatively predicts EBV‐lymphoproliferative disease following T‐cell‐depleted SCT. Blood 2001; 98: 972–978. [DOI] [PubMed] [Google Scholar]
- 14. Van Esser JW, Niesters HG, Thijsen SF et al. Molecular quantification of viral load in plasma allows for fast and accurate prediction of response to therapy of Epstein–Barr virus‐associated lymphoproliferative disease after allogeneic stem cell transplantation. Br J Haematol 2001; 113: 814–821. [DOI] [PubMed] [Google Scholar]
- 15. Niesters HG, Van Esser J, Fries E, Wolthers KC, Cornelissen JJ, Osterhaus AD. Development of a real‐time quantitative assay for detection of Epstein–Barr virus. J Clin Microbiol 2000; 38: 712–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Emery VC, Sabin CA, Cope AV, Gor D, Hassan‐Walker AF, Griffiths PD. Application of viral‐load kinetics to identify patients who develop cytomegalovirus disease after transplantation. Lancet 2000; 355: 2032–2036. [DOI] [PubMed] [Google Scholar]
- 17. Emery VC, Griffiths PD. Prediction of cytomegalovirus load and resistance patterns after antiviral chemotherapy. Proc Natl Acad Sci USA 2000; 97: 8039–8044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Humar AD, Kumar D, Boivin G, Caliendo AM. Cytomegalovirus (CMV) virus load kinetics to predict recurrent disease in solid‐organ transplant patients with CMV disease. J Infect Dis 2002; 186: 829–833. [DOI] [PubMed] [Google Scholar]
- 19. EASL. International Consensus Conference on hepatitis C. Consensus statement. J Hepatol 1999; 31(suppl 1): 3–8. [PubMed] [Google Scholar]
- 20. Sugauchi F, Orito E, Ichida T et al. Hepatitis B virus of genotype B with or without recombination with genotype C over the precore region plus the core gene. J Virol 2002; 76: 5985–5992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schuurman R, Demeter L, Reichelderfer P, Tijnagel J, De Groot T, Boucher C. Worldwide evaluation of DNA sequencing approaches for identification of drug resistance mutations in the human immunodeficiency virus type 1 reverse transcriptase. J Clin Microbiol 1999; 37: 2291–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stuyver L, Wyseur A, Rombout A et al. Line probe assay for rapid detection of drug‐selected mutations in the human immunodeficiency virus type 1 reverse transcriptase gene. Antimicrob Agents Chemother 1997; 41: 284–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stuyver L, Van Geyt C, De Gendt S et al. Line probe assay for monitoring drug resistance in hepatitis B virus‐infected patients during antiviral therapy. J Clin Microbiol 2000; 38: 702–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pas SD, De Man RA, Fries E, Osterhaus AD, Niesters HG. The dynamics of mutations in the YMDD motif of the hepatitis B virus polymerase gene during and after lamivudine treatment as determined by reverse hybridisation. J Clin Virol 2002; 25: 63–71. [DOI] [PubMed] [Google Scholar]
- 25. Allen MI, Gauthier J, Deslauriers M et al. Two sensitive PCR‐based methods for detection of hepatitis B virus variants associated with reduced susceptibility to lamivudine. J Clin Microbiol 1999; 37: 3338–3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Niesters HG, De Man RA, Pas SD, Fries E, Osterhaus AD. Identification of a new variant in the YMDD motif of the hepatitis B virus polymerase gene selected during lamivudine therapy. J Med Microbiol 2002; 51: 695–699. [DOI] [PubMed] [Google Scholar]
- 27. Savolainen CS, Blomqvist S, Mulders MN, Hovi T. Genetic clustering of all 102 human rhinovirus prototype strains: serotype 87 is close to human enterovirus 70. J Gen Virol 2002; 83: 333–340. [DOI] [PubMed] [Google Scholar]
- 28. Oberste MS, Maher K, Flemister MR, Marchetti G, Kilpatrick DR, Pallansch MA. Comparison of classic and molecular approaches for the identification of untypeable enteroviruses. J Clin Microbiol 2000; 38: 1170–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Espy MJ, Uhl JR, Mitchell PS et al. Diagnosis of herpes simplex virus infections in the clinical laboratory by LightCycler PCR. J Clin Microbiol 2000; 38: 795–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Van Den Hoogen BG, De Jong JC, Groen J et al. A newly discovered human pneumovirus isolated from young children with respiratory tract disease. Nat Med 2001; 7: 719–724. [DOI] [PMC free article] [PubMed] [Google Scholar]