

Abstract
Severe acute respiratory syndrome coronavirus (SARS-CoV) proteins belong to a large group of proteins that is difficult to express in traditional expression systems. The ability to express and purify SARS-CoV proteins in large quantities is critical for basic research and for development of pharmaceutical agents. The work reported here demonstrates: (1) fusion of SUMO (small ubiquitin-related modifier), a 100 amino acid polypeptide, to the N-termini of SARS-CoV proteins dramatically enhances expression in Escherichia coli cells and (2) 6× His-tagged SUMO-fusions facilitate rapid purification of the viral proteins on a large scale. We have exploited the natural chaperoning properties of SUMO to develop an expression system suitable for proteins that cannot be expressed by traditional methodologies. A unique feature of the system is the SUMO tag, which enhances expression, facilitates purification, and can be efficiently cleaved by a SUMO-specific protease to generate native protein with a desired N-terminus. We have purified various SARS-CoV proteins under either native or denaturing conditions. These purified proteins have been used to generate highly specific polyclonal antibodies. Our study suggests that the SUMO-fusion technology will be useful for enhancing expression and purification of the viral proteins for structural and functional studies as well as for therapeutic uses.
Keywords: SARS-CoV 3CL protease, SARS-CoV Nucleocapsid, SARS-CoV Spike protein, SUMO, SUMO-fusion system, SUMO protease, Protein expression, Ni–NTA affinity purification, Escherichia coli culture
Severe acute respiratory syndrome (SARS) 1 is a respiratory illness that has only recently been reported in Asia, North America, and Europe. After the first case of the disease in humans was found in Southern China late 2002, the outbreak spread quickly to about 35 countries on five continents, resulting in more than 8000 cases and 800 deaths. At present, there is no efficacious treatment regime for SARS. The need for both a reliable diagnostic assay and a therapeutic agent (antiviral or vaccine) is obvious. A previously unknown coronavirus has been identified as the causative agent of SARS. Scientists at the CDC and other laboratories determined the genomic sequence of this coronavirus and named it SARS-CoV [1], [2], [3].
Coronavirus, a genus within the family Coronaviridae, contains a group of large, positive stranded, enveloped, pathogenic RNA viruses that infect many species of animals, including humans. They cause respiratory, enteric, and central nervous system diseases [4]. The genomic sequence of SARS-CoV provides important information for the development of diagnostic tests and vaccines. This information affords the opportunity to express any SARS-CoV protein of choice for recombinant subunit vaccines. Development of protein-based diagnostic and therapeutic methods would be greatly facilitated by the ability to produce viral proteins of high quality in tractable amounts, which requires protein engineering, expression, and purification. Six proteins of SARS-CoV, namely Spike (S), Nucleocapsid (Nc), Envelope (E), SARS polymerase (RdRp), SARS protease (3CL), and membrane (M), have become the focus of efforts to produce antiviral agents and vaccines against SARS. The SARS-CoV proteins investigated in this study are described briefly below.
SARS-CoV 3CL protease (3CL, 3CLpro or Mpro) is the principal coronavirus protease utilized by the virus to process its replicase proteins into mature forms. The full length of the 3CL has 306 amino acids (molecular weight ∼33.8 kDa). The protease cleaves the replicase polyproteins (pp1a and pp1ab) to generate RNA-dependent polymerase (RdRp), 3CL, and helicase, all crucial for viral replication [5], [6], [7]. Therefore, 3CL represents an attractive target for the design and discovery of coronavirus antiviral agents, as does the polymerase [8]. SARS-CoV Nucleocapsid protein (N or Nc) is a phosphoprotein containing 423 amino acids (molecular weight ∼46 kDa) [9]. Large quantities of the protein are translated on free polysomes in the cytoplasm, where some molecules are rapidly phosphorylated. It is known that the protein binds the viral RNA and forms the nucleocapsid, but its exact mechanisms and role in replication are not yet clear. The Nc protein is known to have B and T cell epitopes and to elicit host protective immune responses [10], [11]. Spike protein (S or Spk) is a glycoprotein containing 1255 amino acids [12]. Upon translation, it is inserted into the rough endoplasmic reticulum and glycosylated with N-linked glycans [13]. Some of the proteins accumulate in the Golgi apparatus, and a fraction of oligomeric spike protein is transported to the membrane, where it mediates cell–cell fusion. Like those of other coronaviruses, the SARS-CoV spike protein likely contains many of the neutralizing antibody epitopes as well as T cell epitopes [14].
A supply of purified SARS-CoV proteins would be valuable for both clinical and investigational purposes. Although several strategies have been developed over the years to express heterologous recombinant proteins in bacterial, yeast, mammalian, and insect cells, the expression of heterologous genes in bacteria is by far the simplest and most inexpensive means available for research or commercial purposes. However, heterologous gene products often fail to attain their correct three-dimensional (3-D) conformation, or are simply expressed very poorly in Escherichia coli. Selection of ORFs for structural genomics projects has shown that only ∼20% of all heterologous genes expressed in E. coli render soluble or correctly folded proteins [15], [16]. Several gene-fusion systems, such as NusA, maltose binding protein (MBP), glutathione-S-transferase (GST), ubiquitin (UB), and thioredoxin (Trx), have been developed [17], [18]. All of these conventional methods have shortcomings, primarily inefficient expression and/or inconsistent cleavage.
Small ubiquitin-related modifier (SUMO) is a ubiquitin-related protein that functions by covalent attachment to other proteins. SUMO and its associated enzymes are present in all eukaryotes and are highly conserved from yeast to humans [19], [20], [21]. SUMO has 18% sequence identity with ubiquitin [22]. The yeast Saccharomyces cerevisiae has only a single SUMO gene (SMT3) that is essential for viability [20]. In contrast to yeast SMT3, three members of SUMO have been described in vertebrates: SUMO-1, SUMO-2, and SUMO-3. Human SUMO-1, a 101 amino acid polypeptide, shares 50% sequence identity with human SUMO-2/SUMO-3 [23], which are close homologues. Yeast SUMO shares 47% sequence identity with mammalian SUMO-1. Although overall sequence identity between ubiquitin and SUMO is only 18%, structure determination by NMR reveals that they share a common three-dimensional structure characterized by a tightly packed globular fold with β-sheets wrapped around a single α-helix [24], [25]. It is known that SUMO, fused at the N-terminus with other proteins, can fold and protect the protein by its chaperoning properties, making it a useful tag for heterologous expression [26]. All SUMO genes encode precursor proteins with a short C-terminal sequence that extends from the conserved C-terminal Gly–Gly motif. SUMO proteases remove SUMO from proteins, by cleaving the C-termini of SUMO (-GGATY) in yeast to the mature form (-GG) or deconjugating it from lysine side chains [27], [28]. The former activity (protease) is useful for removal of SUMO as an expression tag. There are 2 SUMO proteases in yeast [27], [28] and at least 6 in humans, the human enzymes ranging from 238 to 1112 amino acid residues [22], [29], [30], [31].
We have developed a novel SUMO-fusion system that provides increased levels of expression of heterologous proteins in E. coli and allows rapid purification of proteins of interest [26], [32]. We report here the application of SUMO-fusion technology to the expression and purification of major SARS-CoV proteins.
Materials and methods
SARS-CoV 3CL Protease (3CL), SARS-CoV Nucleocapsid (Nc), and SARS-CoV Spike C-terminal fragment protein (Spk C) were fused with SUMO and expressed in E. coli. For expression of the proteins, SARS-CoV cDNA was derived from infected cell RNA, provided by the CDC, Atlanta, to S.R.W. (University of Pennsylvania).
Construction of SUMO-SARS-CoV-fusion protein expression vectors
Expression constructs encoding the SUMO-fusion proteins all utilized the pSUMO plasmid (LifeSensors, Malvern, PA) as the backbone. The pET24 derivative carrying the SMT3 gene of S. cerevisiae, which encodes the yeast SUMO protein, has been described previously pSUMO [26]. It contains an N-terminal hexahistidine (6× His) tag, introduced by PCR into the SUMO coding sequence, as well as a unique BsaI site at the C-terminus. The cloning strategy to express fusion proteins employed this BsaI site to insert the SARS-CoV protein coding sequences in frame with SUMO. PCR primers (Table 1 ) incorporating this site or Esp 3I were used to amplify the SARS-CoV coding sequences from cDNA clones carried in pTOPO vectors. The 3′ primers carried a BamHI site for insertion into the multiple cloning site of pET24d. The primer pairs used to PCR amplify the SARS-CoV protein genes are listed in Table 1. Because of its large size, Spike protein was designed as two half-molecules, S1 (N-terminal fragment, amino acids 1–667, SpK N) and S2 (C-terminal fragment, amino acids 668–1193, SpK C) domains and the Spk C was tested for expression and purification in this study. For PCR amplification of the genes of interest, a proofreading polymerase was used (Platinum Taq, Invitrogen, Carlsbad, CA). PCR fragments were subcloned into pET24-6× His-SUMO or pET24-6× His (a parallel vector that does not carry the SUMO sequence) to produce parallel sets of constructs encoding 6× His-SUMO and 6× His fused versions of the proteins of interest. All plasmids were routinely sequenced.
Table 1.
PCR primers for amplifying the SARS-CoV protein genes
| Proteins | Region of genes | Primers |
|---|---|---|
| Spike protein—C terminal fragment | AA 668–1193 | tttGGTCTCaaggtatgagtactagccaaaaatctattgtggc |
| cgcGGATCCtcatttaatatattgctcatattttc | ||
| Nucleocapsid protein | Entire gene | tttGGTCTCaaggtatgtctgataatggaccccaatc |
| cgcGGATCCtcatgcctgagttgaatcagcag | ||
| CL Protease | Entire gene | tttCGTCTCaaggtagtggttttaggaaaatggcattcccg |
| cgcGGATCCtcattggaaggtaacaccagagc |
Restriction enzyme recognition sites used for cloning are indicated in uppercase letters. Owing to the presence of a BsaI site within the 3CL protease coding sequences, a different restriction enzyme, Esp3I, was used at the 5′ end to join the BsaI site in the pET-SUMO vector.
Expression of SARS proteins using SUMO-fusion
To test and compare expression of the SARS proteins, a single colony of the E. coli strain BL21 (DE3) containing each of the plasmids described above was inoculated into 5 ml of either Luria–Bertani (LB) or M9 minimal (MM) media. The antibiotic kanamycin was also included at 30 μg/ml in all media. The cells were grown at 37 °C overnight with shaking at 250 rpm. The next morning the overnight culture was transferred into 50 ml fresh medium to permit exponential growth. When the OD600 value reached ∼0.6–0.7, protein expression was induced by addition of 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside), followed by prolonged growth at either 37 or 20 °C to determine optimal induction conditions. For protein purification, cultures were scaled up to 0.5–1.0 L LB medium.
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) was used to verify expression of the protein. Briefly, 1.5 ml samples of culture were removed just before expression was induced and after induction, and cells were collected by centrifugation at 6000 rpm for 5 min. The cell pellets were suspended in 50 μl of distilled water, and the samples were freeze–thawed once to facilitate disruption of the cells. The cell suspensions were treated with RNAse and DNAse (both at 40 μg/ml) to digest nucleic acids. After mixing with SDS–PAGE sample buffer containing SDS and β-mercaptoethanol, samples were heated at 95 °C for 5 min to facilitate denaturation and reduction of proteins. Proteins were detected using SDS–polyacrylamide gels with Tris–glycine running buffer and Coomassie blue staining.
Western blots
Proteins separated by SDS–PAGE were transferred onto nitrocellulose membranes at 42 V (∼150 mA) for 2.5 h. Membranes were then incubated with 30 ml of TTBS buffer (pH 8.0), containing 5% nonfat dry milk for 1 h at room temperature. The expressed proteins were probed with either monoclonal anti-His-tag or polyclonal antibodies obtained from rabbits immunized against individual SUMO-SARS-CoV-fusion proteins (Rockland Immunochemicals) by incubating overnight at 4 °C with 1: 1000 dilution of the primary antibodies. After the membranes were washed with TTBS buffer for 5 min, they were incubated with a secondary antibody (Peroxidase-conjugated goat anti-rabbit IgG, Rockland Immunochemicals, diluted 1000-fold) for 45 min. The membranes were finally washed with TTBS for 10 min before the chemiluminescent Western blot substrates were applied (Roche, Mannheim, Germany), and visualized on films (Kodak BioMax).
Purification of SARS-CoV proteins
Because the SUMO constructs bear an N-terminal 6× His tag, expressed SARS-CoV proteins fused with SUMO can be rapidly purified by Ni–NTA affinity chromatography. In this study, the soluble proteins from E. coli cell lysates and the insoluble proteins from the cell inclusion bodies were purified under native and denaturing conditions, respectively. A typical procedure for purification of the SARS-CoV proteins is illustrated in Fig. 1 . Protein concentrations were determined using the Bradford color-reaction assay (Bio-Rad) measured spectrophotometrically at 595 nm with bovine serum albumin as a standard, according to the manufacturer’s instructions. SDS–PAGE and Coomassie blue staining were used to evaluate the effectiveness of the purifications and cleavage of SUMO-SARS-CoV protein fusions.
Fig. 1.
The procedure for purification of SARS-CoV proteins expressed with SUMO-fusion system in E. coli and cleavage of 6× His-SUMO-tagged proteins.
Preparation of soluble and insoluble protein samples from E. coli cells
The E. coli cells expressing the SARS-CoV proteins were harvested from LB medium (typically, 1.0 L) by centrifugation (8000g for 10 min at 4 °C). Typically, the wet weights of the E. coli cells harvested from 1 L culture were 10–15 g. The cell pellets were resuspended in lysis buffer (PBS containing additional 150 mM NaCl, 10 mM imidazole, 1% Triton X-100, and 1 mM PMSF, pH 8.0) at 3 ml for 1 g of the cells, resulting in ∼4 mg protein per ml after the proteins were extracted. The cells were lysed by sonication (50% output for 5 × 30 second pulses). Sonication was conducted with the tube jacketed in wet ice and observing 1 min intervals between pulse cycles to prevent heating. After the lysates were incubated with DNase and RNase (each at 40 μg/ml) for 20 min, they were centrifuged at 20,000g for 30 min at 4 °C, and supernatants (soluble protein fractions) were collected. The pellets containing inclusion bodies were washed three times in buffer (PBS containing 25% sucrose, 5 mM EDTA, and 1% Trition X-100, pH 7.5) followed by centrifugation, as described above. The washed inclusion bodies were resuspended in denaturing solubilization buffer (Novagen), which contained 50 mM Caps (pH 11.0), 0.3% N-lauryl sarcosine, and 1 mM DTT, and incubated for 30 min at room temperature with shaking to extract the insoluble proteins. Because debris from inclusion bodies was much smaller than that in the cell lysate, the extract for the insoluble proteins was obtained by high-speed centrifugation (80,000g for 30 min at 4 °C).
Purification of 6× His-tagged SUMO-SARS-CoV proteins
The soluble proteins extracted from E. coli cells were purified under native conditions and a BioLogic Duo-Flow FPLC system (Bio-Rad) was used for fractionations. Briefly, the cell lysate (typically, 20–40 ml containing 0.2–0.5 g proteins) was loaded onto a column containing ∼10 ml Ni–NTA superflow resin (Qiagen, Valencia, CA) and the samples of flow-through containing unbound proteins were collected for subsequent analysis. The resin was extensively washed with ∼50–100 ml of wash buffer (PBS containing 20 mM imidazole and additional 150 mM NaCl, pH 8.0) until OD280 reached or fell below the base line (UV value = 0). Finally, the 6× His-tagged SUMO-fusion proteins were eluted with elution buffer (PBS containing 300 mM imidazole and additional 150 mM NaCl, pH 8.0). The purified SUMO-fused proteins eluted as a single isolated UV peak. The proteins with high OD280 values were collected in 4 ml fractions that were checked on SDS-gels and pooled.
The insoluble proteins extracted from the E. coli inclusion bodies were purified under denaturing conditions, which were similar to the native conditions described above except for the use of highly alkaline pH buffer containing detergent. Briefly, an insoluble protein sample (∼20–40 ml) prepared in the denaturing buffer (50 mM Caps, 0.3% N-lauryl sarcosine, and 1 mM DTT, pH 11.0) was incubated with ∼10 ml of Ni–NTA superflow resin at 4 °C for 1 h with shaking for effective binding of the 6× His-tagged proteins to the resin. The mixture was then loaded into an empty column and the flow-through sample was collected. Subsequently, the resin was continually washed with denaturing wash buffer that contained 20 mM imidazole, 0.3% N-lauroyl sarcosine, 0.3 M NaCl, and 50 mM Caps, pH 11, until OD280 fell below the base line. The 6× His-tagged SUMO-fusion proteins were finally eluted using denaturing elution buffer that contained the same components as in the denaturing wash buffer, except that the concentration of imidazole was increased to 300 mM.
Cleavage of SUMO-fusion by the SUMO protease
The SUMO protease used in this study was produced in our laboratory as described [26], and a unit of the protease activity was defined as the amount of SUMO protease that cleaves 100 μg of SUMO-Met-GFP-fusion substrate at 25 °C in 1 h in buffer containing 20 mM Tris–HCl, pH 8.0, and 5 mM β-mercaptoethanol [26]. Before adding the enzyme for cleavage, the purified SUMO-fusion proteins (soluble fraction) were dialyzed with 3.5 kDa cutoff membranes against PBS (pH 7.4) for 12–15 h at 4 °C to remove high salt and imidazole, while the purified sample in denaturing buffer were refolded by extensive dialysis for at least 24 h against 20 mM Tris–HCl (pH 8.0) containing 10% glycerol. No protein precipitation was observed during the dialysis. The minimum amount of SUMO protease required for complete cleavage of a given SUMO-fusion was variable. Typically, for most of the purified SUMO-SARS-CoV proteins we added the enzyme at a ratio of 1 U to 15 μg substrates and incubated in either PBS (pH 7.4) or 20 mM Tris buffer (pH 8.0), containing 5 mM β-mercaptoethanol, at 30 °C for 1 h. In this study, cleavage of the SUMO-SARS-CoV Nc protein was achieved with a lower amount of the SUMO protease after checking effectiveness of the enzyme in serial dilution (see Fig. 8).
Fig. 8.
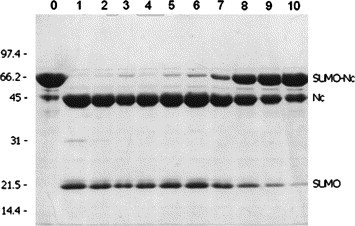
Effect of different amounts of SUMO protease on cleavage of SUMO-Nc-fusion. Serially diluted SUMO protease was added to 10 μg of purified SUMO-Nc-fusion protein and incubated in 30 μl PBS containing 5 mM β-mercaptoethanol at 30 °C for 1 h. The protease used was 20 U/μg. Aliquots (∼12 μl) from the incubation mixture were resolved on a 12% SDS–polyacrylamide gel and stained with Coomassie blue. Lanes: 0, uncleaved fusion (control); 1, 2 U of the protease; 2, 1 U; 3, 0.5 U; 4, 0.25 U; 5, 0.125 U; 6, 0.063 U; 7, 0.032 U; 8, 0.016 U; 9, 0.008 U; 10, 0.004 U.
Removal of SUMO and SUMO protease for final purification of SARS-CoV proteins
Since both SUMO and SUMO protease had 6× His tags, but SARS-CoV proteins did not, the cleaved SUMO-fusion samples could be re-applied to the nickel column to obtain the purified membrane proteins by subtracting the 6× His-tagged proteins. Briefly, after the SUMO-fusions were cleaved by the SUMO protease, the sample was loaded onto a nickel column with Ni–NTA resin. Most of the SARS-CoV protein without 6× His tags was eluted in the flow-through (unbound) fractions, and the rest was recovered by washing the resin with PBS. The eluted and washed proteins appearing in fractions with high-UV values at OD280 were pooled as the final purified sample. The purified proteins were checked on SDS-gels and the samples were stored at −80 °C after glycerol was added to 10%.
Results
Enhanced expression of SARS-CoV proteins with SUMO-fusion
SARS-CoV proteins 3CL, Nc, and Spike C, in versions fused to either 6× His-SUMO or 6× His, were expressed in E. coli cells under various conditions. The expressed proteins were readily identified by their migration positions in SDS-gels based on their molecular weights, and were further confirmed by immunological reactions with their respective antibodies on Western blots.
The expressed SARS-CoV 3CL protease (3CL) was detected in lysates of E. coli cells under several culture and induction conditions (Fig. 2 ); induced cell lysate samples showed appropriate protein bands (approximately 35 kDa for 3CL and 47 kDa for SUMO-3CL-fusion) on the SDS-gels (the sequence-predicted sizes of 3CL and SUMO-3CL are 33.8 and 45.8 kDa, respectively). When fused to the 3CL, SUMO significantly enhanced expression of its partner protein in both LB and MM media under all the conditions tested, compared to the 3CL expressed without SUMO-fusion (Fig. 2). Overnight growth (∼15 h) at 20 °C resulted in an increased yield of SUMO-fused 3CL compared to a 6 h culture at the same temperature and a 3 h culture at 37 °C (Fig. 2).
Fig. 2.
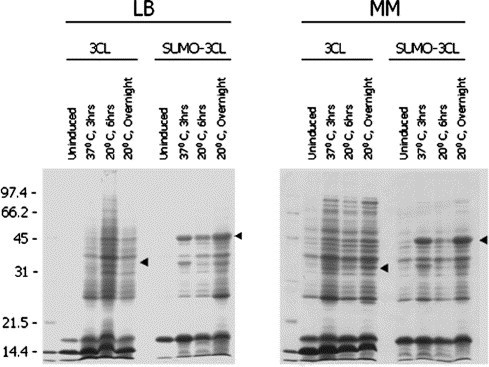
Enhanced expression of SARS-CoV 3CL protease (3CL) by SUMO-fusion in E. coli. Cells grown in either Luria–Bertani (LB) or M9 minimal (MM) medium were induced at the temperatures and for the lengths of time indicated. Just before expression was induced and after induction was completed the cells from a 1.5 ml aliquot of culture were lysed. Samples of whole cell lysates (∼7.5 μl) from the various expression conditions were resolved in 12% SDS-gels and stained with Coomassie blue. Molecular weights were as indicated, and arrowheads highlight expected/observed positions of respective expressed protein bands.
Expressed SARS-CoV Nucleocapsid (Nc) was detected in either unfused (∼46 kDa) or SUMO-fused (∼60 kDa) versions from IPTG-induced E. coli cells under various culture and induction conditions (Fig. 3 ). Notably, much higher yields of the expressed proteins were observed from rich medium (LB) than from minimal medium (MM) (Fig. 3), suggesting the former should be better for large-scale production and purification of the proteins. Similar to the 3CL results, expression enhancement was seen when Nc was fused to SUMO and expressed in minimal medium, but in LB medium there were no significant differences between the expression of Nc without SUMO and Nc fused with SUMO (Fig. 3).
Fig. 3.
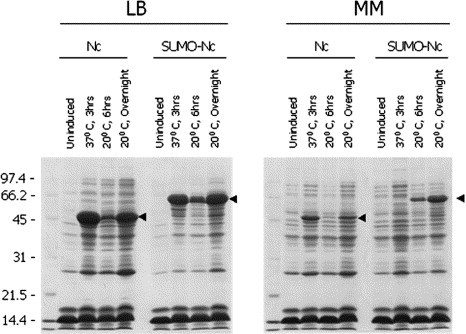
Enhanced expression of SARS-CoV Nucleocapsid protein (Nc) by SUMO-fusion in E. coli. The conditions for cell culture, protein expression, and gel detection were the same as in Fig. 2. The yields of expressed SUMO-Nc proteins were higher than the Nc expressed without SUMO in minimal media, but there were no significant differences in their expression in LB media.
The SUMO-fusion also greatly increased the level of expression of the C-terminal half of the SARS-CoV Spike protein (Spk C) compared to that of unfused Spk C in LB media (Fig. 4 ). Only a very weak protein band (∼58 kDa) of unfused Spk C could be seen in the SDS-gel and no band was seen in the Western blot probed with anti-His-tag antibodies, indicating that Spk C was poorly expressed without SUMO-fusion under the conditions tested. In contrast, an intense protein band was observed at the SUMO-Spk C migration position (∼68 kDa) on the SDS-gel (Fig. 4, left panel) when SpK C was fused with SUMO and the identity of the fusion protein was confirmed by reactions with anti-His-tag antibody (Fig. 4, right panel).
Fig. 4.
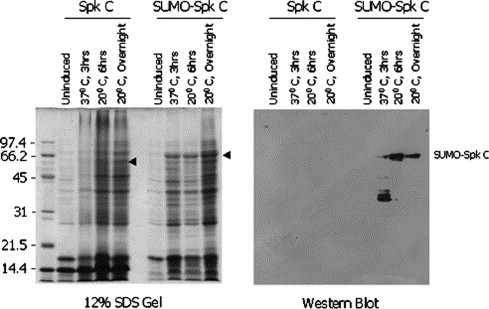
Enhanced expression of SARS-CoV Spike C protein (Spk C) by SUMO-fusion in E. coli. The cells were cultured in LB media and the conditions for cell culture, protein expression, and gel detection were the same as in Fig. 2. The Western blot (right panel) was performed with anti-His-tag primary antibody.
Purification of SARS proteins
Purification of SARS-CoV 3CL protease
Fig. 5 shows detection of the proteins from a representative purification of soluble SARS-CoV 3CL under native conditions. The cell lysate containing soluble SUMO-3CL was used for this purification, because a majority of the expressed protein (>80%) was present in the soluble fraction (data not shown). Proteins without 6× His tags were removed from the Ni–NTA resin using wash buffer containing 20 mM imidazole, and the 6× His-tagged SUMO-3CL-fusion was eluted using elution buffer containing 300 mM imidazole. After the SUMO-3CL fractions were pooled, the sample was dialyzed extensively against PBS (pH 7.4) at 4 °C to remove high salt and imidazole, which would interfere with the cleavage reaction. The SUMO-fusion was cleaved by addition of SUMO protease at 30 °C for 1 h under the conditions described in Materials and methods. The completeness of cleavage was confirmed by checking the proteins on a 12% SDS-gel, since the band of the SUMO-3CL disappeared and two new bands corresponding to the expected molecular weights of SUMO and 3CL were detected. After the cleaved sample was re-applied to a Ni–NTA column to subtract 6× His-tagged SUMO and SUMO protease, final purified 3CL was obtained (Fig. 5); the protein from the subtracted sample ran as a single, intense band (∼34 kDa), indicating that 3CL had been purified successfully (>95% purity). In this experiment, a high yield (totally ∼56 mg) of the pure 3CL was achieved from 1 L of E. coli cultured and induced at 20 °C overnight (Table 2 ). We used the anti-SUMO-3CL-fusion antibody to identify the purified 3CL protein, since the antibody could react with the SUMO-3CL-fusion and their cleaved partners. The purified protein was confirmed to be the SARS-CoV 3CL by the Western blot probed with the anti-SUMO-3CL antibody (see below and Fig. 7).
Fig. 5.
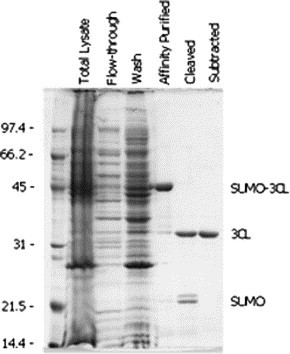
Detection of proteins in samples from various steps of a typical purification of SARS-CoV 3CL protease. Aliquots of the samples (each containing ∼5 μg protein) were separated on a 12% SDS-gel and stained with Coomassie blue. The migration positions of the SUMO-fusion and the proteins resulting from the cleavage are as indicated.
Table 2.
Summary of the SARS-CoV proteins resulting from representative purifications of 1 L E. coli culture
| Proteins | 3CL | Nc | Spk C |
|---|---|---|---|
| Starting samples for purification | Soluble fraction (224 mg) | Soluble fraction (189 mg) | Insoluble fraction (66 mg) |
| Purified SUMO-fusions | 101 mg | 66 mg | 24 mg |
| Purified SARS-CoV proteins | 56 mg | 26 mg | 12 mg |
| Purity | >95% | >95% | ∼30% |
The SARS-CoV proteins fused with SUMO were expressed in E. coli and induced at 20 °C overnight. The wet weights of the cells harvested from 1 L of E. coli culture for the 3CL, Nc, and Spk C were 14, 13, and 10 g, respectively. The samples were prepared and purified as described in Materials and methods.
Fig. 7.
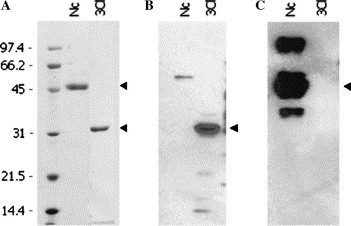
Detection of reactions of the purified Nc and 3CL proteins with the respective SUMO-fusion antibodies. The purified SARS-CoV proteins were detected on the Coomassie blue stained 12% SDS-gel (A) and the Western blots were probed with the SUMO-3CL antibody (B) and with the SUMO-Nc antibody (C). The amounts of the purified proteins loaded for the SDS-gel and the Western blots were 4 and 2 μg, respectively. Arrowheads highlight observed positions of respective SARS protein bands.
Purification of SARS-CoV Nucleocapsid protein
Similar to the SARS-CoV 3CL protease, most of the expressed SUMO-Nc protein was found in the soluble fraction from E. coli cells, and therefore the supernatant of the cell lysate was used for purification of the SARS-CoV Nc protein. The proteins resulting from various steps in the purification procedure were detected using SDS–PAGE (Fig. 6 ). Using Ni–NTA affinity to purify the 6× His-tagged SUMO-fusion was an efficient method, since only a single, high-density protein band was detected in the eluted fractions (Fig. 6). After the purified sample was dialyzed and the SUMO protease added under the conditions described above, complete cleavage of the fusion was achieved. A single, highly intense band (∼46 kDa) was detected in the final purified sample, indicating that >95% pure SARS-CoV Nc was obtained (Fig. 6). In this experiment, approximately 26 mg of the Nc was purified from the 1 L E. coli culture (Table 2). The protein’s identity was confirmed by its reaction with the anti-SUMO-Nc antibody (see Fig. 7 ).
Fig. 6.
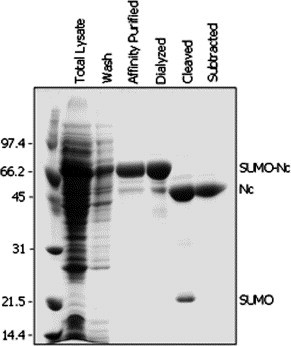
Detection of proteins from various steps of a typical purification of SAR-CoV Nucleocapsid proteins. Aliquots (∼5 μg proteins) of the samples were resolved in a 12% SDS-gel and stained with Coomassie blue. The migration positions of molecular weight markers, SUMO-fusion, and proteins resulting from the cleavage are as indicated.
Detection of the purified SARS-CoV 3CL and Nc proteins using Western blots
Fig. 7 shows that the SUMO-3CL-fusion antibody reacted specifically with the purified 3CL, with a little cross-reactivity with Nc; likewise, SUMO-Nc-fusion antibody had a highly specific reaction to purified Nc, without any cross-reaction with 3CL. The results not only confirmed the identities of the SARS-CoV proteins but also suggested that the purified SARS proteins maintained their immunity response properties.
Effects of variations in the amount SUMO protease on cleavage of SUMO-Nc-fusion proteins
To evaluate the effectiveness of SUMO protease on the cleavage of SUMO-SARS-CoV proteins, serial 1:1 dilutions of the enzyme (starting at 2.0 U) were used to digest aliquots (10 μg) of purified SUMO-Nc in PBS (pH 7.4) containing 5 mM β-mercaptoethanol at 30 °C for 1 h (Fig. 8 ). Since it is known that SUMO has a molecular mass of 11.5 kDa (although it migrates ∼20 kDa in an SDS–polyacrylamide gel), and the Nc band is ∼46 kDa, cleavage is judged to be successful if the protein band representing full-length substrate fusion (e.g., 20 + 46 = 66 kDa in the case of SUMO-Nc) disappears and new bands corresponding to the expected molecular weights of the hydrolysis products are detected. Fig. 8 shows that as little as 0.063 U of the enzyme cleaved >95% of 10 μg of SUMO-Nc-fusion (lane 6) and 0.008 U cleaved ∼50% of the substrate (Lane 9) under the tested conditions.
Purification of SARS-CoV Spike C protein
When fused with SUMO, the C-terminal half of SARS-CoV Spike protein (Spk C) was expressed at high levels in E. coli (Fig. 9 A). Because approximately 60% of the total fusion protein expressed was in the bacterial inclusion bodies, the insoluble protein sample extracted from the inclusion bodies (Fig. 9A, lane 3) was used for purification of the Spk C with Ni–NTA affinity chromatography under denaturing conditions. Briefly, the 6× His-tagged SUMO-Spike C-fusion was eluted by elution buffer containing 300 mM imidazole, but a few other minor proteins that were without 6× His tags but possibly rich in histidine and/or cysteine were also bound to the resin, resulting in impurities of the sample. The unwanted proteins did not interfere with the cleavage of the SUMO-fusion proteins, but reduced the purity of the sample (Fig. 9B, lane 1). After the purified SUMO-Spike C protein was extensively dialyzed, the fusion was effectively cleaved by addition of SUMO protease (>95% cleavage was achieved, see Fig. 9B, lane 2). Finally, the 6× His-tagged SUMO and SUMO protease were removed by applying the cleaved sample to the Ni–NTA column to purify the Spk C. An SDS-gel of the resulting sample showed unfused Spk C (∼58 kDa) along with three minor proteins (see Fig. 9B, lane 3), indicating that partially purified Spk C was obtained. Alternative purification approaches can be used after the Ni–NTA purification to get rid of the impurities if >90% purity is required. In this study, approximately 12 mg of the partially purified SpK C sample was obtained from the 1 L E. coli culture (Table 2).
Fig. 9.
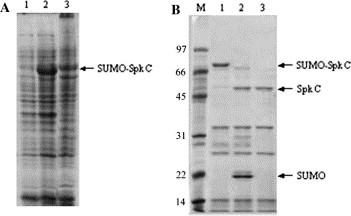
Detection of SUMO-SARS CoV Spike C proteins from expressed E. Coli cells (A) and various steps during a representative purification of Spike C proteins (B). Aliquots (∼ 5 μg protein) of samples were resolved in 12% SDS-gels and stained with Coomassie blue. Lanes in (A): 1, uninduced cells (control); 2, induced cells; 3, insoluble fraction (the starting material for purification). Lanes in (B): M, molecular weight markers; 1, purified SUMO-Spike C-fusion; 2, cleaved SUMO-Spike C-fusion; 3, purified Spike C sample. The migration positions of the respective proteins are as indicated.
Discussion
At least six types of protein are encoded by the SARS coronavirus (SARS-CoV) genome. Large-scale production of these proteins in pure, functionally active form is critical to meet urgent needs in the development of diagnostic and therapeutic methods for SARS, such as antiviral drugs and vaccines, as well as for basic research purposes. Such a task is difficult using conventional expression systems.
Several major protein fusion technologies have been developed to improve expression and purification of heterologous recombinant proteins in bacterial, yeast, mammalian, and insect cells. These include maltose binding protein (MBP), glutathione-S-transferase (GST), and thioredoxin (Trx) gene fusion systems [17], [33]. However, many proteins are not expressed well with these fusion systems in commonly utilized hosts. Fusion of an unstable or misfolded protein with proteins such as ubiquitin and ubiquitin-like proteins, which have a highly evolved structure, can stabilize the candidate protein. We have conducted a systematic comparison of the effectiveness of various fusion tags (MBP, GST, Trx, NusA, and SUMO) when used as GFP fusions expressed in E. coli, and have found SUMO to be superior to the other tags for expression of the protein.
GST and MBP domains have been used as tags to enhance production and purification of proteins of interest [33]. Problems are encountered, however, when these tags must be removed to study the protein’s structure by X-ray crystallography or NMR. Although several proteases such as thrombin, Factor Xa, and AcTEV protease are used for these purposes, all of these enzymes recognize short degenerate sequences, and, thus, cleavage can occur within the proteins of interest. Another problem encountered is inaccessibility of the cleavage site within the fusion due to steric constraints, which could reduce the effectiveness of enzymic cleavage. The SUMO tag, by contrast, is accurately and efficiently removed from the protein of interest [26]. Comparing the cleavage of SUMO-GFP by SUMO protease to the cleavage of NusA-GFP by AcTEV protease, we found that SUMO protease had a 64-fold higher activity than AcTEV protease when the same amount of enzyme was used (unpublished results).
Ubiquitin has been reported to exert chaperoning effects on fused proteins, thus increasing expression of proteins in E. coli and yeast [34], [35], [36]. The fused proteins can be cleaved by Ub-proteases (both UCH and UBP classes), but the enzymes are unstable, difficult to produce, and often must be used in large quantities (an enzyme to substrate ratio of 1:1), making this technology impractical for large-scale protein production [18]. Our laboratory has exploited the chaperoning properties of several ubiquitin-like proteins including SUMO, and the extreme robustness of SUMO protease has allowed us to develop a technology that provides both enhanced expression and cleavage of the fusion protein. A number of difficult proteins have been expressed in our laboratory in both unfused and SUMO-fused forms and compared side-by-side to demonstrate that SUMO-fusion dramatically enhances the expression of many types of proteins, including membrane proteins, and that SUMO protease cleaves a variety of SUMO-fusions with high specificity and efficiency over a wide range of various conditions, including pH (5.5–10.5) and temperature (4–37 °C) [26]. Non-specific cleavage of the substrate was not observed, even when the amount of enzyme was deliberately increased to a 1:1 ratio [26]. In this study, titration of the hydrolytic capacity of SUMO protease on the purified SARS-CoV Nc proteins confirmed that SUMO protease is an extremely potent enzyme (Fig. 8). The predicted molecular weight of the SUMO protease is 26.7 kDa, though it usually runs at ∼31 kDa position on a SDS-polyacrylamide gel.
After evaluating and comparing various SARS-CoV proteins expressed with or without SUMO-fusion in E. coli under several culture and induction conditions, we found that SUMO-fusion significantly increased expression of SARS proteins under nearly all conditions tested. We established a batch production protocol employing 20 °C overnight growth for large-scale expression of SUMO-SARS-CoV proteins, since a shorter time (e.g., 6 h) or higher temperature (37 °C) resulted in lower yields, especially for soluble proteins (data not shown). Although in most cases, cells growing in rich medium (LB) produced more SUMO-fused protein than cells growing in minimal medium (MM), we will use MM to investigate secreted SARS-CoV proteins in future studies, since rich medium contains a large number of interfering proteins.
In addition to producing SARS-CoV proteins in large quantities for basic research and for development of anti-SARS pharmaceutical agents, it is important to produce pure proteins that retain biological activity. The expressed and purified SARS-CoV proteins had immunological activity, but the question remains concerning their functional activities. It appears, at least in the case of one SARS-CoV protein, that SUMO-enhanced expression and purification from E. coli results in active protein. In a study to be published, SUMO-fusion enhanced expression of the SARS-CoV RNA-dependent RNA polymerase (RdRp), and the purified soluble RdRp was biologically active (unpublished results). Finally, we recently observed that SUMO-fusion significantly enhanced expression and purification of SARS-CoV membrane protein (M) as well. Using the SUMO-fusion technology described here, the expression level of SARS-CoV M protein in E. coli was greatly improved, and the insoluble proteins extracted from the bacterial inclusion bodies were purified [32]. Application of the various purified SARS-CoV proteins to the development of SARS vaccines and functional assays are underway.
Acknowledgments
The work described here was supported in part by a grant (R43 GM067271-01) from the NIH/NIGMS to T.R.B. and a grant (RO1-AI 17418) from NIH/NIAID to S.R.W.
Footnotes
Abbreviations used: SARS, severe acute respiratory syndrome; SARS-CoV, SARS coronavirus; DUB, deubiquitinating enzyme; IPTG, isopropyl-β-d-thiogalactopyranoside; Ni–NTA, nickel–nitrilotriacetic acid; SDS–PAGE, sodium dodecyl sulfate–polyacrylamide gel electrophoresis; Ub, ubiquitin; SUMO, small ubiquitin-related modifier; PMSF, phenylmethylsulfonyl fluoride; FPLC, fast performance liquid chromatography.
References
- 1.Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S., Urbani C., Comer J.A., Lim W., Rollin P.E., Dowell S.F., Ling A.E., Humphrey C.D., Shieh W.J., Guarner J., Paddock C.D., Rota P., Fields B., DeRisi J., Yang J.Y., Cox N., Hughes J.M., LeDuc J.W., Bellini W.J., Anderson L.J. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 2.Rota P.A., Oberste M.S., Monroe S.S., Nix W.A., Campagnoli R., Icenogle J.P., Penaranda S., Bankamp B., Maher K., Chen M.H., Tong S., Tamin A., Lowe L., Frace M., DeRisi J.L., Chen Q., Wang D., Erdman D.D., Peret T.C., Burns C., Ksiazek T.G., Rollin P.E., Sanchez A., Liffick S., Holloway B., Limor J., McCaustland K., Olsen-Rasmussen M., Fouchier R., Gunther S., Osterhaus A.D., Drosten C., Pallansch M.A., Anderson L.J., Bellini W.J. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 3.Snijder E., Bredenbeek P.J., Dobbe J.C., Thiel V., Ziebuhr L.L., Poon Y., Guan Y., Rozanov M., Spaan W.J., Gorbalenya A.E. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J. Mol. Biol. 2003;331:991–1004. doi: 10.1016/S0022-2836(03)00865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McIntosh K. Coronaviruses: a comparative review. Curr. Top. Microbiol. Immunol. 1974;63:85–129. [Google Scholar]
- 5.Herold J., Raabe T., Schelle-Prinz B., Siddell S.G. Nucleotide sequence of the human coronavirus 229E RNA polymerase locus. Virology. 1993;195:680–691. doi: 10.1006/viro.1993.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herold J., Raabe T., Siddell S. Molecular analysis of the human coronavirus (strain 229E) genome. Arch. Virol. Suppl. 1993;7:63–74. doi: 10.1007/978-3-7091-9300-6_6. [DOI] [PubMed] [Google Scholar]
- 7.Herold J., Raabe T., Siddell S.G. Characterization of the human coronavirus 229E (HCV 229E) gene 1. Adv. Exp. Med. Biol. 1993;342:75–79. doi: 10.1007/978-1-4615-2996-5_12. [DOI] [PubMed] [Google Scholar]
- 8.Xu X., Liu Y., Weiss S., Arnold E., Sarafianos S.G., Ding J. Molecular model of SARS coronavirus polymerase: implications for biochemical functions and drug design. Nucleic Acids Res. 2003;31:7117–7130. doi: 10.1093/nar/gkg916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Surjit M., Liu B., Kumar P., Chow V.T., Lal S.K. The nucleocapsid protein of the SARS coronavirus is capable of self-association through a C-terminal 209 amino acid interaction domain. Biochem. Biophys. Res. Commun. 2004;317:1030–1036. doi: 10.1016/j.bbrc.2004.03.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Y.D., Sin W.Y., Xu G.B., Yang H.H., Wong T.Y., Pang X.W., He X.Y., Zhang H.G., Ng J.N., Cheng C.S., Yu J., Meng L., Yang R.F., Lai S.T., Guo Z.H., Xie Y., Chen W.F. T-cell epitopes in severe acute respiratory syndrome (SARS) coronavirus spike protein elicit a specific T-cell immune response in patients who recover from SARS. J. Virol. 2004;78:5612–5618. doi: 10.1128/JVI.78.11.5612-5618.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiong S., Wang Y.F., Zhang M.Y., Liu X.J., Zhang C.H., Liu S.S., Qian C.W., Li J.X., Lu J.H., Wan Z.Y., Zheng H.Y., Yan X.G., Meng M.J., Fan J.L. Immunogenicity of SARS inactivated vaccine in BALB/c mice. Immunol. Lett. 2004;95:139–143. doi: 10.1016/j.imlet.2004.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xiao X., Chakraborti S., Dimitrov A.S., Gramatikoff K., Dimitrov D.S. The SARS-CoV S glycoprotein: expression and functional characterization. Biochem. Biophys. Res. Commun. 2003;312:1159–1164. doi: 10.1016/j.bbrc.2003.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwegmann-Wessels C., Al-Falah M., Escors D., Wang Z., Zimmer G., Deng H., Enjuanes L., Naim H.Y., Herrler G. A novel sorting signal for intracellular localization is present in the S protein of a porcine coronavirus but absent from severe acute respiratory syndrome-associated coronavirus. J. Biol. Chem. 2004;279:43661–43666. doi: 10.1074/jbc.M407233200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang H., Wang G., Li J., Nie Y., Shi X., Lian G., Wang W., Yin X., Zhao Y., Qu X., Ding M., Deng H. Identification of an antigenic determinant on the S2 domain of the severe acute respiratory syndrome coronavirus spike glycoprotein capable of inducing neutralizing antibodies. J. Virol. 2004;78:6938–6945. doi: 10.1128/JVI.78.13.6938-6945.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Waldo G.S., Standish B.M., Berendzen J., Terwilliger T.C. Rapid protein-folding assay using green fluorescent protein. Nat. Biotechnol. 1999;17:691–695. doi: 10.1038/10904. [DOI] [PubMed] [Google Scholar]
- 16.Wright L.C., Seybold J., Robichaud A., Adcock I.M., Barnes P.J. Phosphodiesterase expression in human epithelial cells. Am. J. Physiol. 1998;275:L694–L700. doi: 10.1152/ajplung.1998.275.4.L694. [DOI] [PubMed] [Google Scholar]
- 17.Nilsson J., Stahl S., Lundeberg J., Uhlen M., Nygren P.A. Affinity fusion strategies for detection, purification, and immobilization of recombinant proteins. Protein Expr. Purif. 1997;11:1–16. doi: 10.1006/prep.1997.0767. [DOI] [PubMed] [Google Scholar]
- 18.Catanzariti A.M., Soboleva T.A., Jans D.A., Board P.G., Baker R.T. An efficient system for high-level expression and easy purification of authentic recombinant proteins. Protein Sci. 2004;13:1331–1339. doi: 10.1110/ps.04618904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muller S., Hoege C., Pyrowolakis G., Jentsch S. SUMO, ubiquitin’s mysterious cousin. Nat. Rev. Mol. Cell Biol. 2001;2:202–210. doi: 10.1038/35056591. [DOI] [PubMed] [Google Scholar]
- 20.Jentsch S., Pyrowolakis G. Ubiquitin and its kin: how close are the family ties? Trends Cell Biol. 2000;10:335–342. doi: 10.1016/s0962-8924(00)01785-2. [DOI] [PubMed] [Google Scholar]
- 21.Melchior F. SUMO—nonclassical ubiquitin. Annu. Rev. Cell Dev. Biol. 2000;16:591–626. doi: 10.1146/annurev.cellbio.16.1.591. [DOI] [PubMed] [Google Scholar]
- 22.Yeh E.T., Gong L., Kamitani T. Ubiquitin-like proteins: new wines in new bottles. Gene. 2000;248:1–14. doi: 10.1016/s0378-1119(00)00139-6. [DOI] [PubMed] [Google Scholar]
- 23.Saitoh H., Hinchey J. Functional heterogeneity of small ubiquitin-related protein modifiers SUMO-1 versus SUMO-2/3. J. Biol. Chem. 2000;275:6252–6258. doi: 10.1074/jbc.275.9.6252. [DOI] [PubMed] [Google Scholar]
- 24.Goettsch S., Bayer P. Structural attributes in the conjugation of ubiquitin, SUMO and RUB to protein substrates. Front Biosci. 2002;7:a148–a162. doi: 10.2741/goet. [DOI] [PubMed] [Google Scholar]
- 25.Bayer P., Arndt A., Metzger S., Mahajan R., Melchior F., Jaenicke R., Becker J. Structure determination of the small ubiquitin-related modifier SUMO-1. J. Mol. Biol. 1998;280:275–286. doi: 10.1006/jmbi.1998.1839. [DOI] [PubMed] [Google Scholar]
- 26.Malakhov M.P., Mattern M.R., Malakhova O.A., Drinker M., Weeks S.D., Butt T.R. SUMO fusions and SUMO-specific protease for efficient expression and purification of proteins. J. Struct. Funct. Genom. 2004;5:75–86. doi: 10.1023/B:JSFG.0000029237.70316.52. [DOI] [PubMed] [Google Scholar]
- 27.Li S.J., Hochstrasser M. A new protease required for cell-cycle progression in yeast. Nature. 1999;398:246–251. doi: 10.1038/18457. [DOI] [PubMed] [Google Scholar]
- 28.Li S.J., Hochstrasser M. The yeast ULP2 (SMT4) gene encodes a novel protease specific for the ubiquitin-like Smt3 protein. Mol. Cell. Biol. 2000;20:2367–2377. doi: 10.1128/mcb.20.7.2367-2377.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Suzuki T., Ichiyama A., Saitoh H., Kawakami T., Omata M., Chung C.H., Kimura M., Shimbara N., Tanaka K. A new 30-kDa ubiquitin-related SUMO-1 hydrolase from bovine brain. J. Biol. Chem. 1999;274:31131–31134. doi: 10.1074/jbc.274.44.31131. [DOI] [PubMed] [Google Scholar]
- 30.Kim K.I., Baek S.H., Chung C.H. Versatile protein tag, SUMO: its enzymology and biological function. J. Cell Physiol. 2002;191:257–268. doi: 10.1002/jcp.10100. [DOI] [PubMed] [Google Scholar]
- 31.Kim K.I., Baek S.H., Jeon Y.J., Nishimori S., Suzuki T., Uchida S., Shimbara N., Saitoh H., Tanaka K., Chung C.H. A new SUMO-1-specific protease, SUSP1, that is highly expressed in reproductive organs. J. Biol. Chem. 2000;275:14102–14106. doi: 10.1074/jbc.275.19.14102. [DOI] [PubMed] [Google Scholar]
- 32.X. Zuo, S. Li, J. Hall, M.R. Mattern, H. Tran, J. Shoo, R. Tan, S.R. Weiss, T.R. Butt, Enhanced expression and purification of membrane proteins by SUMO fusion in Escherichia coli, J. Struct. Funct. Genom. (2005) (in press) [DOI] [PMC free article] [PubMed]
- 33.Jonasson P., Liljeqvist S., Nygren P.A., Stahl S. Genetic design for facilitated production and recovery of recombinant proteins in Escherichia coli. Biotechnol. Appl. Biochem. 2002;35:91–105. doi: 10.1042/ba20010099. [DOI] [PubMed] [Google Scholar]
- 34.McDonnell D.P., Pike J.W., Drutz D.J., Butt T.R., O’Malley B.W. Reconstitution of the vitamin D-responsive osteocalcin transcription unit in Saccharomyces cerevisiae. Mol. Cell. Biol. 1989;9:3517–3523. doi: 10.1128/mcb.9.8.3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ecker D.J., Stadel J.M., Butt T.R., Marsh J.A., Monia B.P., Powers D.A., Gorman J.A., Clark P.E., Warren F., Shatzman A. Increasing gene expression in yeast by fusion to ubiquitin. J. Biol. Chem. 1989;264:7715–7719. [PubMed] [Google Scholar]
- 36.Butt T.R., Jonnalagadda S., Monia B.P., Sternberg E.J., Marsh J.A., Stadel J.M., Ecker D.J., Crooke S.T. Ubiquitin fusion augments the yield of cloned gene products in Escherichia coli. Proc. Natl. Acad. Sci. USA. 1989;86:2540–2544. doi: 10.1073/pnas.86.8.2540. [DOI] [PMC free article] [PubMed] [Google Scholar]