

Abstract
Developing antiviral drugs, vaccines and diagnostic markers is still the most ambitious challenge in clinical virology. In the past few decades, data from high‐throughput technologies have allowed for the rapid development of new antiviral therapeutic strategies, thus making a profound impact on translational research. Most of the current preclinical studies in virology are aimed at evaluating the dynamic composition and localization of the protein platforms involved in various host–virus interactions. Among the different possible approaches, mass spectrometry‐based proteomics is increasingly being used to define the protein composition in subcellular compartments, quantify differential protein expression among samples, characterize protein complexes, and analyse protein post‐translational modifications. Here, we review the current knowledge of the most useful proteomic approaches in the study of viral persistence and pathogenicity, with a particular focus on recent advances in hepatitis C research.
Keywords: Biomarker discovery, HCV, mass spectrometry, post‐translational modifications, proteomics
Introduction
Our understanding of host–virus interaction has recently been advanced by system‐wide analyses. ‘Omics’ technologies such as genomics and transcriptomics have greatly enhanced the systemic evaluation of gene expression. Despite their usefulness, genomics and transcriptomics approaches have certain limitations. First, gene mutations and mRNA expression levels determined quantitatively often do not mirror the real situation in terms of mature proteins. Moreover, translational and post‐translational regulation affect not only the level of a mature protein but also its function. In fact, the effects on protein structure and biological function of alternative splicing, post‐translational modifications and virus–host interactions cannot be easily predicted by genomics and transcriptomics studies. In light of this, mass spectrometry (MS)‐based proteomics is a unique and necessary tool for system‐wide, structural and functional analysis of the effectors of biological functions.
MS‐based Proteomics
Proteomics offers a great variety of applications. Each experimental proteomic analysis can be summarized in three major steps: (i) sample preparation; (ii) protein separation; and (iii) identification. A downstream bioinformatic analysis can also be performed for data processing (Fig. 1).
Figure 1.
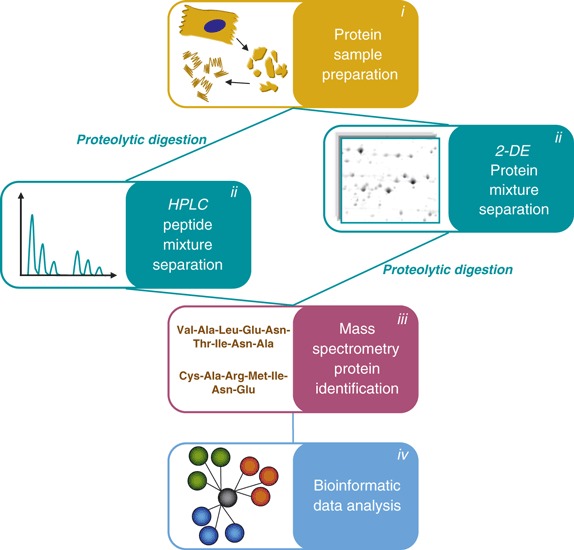
MS‐based proteomic workflow. Experimental proteomic analysis consists of several steps, listed in the different panels: sample preparation (i), protein or peptide mixture separation (ii), protein identification by MS (iii), and bioinformatic analysis (iv). 2‐DE, two‐dimensional gel electrophoresis.
(i) The first requirement for successful proteomic analysis is appropriate and specific protein sample enrichment by fractionation techniques. Indeed, many of the crucial viral life‐cycle steps are associated with defined subcellular complexes; moreover, the more attractive therapeutic targets or prognostic markers are either specific cell membrane components, or circulate in biological fluids at low concentrations [1, 2].
(ii) The sample preparation step leads to a complex mixture of different proteins. Proper separation of these mixtures is essential for the accurate identification and quantification of proteins by MS. Traditionally, two‐dimensional gel electrophoresis (2‐DE) has been considered to be a suitable protein separation method. In the past few years, this technique has been complemented by a quantitative application (two‐dimensional difference in‐gel electrophoresis (DIGE)) that allows for analysis of the relative changes in protein abundance among different samples [3]. However, owing to recent technical advances, high‐resolution nano‐liquid chromatography is gradually replacing both 2‐DE and DIGE. This is because the low volume and low flow rate of nano‐liquid chromatography allows for analysis of proteins on an attomole level, significantly reducing solvent waste and enhancing MS‐based protein identification [4].
(iii) MS is the core of proteomics. The biological applications of MS have grown exponentially since the discovery of matrix‐assisted laser desorption ionization and electrospray ionization techniques [5]. Protein identification is the primary aim of MS in proteomic applications. Because of the difficulties in analysing the mass of entire proteins, identification is performed on peptides obtained from them by proteolytic cleavage.
The development of quantitative techniques has extended MS applications in proteomics. In fact, high‐throughput relative and absolute protein abundance measurements are now possible in liquid chromatography/MS configurations with a variety of isotope‐mediated approaches, such as isotope‐coded affinity tags, isotope‐coded protein labelling, the accurate mass and time tag approach, isobaric tags for relative and absolute quantitation, and stable isotope labelling of amino acids in cell culture [6].
Proteomic Applications in Preclinical and Clinical Virology
Viral replication and propagation cause host cellular proteome variations. The identification of proteins whose presence is affected by the presence of a virus may serve for either diagnostic or prognostic purposes, or for the discovery of novel therapeutic targets (Fig. 2). Biomarker investigations are normally performed on biological fluids such as serum, plasma, and saliva. An excellent example of this can be observed in an investigation by Wiederin et al. [7] on sera of human immunodeficiency virus‐1‐associated dementia patients that allowed for identification of two differentially expressed proteins (prealbumin and gelsolin) as early biomarkers in human immunodeficiency virus‐induced neurodegenerative disorders.
Figure 2.
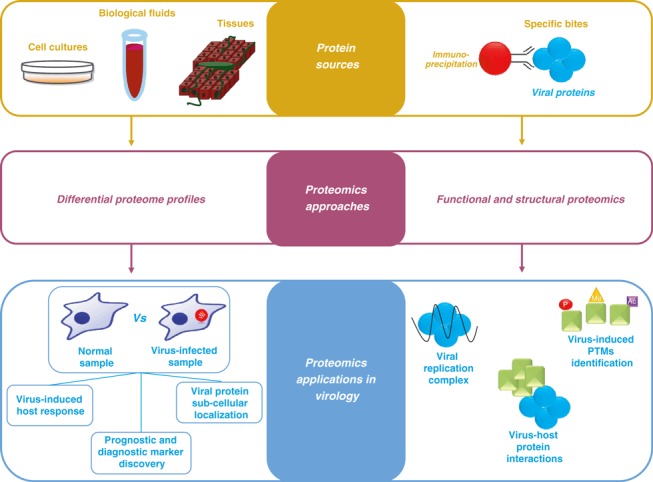
Proteomic applications in virology research. Differential proteomics allows comparison of the abundance profiles of proteins isolated from different protein sources (left workflow). Structural and functional proteomics are performed to analyse viral‐induced protein–protein interaction and post‐translational modifications (PTMs) (right workflow). Specific bites: specific bound proteins; Vs: versus.
Interestingly, biomarker identification can also be obtained with tissues or cell cultures as a protein source. For example, by analysing the hepatitis B virus‐replicating hepatoma HepG2 cell line with an iTRAQ approach, Feng et al. [8] identified apolipoprotein (Apo)A‐I and a2‐HS glycoprotein as prognostic biomarker candidates for hepatitis B virus‐associated hepatocellular carcinoma (HCC).
The efficacy of antiviral agents often depends on their ability to reach viral subcellular niches in the host cell. Differential proteomics has also proved to be a suitable tool for the subcellular localization of viruses. Emmott et al. [9] used this approach in the study of coronavirus infectious bronchitis virus infection. In particular, a SILAC‐based proteomic analysis allowed for identification of the viral protein N localized in the nucleolus, thus opening new avenues for the development of compounds targeted to the infectious bronchitis virus life cycle.
The potential of MS‐based proteomics is well established in functional and structural studies [10]. The complete description of viral proteins forming the infectious particles and the characterization of the virus–host protein interactions are current challenges in molecular virology with great implications for translational and clinical research. Approaches for protein–protein interaction analysis, such as tandem affinity purification and co‐immunoprecipitation, coupled with MS‐based proteomics have elucidated several mechanisms by which viruses exploit host molecular machineries for their benefit, as well as the host countermeasures (Fig. 2). Notably, in one of these proteomic studies, very low‐density lipoprotein‐associated proteins were identified during hepatitis C virus (HCV) replication complex purification [11], thus revealing that assembly and secretion of very low‐density lipoprotein particles are required for virus production and propagation.
Proteomic Applications to Hepatitis C
Up to 200 million persons worldwide are infected with HCV; this infection causes chronic liver disease, eventually leading to cirrhosis and HCC [12].
The current standard therapy for chronic hepatitis C is the combination of peginterferon and ribavirin, which results in a sustained virological response (SVR) in only half of the patients [13].
This therapy, besides being relatively inefficient, is also often associated with serious side effects. Novel direct‐acting antiviral treatments, which target viral proteins, have recently been introduced, and should allow an increased SVR rate to be achieved; however, their efficacy still depends on their combination with peginterferon and ribavirin. Therefore, it is crucial to identify both pretreatment and early treatment outcome predictors, in order to provide consistent estimates of the SVR. Moreover, although these drugs represent a major step towards HCV eradication, they have the limitation of selecting for drug‐resistant viruses [14]. For this reason, identification of novel drugs that target host factors necessary for the virus life cycle remains an important goal in the near future to halt HCV infection.
The progress in proteomics has led to improvements in the field of molecular mechanisms of HCV‐induced pathogenesis and the identification of markers valuable for: (i) discriminating between patients who respond (SVR) and do not respond to the current antiviral therapy; and (ii) for fibrosis, cirrhosis or HCC diagnosis. In this section, we provide a few examples of these recent findings.
Recently, when comparing the circulating lipoprotein proteomes of HCV‐infected patients and healthy donors, we observed that HCV induces impaired association of ApoA‐I with the low‐density lipoprotein particles [15]. Using HCV cellular models, we demonstrated that ApoA‐I–low‐density lipoprotein association impairment occurs during lipoprotein generation, and is caused by viral replication. Interestingly, ApoA‐I downregulation induces significant impairment of HCV production [15], thus reinforcing the notion that HCV requires the host lipoprotein machineries [11].
To understand the molecular mechanism of HCV‐induced lipoprotein generation defects, we performed quantitative proteomics analysis of SILAC‐labelled ApoB‐100‐containing subcellular fractions. We demonstrated, in vitro, that HCV induces ferritin heavy chain (Fth) upregulation, and that Fth is the cellular determinant for HCV‐induced ApoB‐100 secretion inhibition. Importantly, a negative correlation between ferritin and ApoB‐100 serum levels was found in patients. Moreover, Fth expression was found to be required for robust HCV infection. Altogether, our results provide a novel molecular explanation for HCV‐induced hypobetalipoproteinaemia and liver steatosis, which could be useful for developing a new antiviral strategy [16].
These data, together with other results not mentioned here, underline how HCV–lipoprotein interactions represent attractive targets for indirect‐acting antiviral development.
Elegant studies performed by Katze’s laboratory shed light on the early steps of HCV pathogenesis. By means of an Accurate Mass and Time tag (AMT) approach, they analysed the proteome of sequential liver biopsy specimens of HCV‐infected patients (before, and 6 and/or 12 months after, liver transplantation).
They generated a proteomic database of the highly permissive Huh‐7.5 cells containing a full‐length HCV replicon, which allowed for the identification of many proteins from only 1 μg of the total liver biopsy extracts. Using liver biopsy specimens from patients with different grades of fibrosis, the authors identified a subset of proteins that are able to indicate whether a patient will progress to severe liver disease post‐transplantation. These included proteins involved in: (i) amino acid, carbohydrate and lipid metabolism pathways [17]; (ii) immune response (i.e. proinflammatory; acute‐phase response; cellular recruitment); and (iii) hepatoprotective (i.e. associated with superoxide, cysteine, glutathione and xenobiotic metabolism) and fibrogenic (i.e. linked to epithelial‐to‐mesenchymal transition) processes [18]. The observed an increase in proinflammatory activity and an impairment in antioxidant defence, suggesting that patients who develop significant liver injury experience elevated oxidative stress. Importantly, these data were confirmed by serum metabolomics of an independent cohort of HCV patients [18].
Overall, these findings demonstrate the importance of using in vitro observation for assisting the identification of proteins/pathways involved in HCV‐induced liver disease progression.
Proteomics, together with other ‘omics’ approaches, has been largely applied to identify prognostic markers of anti‐HCV therapy outcome. The main breakthroughs of genomic and trascriptomic studies were the identification of: (i) a single‐nucleotide polymorphism in the IL28B gene (coding for interferon (IFN)‐λ3) [19]; and (ii) the dysregulation of liver expression levels of IFN‐inducible genes [20].
In this regard, we recently performed a proteomic analysis of pretreatment liver biopsy specimens from patients with SVRs and non‐responding patients by 2D‐DIGE/MS [21]. Our study confirmed the presence of an inverse correlation between the levels of IFN‐related genes and the rate of the SVR, and also identified proteins involved in stress response and energy metabolism as putative prognostic factors.
Proteomic studies from our and other laboratories have also shown that post‐translational modifications are another source of prognostic markers of treatment outcome, such as the charge modification of dihydroxyacetone kinase, a negative regulator of the MDA5‐mediated antiviral pathway, detected in liver biopsy specimens, and the phosphorylation of AKT, JAK1, p70 S6 kinase, protein kinase C‐ζ/λ, TYK2 and ZAP‐70 observed in peripheral blood mononuclear cells [21, 22].
Many efforts to identify prognostic and diagnostic markers have been made with serum, as it is an easily accessible biological fluid that may reflect immunological and pathological events during chronic HCV infection. Despite these advantages, there are some drawbacks, of which the difficulty in finding low‐abundance proteins, owing to the presence of high‐abundance proteins (albumin, immunoglobulin, etc.), is one of the most challenging. Many efforts have been also made to overcome this problem.
In this regard, SELDI‐TOF, a technology that combines chromatography separation and MS analysis, has represented an important advance in identifying blood‐circulating markers. For instance, different pretreatment serum proteins, whose levels significantly differ between patients with SVR and non‐responding patients, were identified with this approach [23]. Importantly, the association of serum protein levels with clinical and biochemical characteristics, such as the stage of fibrosis, IL28B polymorphisms, and viral genotype, has led to a more accurate prediction of treatment response, indicating that the combinatory analysis of clinical and molecular data is required to achieve a reliable prognostic test [23, 24].
Moreover, Gangadharan et al. [25, 26] identified low‐abundance plasma proteins (narrowing the pH range of 2‐DE‐based proteomic analyses) of HCV patients with different degree of liver fibrosis. Among new candidate biomarkers, the levels of six proteins (lipid transfer inhibitor protein, complement C3d, complement C4, corticosteroid‐binding globulin, ApoJ, and ApoL1) were found to decrease inversely with the patient’s fibrosis grade [25, 26].
In Beretta’s laboratory, extensive MS analysis of a large cohort of HCC patients was performed after immunodepletion of the most abundant plasma proteins and multidimensional protein separation by 2D‐HPLC, followed by SDS‐PAGE [27]. They found that osteopontin is a sensitive HCC prognostic marker [27].
A further approach to identify HCV‐related HCC biomarkers has been attempted by Nomura’s laboratory [28]. Downstream of a proteomics analysis on liver biopsy specimens of HCC patients, the authors investigated the possibility of detecting serum autoantibodies directed against the proteins found to be overexpressed. On the basis of their findings with this approach, they propose the serum level of anti‐Ku86 antibodies as a potential biomarker for early detection of HCC in HCV‐infected patients [28].
Altogether, these studies provide initial evidence that the recent development of reliable proteomic approaches may (in the near future) allow for complete characterization of HCV patient dysfunctions, in order to prevent and monitor disease progression, and achieve virus eradication.
Acknowledgements
We sincerely apologize to all those colleagues whose important work was not cited, owing to limitations of space. We are very grateful to A. Baker for the editing. This study was supported by grants from the Italian Ministry of Health (‘Ricerca Corrente’ and ‘Ricerca Finalizzata’).
Transparency Declaration
All authors declare no potential conflicts of interest.
References
- 1. Peng X, Chan EY, Li Y, Diamond DL, Korth MJ, Katze MG. Virus–host interactions: from systems biology to translational research. Curr Opin Microbiol 2009; 12: 432–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Miller S, Krijnse‐Locker J. Modification of intracellular membrane structures for virus replication. Nat Rev Microbiol 2008; 6: 363–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhou G, Li H, DeCamp D et al. 2D differential in‐gel electrophoresis for the identification of esophageal scans cell cancer‐specific protein markers. Mol Cell Proteomics 2002; 1: 117–124. [DOI] [PubMed] [Google Scholar]
- 4. Walther TC, Mann M. Mass spectrometry‐based proteomics in cell biology. J Cell Biol 2010; 190: 491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aebersold R, Mann M. Mass spectrometry‐based proteomics. Nature 2003; 422: 198–207. [DOI] [PubMed] [Google Scholar]
- 6. Ong SE, Mann M. Mass spectrometry‐based proteomics turns quantitative. Nat Chem Biol 2005; 1: 252–262. [DOI] [PubMed] [Google Scholar]
- 7. Wiederin J, Rozek W, Duan F, Ciborowski P. Biomarkers of HIV‐1 associated dementia: proteomic investigation of sera. Proteome Sci 2009; 7: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Feng H, Wang M, Chen WN. iTRAQ‐coupled 2D LC‐MS/MS analysis of secreted proteome of HBV‐replicating HepG2 cells: potential in biomarkers for prognosis of HCC. Curr Microbiol 2010; 61: 280–284. [DOI] [PubMed] [Google Scholar]
- 9. Emmott E, Rodgers MA, Macdonald A, McCrory S, Ajuh P, Hiscox JA. Quantitative proteomics using stable isotope labeling with amino acids in cell culture reveals changes in the cytoplasmic, nuclear, and nucleolar proteomes in Vero cells infected with the coronavirus infectious bronchitis virus. Mol Cell Proteomics 2010; 9: 1920–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zheng J, Sugrue RJ, Tang K. Mass spectrometry based proteomic studies on viruses and hosts—a review. Anal Chim Acta 2011; 702: 149–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huang H, Sun F, Owen DM et al. Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low‐density lipoproteins. Proc Natl Acad Sci USA 2007; 104: 5848–5853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lauer GM, Walker BD. Hepatitis C virus infection. N Engl J Med 2001; 345: 41–52. [DOI] [PubMed] [Google Scholar]
- 13. Casey LC, Lee WM. Hepatitis C therapy update. Curr Opin Gastroenterol 2012; 28: 188–192. [DOI] [PubMed] [Google Scholar]
- 14. Barritt AS IV, Fried MW. Maximizing opportunities and avoiding mistakes in triple therapy for hepatitis C virus. Gastroenterology 2012; 142: 1314–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mancone C, Steindler C, Santangelo L et al. Hepatitis C virus production requires apolipoprotein A‐I and affects its association with nascent low‐density lipoproteins. Gut 2011; 60: 378–386. [DOI] [PubMed] [Google Scholar]
- 16. Mancone C, Montaldo C, Santangelo L et al. Ferritin heavy chain is the host factor responsible for HCV‐induced inhibition of apoB‐100 production and is required for efficient viral infection. J Proteome Res 2012; 11: 2786–2797. [DOI] [PubMed] [Google Scholar]
- 17. Diamond DL, Jacobs JM, Paeper B et al. Proteomic profiling of human liver biopsies: hepatitis C virus‐induced fibrosis and mitochondrial dysfunction. Hepatology 2007; 46: 649–657. [DOI] [PubMed] [Google Scholar]
- 18. Diamond DL, Krasnoselsky AL, Burnum KE et al. Proteome and computational analyses reveal new insights into the mechanisms of hepatitis C virus‐mediated liver disease post‐transplantation. Hepatology 2012; 56: 28–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ge D, Fellay J, Thompson AJ et al. Genetic variation in IL28B predicts hepatitis C treatment‐induced viral clearance. Nature 2009; 461: 399–401. [DOI] [PubMed] [Google Scholar]
- 20. Chen L, Borozan I, Feld J et al. Hepatic gene expression discriminates responders and non‐responders in treatment of chronic hepatitis C viral infection. Gastroenterology 2005; 128: 1437–1444. [DOI] [PubMed] [Google Scholar]
- 21. Perdomo AB, Ciccosanti F, Iacono OL et al. Liver protein profiling in chronic hepatitis C: identification of potential predictive markers for interferon therapy outcome. J Proteome Res 2012; 11: 717–727. [DOI] [PubMed] [Google Scholar]
- 22. Younossi ZM, Limongi D, Stepanova M et al. Protein pathway activation associated with sustained virologic response in patients with chronic hepatitis C treated with pegylated interferon (PEG‐IFN) and ribavirin (RBV). J Proteome Res 2011; 10: 774–779. [DOI] [PubMed] [Google Scholar]
- 23. Paradis V, Asselah T, Dargere D et al. Serum proteome to predict virologic response in patients with hepatitis C treated by pegylated interferon plus ribavirin. Gastroenterology 2006; 130: 2189–2197. [DOI] [PubMed] [Google Scholar]
- 24. Patel K, Lucas JE, Thompson JW et al. High predictive accuracy of an unbiased proteomic profile for sustained virologic response in chronic hepatitis C patients. Hepatology 2011; 53: 1809–1818. [DOI] [PubMed] [Google Scholar]
- 25. Gangadharan B, Antrobus R, Chittenden D et al. New approaches for biomarker discovery: the search for liver fibrosis markers in hepatitis C patients. J Proteome Res 2011; 10: 2643–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gangadharan B, Bapat M, Rossa J et al. Discovery of novel biomarker candidates for liver fibrosis in hepatitis C patients: a preliminary study. PLoS ONE 2012; 7: e39603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shang S, Plymoth A, Ge S et al. Identification osteopontin as a novel marker for early hepatocellular carcinoma. Hepatology 2012; 55: 483–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nomura F, Sogawa K, Noda K et al. Serum anti‐Ku86 is a potential biomarker for early detection of hepatitis C virus‐related hepatocellular carcinoma. Biochem Biophys Res Commun 2012; 421: 837–843. [DOI] [PubMed] [Google Scholar]