

Abstract
The Spike (S) glycoprotein of coronaviruses (CoV) mediates viral entry into host cells. It contains two hydrophobic heptad repeat (HR) regions, denoted HRN and HRC, which oligomerize the S glycoprotein into a trimer in the native state and when activated collapse into a six-helix bundle structure driving fusion of the host and viral membranes. Previous studies have shown that peptides of the HR regions can inhibit viral infectivity. These studies imply that the HR regions are accessible and that agents which can interact with them may prevent viral entry. In the present study, we have investigated an approach to generate antibodies that specifically recognize the HRN and HRC regions of the SARS-CoV spike (S) glycoprotein in order to evaluate whether these antibodies can inhibit viral infectivity and thus neutralize the SARS-CoV. In this regard, we incorporated HRN and HRC coiled-coil surface residues into a de novo designed two-stranded α-helical coiled-coil template for generating conformation-specific antibodies that recognize α-helices in proteins (Lu, S.M., Hodges, R.S., 2002. J. Biol. Chem. 277, 23515–23524). Eighteen surface residues from two regions of HRN and HRC were incorporated into the template and used to generate four anti-sera, HRN1, HRN2, HRC1, and HRC2. Our results show that all of the elicited anti-sera can specifically recognize HRN or HRC peptides and the native SARS-CoV S protein in an ELISA format. Flow cytometry (FACS) analysis, however, showed only HRC1 and HRC2 anti-sera could bind to native S protein expressed on the cell surface of Chinese hamster ovary cells, i.e., the cell surface structure of the S glycoprotein precluded the ability of the HRN1 or HRN2 anti-sera to see their respective epitope sites. In in vitro viral infectivity assays, no inhibition was observed for either HRN1 or HRN2 anti-serum, whereas both HRC1 and HRC2 anti-sera could inhibit SARS-CoV infection in a dose-dependent manner. Interestingly, the HRC1 anti-serum, which was a more effective inhibitor of viral infectivity compared to HRC2 anti-serum, could only bind the pre-fusogenic state of HRC, i.e., the HRC1 anti-serum did not recognize the six-helix bundle conformation (fusion state) whereas HRC2 anti-serum did. These results suggest that antibodies that are more specific for the pre-fusogenic state of HRC may be better neutralizing antibodies. Overall, these results clearly demonstrate that the two-stranded coiled-coil template acts as an excellent presentation system for eliciting helix-specific antibodies against highly conserved viral antigens and HRC1 and HRC2 peptides may represent potential candidates for use in a peptide vaccine against the SARS-CoV.
Keywords: SARS, SARS-CoV, Coronavirus, Spike glycoprotein, Neutralizing antibodies, Coiled-coils, Fusion protein, Heptad repeat regions
1. Introduction
The etiologic agent responsible for the global outbreak of SARS (severe acute respiratory syndrome) which occurred in the fall of 2002–2003 has since been identified as a novel coronavirus (SARS-CoV) (Drosten et al., 2003, Ksiazek et al., 2003, Kuiken et al., 2003, Peiris et al., 2003, Poon et al., 2003). Coronaviruses are a diverse group of large enveloped, positive-sense single-stranded RNA viruses that cause respiratory and enteric diseases in humans and domestic animals (McIntosh, 1974, Siddell et al., 1983, Lai and Holmes, 2001). Sequencing of the SARS-CoV genome has shown that it is genetically distinct from previously known members of the Coronaviridae family (Marra et al., 2003, Rota et al., 2003) and that its nucleotide sequence is >90% identical to novel CoV strains now found in the Himalayan masked palm civet (Paguma larvata) and the Chinese horseshoe bat (Rhinolophus sinicus) (Guan et al., 2003, Martina et al., 2003, Lau et al., 2005). The close proximity of these mammals in live animal meat markets in southern China has suggested that the ancestral origin of the SARS-CoV may have come from a cross-species transmission of a coronavirus harbored in one of these mammals to humans. Although there have been no new reported human cases of SARS since 2004, the observation of closely related SARS-CoV-like strains still existing in animal reservoirs raises the possibility of a re-emergence of the SARS-CoV in humans. Due to this threat, development of prophylactics and viable vaccine candidates providing protection from the SARS-CoV are clearly needed.
Similar to other coronaviruses, the SARS-CoV enters target cells by inducing fusion between the viral and cellular membranes, a process mediated by the viral spike (S) glycoprotein (Spaan et al., 1988, De Groot et al., 1989, Bos et al., 1995, Luo and Weiss, 1998, Gallagher and Buchmeier, 2001, Simmons et al., 2004, Yang et al., 2004a). The spike protein is a type-1 transmembrane protein. The spike protein is synthesized as a single polypeptide precursor which for several CoV members is cleaved by a cellular endo-protease to generate two non-covalently bound subunits S1 and S2 (Frana et al., 1985, Sturman et al., 1985). In the case of the SARS-CoV S protein, it is believed that the S polypeptide chain is not cleaved upon release from infected cells but is cleaved during virus entry (Follis et al., 2005, Simmons et al., 2005). The S1 subunit of the spike protein forms a globular head and is responsible for recognition of the host cell receptor(s) (Taguchi, 1995), which for the SARS-CoV has been identified as angiotensin converting enzyme 2 (ACE2) and CD209L (Li et al., 2003, Jeffers et al., 2004). The S2 subunit constitutes the stalk-like region of the spike and functions in membrane fusion. The sequence of the S2 subunit contains a hydrophobic fusion peptide sequence, two characteristic 4–3 hydrophobic (heptad) repeat regions (denoted HRN and HRC herein), a transmembrane domain and cytoplasmic tail. These regions are responsible for membrane anchoring, oligomerization of the S protein into a trimeric state and fusion between viral and cellular membranes (Luo et al., 1999, Bosch et al., 2003, Bosch et al., 2004, Ingallinella et al., 2004, Liu et al., 2004, Tripet et al., 2004, Zhu et al., 2004, Guillen et al., 2005). In the current model describing S protein function, S is thought to drive membrane fusion by coupling irreversible protein refolding with membrane juxtaposition. After binding to the host cell receptor, a conformational change is transmitted from S1 to S2 which causes the fusion peptide (a region of ∼20 hydrophobic residues located N-terminal to the first heptad repeat region, HRN in S2) to be released from the interior of the molecule and inserted into the target cell bilayer (Hernandez et al., 1997, Damico et al., 1998). This “meta-stable” fusion intermediate state is then followed by a further conformational change to a new low energy post-fusion state. Conversion to the post-fusion state involves a collapse of the two heptad repeat regions such that they now form a six-helix bundle, where three HRN helices fold into a central parallel triple stranded α-helical coiled-coil, and wrapped on the outside of this core is an outer layer of three anti-parallel HRC strands (Epand, 2003, Bosch et al., 2004, Ingallinella et al., 2004, Liu et al., 2004, Tripet et al., 2004, Zhu et al., 2004). This post-fusion conformation displays the trimer of hairpins organization characteristically seen for other class I viral fusion protein domains such as HIV-1 gp41, influenza virus HA, and Ebola virus gp21 proteins (Bullough et al., 1994, Chan et al., 1997, Weissenhorn et al., 1997, Weissenhorn et al., 1998, Chen et al., 1999, Malashkevich et al., 1999). The structures of the S2 ectodomain in the six-helix conformation for both SARS-CoV and murine coronavirus MHV (a related CoV strain) have recently been reported (Ingallinella et al., 2004, Supekar et al., 2004, Xu et al., 2004, Xu et al., 2004). The refolding of the HR regions into the six-helix bundle structure places the fusion peptide and the transmembrane domain in close proximity, thereby facilitating membrane pore formation and delivery of the nucleocapsid into the cell (Eckert and Kim, 2001, Epand, 2003).
Since the spike protein is recognized to play a critical role in viral entry into host cells, it is understandable that the S protein represents a major target for generating inhibitory drugs and antibodies which can neutralize the virus. Indeed several groups have shown that immunization with the spike glycoprotein of several animal CoVs induces anti-S antibodies which provide protective immunity against the corresponding virus (Holmes, 2003, Navas-Martin and Weiss, 2003). More recently, several groups have now shown that immunization with the SARS-CoV S protein via inactivated SARS-CoV pseudovirus, vaccinia virus expressed S, recombinant S protein, or cDNA which encodes S, all elicit S-specific antibodies which appear to be neutralizing in small animal testing (Bisht et al., 2004, Bukreyev et al., 2004, Yang et al., 2004b, Zang et al., 2005 and references therein). Despite their promising success, care and concern regarding the use of full-length S protein has also been advised. Although the sera of SARS patients have been shown to contain neutralizing IgGs against the S protein (attesting to the usefulness of a strong humoral response), other non-neutralizing S protein epitope-specific antibodies in these patients appear to have caused an auto-immune response, leading to enhancement of viral infection. When these auto-antibodies were analyzed in an in vitro system, they cross-reacted with lung epithelial cells and induced cytotoxicity, an event which correlates well with lung cytotoxicity observed in SARS patients (Lin et al., 2005). Also, immunization of SARS-infected ferrets with a full-length S protein vaccine exacerbated the liver damage caused by the virus (Czub et al., 2005). Additionally, the problem associated with antibody induced enhancement of disease has previously been observed for a feline coronavirus vaccine using the S protein of that virus as an antigen (Olsen, 1993). Thus, for these reasons, a SARS vaccine approach whereby full-length S protein is utilized may not be a safe and viable vaccine for humans.
An alternative to using full-length S protein to generate SARS-CoV neutralizing antibodies is the use of small well characterized S protein epitopes. Small epitopes can be synthetically prepared, thus eliminating the problems associated with large volumes of live or recombinant virus. Any epitopes identified with adverse side-effects can be easily eliminated, producing a safer vaccine. Support for such an approach can be gained from recent studies of Zhou et al., 2004, He and Jiang, 2005 which have shown that monoclonal antibodies generated against a fragment of the SARS-CoV S1 domain can recognize epitopes in the receptor-binding domain and can inhibit viral entry by blocking association of the virus and receptor. In a similar vein, we hypothesized that antibodies raised against the heptad repeat regions (HRN or HRC) could also represent ideal epitope sites to produce neutralizing antibodies. These anti-HR antibodies could potentially target the exposed trimeric coiled-coils in the pre-hairpin intermediate state of S in a manner analogous to peptide inhibitors which have been shown to bind to type 1 fusion proteins and thus block the transition of the S2 domain into the post-fusogenic state preventing the formation of the six-helix bundle structure. Moreover, the amino acid residues on the surface of these coiled-coil regions appear to be highly conserved, a likely consequence of their importance in assembly of the fusion-competent structure, which suggests these amino acids may be more constrained in their ability to undergo antibody resistant mutations preserving the usefulness of the vaccine for recognition of both primary isolates and adapted SARS coronavirus strains.
A critical aspect in using peptides to generate high affinity and/or protective antibodies against epitopes within native proteins relies on the ability of the immunogenic peptides to mimic the three-dimensional structure of the respective B-cell epitope in the native protein. In the case of the SARS-CoV HR regions, each has been shown to exist in an α-helical coiled-coil structure (Bosch et al., 2003, Ingallinella et al., 2004, Liu et al., 2004, Tripet et al., 2004), and therefore the most effective display of the respective epitopes will be in an α-helical structure. To construct the HRN and HRC epitopes in a defined α-helical orientation, a novel two-stranded coiled-coil template strategy developed previously in our laboratory was used (Lu and Hodges, 2002). In this strategy, one selects the specific surface-oriented residues that constitute the epitope site and then incorporates these into the de novo designed two-stranded coiled-coil template sequence. This process is akin to grafting the molecular surface of one protein onto another. The template is designed with very high stability which ensures correct presentation, high solubility which facilitates peptide handling, and contains a small well defined epitope site making the elicited polyclonal anti-serum focused to a specific site on the protein. To date, the template strategy has been tested and shown to induce conformation-specific antibodies to several epitopes in heptad repeat regions which are reactive against their native proteins (unpublished data, S. Lu and R.S. Hodges).
Thus, in the present study, we describe the comparative analysis of several B-cell immunogens spanning the HRN and HRC coiled-coil regions of S2 of the SARS-CoV. We have investigated their structural and immunological properties and determined the potential of the anti-sera to neutralize SARS-CoV infectivity. Our results show that all of the elicited anti-sera can specifically recognize HRN or HRC peptides and the native SARS-CoV S protein in an ELISA format. FACS analysis, however, showed only HRC1 and HRC2 anti-sera could bind to native S protein expressed on the cell surface, i.e., the cell surface structure of S precluded the ability of the HRN1 or HRN2 anti-sera to see their respective epitope sites. In in vitro viral infectivity assays, no inhibition was observed for either HRN1 or HRN2 anti-sera, whereas both HRC1 and HRC2 anti-sera could inhibit SARS-CoV infection. Interestingly, the HRC1 anti-serum, which was a more effective inhibitor of viral infectivity compared to HRC2 anti-serum, could only bind the pre-fusogenic state of HRC, whereas the HRC2 anti-serum also recognized the six-helix bundle conformation of the fusion state. These results suggest that antibodies that are more specific for the pre-fusogenic state of HRC may be better neutralizing antibodies. Overall, these results clearly demonstrate that the two-stranded coiled-coil template acts as an excellent presentation system for eliciting helix-specific antibodies against highly conserved viral antigens, and show that HRC1 and HRC2 peptides may represent potential candidates for use in a peptide vaccine against SARS-CoV.
2. Methods
2.1. Peptide synthesis
Peptides were prepared by solid-phase synthesis methodology using 4-benzylhydrylamine hydrochloride resin with conventional N α-t-butyloxycarbonyl (Boc) chemistry (Chan and White, 2000). Following synthesis the peptides were deprotected and N-terminally acylated using either acetic anhydride (Ac), benzoylbenzoic acid (BB) anhydride or Boc-p-amino-benzoic acid (Abz) anhydride. The N-terminally acetyl and Abz peptides were cleaved from the resin with hydrogen fluoride (10 ml/g of resin) containing 10% anisole (v/v) and 2% 1,2-ethanediol at −4 °C for 1 h. For peptides that contained BB, thioanisole was used in place of anisole and 1,2-ethandiol was omitted. Following cleavage and removal of hydrogen fluoride, the crude peptides were washed several times with ethyl ether and extracted with 50% acetonitrile (v/v) and lyophilized. Crude peptides were resuspended in a buffer containing 20 mM trifluoroacetic acid (TFA), 5 mM Tris(2-carboxyethyl)-phosphine hydrochloride (TCEP) and purified using reversed-phase high-performance chromatography (RP-HPLC) (Zorbax SB-C8 300 Å, 6.5 μ, 9.4 mm I.D. × 250 mm column) to homogeneity using a AB gradient where eluent A is 0.2% aqueous TFA and eluent B is 0.2% TFA in acetonitrile and a shallow gradient approach (0.1% acetonitrile/min). Each peptide was characterized by amino acid analysis and electrospray mass spectrometry.
2.2. Formation of disulfide-bridged hetero two-stranded peptides
Preferential disulfide bridge formation between the acetylated and the BB containing peptide strands (see Fig. 1 ) of the immunogen peptides was performed as described by Tripet et al. (2004). In brief, 10 mg of 2,2′-dithiopyridine (DTDP) was dissolved in 100 μl of dimethylformamide with sonication. A 10 μl (3.4 μmol) aliquot of this solution was added to a 3:1 (v/v) acetic acid/H2O solution containing 2 mg (0.8 μmol) of HRN or HRC peptide which did not contain the BB moiety and stirred for 6 h. Distilled water (1 ml) was added and the solution was then extracted 3× with 500 μl of ether and the aqueous layer applied to a Sephadex G-25 desalting column conditioned with 50 mM NH4Ac, pH 5.5 running buffer. Fractions (1 ml) eluted from the column were collected and those with 220 nm absorbance pooled. HRN or HRC peptide containing the BB moiety (2 mg dissolved in 1 ml of 8 M urea, and 50 mM NH4Ac, pH 5.5, buffer) was added in 100 μl aliquots over 30 min. The reaction were then stirred for 1 h and the final complex purified by RP-HPLC and freeze dried.
Fig. 1.

Top panel: the two-stranded α-helical coiled-coil template sequence. The 18 residue positions which can be substituted with native S protein residues are indicated with an asterisk (*). Residues which form the 4–3 hydrophobic repeat of the coiled-coil structure are underlined. The relative position of the * residues when in a two-stranded coiled structure are shown in an end cross-sectional view (below left) and cartoon (below right). In the cross-sectional view, the direction of the helices is into the page from NH2 to COOH terminus with the polypeptide chains parallel and in-register. Heptad positions are labeled a–g, with the prime indicating corresponding positions on the opposing helix. Arrows depict the hydrophobic interactions that occur between residues in the “a” and “d” positions. In the cartoon model, the shaded circles denote the substituted positions on the front helix of the dimer. Middle and Bottom panels show the amino acid sequence of the HRN and HRC peptides used in this study. Peptide names are indicated on the left. Numbers in parentheses indicate the amino acid sequence region of the native S protein which the peptide spans. The location of the disulfide bridge between cysteine residues is denoted by a solid line. Core a and d residues in template are underlined. Ac, denotes Nα-acetyl; -amide, denotes Cα-amide; BB, denotes Nα-benzoylbenzoyl; nL, denotes norleucine; and X, denotes either pAbz-Cys or Ac-Cys depending on the use of the peptide.
2.3. Formation of trimeric HRN and HRC peptides
Covalently linked homo-trimeric HRN coiled-coil molecules were prepared by dissolving 1 mg of HRN peptide (Abz-Cys-HRN(902–950)) in 1 ml of a 10 mM Na2HPO4, 2 mM EDTA, 10% (v/v) N,N-dimethylformamide, pH 7.5, buffer and adding 0.33 mol equivalents (to 1 equivalent peptide) of a Tris–maleimido crosslinker (see below). The reaction was stirred for 2 h and then applied to a RP-HPLC analytical column (4.6 mm I.D. × 250 mm Zorbax SB-C8 300 Å, 5 μ) and purified using a linear AB gradient and a gradient rate of 2% acetonitrile/min, where A is aqueous 0.2% TFA and B is 0.2% TFA in acetonitrile. Fractions deemed >95% pure where pooled and lyophilized. The Tris–maleimido crosslinker was prepared by mixing 105 mg (0.5 mmol) of ε-maleimido-caproic acid with 23 μl (0.15 mmol) of Tris(2-aminoethyl)amine and 78 μl (0.5 mmol) of diisopropylcarbodiimide in 25 ml of dichloromethane overnight. The dichloromethane was removed by rotary evaporation and the Tris–maleimido crosslinker purified by flash chromatography using mesh 60 Å silica and a 3:1 (v/v) chloroform/methanol. The same method was used to prepare the HRC trimeric molecule with the exception that the peptide used for coupling was Abz-Cys-HRC(1150–1185). Note: this peptide corresponds to HRC residues 1150–1185 and the former peptide corresponds to HRN residues 902–950 of the SARS-CoV S sequence with an additional cysteine residue added to its amino-terminus and the final N-terminal group acylated with p-aminobenzoic acid.
2.4. Preparation of peptide-carrier protein conjugates
Peptides used for immunization were conjugated to keyhole limpet hemocyanin (KLH) and peptides used for analysis of specific anti-sera were conjugated to bovine serum albumin (BSA). For peptide-KLH conjugates, KLH was dissolved in 8 M urea, 50 mM sodium bicarbonate, pH 8.9, at 25 mg/ml. To 100 μl of the KLH solution was added ∼2 mg of the disulfide linked two-stranded (BB containing) HR peptide such that the molar ratio was ∼8:1 peptide:carrier. The peptide carrier solutions were put in quartz tubes and placed in a Rayonet photoreaction chamber (Southern New England Ultraviolet Company, Bradford, CT) and irradiated with UV light (350 nm) for 2 h. 4-Benzoylbenzoic acid (added during synthesis) served as a photo-activated linker to crosslink the peptide to the carrier protein.
After photolysis, the conjugation mixture was diluted to 2 ml with 2 M urea, 50 mM sodium bicarbonate, pH 8.5, and dialyzed against 10 mM Na2HPO4, 150 mM NaCl, pH 7.4 buffer (PBS) overnight at 4 °C. The number of peptide molecules per carrier molecule was determined by amino acid analysis using the norleucine (peptide) to phenylalanine (carrier) molar ratio. In general, the concentrations of the conjugates ranged from 0.6 to 0.9 mg/ml with an average peptide to KLH molar ratio of 4:1. Peptide-BSA conjugates where prepared by the following methods. For monomeric HRN- or HRC-BSA conjugates, 2 mg BSA dissolved in 1 ml of PBS was mixed with 0.2 mg of m-maleimidobenzyl-N-hydroxysulfosuccinimide ester (Sulfo-MBS, Pierce, Rockford, IL, USA) and stirred for 1 h. The solution was then applied to a Sephadex G-25 desalting column conditioned with PBS running buffer. Fractions (1 ml) eluted from the column were collected and those with 280 nm absorbance pooled. To one half of the pooled solution was added either 1 mg of Ac-Cys-HRN(916-950) peptide or Ac-Cys-HRC(1150–1185) peptide. The solution was stirred for 2 h at room temperature and then dialyzed against several changes of PBS buffer at 4 °C. Trimeric HRN- or HRC-BSA conjugates where prepared by dissolving 1 mg of the covalently linked homo-trimeric HRN or HRC peptides (see above) in 1 ml of PBS buffer, pH 6.0, and then adding 0.1 mg of sulfosuccinimidyl(4-iodoacetyl)aminobenzoate (Sulfo-SIAB, Pierce, Rockford, IL). The solution was stirred for 4 h at room temperature. The solution was then applied to a Sephadex G-25 desalting column conditioned with PBS running buffer. Fractions (1 ml) eluted from the column were collected and those with 210 nm absorbance pooled. To one half of the pooled solution was added 1 ml of a 1 mg/ml solution of BSA which had been pre-treated with the Traut’s reagent according to standard protocols described by Pierce. The solution was stirred for 2 h at room temperature and then dialyzed against several changes of PBS buffer at 4 °C.
2.5. Immunization protocol
All of the animal work was carried out at the University of Colorado Health Sciences Center laboratory in accordance with established protocols on file. Briefly, for each immunogen, three New Zealand white rabbits were immunized at two intramuscular sites. Primary immunization contained 50 μg of the keyhole limpet hemocyanin-peptide conjugate (in PBS, pH 7.4) mixed 1:1 with Freund’s complete adjuvant. Secondary, tertiary and booster immunizations (at days 7, 28, and 50) also contained 50 μg of conjugate but were mixed with Freund’s incomplete adjuvant. After exsangination on day 58, serum was collected and stored at −20 °C.
2.6. Purification of IgG
Polyclonal antibodies were precipitated from sera using caprylic acid. Sera were diluted 5 fold with 60 mM sodium acetate, pH 4.5. Caprylic (octanoic) acid was added dropwise to 2.5% (v/v) with vigorous stirring at room temperature for 30 min and then centrifuged at 10 000g. PBS stock buffer (10×) was added to the supernatant to give a buffer concentration of 1× and the pH was adjusted to 7.4 with 5 N sodium hydroxide. Crystalline ammonium sulfate was added to 45% saturation (0.277 g/ml) while stirring in an ice bath to precipitate the immunoglobulins. Centrifugation at 7000g was used to collect the precipitated antibodies. The pellet was resuspended in 2.5 ml of PBS and dialyzed overnight against PBS, pH 7.4. The antibody solution was then further purified on a protein G affinity column (1.5 cm I.D. × 10 cm, protein G–Sepharose 4 Fast Flow, Amersham Biosciences, Piscataway, NJ). The bound antibody was eluted from the column using a 0.5 M ammonium acetate, pH 3 buffer after which the solution was immediately adjusted to pH 7–8 with ammonium hydroxide and dialyzed against PBS overnight. Subsequently, the antibody solution was concentrated to <5 ml in an Amicon concentration unit using YM30 ultrafiltration discs (Millipore Corp., Bedford, MA). The concentration of each antibody solution was determined by amino acid analysis, assuming a molecular mass of 150 000 Da. Finally, the antibody solution was stored at −20 °C until use.
2.7. Circular dichroism spectroscopy
Circular dichroism (CD) spectra were recorded on a Jasco J-810 spectropolarimeter (Jasco Inc., Easton, MD). The CD wave scans were measured from 190 to 255 nm in benign buffer (0.1 M potassium chloride, 0.05 M potassium phosphate, pH 7). Temperature denaturation midpoints (T 1/2) for the peptides were determined by following the change in molar ellipticity at 222 nm from 4 to 95 °C in a 1 mm path length cell and a temperature increase rate of 1 °C/min. Ellipticity readings were normalized where 1 represent the molar ellipticity values for the fully folded species and zero equals the fully unfolded species.
2.8. ELISA protocol
High-binding polystyrene 96-well ELISA plates (Costar 3590 from Fisher Scientific, Pittsburg, PA) were used. BSA-peptide conjugate or soluble S protein (0.2 μg/well) was adsorbed to the bottom of each well in 50 mM sodium carbonate, pH 9.6 or PBS, pH 7.4 overnight at 4 °C. After washing with PBS (pH 7.4) each well was blocked with a 5% BSA solution (37 °C, 1 h). After washing with TPBS (PBS + Tween 20) (0.05% v/v) pH 7.4 three times, crude serum or purified antibody (diluted 1:5000 for specificity analysis or serially diluted 1:4 from a starting serum dilution of 1:1000 for titer analysis) was added to each well and incubated at 37 °C for 1 h. After washing away unbound primary (rabbit) antibody goat anti-rabbit horseradish peroxidase secondary antibody (Jackson Immunolaboratory, West Grove, PA) (diluted 1:5000) was incubated in each well at 37 °C for 1 h. After washing, 2,2′-azino-bis-3-ethylbenthiazoline-6-sulfonic acid in 10 mM sodium citrate, pH 4.2 with 0.03% H2O2 was incubated for 30 min. The plates were read on a SpectraMax 386 Plus plate reader (Molecular Devices, Sunnyvale, CA) at 450 nm.
2.9. Western analysis
The protein samples were loaded onto a 8% SDS–acrylamide gel, separated using a Bio-Rad Mini-Protean II electrophoresis system, and transferred electrophoretically to a nitrocellulose or PVDF membrane using standard Bio-Rad protocols. After several washes with PBS (with 0.05% Tween 20), non-specific protein-binding sites were blocked with 5% non-fat milk for 1 h at 25 °C with gentle agitation. After washing, HR-specific anti-sera (diluted 1:2000) was allowed to bind for 1 h. Goat anti-rabbit horseradish peroxidase (Jackson Immunolaboratory, West Grove, PA) (diluted 1:2500) or goat anti-rabbit alkaline phosphatase secondary antibody was then added after washing away the primary rabbit sera. The reactive protein bands were visualized using either a chemiluminescent substrate (horseradish peroxidase LumiBlot; Novagen, Madison, WI) according to the included instructions or nitrobluetetrazoliumchloride/5-bromo-4-cloro-3-indolyl-phosphate, toluidine-salt (NBT/BCIP ready-to-use tablets, Boehringer Mannheim, Indianapolis IN).
2.10. Surface plasmon resonance (SPR) analysis
All kinetic experiments were performed on a BIAcore 3000 instrument at 25 °C. The instrument was programmed for iterative cycles in which each kinetic cycle consisted of (1) a 180 s association phase, (2) a 240 s dissociation phase, and (3) a 10 s regeneration phase. A flow rate of 70 μl/min was used for the association and dissociation phases and 40 μl/min was used for the regeneration step. Immobilization of the HRN and HRC peptides (which contained an N-terminal cysteine residue) to the biosensor surface was carried out using the ligand thiol method as described by (Johnsson et al., 1991). Briefly, the dextran surface of the sensor chip (CM5 from BIAcore Inc. Piscataway, NJ) was first activated with N,N-(3-dimethylaminopropyl)-N′-ethyl-carbodiimide hydrochloride (EDC) and N-hydroxysuccinimide (NHS/EDC) (35 μl) followed by addition of 2-(2-pyridinyldithiol)-ethaneamine (PDEA) (20 μl). HRN peptide (50 μg/ml) in 10 mM sodium acetate buffer, pH 4.3, or HRC peptide (50 μg/ml) in PBS, pH 6.1, were injected and allowed to react to give a surface density of approximately 50 resonance units. Remaining activated groups were then blocked by injection (10 μl) of a 50 mM cysteine, 1 M NaCl , 0.1 M formate, pH 4.3 deactivation solution. Immobilization of the HRN and HRC covalently linked trimer peptides to the biosensor surface was carried out using the amine coupling method as described by (Johnsson et al., 1991). The dextran surface of the sensor chip was first activated with NHS/EDC (as above) followed by addition of the peptide (10 μg/ml) dissolved in 10 mM sodium acetate buffer, pH 4.3, until sufficient peptide reacted to give a surface density of approximately 50 resonance units. Remaining activated groups were then blocked by injection (10 μl) of 1 M ethanolamine, pH 8.5, deactivation solution. All kinetic experiments were carried out using a running buffer which contained 10 mM sodium phosphate, pH 7.4, 100 mM NaCl and 0.005% P20, and a regeneration buffer containing 20 mM phosphoric acid, pH 2. Binding data were processed using the Scrubber (BioLogic Software Pty., Australia) software package. The binding curves were fit to a 1:1 (Langmuir) binding model using Biaevaluation software version 4.1 (Biacore AB, Sweden).
2.11. Cell-binding assay
Chinese hamster ovary (CHO) cells were grown in high glucose Dulbecco modified Eagle’s medium supplemented with 10% fetal bovine serum and 2% antibiotic/anti-mycotic, and then transfected with the plasmid expression vector pcDNA3.1 SARS-S Δ19 which encodes the native SARS-CoV S protein minus the C-terminal 19 residues. Cells were incubated in a CO2 incubator at 37 °C and 5% CO2 in a humidified atmosphere for 48 h. Cell suspensions were prepared by trypsinization of the cell cultures, counted, and then placed into several wells of a 96-well plate (round bottom) with approximately 1 × 105 cells/well. Cells were washed briefly in FACS buffer (phosphate-buffered saline (PBS) with 2% BSA and 2% normal goat serum). Antibodies (100 μl diluted 1:250) were then incubated with the cells at 4 °C for 1 h. After being washed three times with FACS buffer, cells were incubated with 100 μl of phycoerythrin (PE)-conjugated goat anti-rabbit IgG (diluted 1:2000) (Jackson Immuno Research Laboratories, Inc, West Grove, PA, USA) at 4 °C for 1 h and washed again with FACS buffer 3 times. Cells were fixed 1% with paraformaldehyde in PBS at room temperature for 10 min. The cell-binding activity of the sera antibodies was analyzed by flow cytometry (FACSCalibur, BD Biosciences, San Jose, CA, USA) with excitation at 488 nm at the UCHSC Flowcytometry core facility.
2.12. Viral neutralization assays
The neutralization activity of the HRN and HRC anti-sera on SARS-CoV infectivity of Vero E6 cells was assessed. 50 plaque forming units (PFU) of SARS-CoV Urbani strain (Accession No. AY278741) was mixed with an equal volume of HRN or HRC anti-sera at graded concentrations and incubated at room temperature for 30 min. The mixture was added to monolayers of Vero E6 cells in 6-well tissue-culture plates and incubated at 37 °C for 1 h. The supernatants were removed and the cells were overlaid with Seakem agar containing 2× modified Eagle’s medium. On day 3 after inoculation the cells were overlaid with Seakem agar containing 2× modified Eagle’s medium and neutral red. Twenty-four hours after the overlay the number of plaques were counted and plotted.
3. Results
3.1. The two-stranded α-helical coiled-coil template
To elicit antibodies which would specifically recognize the α-helical structure of the HRN and HRC hydrophobic (heptad) repeat regions of the SARS-CoV S protein, we utilized a novel two-stranded α-helical coiled-coil template system developed previously in our laboratory (Lu and Hodges, 2002). In brief, the coiled-coil template (Fig. 1 top) consists of two peptide strands that are covalently linked by a disulfide bridge via cysteine residues located at their N-termini. The disulfide link serves to stabilize the coiled-coil structure by maintaining the parallel in-register orientation of the two strands while removing the concentration dependence of the molecule. Each sequence contains Ile and Leu residues, respectively, in the corresponding a and d heptad repeat positions (denoted (abcdefg)n). The Ile/Leu residues are known to maximize the helical character and stability of a coiled-coil in these positions (Tripet et al., 2000). Additionally, the residues in these positions have been shown to determine the oligomerization state of a coiled-coil, and the Ile/Leu residue pattern is known to favor dimer formation (the oligomerization state desired in this case) (Harbury et al., 1993). Two arginine residues were added at the C-terminus of each strand to aid in the overall solubility of the molecule. The total length of the coiled-coil is 31 residues, of which 18 residues positions are available to be utilized by the epitope/antigenic sequence. These positions occur in the surface exposed b, c, e, f, and g heptad positions. Utilizing all 18 positions then allows for display of an α-helical epitope which encompasses seven helical turns in each helix. Finally, the N-terminus of one strand is acetylated while the second strand is extended two additional residues with norleucine and glycine residues. The norleucine/glycine residues act as a spacer between the coiled-coil immunogen and the site of conjugation to the carrier protein, while norleucine allows for easy quantitation of the peptide/carrier ratio after conjugation. At the very N-terminus of the extended strand was added benzoylbenzoic acid (BB). BB is a very efficient photo-activated cross-linker of synthetic peptides to carrier proteins (Parker and Hodges, 1985a, Parker and Hodges, 1985b, Ngai et al., 1994).
3.2. The immunogenic peptides
Four epitope sites were chosen to be incorporated into the coiled-coil template peptide. Two sites were selected from within the HRN coiled-coil region. The first, HRN1 (see Fig. 1 middle) corresponds to residues 921–923, 925–926, 928–930, 932–933, 935–937, 939–940, and 942–944 (TTT-TA-GKL-DV-NQN-QA-NTL) of the SARS-CoV S protein. These residues are located along the exterior surface of HRN (Fig. 2 A) and within its hydrophobic core which are now exposed on the surface of the template (T923, L930, N937, and L944). The choice for these residues coincides with those involved in forming, and flanking, the HRC-binding site on HRN as seen in the high resolution six-helix bundle crystal structure (Fig. 2 center). The second epitope site, HRN2, corresponds to residues 917–919, 921–922, 924–926, 928–929, 931–933, 935–936, and 938–940 (QES-TT-STA-GK-QDV-NQ-AQA) of the S protein. This epitope site is similar to the HRN1 site, but contains only residues which are observed in the exterior surface positions of HRN (Fig. 2C) (i.e., no residues located within the hydrophobic core positions) (Fig. 1). These residues only flank the HRC-binding site on HRN. Thus analysis of the elicited anti-sera against these epitopes should evaluate not only the accessibility of the HRN coiled-coil region in the S protein but also whether HRN specific antibodies must directly compete for the same residues on HRN where HRC binds in order to block the conformational change from a pre-fusogenic to a post-fusogenic state or whether binding within the general area is sufficient for inhibition.
Fig. 2.

Molecular graphics representation of the SARS-CoV S HRN (residues 896–972, blue) and molecular modeling representation of HRC (residues 1150–1185, red) trimeric coiled-coil domains. Residues chosen to be incorporated into the coiled-coil template sequence are colored in yellow (A, C, E, and G). Accessibility of the chosen surface residues when in the six-helix bundle conformation of HRN/HRC is also shown (B, D, F, and H). The molecular graphics of the HRN trimeric coiled and six-helix bundle structure are based on the X-ray crystallographic structure (pdb Accession No. 1WNC ) shown in ribbon representation (center). The HRC trimeric coiled-coil model was based on the X-ray structure of a trimeric GCN4 coiled-coil (pdb Accession No. 1GCM ). Models were prepared using the molecular graphics program VMD, version 1.8.3 (Humphrey et al., 1996). In the six-helix bundle structures (B, D, F, and H) the HRC helices are antiparallel to the HRN helices as shown (center).
Similarly, two sites were chosen within the HRC coiled-coil region of the SARS-CoV S protein for incorporation into the template peptide. HRC1 (see Fig. 1 for sequence details) corresponds to residues 1155–1157, 1159–1160, 1162–1164, 1166–1167, 1169–1171, 1173–1174, and 1176–1178 (residues NAS-VN-QKE-DR-NEV-KN-NES), while HRC2 corresponds to residues 1162–1164, 1166–1167, 1169–1171, 1173–1174, 1176–1178, 1180–1181, and 1183–1185 (residues QKE-DR-NEV-KN-NES-ID-QEL). Both of these epitope sites include the centrally located small helical structure observed in the HRC region when bound to the HRN coiled-coil (Figs. 2 center, F, and H) as well as residues which extend further either towards the N-terminus (for HRC1) or the C-terminus (for HRC2) of this site. In both cases, the residues chosen in the HRC region represent those expected to exist in the surface projecting b, c, e, f and g heptad positions of the trimeric coiled-coil. Thus analysis of both of these anti-sera will allow us to evaluate whether antibodies elicited to different locations within HRC are different in either their ability to bind to the pre- or post-fusogenic state of S-protein or cause an inhibitory effect.
3.3. CD analysis of the immunogenic peptides
The structures of the four immunogen peptides were assessed by circular dichroism (CD) spectroscopy (Fig. 3 A). The CD spectra showed characteristic double minima at 208 and 222 nm and a maximum at ∼196 nm, typical of α-helical structure. The molar ellipticity at 222 nm ranged from −31 000° to −35 000° (Table 1 ) indicating the peptides exist in a fully α-helical state. The theoretical molar ellipticity for a fully helical 31-residue peptide is −34 070 (Gans et al., 1991). To assess the stability of the HR immunogen peptides, each peptide was thermally denatured and its change in structure from a fully folded α-helical state to an unfolded state monitored at 222 nm. As shown in Fig. 3B, all of the peptides displayed very stable thermal denaturation profiles. The temperature denaturation midpoints for the four disulfide bridged peptides were calculated to be >95 °C in benign buffer conditions (Table 1). Only when the disulfide bridge between the two-strands of the template peptides was reduced using dithiothreitol (DTT) could full temperature denaturation profiles be observed. Thus, the de novo designed α-helical coiled-coil template is more than adequate for presenting the incorporated HRN and HRC residues in an α-helical structure for immunization.
Fig. 3.

(A) Far UV CD spectra of the SARS-CoV S immunogen peptides. Spectra were recorded in a 0.1 M KCl, 0.05 M K2HPO4, pH 7 buffer. Peptide concentrations were 100 μM. (B) Temperature denaturation profiles of the helical template immunogen peptides. Denaturations were monitored by CD at 222 nm in a 0.1 M KCl, 0.05 M K2HPO4, pH 7 buffer. For analysis of reduced peptides, the above buffer also contained 2 mM dithiothreitol (DTT). Peptide concentrations were 100 μM.
Table 1.
Ellipticity and stability of the immunogen peptides studied
| Peptide namea | [θ]222b (degrees cm2 dmol−1) | T1/2c (°C) |
|---|---|---|
| HRN1 | −36 500 | >95 (82) |
| HRN2 | −35 100 | >95 (76) |
| HRC1 | −30 000 | >95 (71) |
| HRC2 | −31 200 | >95 (56) |
Name of each peptide studied. The sequence of each peptide is shown in Fig. 1.
The mean residue molar ellipticity at 222 nm was measured at 22 °C in benign buffer (0.1 M KCl, 0.05 M K2PO4, pH 7).
T1/2 is the transition midpoint temperature at which there is a 50% decrease in molar ellipticity [θ]222 compared with the fully folded peptide determined by CD at 5 °C. Values in brackets represent the T1/2 temperature for the reduced form of the peptide.
3.4. Polyclonal rabbit HRN and HRC anti-sera
To generate antibodies against the HR peptides, each of the immunogen peptides (HRN1, HRN2, HRC1, and HRC2) was immunized into three New Zealand White rabbits each according to standard animal protocols. Initial injections were done in complete Freund’s adjuvant while subsequent booster shots (3) were done in incomplete Freund’s adjuvant. After day 58, final sera was collected and analyzed for specificity, affinity and its ability to neutralize the SARS-CoV.
3.5. Specificity of the HRN and HRC anti-sera
Initially each of the crude HR anti-sera was screened for its ability to bind to plated HRN(916–950)-BSA or HRC(1150–1185)-BSA native sequence peptide conjugates in a qualitative ELISA. These conjugates represent the linear forms of the native HR regions. As shown in Fig. 4 , each of the anti-sera showed good signal response at a 1:5000 dilution for binding to its respective native SARS-CoV S protein sequence indicating that the peptides were highly immunogenic. HRN1 and HRN2 elicited antibodies that bound specifically to the HRN(916–950)-BSA conjugate and not to the HRC(1150–1185)-BSA conjugate. Correspondingly, HRC1 and HRC2 antibodies bound to the native HRC(1150–1185)-BSA conjugate and not to the HRN(916–950)-BSA conjugate peptide. No antibody binding was observed from pre-immune sera, and BSA binding was negligible (absorbance less than 0.05, data not shown).
Fig. 4.
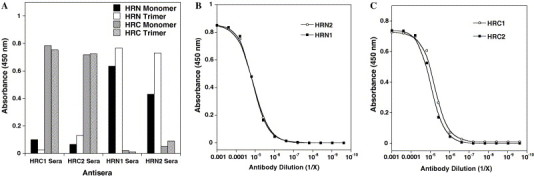
(A) ELISA reactivity of anti-HR sera with a panel of HR synthetic peptides. HRN monomer denotes single stranded HRN(916–950) peptide conjugated to BSA. HRN trimer denotes covalently cross-linked trimeric HRN(902–950) peptide conjugated to BSA. HRC monomer denotes HRC(1150–1185) peptide conjugated to BSA. HRC trimer denotes covalently cross-linked trimeric HRC(1150–1185) peptide conjugated to BSA. Sera were diluted 1:5000 and 0.2 μg/well of peptide conjugate plated. (B) HRN1 and HRN2 sera binding to covalently cross-linked trimeric HRN(902–950) peptide. (C) HRC1 and HRC2 sera binding to covalently cross-linked trimeric HRC(1150–1185) peptide. For (B and C), serial (fourfold) dilutions of the sera were applied to the peptide (0.2 μg/well) and the amount of bound antibodies measured by an ELISA assay as described in Section 2. The background was estimated by the amount of antibody bound to BSA and subtracted.
To test the ability of the anti-sera to bind to the trimeric state of the HR peptides (which is presumed to be the oligomeric state of the HR regions in the S protein in the pre-fusogenic state), covalently linked HRN(902–950)-trimeric-BSA and HRC(1150–1185)-trimeric-BSA conjugates were plated and analyzed as above. Similar to that observed above, the HRN1, HRN2, HRC1, and HRC2 anti-sera demonstrated good specificity in recognizing the corresponding native sequence peptide which contained the epitope site versus the alternate HR region. The HRN1 and HRN2 anti-sera also showed a slightly greater ability to bind to the trimeric state of the HRN peptide (plated at a similar concentration to the monomeric HRN-BSA conjugate above) suggesting that conformation of this epitope site is indeed important for these antibodies to bind. The HRC anti-sera appeared to bind equally well to both HRC peptide conjugates (i.e., the monomeric and trimeric states). Titer analysis of each of the anti-sera also showed that each of the HR peptides elicited equally good IgG responses against the antigens and that all animals responded equally (data not shown). The estimated 50% titers (i.e., the dilution at which 50% of the antigen is bound using a concentration of 0.2 μg/well of antigen) for antibodies directed against HRC- and HRN-trimeric peptide conjugates (Fig. 4, panels B and C) were 0.6 × 105, 0.5 × 105, 1.9 × 105 and 1.2 × 105 for HRN1, HRN2, HRC1, and HRC2 sera, respectively. Finally, it is interesting to note that in the analysis of the specificity of the sera, we did see a very low level amount of cross-reactivity between the HRN and HRC sera for the different peptides. Because there is no sequence identity between the two HR regions nor the template peptide other than the 3–4 hydrophobic repeat, we can only surmise that despite the high stability of the template immunogen peptides in aqueous solution, a small amount of the peptides may have unfolded in the oil associated with the use of the Freund’s adjuvant facilitating antibodies to be elicited against the coiled-coil core.
3.6. Kinetic analysis of the anti-sera
To determine the relative-binding affinity of the individual anti-sera for the native HR regions, each of the anti-sera was passed over biosensor surfaces immobilized with either single stranded HRN(916–950) or HRC(1150–1185) peptides or trimeric HRN(902–950) or HRC(1150–1185) peptides and binding monitored by changes in the surface plasmon resonance signal on a BIAcore 3000. In these experiments, biosensor surfaces were prepared with a surface density of ∼50–100 RU (response units) of peptide and a flow rate of 70 μl/min was used during analysis. Figs. 5 A and B, display representative sensorgrams of HRN1 and HRC1 purified anti-sera binding to HRN and HRC trimeric peptides, respectively. Passage of the HRN1 and HRN2 anti-sera over the biosensor surface immobilized with single stranded HRN(916–950) peptide showed no binding interactions at any of the sera concentrations analyzed (1.6 nM to 1 μM). The apparent absence of binding appears to be a consequence of the conformational specificity of the anti-sera, since the same sera displayed positive-binding sensorgrams when passed over a surface immobilized with covalently linked trimeric HRN(902–950) peptide (Fig. 5A). The latter sensorgrams for each sera were globally fit using a simple 1:1 (Langmuir) binding model and the apparent rate constants (k on and k off) and equilibrium dissociation constant (K d) for the binding interactions derived (Table 2 ). For the HRN2 sera, fitting of the data showed a high-binding affinity (K d = 12 × 10−9 M) derived from a relatively fast association rate and a slow dissociation rate (k on = 2.9 × 104 M−1 s−1 and k off = 3.4 × 10−4 s−1). The ability of the antibodies to bind quickly to the trimeric HRN coiled-coils shows that the HRN2 epitope site is easily accessible and recognizable. In contrast, binding of the HRN1 sera showed a lower affinity (K d = 1.5 × 10−5 M), arising from a slower association rate (k on = 1.4 × 102 M−1 s−1) and faster off rate (2.1 × 10−3 s−1). Since several of the HRN1 epitope residues selected were residues located in the hydrophobic core of the HRN coiled-coil (observed in the high resolution six-helix crystal structure; Fig. 2) it is tempting to speculate that the lower association rate arises as a result of these residues not being accessible in the folded trimeric coiled-coil structure but being accessible in the template coiled-coil immunogen. If so, this would suggest that the trimeric HRN peptide coiled-coil structure is similar to the HRN coiled-coil structure observed in the six-helix bundle structure (post-fusion state) and that this structure exists even in the absence of the HRC peptide (pre-fusion state). Regardless of the reason, the data clearly indicates that the HRN1 residues chosen are not as ideal as the HRN2 residues for generating high affinity antibodies against the correctly folded trimeric HRN coiled-coil.
Fig. 5.
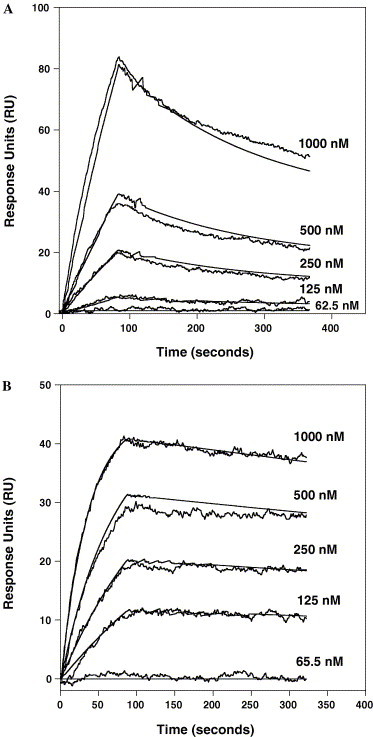
Kinetic analysis of HRN1 and HRC1 purified sera binding to immobilized HRN(902–950)-trimer peptide (A) and immobilized HRC(1150–1185)-trimer (B) at various IgG concentrations, respectively. The binding reactions are expressed as response units (RU) as a function of time. Total IgG concentration is given to the right of each trace in nM. Each analysis consisted of a 110 μl injection association phase in PBS buffer at a flow rate of 70 μl/min, followed by a 240 s dissociation phase in PBS buffer. The fit lines (smooth lines) obtained from global analysis of the data set are shown overlaid with each curve.
Table 2.
Summary of affinities and rate constants for anti-HR IgG binding to immobilized HR peptides
| Anti-seraa | Immobilized peptideb | kon (M−1s−1)c | koff (s−1)c | Kd (M)d |
|---|---|---|---|---|
| HRN1 | HRN(916–950)monomer | —e | — | — |
| HRN2 | HRN(916–950)monomer | — | — | — |
| HRN1 | HRN(902–950)trimer | 1.4 × 102 | 2.1 × 10−3 | 1.5 × 10−5 |
| HRN2 | HRN(902–950)trimer | 2.9 × 104 | 3.4 × 10−4 | 12.0 × 10−9 |
| HRC1 | HRC(1150–1185)monomer | 5.8 × 104 | 5.6 × 10−4 | 9.7 × 10−9 |
| HRC2 | HRC(1150–1185)monomer | 4.0 × 104 | 2.0 × 10−4 | 5.0 × 10−9 |
| HRC1 | HRC(1150–1185)trimer | 6.5 × 104 | 7.5 × 10−4 | 12.0 × 10−9 |
| HRC2 | HRC(1150–1185)trimer | 5.2 × 104 | 3.4 × 10−4 | 6.5 × 10−9 |
Name of the anti-sera passed over the biosensor surface.
Name of the peptide attached to the biosensor surface. The sequence of each peptide is shown in Fig. 1; (monomer) indicates the peptide is immobilized as a single strand; (trimer) indicates the peptide is immobilized as a covalently linked three-stranded (trimer); see Section 2.
kon and koff constants were obtained from non-linear least squares global fitting of the respective sensorgrams using Biaevaluation software version 4.1 and a simple bimolecular binding model.
Kd (M) is the equilibrium dissociation constant derived from the rate constants koff and kon by the equation Kd = koff/kon.
(—) denotes no and/or low binding which precluded accuracy of rate constant determination and calculation of an equilibrium dissociation constant.
The same experiments were repeated for HRC1 and HRC2 sera binding to the monomeric and trimeric HRC(1150–1185) peptide surfaces. Both sera displayed strong binding interactions with both the monomeric and trimeric surfaces (trimer binding shown in Fig. 5B). Global fit analysis of the sensorgrams showed that HRC1 and HRC2 antibodies could bind with almost equal affinity to the two different surfaces (Table 1). The apparent equilibrium constants were K d(monomeric) = 9.7 × 10−9 M and K d(trimeric) = 12.0 × 10−9 M for HRC1 and K d(monomeric) = 5.0 × 10−9 M and K d(trimeric) = 6.5 × 10−9 M for HRC2. Each of the sera showed that the high affinity equilibrium value was generated from fast on rates (k on = 4–6.5 × 104 M−1 s−1) and slow off rates (k off = 2–7.5 × 10−4 s−1). The fact that the HRC anti-sera can bind to the monomeric HRC state as well as it can bind to the trimeric state likely reflects the affinity of the antibodies and the propensity of the peptide to be helical. That is, an antibody with high affinity can induce monomeric peptides which are unstructured into a helical conformation (Lu and Hodges, 2002). Second, the HRC peptide (1151–1185) has a high propensity to form a helical structure as demonstrated by its ability to form a trimeric structure on its own (Tripet et al., 2004). This is in contrast to the HRN(916–950) peptide which was observed to be in a completely random coil structure at all concentrations analyzed ((Tripet et al., 2004) and data not shown).
3.7. Ability of the anti-sera to bind the six-helix bundle fusogenic state
We next tested whether the elicited HRN and HRC antibodies could bind to their respective epitope sites after the two coiled-coil regions (HRN and HRC) were allowed to interact to form the anti-parallel hetero-six-helix bundle structure (i.e., the fusogenic state, Fig. 2, center). To determine this, we carried out comparative binding studies. For the HRN anti-sera, we first immobilized trimeric HRN(902–950) molecules to the surface of a CM5 biosensor chip and then passed HRC peptide over the surface to allow binding and formation of the six-helix bundle structure. Next, HRN1 and HRN2 anti-sera were individually passed over the surface and the amount of binding after a 70 s association phase recorded. Positive-binding sensorgrams were observed for both HRN1 and HRN2 anti-sera (data not shown). When the individual sensorgrams were compared to sensorgrams observed for the same sera binding the surface in the absence of HRC peptide, the data showed that both anti-sera retained the ability to achieve ∼90–95% of HRN binding in the presence of the HRC peptide. Alternatively, when the anti-sera were allowed to bind first to the trimeric HRN(902–950) molecule and then followed by addition of the HRC peptide, neither HRN1 nor HRN2 antibodies was able to be effectively competed by the free peptide (i.e., only a negligible drop, 5–10% in RU units, was observed upon addition of the HRC peptide).These same results were also observed in an ELISA format where trimeric HRN(902–950) molecules were plated and HRC peptide added. The titration curves for HRN1 and HRN2 anti-sera showed only small shifts to higher antibody concentration for similar binding in the presence of the HRC peptide relative to titration curves observed in the absence of HRC peptide (Fig. 6 A). Thus, despite the close proximity of the two epitope sites to the HRC binding site on HRN, the antibodies raised from these two epitopes appear to be utilizing mostly the exterior position residues on HRN for binding which are still exposed even after HRC binding (Figs. 2B and D). Further, these results suggest that these antibodies will be able to bind both the pre- and post-fusogenic states of the SARS-CoV S protein (if accessible).
Fig. 6.

ELISA reactivity of anti-HR sera against plated HRN(902–950)/HRC(1150–1185) complex (six-helix bundle state). The average of duplicate runs is reported.
The HRC sera were screened in an analogous manner. For HRC1 anti-sera binding on the BIAcore, HRC1 displayed strong binding sensorgrams to the covalently cross-linked trimeric HRC(1150–1185) peptide. However, when HRC peptide was loaded onto an immobilized HRN(902–950) trimer surface (as described above to produce the six-helix bundle fusogenic state), no binding of the anti-sera was observed. This observation was also seen in the ELISA format experiment where HRN(902–950) trimeric molecules were plated, HRC peptide was added, and the anti-sera titered in these wells. All of the wells in which HRC peptide was added to HRN peptide showed little if any binding compared to native HRC trimer plated wells (Fig. 6B). Thus, the HRC1 anti-serum is specific only for the pre-fusogenic state of HRC.
In the case of HRC2 sera, these antibodies showed almost equal binding to the covalently cross-linked trimeric HRC(1150–1185) peptide as to the HRC peptide when added to the immobilized HRN(902–950)-trimeric molecule (to generate the six-helix bundle state). Similar results were also seen in ELISA format. The titration curves for HRC2 showed only a small shift to higher antibody concentration for similar binding for HRC peptide bound to HRN relative to titration curves observed for HRC trimeric peptide alone (Fig. 6B). Therefore, in contrast to anti-HRC1 serum, anti-HRC2 serum is not sensitive to any conformational changes in the HRC region and can bind to HRC in both the pre- and post-fusogenic states.
3.8. Ability of the antibodies to bind the SARS-CoV S protein
To test the ability of the anti-sera to bind full length denatured S protein, whole cell lysates of CHO cells with (or without) expressed S protein were separated by gel electrophoresis and transferred to nitrocellulose membranes for Western blotting. For all anti-sera, no bands were observed in the blank and control well lanes (CHO cell lysates and mock transfected CHO cell lysates) whereas individual bands were observed in lanes which contain expressed whole S protein (data not shown). The same band was located by all anti-sera and the location of the band corresponded to a molecular weight of ∼190,000 Da. The diffuse nature of the band supports the prediction of putative glycosylation sites within the S protein but this did not appear to affect binding by the anti-sera.
To test whether the HR specific anti-sera can recognize native soluble SARS-CoV S protein, soluble S protein 1180K (which corresponds to the S-protein residues 1–1180, described previously (Jeffers et al., 2004)) was plated in 96 wells and binding by HR anti-sera analyzed in an ELISA format. Both HRN1 and HRN2 anti-sera showed almost equal abilities to bind to the S protein with titer dilution midpoints equal to ∼1 × 104 (Fig. 7 A). Similarly, both of the HRC anti-sera (HRC1 and HRC2) demonstrated strong binding interactions with the S protein. Titration analysis (Fig. 7B) showed at least a 10-fold greater titer dilution midpoint for the HRC anti-sera compared to the HRN anti-sera, and the HRC1 anti-sera appeared to bind better to the S protein than HRC2 serum. It should be noted that the slightly lower midpoint titer of the HRC2 serum could be a result of the absence of the last five residues of its epitope in this construct of S. Taken together, these results indicate both of the HRN and HRC epitope sites are accessible to antibody interaction within the denatured and soluble conformations of S.
Fig. 7.

ELISA reactivity of anti-HR sera against plated soluble S protein 1180K. The average of triplicate runs is reported.
3.9. Ability of the antibodies to bind to SARS-CoV S protein on surface of CHO cells
We next tested whether the antibodies raised against the HR immunogens were able to recognize native SARS-CoV S protein displayed on the surface of Chinese hamster ovary (CHO) cells. These experiments were carried out by performing indirect immunofluorescence experiments under non-permeabilized conditions. CHO cells were grown till confluent and then transfected with plasmid pcDNA3.1-SARS-S Δ19 (which encodes for full length SARS-CoV S protein but lacking the C-terminal 19 residues) 24–36 h was allowed for the S protein to be expressed and transported to the cell surface following which the cells were released from the plates, divided and incubated with HR specific anti-sera (1°) and then PE-conjugated goat anti-rabbit antibody (2°). The ability of the HR sera to bind the cell surface S protein was determined by fluorescence activated cell sorting (FACS) and the results calculated as a percent increase in cell fluorescence relative to 2° binding only of the transfected cells. Interestingly, despite the HRN1 and HRN2 anti-sera indicating an ability to bind to the soluble S protein and full length denatured S protein expressed in CHO cells for Western analysis, little or no binding was observed by the anti-HRN1 and anti-HRN2 antibodies to S protein expressed on the cell surface of CHO cells (i.e., no significant increase in the PE fluorescence intensity was observed relative to the mock transfected cells, Fig. 8 ). These results indicate that the native S protein conformation is such that the HRN coiled-coil, at least in the vicinity of the HRC binding site, is not accessible to antibody. In contrast, the HRC1 and HRC2 anti-sera clearly detected the native SARS-CoV S protein on the cell surface. Further, comparison between the two anti-sera showed that the HRC1 anti-sera was ∼1.5-fold more effective at binding to the trimeric membrane anchored S protein. Since our earlier analysis had shown that the HRC1 and HRC2 anti-sera are almost equal in affinity and concentration, we would have to surmise that this difference in binding may be a function of slight differences in accessibility of the two sites within the HRC coiled-coil.
Fig. 8.
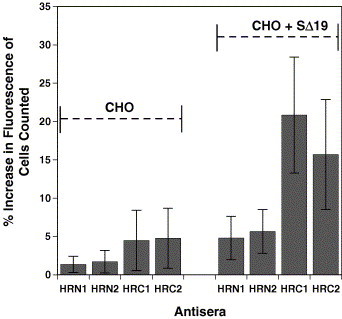
Flow cytometric analysis of cell surface S glycoprotein. CHO cells were stained using HRN1, HRN2, HRC1 and HRC2 sera and a secondary phycoerythrin (PE)-conjugated anti-rabbit antibody. Mock transfected CHO cells are shown on the left (first four columns) and CHO cells transiently expressing the wild type S protein (SΔ19) are shown on the right (columns five to eight). Cells were analyzed using a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA), and propidium iodide-staining (dead) cells were excluded. Five thousand cells were counted per analysis. The percentage of cells with an increase in fluorescence relative to mock cells stained with secondary antibody only (representing background) is shown. The average of triplicate analysis with error bars indicating the standard deviation is reported.
3.10. Ability of the antibodies to neutralize SARS-CoV infectivity
Finally, after determining the relative affinity and specificity of all of the anti-sera against the native form of the SARS-CoV S protein, all sera were further assessed for the ability to inhibit virus entry. For this assay, 50 PFU (plaque forming units) of SARS-CoV stock (Urbani strain, Accession No. AY278741) was mixed with serially diluted serum (31 μg/ml to 2 mg/ml total IgG) for 1 h at 37 °C and then added to Vero E6 target cells in 96 well tissue culture plates. After incubation at 37 °C for 1 h , the supernants were removed and fresh medium was added. On day 3 after infection, the cytopathic effects (plaques) were recorded. As seen in Fig. 9 , no virus neutralization was observed for either HRN1 or HRN2 anti-sera compared to pre-immune rabbit sera. This result is in agreement with the cell surface binding assay which also showed no ability of the anti-sera to bind to the surface expressed S protein. The neutralization result further supports the idea that the HRN coiled-coil (in the vicinity of the HRC binding site) is not accessible to antibody binding in its native pre-fusion state. In contrast, both of the HRC sera which did demonstrate S-protein binding on the cell surface did neutralize the infectivity of SARS-CoV. Moreover, titering of the individual anti-sera showed that the HRC1 antiserum was a far more effective inhibitor of SARS-CoV entry compared to the HRC2 antiserum, i.e., the HRC1 antiserum reduced the viral infectivity level to almost 0 above an IgG concentration of 0.5 mg/ml compared to 2.0 mg/ml for HRC2 antiserum (Fig. 9). Since previous analysis had shown that the two HRC sera were almost equal in affinity and concentration, the difference in virus neutralization activity must result from either a difference in accessibility of HRC in the native state S protein or the ability to inhibit progression to the fusion competent six-helix bundle state.
Fig. 9.

Neutralization activity of anti-HR sera on in vitro SARS-CoV infection of Vero E6 cells. Each point represent the mean of triplicate determinant, error bars represent variance of data. HRC1 and HRC2 significantly inhibited the infection of SARS-CoV into Vero E6 cells and the inhibition was effected in a dose-dependent manner, whereas the control sera and HRN1 and HRN2 anti-sera did not cause any effect under the same conditions.
4. Discussion
In the present study, we have investigated whether antibodies directed against the two coiled-coil domains in the spike protein of SARS-CoV can arrest virus entry. To elicit antibodies which would specifically recognize the α-helical structure of the HR regions, we employed the use of a novel two-stranded α-helical coiled-coil template strategy. Four antigenic sites were chosen and displayed in the coiled-coil template system, two from within the HRN region, and two from within the HRC region. We showed that the four engineered HR peptides were highly immunogenic and raised high titer antibody responses in immunized rabbits. Qualitative analysis showed that each of the anti-sera to the HRN and HRC regions could specifically react with its corresponding trimeric coiled-coil S peptide and the soluble S protein in ELISA and Western assays. Thus, despite the antigens being presented to the immune system as two-stranded coiled-coils, the antibodies elicited do not appear to be oligomer specific. Importantly, BIAcore analysis demonstrated clearly that the HRN1 and HRN2 antibodies were conformation (helix)-specific since positive binding sensorgrams were obtained when HRN1 and HRN2 sera were passed over the immobilized trimeric HRN peptide surface, but showed no binding interactions for the linear monomeric single strand surface. Although the HRC anti-sera did not show this same specificity, the ability of the HRC1 and HRC2 sera to react equally with different states (monomeric and trimeric) of HRC more likely reflects the ability of HRC peptide to readily adopt an α-helical structure. As seen in our earlier study, the HRC peptide (1151–1185) folded into a partially helical structure versus HRN peptide (916–950) which was unfolded (Tripet et al., 2004). BIAcore analysis also demonstrated that each antiserum, with the exception of HRN1, binds with high affinity (5–12 nanomolar) to the HRN or HRC peptide trimer in its coiled-coil state. The significantly lower affinity for HRN1 (15 μM) is attributed to the fact that some of its antigenic residues are not readily accessible to the antibodies.
4.1. Anti-sera binding to the hexameric state
We also investigated the ability of the elicited anti-sera to bind to the six-helix bundle structure of HRN/HRC (generated from interacting peptides HRN 902–950 with HRC 1150–1185). The results showed that HRN1 and HRN2 anti-sera could bind concomitantly with the HRC peptide bound to HRN. It did not matter whether the HRC peptide was added before or after binding of the HRN anti-sera to HRN. These observations indicate that the binding sites on HRN for the anti-HRN antibodies and the HRC peptide are distinct, and that the presence of HRC bound to HRN does not even sterically affect binding of the HRN specific antibodies. In choosing the residues for the HRN antigens, we chose residues which existed in the exterior surface positions of the coiled-coil (Figs. 2A and C), and these residues are still largely exposed even in the presence of bound HRC (Figs. 2B and D) (Ingallinella et al., 2004, Supekar et al., 2004, Xu et al., 2004, Xu et al., 2004). Although the HRN1 antigen did contain some residues which were located in the hydrophobic core of the HRN coiled-coil which would definitely be buried by the binding of HRC, it would appear that the anti-HRN1 antibodies do not utilize these residues for binding. Similarly, the HRC2 antiserum also bound to the six-helix bundle structure. The HRC2 epitope spans the middle and C-terminus of the HRC region (Fig. 1, Fig. 2G). The observation of continued binding of the anti-HRC2 antibodies to the HRC peptide in the six-helix bundle structure where the HRC peptide partially unwinds at either end (Fig. 2 center) indicates that these antibodies must be binding to the small centrally located helical region within HRC. Thus, despite the larger size of the antigenic peptide most of the polyclonal anti-HRC2 serum would seem to be focused only to the small central portion within the antigen site. In contrast, the HRC1 antiserum did not bind to the six-helix bundle state. The HRC1 epitope site spans the middle and N-terminus of the HRC region (Fig. 1, Fig. 2E). The absence of binding of this antiserum to the six-helix bundle indicates that this antiserum is clearly dependent upon the α-helical conformation of the N-terminus of the HRC region. This region of HRC adopts an extended conformation when bound to HRN. Thus, analysis shows that only the HRC1 antiserum appears to be uniquely sensitive to the fusogenic state of the S2 domain.
4.2. Structural properties of S
The reactivity of the various anti-sera against full length S raises interesting points regarding the native structure of the SARS-CoV S protein. Despite the observation that both HRN1 and HRN2 anti-sera bind to the trimeric coiled-coil HRN peptide, neither antiserum was able to bind to the cell surface-exposed trimeric S protein on CHO cells. This suggests that the HRN site (corresponding to residues 916–950) is not accessible in the native pre-fusion state of S. In Western analysis, we observed the anti-sera could bind to the same S-protein produced in CHO cells when denatured, indicating that modification of the epitopes sites (e.g., glycosylation) was not a factor, but rather accessibility must be preventing binding. Interestingly, although we did see binding by the HRN anti-sera to the soluble S protein in an ELISA format, this binding could have been a result of a conformational change in S, (i.e., it is uncertain to what extent the S protein maintains its native state structure when immobilized on plastic). It should also be noted that the soluble S protein contained a lower amount of glycosylation than that observed for the S-protein produced in CHO cells, and the level of neighboring glycosylation could affect antibody recognition. Steric blocking by either glycosylation, the interhelical domain, the S1 domain or any combination of these regions could be responsible for the inaccessibility of the HRN site. It would seem appropriate that the S protein would hide the HRC binding site on HRN in order to prevent premature conversion from a pre-fusogenic to post-fusogenic state prior to binding of the ACE2/CD209L receptor. In contrast, both of the HRC epitopes elicited anti-sera which could react with the S-protein in the pre-fusogenic state. Thus, the HRC coiled-coil region of S is accessible. The binding of the two HRC anti-sera also suggests that the HRC region is not glycosylated. (i.e., there are two potential N-glycosylation sites within HRC (Asn-1155 and Asn-1176) which would have blocked binding had they been glycosylated). Further, the inhibitory effect observed for these sera also implies that the HRC region is readily accessible even on the virus envelope surface despite its close proximity to the viral membrane.
4.3. Virus neutralization assays
To determine if any of the HR specific anti-sera could block SARS-CoV infection, in vitro virus neutralization assays were carried out in Vero E6 cells. Our results showed that neither HRN1 nor HRN2 specific antiserum could inhibit virus entry. Thus the HRN coiled-coil regions analyzed did not show the potential for eliciting neutralizing antibodies. These results are in good agreement with recent studies by Duan et al., 2005, Keng et al., 2005, Lai et al., 2005, Lip et al., 2006 which have mapped the location of neutralizing antibodies generated from mice immunized with truncated sections of the S2 domain. These studies show no neutralizing antibodies corresponding to the HRN region. One possibility for the inability of the anti-HRN1 and HRN2 sera to neutralize virus infectivity is that the anti-sera were unable to bind to HRN because this site is inaccessible on the trimer (since we also observed no binding by these same anti-sera to the S protein in the cell surface binding assays). However, because binding assays on the live SARS virus were not carried out, we cannot conclusively rule out the possibility that the HRN specific antibodies do bind to the HRN region of S in the viral envelope but are just unable to block binding of the HRC region to HRN and thus prevent the S protein from switching to the fusion competent six-helix bundle state which drives membrane fusion and viral infectivity. It should be noted that two linear peptides, D07 and D08 which correspond to HRN residues 927–937 and 942–951, respectively, of the S protein, which overlap in sequence with the HRN1 and HRN2 antigens were shown to compete with cross-reactive antibodies in convalescent serum of SARS infected patients which induce a cytotoxic effect on A549 lung epithelial cells and are implicated in the auto-immune response leading to enhancement of SARS-CoV infection (Lin et al., 2005). Thus, the HRN region is not a viable region for vaccine development.
In contrast, both of the HRC specific anti-sera (HRC1 and HRC2) inhibited SARS-CoV entry in a dose dependent manner. Thus, the HRC region is a site capable of eliciting neutralizing antibodies. These results are in agreement with the findings of two other groups which have recently described the mapping of neutralizing monoclonal antibodies generated from immunization of recombinant r268 (corresponding to residues 268–1255 of SARS-CoV S) or truncated S fragment 1029–1192 in mice.
Using r268 as an immunogen, Lai et al. (2005) identified that all of the neutralizing monoclonal antibodies recognized a 15-residue peptide (CB-119) corresponding to residues 1143–1157 (SPDVDLGDISGINAS). The last three residues exist within the HRC1 antigen site (the rest of the sequence is slightly N-terminal to the HRC1 antigen site). Lip et al. (2006) identified four neutralizing monoclonal antibodies which mapped to residues 1091–1130, 1111–1130, 1151–1170, and 1151–1192 of the S protein. The first two are N-terminal in sequence to the HRC1 antigen sequence, while the latter two regions directly overlap the HRC1 and HRC2 antigen sequences used herein. Thus, it would appear that the HRC region and residues slightly N-terminus to it represent the principle site for eliciting neutralizing antibodies within the S2 domain of the S protein.
We also observed that the HRC1 antiserum was a more effective inhibitor of viral infectivity than anti-HRC2. We have shown that HRC1 and HRC2 anti-sera displayed similar titers and relative binding affinities, thus eliminating obvious concentration and affinity differences. Interestingly, the HRC1 antiserum can only bind the pre-fusogenic state of HRC, i.e., the HRC1 antiserum did not recognize the six-helix bundle conformation (fusion state) whereas HRC2 antiserum could bind to both states. It is tempting to speculate that the greater virus neutralization activity of HRC1 antiserum reflects its ability to arrest the S protein in the pre-fusion state, whereas the weaker virus neutralization activity observed for HRC2 antiserum reflects it inability to prevent progression to the six-helix bundle fusion-state because it can also recognize and bind to this structural state. However, we did see a slight difference in the ability of the two anti-sera to bind to the cell surface displayed S protein, suggesting that accessibility difference could also be the cause of this inhibitory difference.
In summary, we have shown that SARS-CoV S sequences incorporated into an α-helical two-stranded coiled-coil template can effectively raise conformation-dependent antibodies which recognize the native S protein. Since HRC1 antiserum showed the greatest virus neutralization activity, the HRC1 peptide antigen will now be further investigated as a potential immunogen for the development of therapeutic neutralizing antibodies against SARS-CoV and for vaccine development to elicit protective humoral immunity.
Acknowledgments
This work was support by Program Project Grant P01-A1059576 from the National Institutes of Health and the John Stewart Chair in Peptide Chemistry (to R.S.H). We thank the Flow cytometry and Biophysical core facilities at the University of Colorado at Denver and Health Sciences Center and Eric Mossel from Colorado State University. SAJ was supported by NIH postdoctoral training grant 1 T32 AI07587.
References
- Bisht H., Roberts A., Vogel L., Bukreyev A., Collins P.L., Murphy B.R., Subbarao K., Moss B. Severe acute respiratory syndrome coronavirus spike protein expressed by attenuated vaccinia virus protectively immunizes mice. Proc. Natl. Acad. Sci. USA. 2004;101:6641–6646. doi: 10.1073/pnas.0401939101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos E.C., Heijnen L., Luytjes W., Spaan W.J. Mutational analysis of the murine coronavirus spike protein: effect on cell-to-cell fusion. Virology. 1995;214:453–463. doi: 10.1006/viro.1995.0056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch B.J., Martina B.E., Van Der Zee R., Lepault J., Haijema B.J., Versluis C., Heck A.J., De Groot R., Osterhaus A.D., Rottier P.J. Severe acute respiratory syndrome coronavirus (SARS-CoV) infection inhibition using spike protein heptad repeat-derived peptides. Proc. Natl. Acad. Sci. USA. 2004;101:8455–8460. doi: 10.1073/pnas.0400576101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch B.J., van der Zee R., de Haan C.A., Rottier P.J. The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J. Virol. 2003;77:8801–8811. doi: 10.1128/JVI.77.16.8801-8811.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukreyev A., Lamirande E.W., Buchholz U.J., Vogel L.N., Elkins W.R., St Claire M., Murphy B.R., Subbarao K., Collins P.L. Mucosal immunisation of African green monkeys (Cercopithecus aethiops) with an attenuated parainfluenza virus expressing the SARS coronavirus spike protein for the prevention of SARS. Lancet. 2004;363:2122–2127. doi: 10.1016/S0140-6736(04)16501-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullough P.A., Hughson F.M., Skehel J.J., Wiley D.C. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature. 1994;371:37–43. doi: 10.1038/371037a0. [DOI] [PubMed] [Google Scholar]
- Chan D.C., Fass D., Berger J.M., Kim P.S. Core structure of gp41 from the HIV envelope glycoprotein. Cell. 1997;89:263–273. doi: 10.1016/s0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- Chan W.C., White P.D. Oxford University Press; 2000. Fmoc Solid Phase Peptide Synthesis. [Google Scholar]
- Chen J., Skehel J.J., Wiley D.C. N- and C-terminal residues combine in the fusion-pH influenza hemagglutinin HA(2) subunit to form an N cap that terminates the triple-stranded coiled coil. Proc. Natl. Acad. Sci. USA. 1999;96:8967–8972. doi: 10.1073/pnas.96.16.8967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czub M., Weingartl H., Czub S., He R., Cao J. Evaluation of modified vaccinia virus Ankara based recombinant SARS vaccine in ferrets. Vaccine. 2005;23:2273–2279. doi: 10.1016/j.vaccine.2005.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damico R.L., Crane J., Bates P. Receptor-triggered membrane association of a model retroviral glycoprotein. Proc. Natl. Acad. Sci. USA. 1998;95:2580–2585. doi: 10.1073/pnas.95.5.2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Groot R.J., Van Leen R.W., Dalderup M.J., Vennema H., Horzinek M.C., Spaan W.J. Stably expressed FIPV peplomer protein induces cell fusion and elicits neutralizing antibodies in mice. Virology. 1989;171:493–502. doi: 10.1016/0042-6822(89)90619-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosten C., Gunther S., Preiser W., van der Werf S., Brodt H.R., Becker S., Rabenau H., Panning M., Kolesnikova L., Fouchier R.A., Berger A., Burguiere A.M., Cinatl J., Eickmann M., Escriou N., Grywna K., Kramme S., Manuguerra J.C., Muller S., Rickerts V., Sturmer M., Vieth S., Klenk H.D., Osterhaus A.D., Schmitz H., Doerr H.W. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- Duan J., Yan X., Guo X., Cao W., Han W., Qi C., Feng J., Yang D., Gao G., Jin G. A human SARS-CoV neutralizing antibody against epitope on S2 protein. Biochem. Biophys. Res. Commun. 2005;333:186–193. doi: 10.1016/j.bbrc.2005.05.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert D.M., Kim P.S. Mechanisms of viral membrane fusion and its inhibition. Annu. Rev. Biochem. 2001;70:777–810. doi: 10.1146/annurev.biochem.70.1.777. [DOI] [PubMed] [Google Scholar]
- Epand R.M. Fusion peptides and the mechanism of viral fusion. Biochim. Biophys. Acta. 2003;1614:116–121. doi: 10.1016/s0005-2736(03)00169-x. [DOI] [PubMed] [Google Scholar]
- Follis K.E., York J., Nunberg J.H. Serine-scanning mutagenesis studies of the C-terminal heptad repeats in the SARS coronavirus S glycoprotein highlight the important role of the short helical region. Virology. 2005;341:122–129. doi: 10.1016/j.virol.2005.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frana M.F., Behnke J.N., Sturman L.S., Holmes K.V. Proteolytic cleavage of the E2 glycoprotein of murine coronavirus: host-dependent differences in proteolytic cleavage and cell fusion. J. Virol. 1985;56:912–920. doi: 10.1128/jvi.56.3.912-920.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher T.M., Buchmeier M.J. Coronavirus spike proteins in viral entry and pathogenesis. Virology. 2001;279:371–374. doi: 10.1006/viro.2000.0757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gans P.J., Lyu P.C., Manning M.C., Woody R.W., Kallenbach N.R. The helix-coil transition in heterogeneous peptides with specific side-chain interactions: theory and comparison with CD spectral data. Biopolymers. 1991;31:1605–1614. doi: 10.1002/bip.360311315. [DOI] [PubMed] [Google Scholar]
- Guan Y., Zheng B.J., He Y.Q., Liu X.L., Zhuang Z.X., Cheung C.L., Luo S.W., Li P.H., Zhang L.J., Guan Y.J., Butt K.M., Wong K.L., Chan K.W., Lim W., Shortridge K.F., Yuen K.Y., Peiris J.S., Poon L.L. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science. 2003;302:276–278. doi: 10.1126/science.1087139. [DOI] [PubMed] [Google Scholar]
- Guillen J., Perez-Berna A.J., Moreno M.R., Villalain J. Identification of the membrane-active regions of the severe acute respiratory syndrome coronavirus spike membrane glycoprotein using a 16/18-mer peptide scan: implications for the viral fusion mechanism. J. Virol. 2005;79:1743–1752. doi: 10.1128/JVI.79.3.1743-1752.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbury P.B., Zhang T., Kim P.S., Alber T. A switch between two-, three-, and four-stranded coiled coils in GCN4 leucine zipper mutants. Science. 1993;262:1401–1407. doi: 10.1126/science.8248779. [DOI] [PubMed] [Google Scholar]
- He Y., Jiang S. Vaccine design for severe acute respiratory syndrome coronavirus. Viral Immunol. 2005;18:327–332. doi: 10.1089/vim.2005.18.327. [DOI] [PubMed] [Google Scholar]
- Hernandez L.D., Peters R.J., Delos S.E., Young J.A., Agard D.A., White J.M. Activation of a retroviral membrane fusion protein: soluble receptor-induced liposome binding of the ALSV envelope glycoprotein. J. Cell Biol. 1997;139:1455–1464. doi: 10.1083/jcb.139.6.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes K.V. SARS coronavirus: a new challenge for prevention and therapy. J. Clin. Invest. 2003;111:1605–1609. doi: 10.1172/JCI18819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W., Dalke A., Schulten K. VMD—visual molecular dynamics. J. Mol. Graphics. 1996;14.1:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Ingallinella P., Bianchi E., Finotto M., Cantoni G., Eckert D.M., Supekar V.M., Bruckmann C., Carfi A., Pessi A. Structural characterization of the fusion-active complex of severe acute respiratory syndrome (SARS) coronavirus. Proc. Natl. Acad. Sci. USA. 2004;101:8709–8714. doi: 10.1073/pnas.0402753101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffers S.A., Tusell S.M., Gillim-Ross L., Hemmila E.M., Achenbach J.E., Babcock G.J., Thomas W.D., Jr., Thackray L.B., Young M.D., Mason R.J., Ambrosino D.M., Wentworth D.E., Demartini J.C., Holmes K.V. CD209L (L-SIGN) is a receptor for severe acute respiratory syndrome coronavirus. Proc. Natl. Acad. Sci. USA. 2004;101:15748–15753. doi: 10.1073/pnas.0403812101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnsson B., Lofas S., Lindquist G. Immobilization of proteins to a carboxymethyldextran-modified gold surface for biospecific interaction analysis in surface plasmon resonance sensors. Anal. Biochem. 1991;198:268–277. doi: 10.1016/0003-2697(91)90424-r. [DOI] [PubMed] [Google Scholar]
- Keng C.T., Zhang A., Shen S., Lip K.M., Fielding B.C., Tan T.H., Chou C.F., Loh C.B., Wang S., Fu J., Yang X., Lim S.G., Hong W., Tan Y.J. Amino acids 1055 to 1192 in the S2 region of severe acute respiratory syndrome coronavirus S protein induce neutralizing antibodies: implications for the development of vaccines and antiviral agents. J. Virol. 2005;79:3289–3296. doi: 10.1128/JVI.79.6.3289-3296.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S., Urbani C., Comer J.A., Lim W., Rollin P.E., Dowell S.F., Ling A.E., Humphrey C.D., Shieh W.J., Guarner J., Paddock C.D., Rota P., Fields B., DeRisi J., Yang J.Y., Cox N., Hughes J.M., LeDuc J.W., Bellini W.J., Anderson L.J. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- Kuiken T., Fouchier R.A., Schutten M., Rimmelzwaan G.F., van Amerongen G., van Riel D., Laman J.D., de Jong T., van Doornum G., Lim W., Ling A.E., Chan P.K., Tam J.S., Zambon M.C., Gopal R., Drosten C., van der Werf S., Escriou N., Manuguerra J.C., Stohr K., Peiris J.S., Osterhaus A.D. Newly discovered coronavirus as the primary cause of severe acute respiratory syndrome. Lancet. 2003;362:263–270. doi: 10.1016/S0140-6736(03)13967-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai M.M.C., Holmes K.V. Lippincott Williams and Wilkins; Philadelphia: 2001. Coronaviridae: The Viruses and Their Replication. [Google Scholar]
- Lai S.C., Chong P.C., Yeh C.T., Liu L.S., Jan J.T., Chi H.Y., Liu H.W., Chen A., Wang Y.C. Characterization of neutralizing monoclonal antibodies recognizing a 15-residues epitope on the spike protein HR2 region of severe acute respiratory syndrome coronavirus (SARS-CoV) J. Biomed. Sci. 2005;12:711–727. doi: 10.1007/s11373-005-9004-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau S.K., Woo P.C., Li K.S., Huang Y., Tsoi H.W., Wong B.H., Wong S.S., Leung S.Y., Chan K.H., Yuen K.Y. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl. Acad. Sci. USA. 2005;102:14040–14045. doi: 10.1073/pnas.0506735102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Moore M.J., Vasilieva N., Sui J., Wong S.K., Berne M.A., Somasundaran M., Sullivan J.L., Luzuriaga K., Greenough T.C., Choe H., Farzan M. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y.S., Lin C.F., Fang Y.T., Kuo Y.M., Liao P.C., Yeh T.M., Hwa K.Y., Shieh C.C., Yen J.H., Wang H.J., Su I.J., Lei H.Y. Antibody to severe acute respiratory syndrome (SARS)-associated coronavirus spike protein domain 2 cross-reacts with lung epithelial cells and causes cytotoxicity. Clin. Exp. Immunol. 2005;141:500–508. doi: 10.1111/j.1365-2249.2005.02864.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lip K.M., Shen S., Yang X., Keng C.T., Zhang A., Oh H.L., Li Z.H., Hwang L.A., Chou C.F., Fielding B.C., Tan T.H., Mayrhofer J., Falkner F.G., Fu J., Lim S.G., Hong W., Tan Y.J. Monoclonal antibodies targeting the HR2 domain and the region immediately upstream of the HR2 of the S protein neutralize in vitro infection of severe acute respiratory syndrome coronavirus. J. Virol. 2006;80:941–950. doi: 10.1128/JVI.80.2.941-950.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S., Xiao G., Chen Y., He Y., Niu J., Escalante C.R., Xiong H., Farmar J., Debnath A.K., Tien P., Jiang S. Interaction between heptad repeat 1 and 2 regions in spike protein of SARS-associated coronavirus: implications for virus fusogenic mechanism and identification of fusion inhibitors. Lancet. 2004;363:938–947. doi: 10.1016/S0140-6736(04)15788-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S.M., Hodges R.S. A de novo designed template for generating conformation-specific antibodies that recognize alpha-helices in proteins. J. Biol. Chem. 2002;277:23515–23524. doi: 10.1074/jbc.M201981200. [DOI] [PubMed] [Google Scholar]
- Luo Z., Matthews A.M., Weiss S.R. Amino acid substitutions within the leucine zipper domain of the murine coronavirus spike protein cause defects in oligomerization and the ability to induce cell-to-cell fusion. J. Virol. 1999;73:8152–8159. doi: 10.1128/jvi.73.10.8152-8159.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Z., Weiss S.R. Roles in cell-to-cell fusion of two conserved hydrophobic regions in the murine coronavirus spike protein. Virology. 1998;244:483–494. doi: 10.1006/viro.1998.9121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malashkevich V.N., Schneider B.J., McNally M.L., Milhollen M.A., Pang J.X., Kim P.S. Core structure of the envelope glycoprotein GP2 from Ebola virus at 1.9-A resolution. Proc. Natl. Acad. Sci. USA. 1999;96:2662–2667. doi: 10.1073/pnas.96.6.2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marra M.A., Jones S.J., Astell C.R., Holt R.A., Brooks-Wilson A., Butterfield Y.S., Khattra J., Asano J.K., Barber S.A., Chan S.Y., Cloutier A., Coughlin S.M., Freeman D., Girn N., Griffith O.L., Leach S.R., Mayo M., McDonald H., Montgomery S.B., Pandoh P.K., Petrescu A.S., Robertson A.G., Schein J.E., Siddiqui A., Smailus D.E., Stott J.M., Yang G.S., Plummer F., Andonov A., Artsob H., Bastien N., Bernard K., Booth T.F., Bowness D., Czub M., Drebot M., Fernando L., Flick R., Garbutt M., Gray M., Grolla A., Jones S., Feldmann H., Meyers A., Kabani A., Li Y., Normand S., Stroher U., Tipples G.A., Tyler S., Vogrig R., Ward D., Watson B., Brunham R.C., Krajden M., Petric M., Skowronski D.M., Upton C., Roper R.L. The Genome sequence of the SARS-associated coronavirus. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- Martina B.E., Haagmans B.L., Kuiken T., Fouchier R.A., Rimmelzwaan G.F., Van Amerongen G., Peiris J.S., Lim W., Osterhaus A.D. Virology: SARS virus infection of cats and ferrets. Nature. 2003;425:915. doi: 10.1038/425915a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh K. Coronaviruses: a comparative review. Curr. Top. Microbiol. Immunol. 1974;63:85–129. [Google Scholar]
- Navas-Martin S., Weiss S.R. SARS: lessons learned from other coronaviruses. Viral Immunol. 2003;16:461–474. doi: 10.1089/088282403771926292. [DOI] [PubMed] [Google Scholar]
- Ngai S.M., Sonnichsen F.D., Hodges R.S. Photochemical cross-linking between native rabbit skeletal troponin C and benzoylbenzoyl-troponin I inhibitory peptide, residues 104–115. J. Biol. Chem. 1994;269:2165–2172. [PubMed] [Google Scholar]
- Olsen C.W. A review of feline infectious peritonitis virus: molecular biology, immunopathogenesis, clinical aspects, and vaccination. Vet. Microbiol. 1993;36:1–37. doi: 10.1016/0378-1135(93)90126-R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker J.M.R., Hodges R.S. Photoaffinity probes provide a general method to prepare synthetic peptide-conjugates. J. Protein Chem. 1985;3:465–478. [Google Scholar]
- Parker J.M.R., Hodges R.S. Photoaffinity probes provide a general method to prepare peptide conjugates from native protein fragments. J. Protein Chem. 1985;3:479–489. [Google Scholar]
- Peiris J.S., Lai S.T., Poon L.L., Guan Y., Yam L.Y., Lim W., Nicholls J., Yee W.K., Yan W.W., Cheung M.T., Cheng V.C., Chan K.H., Tsang D.N., Yung R.W., Ng T.K., Yuen K.Y. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet. 2003;361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon L.L., Wong O.K., Chan K.H., Luk W., Yuen K.Y., Peiris J.S., Guan Y. Rapid diagnosis of a coronavirus associated with severe acute respiratory syndrome (SARS) Clin. Chem. 2003;49:953–955. doi: 10.1373/49.6.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rota P.A., Oberste M.S., Monroe S.S., Nix W.A., Campagnoli R., Icenogle J.P., Penaranda S., Bankamp B., Maher K., Chen M.H., Tong S., Tamin A., Lowe L., Frace M., DeRisi J.L., Chen Q. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- Siddell S., Wege H., Ter Meulen V. The biology of coronaviruses. J. Gen. Virol. 1983;64(Pt. 4):761–776. doi: 10.1099/0022-1317-64-4-761. [DOI] [PubMed] [Google Scholar]
- Simmons G., Gosalia D.N., Rennekamp A.J., Reeves J.D., Diamond S.L., Bates P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. USA. 2005;102:11876–11881. doi: 10.1073/pnas.0505577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons G., Reeves J.D., Rennekamp A.J., Amberg S.M., Piefer A.J., Bates P. Characterization of severe acute respiratory syndrome-associated coronavirus (SARS-CoV) spike glycoprotein-mediated viral entry. Proc. Natl. Acad. Sci. USA. 2004;101:4240–4245. doi: 10.1073/pnas.0306446101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaan W., Cavanagh D., Horzinek M.C. Coronaviruses: structure and genome expression. J. Gen. Virol. 1988;69(Pt 12):2939–2952. doi: 10.1099/0022-1317-69-12-2939. [DOI] [PubMed] [Google Scholar]
- Sturman L.S., Ricard C.S., Holmes K.V. Proteolytic cleavage of the E2 glycoprotein of murine coronavirus: activation of cell-fusing activity of virions by trypsin and separation of two different 90K cleavage fragments. J. Virol. 1985;56:904–911. doi: 10.1128/jvi.56.3.904-911.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supekar V.M., Bruckmann C., Ingallinella P., Bianchi E., Pessi A., Carfi A. Structure of a proteolytically resistant core from the severe acute respiratory syndrome coronavirus S2 fusion protein. Proc. Natl. Acad. Sci. USA. 2004;101:17958–17963. doi: 10.1073/pnas.0406128102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi F. The S2 subunit of the murine coronavirus spike protein is not involved in receptor binding. J. Virol. 1995;69:7260–7263. doi: 10.1128/jvi.69.11.7260-7263.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripet B., Howard M.W., Jobling M., Holmes R.K., Holmes K.V., Hodges R.S. Structural characterization of the SARS-coronavirus spike S fusion protein core. J. Biol. Chem. 2004;279:20836–20849. doi: 10.1074/jbc.M400759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripet B., Wagschal K., Lavigne P., Mant C.T., Hodges R.S. Effects of side-chain characteristics on stability and oligomerization state of a de novo-designed model coiled-coil: 20 amino acid substitutions in position “d. J. Mol. Biol. 2000;300:377–402. doi: 10.1006/jmbi.2000.3866. [DOI] [PubMed] [Google Scholar]
- Weissenhorn W., Carfi A., Lee K.H., Skehel J.J., Wiley D.C. Crystal structure of the Ebola virus membrane fusion subunit, GP2, from the envelope glycoprotein ectodomain. Mol. Cell. 1998;2:605–616. doi: 10.1016/s1097-2765(00)80159-8. [DOI] [PubMed] [Google Scholar]
- Weissenhorn W., Dessen A., Harrison S.C., Skehel J.J., Wiley D.C. Atomic structure of the ectodomain from HIV-1 gp41. Nature. 1997;387:426–430. doi: 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- Xu Y., Liu Y., Lou Z., Qin L., Li X., Bai Z., Pang H., Tien P., Gao G.F., Rao Z. Structural basis for coronavirus-mediated membrane fusion. Crystal structure of mouse hepatitis virus spike protein fusion core. J. Biol. Chem. 2004;279:30514–30522. doi: 10.1074/jbc.M403760200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y., Lou Z., Liu Y., Pang H., Tien P., Gao G.F., Rao Z. Crystal structure of severe acute respiratory syndrome coronavirus spike protein fusion core. J. Biol. Chem. 2004;279:49414–49419. doi: 10.1074/jbc.M408782200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z.Y., Huang Y., Ganesh L., Leung K., Kong W.P., Schwartz O., Subbarao K., Nabel G.J. pH-dependent entry of severe acute respiratory syndrome coronavirus is mediated by the spike glycoprotein and enhanced by dendritic cell transfer through DC-SIGN. J. Virol. 2004;78:5642–5650. doi: 10.1128/JVI.78.11.5642-5650.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z.Y., Kong W.P., Huang Y., Roberts A., Murphy B.R., Subbarao K., Nabel G.J. A DNA vaccine induces SARS coronavirus neutralization and protective immunity in mice. Nature. 2004;428:561–564. doi: 10.1038/nature02463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang D.M., Wang G., Lu J. Severe acute respiratory syndrome: vaccine on the way. Chin. Med. J. 2005;118:1468–1476. [PubMed] [Google Scholar]
- Zhou T., Wang H., Luo D., Rowe T., Wang Z., Hogan R.J., Qiu S., Bunzel R.J., Huang G., Mishra V., Voss T.G., Kimberly R., Luo M. An exposed domain in the severe acute respiratory syndrome coronavirus spike protein induces neutralizing antibodies. J. Virol. 2004;78:7217–7226. doi: 10.1128/JVI.78.13.7217-7226.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J., Xiao G., Xu Y., Yuan F., Zheng C., Liu Y., Yan H., Cole D.K., Bell J.I., Rao Z., Tien P., Gao G.F. Following the rule: formation of the 6-helix bundle of the fusion core from severe acute respiratory syndrome coronavirus spike protein and identification of potent peptide inhibitors. Biochem. Biophys. Res. Commun. 2004;319:283–288. doi: 10.1016/j.bbrc.2004.04.141. [DOI] [PMC free article] [PubMed] [Google Scholar]