

Abstract
Recent work carried out in our laboratory showed the existence of a cytokine storm in SARS patients, dominated by Th1-type mediators. We thus hypothesized that IFN-γ may play a major role in the pathology by triggering immune-mediated alveolar damage. As we assessed or re-assessed some effects of IFN-γ on a number of human lung epithelial and fibroblast cell lines, chosen for their wide use in the literature, we found that alveolar epithelial cells were more sensitive to IFN-γ, in terms of proliferation inhibition and enhancement of Fas-mediated apoptosis. While similar effects were obtained on fibroblasts, concentrations of IFN-γ 4–8-fold greater were required. In addition, both epithelial and fibroblastic cell lines were able to secrete large quantities of T cell-targeting chemokines, similar to the ones detected in SARS patients. Based on the clinical data collected previously, the available literature and our in vitro experimentation, we propose that IFN-γ may be responsible for acute lung injury in the late phase of the SARS pathology.
Keywords: Acute respiratory distress syndrome, Apoptosis, Interferon-γ, SARS
1. Introduction
In 2003, a new emerging infectious disease, termed severe acute respiratory disease (SARS), swept across the world, resulting in the death of 774 people [1]. With no precedent, the scientific community was able to identify the causative agent soon after its emergence [2], [3], [4]. Since then, significant advances have been made in understanding this disease and less than 2 years later, vaccine candidates are already under trial [5]. However, the mechanism(s) underlying the severity of the respiratory distress remain(s) unexplained. Indeed, hypotheses are yet to be made to explain the acute lung injury (ALI) and respiratory failure observed in the severe cases, while in the presence of a declining viral load.
While high fever was the most common characteristic of the new illness, in one-third of cases, the patients also developed an atypical form of pneumonia, with acute respiratory distress (ARDS) as a result of lung damage, characterized by infiltrates on chest radiography [4], [6], [7]. Amongst the changes observed in the lungs of SARS patients were epithelial cell proliferation and desquamation, hyaline membrane formation and cell infiltration (lymphocytes, neutrophils and monocytes) during the early stage of the disease, while increased fibrosis and multinucleated epithelial giant cell formation were seen at a later stage [8].
The existence of an abnormally excessive inflammatory response in the lungs has been suggested to explain the development of ALI in SARS. Indeed, patients still manifested lung injury at a time when the viral load was dropping, in support of the immune nature of the lung damage [9]. Our investigations, and the work of Wong et al., support this hypothesis as we determined the presence of several cytokines and chemokines at high concentrations in the plasma of RT-PCR-confirmed SARS patients [10], [11]. Especially, Th1-type cytokine IFN-γ and other related cytokine (IL-18) and chemokines (MIG, IP-10 and MCP-1) were found at unusually high levels. This seemed to place IFN-γ at the center of any cytokine-induced immune response.
Evidence exists to support the importance of the destruction of the alveolar epithelium in the development of conventional ARDS [12], [13]. The presence of soluble Fas ligand (FasL) was reported in ARDS patients and an agonist antibody was shown to be able to induce alveolar epithelial cell injury and lung inflammation in mice [14], [15]. Although the presence of soluble FasL has not been demonstrated in SARS patients, immune cells capable of expressing membrane-form FasL were found infiltrating the lungs, and ARDS observed in late-stage SARS patients resembled other late-stage ARDS, suggesting the potential role of the Fas/FasL system in the development of ALI in SARS [7], [8], [16].
Previous work by other groups has led to the conclusion that human lung epithelial cells, but not lung fibroblasts, were sensitive to Fas-mediated apoptosis, suggesting that phenotypic differences between these two cell types may contribute to the development of fibrosis, by rendering one type as a prompt for cellular damage while protecting the other [17], [18], [19]. As IFN-γ seems to play a role in SARS, we set out to compare some effects of IFN-γ on a number of human lung fibroblast and epithelial cell lines (chosen for their use in numerous publications as models of their primary counterparts) in order to extrapolate how these cell types co-existing in vivo may react to the presence of IFN-γ. For that purpose, identical culture conditions were used for all cell lines. We report here that in substance both cell types were affected similarly, i.e. susceptible to Fas-mediated apoptosis after IFN-γ stimulation, though to different degrees; fibroblastic lines required much larger amounts to become sensitive. These observations led us to hypothesize that specific epithelial sensitivity to IFN-γ may be the basis underlying the development of lung injury in the late phase of SARS.
2. Results
2.1. Increased sensitivity of epithelial cells to cell proliferation inhibition by IFN-γ
All four cell lines, A549, BEAS-2B, MRC-5 and HFL-1, were cultured in the presence of varying concentrations of IFN-γ, for 24, 48 or 72 h. The culture in such conditions resulted in an inhibition of proliferation that ranged from growth downregulation (BEAS-2B, MRC-5 and HFL-1) to complete growth arrest (A549: p > 0.05 for 48 h vs. 72 h IFN-γ-treated groups), as shown in Fig. 1. However, sensitivity varied among cell lines. Indeed, dose-dependent experiments determined that epithelial cell lines were more susceptible to the effect of IFN-γ than fibroblast cell lines (data not shown). Two hundred and fifty IU mL−1 of IFN- γ in A549 and BEAS-2B resulted in very significant differences in growth, while 62.5 IU mL−1 was sufficient to detect some effects. On the contrary, MRC-5 and HFL-1 required high doses of IFN-γ: 2000 IU mL−1 was necessary to observe a significant growth inhibition.
Fig. 1.
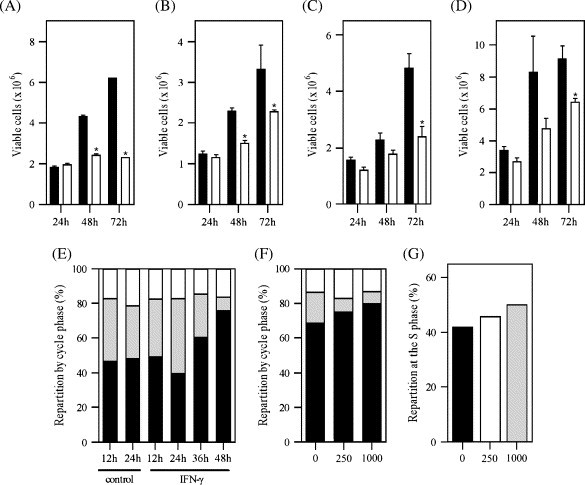
Effect of IFN-γ on cell proliferation. (A–D) Cell growth: epithelial cells and fibroblasts were cultured with (clear) or without (black) IFN-γ. Cells were harvested at 24, 48, or 72 h after seeding, and their number determined with a hemacytometer. (A) A549; seeding density: 1 × 106, IFN-γ concentration: 250 IU mL−1. (B) BEAS-2B; seeding density: 1 × 106, IFN-γ concentration: 250 IU mL−1. (C) MRC-5; seeding density: 2.5 × 105, IFN-γ concentration: 2000 IU mL−1. (D) HFL-1; seeding density: 0.9 × 105, IFN-γ concentration: 2000 IU mL−1. ∗p < 0.05, in comparison to the untreated group. (E–G) Cell cycle: cells were seeded at and cultured in serum-free culture medium, for 24 h (E,F) or 36 h (G). The medium was then replaced by complete medium containing IFN-γ. (E) Arrest in G0/G1; cells were harvested at various time-points after addition of IFN-γ (250 IU mL−1) and their repartition in the G0/G1 (black), S (gray) or G2/M (clear) phase determined. (F) Dose-dependent cell cycle arrest in G0/G1 after 48 h; cells were harvested 48 h after addition of 0, 250 or 1000 IU mL−1 IFN-γ, and their cell cycle phase determined (black, G0/G1; gray, S; clear, G2/M). (G) Dose-dependent delay at the S phase; cells were harvested 18 h after addition of, either, 0, 250 or 1000 IU mL−1 IFN-γ.
2.2. Delay and arrest of the cell cycle in A549 cells
As A549 was the only cell line whose growth was completely arrested within the time-course of our first experiment, we decided to investigate further the effects of IFN-γ on these cells. The only moderate induction of apoptosis by IFN-γ, in the range of 8–10% of the cells, as determined by annexin V/PI and active caspase-8 staining (data not shown), was ruled out as the possible explanation. Cell cycle analysis was on the other hand able to show that, not only was IFN-γ ultimately able to dose-dependently arrest the cells at the G0/G1 phase (Fig. 1E and F), it could also first delay their progression through the S phase, as shown by the accumulation of cells in that phase. This latter effect was dose-dependent (Fig. 1G). The multiple ability of IFN-γ to inhibit the cells from progressing through their cycle probably explains the early growth arrest observed in A549.
2.3. IFN-γ enhanced Fas expression indifferently in all cell lines
We next investigated the expression of Fas by epithelial and fibroblast cell lines. All cells seemed to express CD95, moderately (BEAS-2B) or more significantly (A549, HFL-1 and MRC-5) ( Fig. 2). Interestingly, in the presence of IFN-γ, Fas expression was upregulated on the surface of cells, including fibroblast cell lines. However, epithelial cells were once again more sensitive to the IFN-γ stimulus, as low concentrations enabled Fas expression upregulation (250 IU mL−1), while fibroblast cell lines only responded to high concentrations (data not shown). Note also that in the particular case of MRC-5, surface Fas expression upregulation was only transient, and IFN-γ ceased to have an enhancing effect 72 h after addition (Fig. 2K).
Fig. 2.
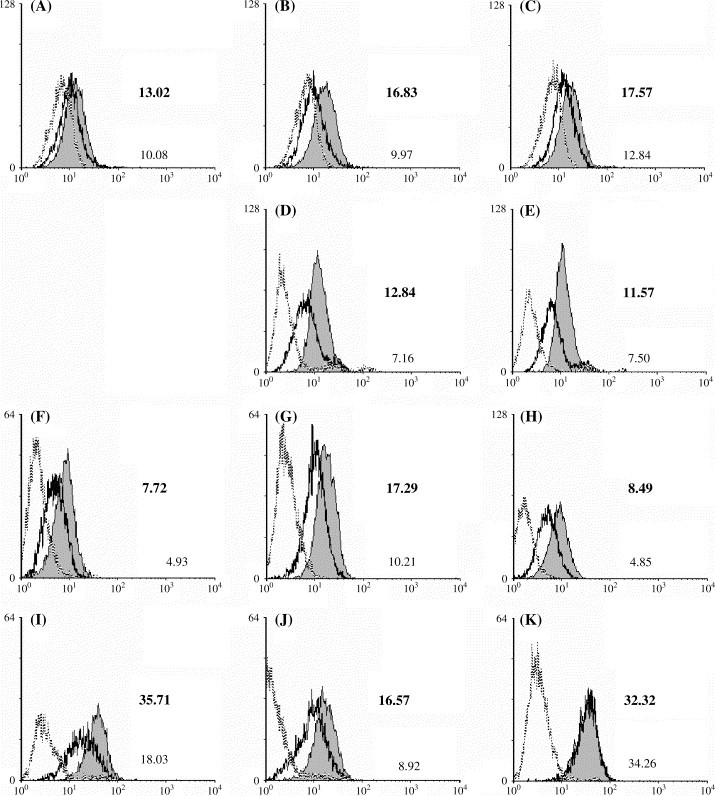
Effect of IFN-γ on Fas (CD95) surface expression. All cells were cultured for 24, 48 and 72 h (with the exception of A549, which was only cultured for 48 and 72 h) in the presence of IFN-γ (250 IU mL−1 for BEAS-2B and A549, 2000 IU mL−1 for HFL-1 and MRC-5). They were then harvested and surface CD95 expression stained before being detected by flow cytometry. (A–C) BEAS-2B; (D–E) A549; (F–H) HFL-1; (I–K) MRC-5. Only one replicate of each staining is presented for each time point. The data are shown in a time sequence from left to right, for each cell line. The gray peak and the black line represent the IFN-γ-treated and the control groups, while the gray line represents the isotype control. On each histogram the fluorescence geometric mean is given for the IFN-γ-treated (bold, top value) and the control (bottom value) groups.
2.4. IFN-γ enhanced Fas-mediated apoptosis of epithelial cells and fibroblasts
Despite the existence of a constitutive expression of Fas on the surface of all four cell lines, triggering of apoptosis by the sole addition of a cross-linking activating anti-Fas antibody was only marginal ( Fig. 3). Induction of apoptosis was only significant in A549, at 36 and 60 h after addition, and in MRC-5 at 12 h, but concerned at any time less than 30% of the viable cell population (Fig. 3B and D). When IFN-γ was added to the culture medium, 12 h before the addition of the anti-Fas antibody, a significant enhancement of apoptosis could be observed in all cell types, although at different times and different extents. In BEAS-2B, an enhanced induction of apoptosis was only observable 72 h after addition of IFN-γ (Fig. 3A). Moreover, this induction was to a lesser degree than in A549, and required the addition of a larger amount of anti-Fas antibody. At the concentration 0.1 μg mL−1, anti-Fas antibody was unsuccessful at inducing apoptosis, even after IFN-γ pre-treatment (data not shown). In A549, IFN-γ-enhanced Fas-mediated apoptosis developed rapidly, as it was already significant by 24 h (Fig. 3B), and successfully, as it extended to the whole monolayer, causing all cells to be dead by 96 h (data not shown). Fibroblasts were also susceptible to the enhancement of Fas-mediated apoptosis by IFN-γ. Like in BEAS-2B, apoptosis appeared enhanced only 72 h after addition of IFN-γ in HFL-1 (Fig. 3C). However, in MRC-5 significant enhancement of Fas-mediated apoptosis was readily observed by 24 h, but declined thereafter to become non-existent by 72 h (Fig. 3D). In this latter cell line, the enhancement of Fas-mediated apoptosis by IFN-γ followed accurately the surface Fas expression profile of MRC-5 cells stimulated by IFN-γ.
Fig. 3.
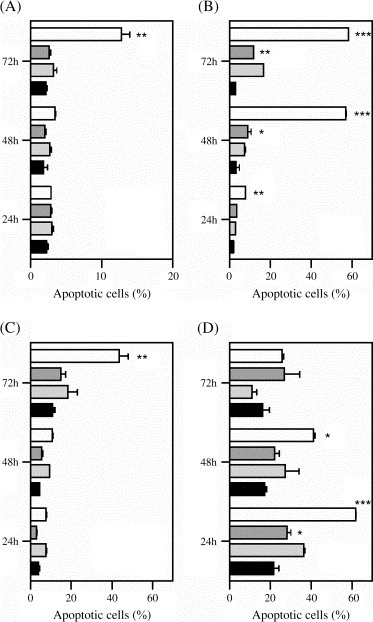
Effect of IFN-γ on Fas-mediated apoptosis. Cells were seeded with or without IFN-γ, and 12 h later anti-CD95 cross-linking antibody was added to the culture supernatant. The cells were finally harvested 24, 48 or 72 h after seeding and the percentage of cells undergoing apoptosis determined by annexin-FITC/PI staining and flow cytometric analysis. (A) BEAS-2B: 250 IU mL−1 IFN-γ and 0.5 μg mL−1 anti-CD95 IgM; (B) A549: 250 IU mL−1 IFN-γ and 0.1 μg mL−1 anti-CD95 IgM; (C) HFL-1: 1000 IU mL−1 IFN-γ and 0.1 μg mL−1 anti-CD95 IgM; (D) MRC-5: 1000 IU mL−1 IFN-γ and 0.1 μg mL−1 anti-CD95 IgM. Black, control group; light gray, IFN-γ-treated only; dark gray, anti-CD95 IgM-treated only; clear, IFN-γ-plus-anti-CD95 IgM-treated group. IFN-γ-plus-anti-CD95 IgM-treated group vs. anti-CD95 IgM-treated only: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; anti-CD95 IgM-treated only vs. control group: ∗p < 0.05, ∗∗p < 0.01.
2.5. Relevance of anti-Fas antibody-mediated apoptosis to cell-to-cell interaction
While the importance of the Fas/FasL system in the development of ARDS is supported by previous work [12], [13], [14], [15], the lack of evidence of the presence of soluble FasL in SARS patients having developed ARDS required us to demonstrate the relevance for cell-to-cell interaction of work that used an anti-Fas activating antibody. A549 cells pre-treated with IFN-γ were thus co-cultured with a lymphocytic T cell line, Jurkat, which had previously been activated in order to increase the expression of surface FasL (data not shown). Stimulation by IFN-γ increased the susceptibility of A549 cells to apoptosis induced by contact with Jurkat cells ( Fig. 4). When the ratio of available Jurkat cells to A549 cells was increased 3-fold, however, the number of unstimulated A549 cells undergoing apoptosis did not significantly increase. But it more than doubled when A549 cells had been stimulated by IFN-γ, clearly indicating the role of Fas in the mediation of this induction of apoptosis by cell contact, and the important role played by IFN-γ in the enhancement of cellular damage.
Fig. 4.

Enhanced apoptosis of IFN-γ-treated A549 cells in co-culture with Jurkat cells. Jurkat cells were activated with 100 ng mL−1 PMA and 500 ng mL−1 ionomycin for 24 h. At the same time, A549 cells were cultured for 24 h, with or without 250 IU mL−1 IFN-γ added 12 h after seeding. Jurkat cells were then centrifuged and counted. The A549 cell number was also determined. Finally, Jurkat cell were resuspended in culture supernatant from individual A549 cultures and the suspension was returned, at a Jurkat:A549 ratio of 1:2 or 3:2. Twenty-four hours later the cells were harvested and stained with annexin V-FITC/PI and anti-CD3 antibody coupled to PerCP. The data shown represents the percentage of A549 cells undergoing apoptosis. Black, control group; clear, IFN-γ-treated only; light gray, co-culture with Jurkat; dark gray, IFN-γ-treated and co-culture with Jurkat. IFN-γ-treated and co-culture with Jurkat vs. co-culture with Jurkat: ∗p < 0.05, ∗∗p < 0.01.
Jurkat cells are T-lymphocyte-like cells of the kind that may be found infiltrating the lungs in SARS patients [8]. T cells are able to infiltrate tissues in response to the release of chemoattractant messengers. It was thus interesting to notice that both A549 and MRC-5 could release, in response to IFN-γ, large amounts of the chemokines that were detected in the blood of SARS patients [8], including Mig and IP-10, some well-known chemoattractants of activated T cells ( Table 1).
Table 1.
IFN-γ-treated A549 and MRC-5 cell-induced chemokines secretion
| IFN-γ (IU mL−1) | A549 |
MRC-5 |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MIG (pg mL−1) | MCP-1 (pg mL−1) | IP-10 (pg mL−1) | RANTES (pg mL−1) | IL-8 (pg mL−1) | MIG (pg mL−1) | MCP-1 (pg mL−1) | IP-10 (pg mL−1) | RANTES (pg mL−1) | IL-8 (pg mL−1) | |
| 0 | 16 | 33,712 | 201 | 304 | 27,040 | <1 | 152 | <1 | <1 | 1336 |
| 62.5 | 393 | 38,416 | 17,688 | 2216 | 33,688 | 4017 | 3240 | 3868 | <1 | 1121 |
| 250 | 1704 | 45,056 | 38,056 | 3024 | 35,512 | 187,856 | 34,288 | 89,312 | <1 | 1024 |
| 1000 | 2128 | 47,168 | 44,944 | 3112 | 32,240 | 200,184 | 36,088 | 69,016 | <1 | 840 |
| 2000 | 157,136 | 29,240 | 45,240 | <1 | 784 | |||||
3. Discussion
A characteristic feature of patients infected with SARS-associated coronavirus is the presence of lung consolidation, lung injury in the form of diffuse alveolar damage and fibrosis in more severe cases. In the latter cases, the development of the lung injury has led to respiratory failure and the need for respiratory assistance at an intensive care unit. Respiratory distress has appeared to be the cause of the majority of deaths related to SARS. One of the hypotheses emerging to explain the severity of the lung inflammation and damage is the development of a “cytokine storm”, as result of the infection, leading to an unbalanced inflammatory response. Cytokine profiling of RT-PCR-confirmed SARS patients has revealed the presence of large amounts of IFN-γ and IFN-γ-related cytokine and chemokines in patients’ blood, and the absence of Th2-type cytokines [10]. IFN-γ is a cytokine secreted mainly, but not only, by activated and cytotoxic T cells, as part of the immune response to eliminate virus-infected cells. Infiltration of immune cells, including lymphocytes, was observed in the lungs of SARS patients [8]. We therefore hypothesized that IFN-γ may be a major contributor to the ALI, for it is known to cause cell proliferation inhibition and apoptosis. We used an array of cell lines to study how IFN-γ could be involved in selective epithelial damage and fibrosis.
The present report showed that human lung epithelial cells (A549 or BEAS-2B) and fibroblasts (HFL-1 or MRC-5) were affected similarly, but not identically, by IFN-γ in terms of Fas-mediated induction of apoptosis. This is in some ways in contradiction to previously available publications. Indeed, Tanaka et al. have reported that they found that human lung fibroblasts were resistant to Fas-mediated apoptosis, by opposition to epithelial cells that are known to be susceptible to Fas cross-linkage [17], [18], [19]. Although we do not know the actual concentration in International Units of bioactive IFN-γ used by these authors to treat fibroblasts, it seems that the dose of IFN-γ, duration of pre-treatment, time-point for detection, or a combination of these is responsible for this apparent resistance to apoptosis. In our work, fibroblasts (HFL-1) required an exposure to high concentrations of IFN-γ to observe any enhancement of surface Fas expression and Fas-mediated apoptosis. MRC-5, the second fibroblast cell line used here, was also sensitive to IFN-γ, although for only a limited period. This could be linked to the phenotypic differences between the two cell lines, MRC-5 possessing some characteristics of myofibroblasts [20]. The possibility that fibroblast differentiation into myofibroblasts protects them from IFN-γ-enhanced Fas-mediated apoptosis is currently under investigation in our laboratory.
Whilst the upregulation of Fas expression on the cell surface may not be the only mechanism underlying the enhanced induction of apoptosis in A549 cells treated with IFN-γ and anti-Fas antibody, a relationship between increased expression and increased apoptosis seems to exist when timing is considered. Indeed, although these cells naturally express Fas proteins on their surface, the density seems insufficient to lead to a major induction of apoptosis (Fig. 3B). However, when IFN-γ increased Fas expression by 48 or 72 h (Fig. 2D or E), more than 50% of the cells underwent apoptosis (Fig. 3B). A similar, yet more convincing, observation could be made with MRC-5 cells. These cells could express Fas, but anti-Fas antibody alone was not able to provoke a clear increase in the number of cells undergoing apoptosis (Fig. 3B). When Fas expression was enhanced by IFN-γ, so was apoptosis, by 24 h (Figs. 2I and 3B). When the Fas expression returned to its basal level by 72 h (Fig. 2K), so had apoptosis (Fig. 3B). The presence of a relationship in time thus seems to support the existence of a correlation between the increased density of Fas protein on the cell surface and the sensitivity of the cell to undergo apoptosis mediated by Fas.
The ability of IFN-γ to enhance Fas-mediated apoptosis of lung fibroblasts is, however, relative, as the amount of cytokine required does not appear to correspond to the levels observed in SARS patients, even though bioactivity of the cytokines detected by ELISA and CBA kits was not determined [10]. In any way, this does not affect the fact that lung epithelial cells are clearly highly sensitive to IFN-γ, while fibroblasts are remotely affected. IFN-γ has thus the ability to prime the destruction of the lung epithelium; indeed, in addition it stimulates epithelial cells, like neighboring fibroblasts, to release significant amounts of chemokines that are known to attract the migration of immune cells. Infiltrating lymphocytes were observed in autopsied cases of SARS [8]. It seems thus plausible that the presence of high amounts of IFN-γ, T cell chemoattractants and infiltrating lymphocytes in the lungs may have led to selective alveolar epithelial damage, as part of an innate immune response, in SARS patients.
With the exception of IL-8, which is not stimulated by IFN-γ in either A549 or MRC-5, and MIG, IFN-γ was able to stimulate the release of chemokines from epithelial cells (A549) at lower concentrations than the one required by fibroblasts (MRC-5). MIG, on the other hand, was more readily produced by fibroblasts, although epithelial cells were also sensitive to the IFN-γ stimulus. This particularity could be the result of the dual effect of IFN-γ on epithelial cells: while it stimulates the expression of proteins, it also inhibits cell growth. The latter effect would undoubtedly downsize any stimulated increase in protein expression. Overall, the data point towards an increased readiness of epithelial cells to respond to IFN-γ by the release of chemokines, in comparison to fibroblasts that require larger concentration of the cytokine. Ultimately, the experiments investigating the production of chemokines were designed to determine if IFN-γ could further contribute to a lung immunopathology by favoring chemoattraction of the necessary actors (i.e. T cells) for immune-mediated cellular damage.
Whilst IFN-γ is essentially regarded as an anti-fibrotic agent, evidence exists to suggest that this cytokine may play an important role in the development of the lung injury that precedes fibrosis [21]. Indeed, the absence of IFN-γ resulted in reduced inflammation in various models of lung injury using IFN-γ-knockout mice [22], [23], [24]. Several studies have also suggested that lymphocytes may play a role in pulmonary injury in rodents. For example, T-cell depletion by anti-CD3 antibody treatment has attenuated pulmonary fibrosis and increased survival in mice treated with bleomycin [25]. A recent publication by Glass et al. indicates that, in mice, the SARS-associated coronavirus successfully replicated and induced an increase in various proinflammatory chemokines, in the lungs, including MIG and IP-10 [26]. However, it failed to stimulate the production of cytokines or the infiltration of leukocytes. In this model, no obvious changes in pulmonary function and little signs of lung injury could be observed. Moreover, the focal necrosis and bronchiolar epithelial damage characterized in SARS-associated coronavirus infected B6 mice were not observed in RAG1−/− mice, which lack T and B lymphocytes. Such observations tend to support the fact that in SARS, cytokines and T cells may be important to the development of ALI.
Although the immune response is thought to contribute significantly to ALI and respiratory distress in SARS patients, it is not clear how a response dominated by Th1-type biological response modifiers may lead to the development of fibrosis. Here we show lung epithelial cells to be more responsive to IFN-γ-induced damage than fibroblasts. Thus, we propose that the Th1-dominant immune-mediated cell death may actually favor the damage of alveolar epithelial cells over the fibroblast layer, leaving the latter relatively intact. In vivo, this would translate into the destruction of the epithelial parenchyma, a basis for the stimulation of repair mechanisms involving fibroblasts, relatively unaffected by the immune response and available for a proliferative response ( Fig. 5). The association of epithelial destruction and fibrosis has been suggested in other situations, and we are currently investigating the possibility that immune-mediated epithelial destruction may directly stimulate proliferation and/or differentiation of fibroblasts [27], [28]. To conclude, these findings are relevant to the understanding of the roles of IFN-γ and the Fas/FasL system in ALI, in which alveolar epithelial damage occurs primarily. We suggest that IFN-γ may play an important role in the development of an immune mechanism that will lead to ALI, fibrosis and ARDS in SARS patients.
Fig. 5.

Representation of the mechanism potentially underlying the lung pathology in SARS.
4. Materials and methods
4.1. Cell lines
A549, a human lung alveolar type II epithelial carcinoma cell line, BEAS-2B, a human lung bronchiolar epithelial cell line, HFL-1 and MRC-5, human embryonic lung fibroblast cell lines, and Jurkat, a human leukemic T cell lymphoblast cell line, were all obtained from American Type Culture Collection (Rockville, MD). They were cultured under standard conditions in the following medium supplemented with 10% heat-inactivated FBS, 2 mM l-glutamine and 1% (v/v) penicillin-streptomycin solution 100×: DMEM for A549 and BEAS-2B, MEM Eagle-Earle's balanced salt solution with non-essential amino acids for MRC-5 and HFL-1, and RPMI 1640 medium for Jurkat. Adherent cells were passaged at 80–90% confluence using 0.25% (w/v) trypsin/1 mM EDTA, while Jurkat cells were subcultured every 3 days. For experimental purposes, A549, BEAS-2B, HFL-1 and MRC-5 were seeded at 1 × 106, 1 × 106, 0.8 × 105 and 2.5 × 105 cells per 60 mm culture dish, respectively, in 3 ml of corresponding growth medium. These seeding densities permitted cell confluence to be reached within 72 h for each cell type.
4.2. Cytokine, antibodies and flow cytometry
Recombinant human IFN-γ was purchased from PreproTech EC (London, UK). Functional grade murine anti-human CD95 (Fas) monoclonal antibody (clone EOS9.1) was obtained from eBioscience (San Diego, CA). Anti-hepatitis B surface antigen culture supernatant from a murine IgM hybridoma was used as the isotype control. Goat anti-mouse IgG (H + L) conjugated to FITC served as secondary antibody (Jackson ImmunoResearch, West Grove, PA). Anti-human CD3 coupled to PerCP and its isotype control murine IgG1κ were purchased from Becton-Dickinson (Mountain View, CA). All flow cytometric data was acquired on a FACSCalibur run with CellQuest (Becton-Dickinson), and later re-analyzed with WinMDI version 2.8 (J. Trotter).
4.3. Cell number and apoptosis staining
Cells were harvested 24, 48, 72 or 96 h after addition of IFN-γ to the culture medium, and counted with a hemacytometer, using eosin Y as an exclusion dye, before any further analysis. Apoptotic cells were stained with an annexin V-FITC/propidium iodide (PI) apoptosis detection kit (BioVision Research, Mountain View, CA) and analyzed by flow cytometry. In some cases, the presence of the active form caspase-8 was also detected using CaspGLOW Fluorescein active caspase-8 staining kit from BioVision Research, and analyzed by flow cytometry.
4.4. Cell cycle analysis
A549 cells were cultured in serum-free culture medium, +37 °C, 5% CO2, for 24 or 36 h, for cell cycle synchronization. The culture supernatant was then replaced by complete medium and IFN-γ added to the medium. Cells were harvested 12, 18, 24, 36 or 48 h later and fixed with cold 70% ethanol. The DNA content was then stained using PI and analyzed by flow cytometry with a FACSCan (Becton-Dickinson). Cell cycle repartition was determined by analysis of the data with ModFit LT (Verity Software House, Topsham, ME).
4.5. A549/Jurkat cells co-culture
Resting Jurkat cells were cultured for 24 h in 100 mm culture plates in complete medium containing 100 ng mL−1 phorbol 12-myristate 13 acetate and 500 ng mL−1 ionomycin. At the same time, A549 cells were seeded and cultured for 24 h. IFN-γ (250 IU mL−1) was added to A549 culture 12 h after seeding. After 24 h of incubation, Jurkat cells were pelleted (400 × g, 5 min) and resuspended with culture supernatant from individual A549 cell cultures. The Jurkat cell suspension was then transferred to A549 cell culture plates from which the culture supernatant originated. Finally whole cell cultures were harvested 24 h later, stained with annexin V-FITC/PI and anti-human CD3 antibody.
4.6. Chemokines secretion
The presence of chemokines MIG, MCP-1, IP-10, RANTES and IL-8, and their concentration, were determined using a human chemokines cytometric bead array (Becton-Dickinson). Samples analyzed were culture supernatants from cells incubated for 48 h with IFN-γ. Each sample was centrifuged after collection (400 × g, 5 min), and only the supernatant fraction was kept for assay.
4.7. Statistical analysis
All experiments were carried out in triplicate and the data are expressed as the mean ± standard error of the mean. Prism (GraphPad Software, San Diego, CA) was used to carry out the Mann–Whitney t-test or one-way analysis of variance (ANOVA) for comparisons. The Newman–Keuls multiple comparisons test was applied as a post-test in ANOVA for individual comparisons. In all analyses, statistical significance was defined as p < 0.05.
Acknowledgements
This work was supported by the SARS Research Project grant NSC92-2751-B006-007 from the National Science Council, Taiwan.
References
- 1.World Health Organization Communicable Disease Surveillance and Response. Summary of probable SARS cases with onset of illness from 1 November 2002 to 31 July 2003. World Health Organization; 2004. <http://www.who.int/csr/country/table2004_04_21/en/>.
- 2.Drosten C., Günther S., Preiser W., van der Werf S., Brodt H.R., Becker S. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 3.Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S. A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 4.Peiris J.S.M., Lai S.T., Poon L.L.M., Guan Y., Yam L.Y.C., Lim W. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet. 2003;361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Denison M.R. Severe acute respiratory syndrome coronavirus pathogenesis, disease and vaccines. Pediatr Infect Dis J. 2004;23:S207–S214. doi: 10.1097/01.inf.0000144666.95284.05. [DOI] [PubMed] [Google Scholar]
- 6.Booth C.M., Matuks L.M., Tomlinson G.A., Rachlis A.R., Rose D.B., Dwosh H.A. Clinical features and short-term outcomes of 144 patients with SARS in the Greater Toronto area. JAMA. 2003;289:2801–2809. doi: 10.1001/jama.289.21.JOC30885. [DOI] [PubMed] [Google Scholar]
- 7.Joynt G.M., Antonio G.E., Lam P., Wong K.T., Li T., Gomersall C.D. Late-stage adult respiratory distress syndrome caused by severe acute respiratory syndrome: abnormal findings at thin-section CT. Radiology. 2004;230:339–346. doi: 10.1148/radiol.2303030894. [DOI] [PubMed] [Google Scholar]
- 8.Ding Y., Wang H., Shen H., Li Z., Geng J., Han H. The clinical pathology of severe acute respiratory syndrome (SARS): a report from China. J Pathol. 2003;200:282–289. doi: 10.1002/path.1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peiris J.S.M., Chu C.M., Cheng V.C.C., Chan K.S., Hung I.F.N., Poon L.L.M. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: a prospective study. Lancet. 2003;361:1767–1772. doi: 10.1016/S0140-6736(03)13412-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang K.J., Su I.J., Theron M., Wu Y.C., Lai S.K., Liu C.C. An interferon-γ-related cytokine storm in SARS patients. J Med Virol. 2004;75:185–194. doi: 10.1002/jmv.20255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wong C.K., Lam C.W.K., Wu A.K.L., Ip W.K., Lee N.L.S., Chan I.H.S. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin Exp Immunol. 2004;136:95–103. doi: 10.1111/j.1365-2249.2004.02415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bachofen M., Weibel E.R. Structural alterations of lung parenchyma in the adult respiratory distress syndrome. Clin Chest Med. 1982;3:35–56. [PubMed] [Google Scholar]
- 13.Matthay M.A., Folkesson H.G., Campagna A., Kheradmand F. Alveolar epithelial barrier and acute lung injury. New Horiz. 1993;1:613–622. [PubMed] [Google Scholar]
- 14.Matute-Bello G., Liles W.C., Steinberg K.P., Keiner P.A., Mongovin S., Chi E.Y. Soluble Fas ligand induces epithelial cell apoptosis in humans with acute lung injury (ARDS) J Immunol. 1999;163:2217–2225. [PubMed] [Google Scholar]
- 15.Matute-Bello G., Winn R.K., Jonas M., Chi E.Y., Martin T.R., Liles W.C. Fas (CD95) induces alveolar epithelial cell apoptosis in vivo. Am J Pathol. 2001;158:153–161. doi: 10.1016/S0002-9440(10)63953-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lew T.W., Kwek T.K., Tai D., Earnest A., Loo S., Singh K. Acute respiratory distress syndrome in critically ill patients with severe acute respiratory syndrome. JAMA. 2003;290:374–380. doi: 10.1001/jama.290.3.374. [DOI] [PubMed] [Google Scholar]
- 17.Coulter K.R., Doseff A., Sweeney P., Wang Y., Marsh C.B., Wewers M.D. Opposing effect by cytokines on Fas-mediated apoptosis in A549 lung epithelial cells. Am J Respir Cell Mol Biol. 2002;26:58–66. doi: 10.1165/ajrcmb.26.1.4285. [DOI] [PubMed] [Google Scholar]
- 18.Tanaka T., Yoshimi M., Maeyama T., Hagimoto N., Kuwano K., Hara N. Resistance to Fas-mediated apoptosis in human lung fibroblast. Eur Respir J. 2002;20:359–368. doi: 10.1183/09031936.02.00252602. [DOI] [PubMed] [Google Scholar]
- 19.Wen L.P., Madani K., Fahrni J.A., Duncan S.R., Rosen G.D. Dexamethasone inhibits lung epithelial cell apoptosis induced by IFN-γ and Fas. Am J Physiol Lung Cell Mol Physiol. 1997;273:921–929. doi: 10.1152/ajplung.1997.273.5.L921. [DOI] [PubMed] [Google Scholar]
- 20.Ohkawa T., Ueki N., Taguchi T., Shindo Y., Adachi M., Amuro Y. Stimulation of hyaluronan synthesis by tumor necrosis factor-alpha is mediated by the p50/p65 NF-kappa B complex in MRC-5 myofibroblasts. Biochim Biophys Acta. 1999;1448:416–424. doi: 10.1016/s0167-4889(98)00155-4. [DOI] [PubMed] [Google Scholar]
- 21.Wynn T.A. Fibrotic disease and the TH1/TH2 paradigm. Nat Rev Immunol. 2004;4:583–594. doi: 10.1038/nri1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen E.S., Greenlee B.M., Wills-Kay M., Moller D.R. Attenuation of lung inflammation and fibrosis in interferon-γ-deficient mice following intratracheal bleomycin. Am J Respir Cell Mol Biol. 2001;24:545–555. doi: 10.1165/ajrcmb.24.5.4064. [DOI] [PubMed] [Google Scholar]
- 23.Segel M.J., Izbicki G., Cohen P.Y., Or R., Christensen T.G., Wallach-Dayan S.B. Role of interferon-gamma in the evolution of murine bleomycin lung fibrosis. Am J Physiol Lung Cell Mol Physiol. 2003;285:1255–1262. doi: 10.1152/ajplung.00303.2002. [DOI] [PubMed] [Google Scholar]
- 24.Yamada M., Kubo H., Kobayashi S., Ishizawa K., Sasaki H. Interferon-γ: a key contributor to hyperoxia-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2004;287:1042–1047. doi: 10.1152/ajplung.00155.2004. [DOI] [PubMed] [Google Scholar]
- 25.Sharma S.K., MacLean J.A., Pinto C., Kradin R.L. The effect of an anti-CD3 monoclonal antibody on bleomycin-induced lymphokine production and lung injury. Am J Respir Crit Care Med. 1996;154:193–200. doi: 10.1164/ajrccm.154.1.8680680. [DOI] [PubMed] [Google Scholar]
- 26.Glass W.G., Subbarao K., Murphy B., Murphy P.M. Mechanisms of host defense following severe acute respiratory syndrome-coronavirus (SARS-CoV) pulmonary infection in mice. J Immunol. 2004;173:4030–4039. doi: 10.4049/jimmunol.173.6.4030. [DOI] [PubMed] [Google Scholar]
- 27.Hagimoto N., Kuwano K., Miyazaki H., Kunitake R., Fujita M., Kawasaki M. Induction of apoptosis and pulmonary fibrosis in mice in response to ligation of Fas antigen. Am J Respir Cell Mol Biol. 1997;17:272–278. doi: 10.1165/ajrcmb.17.3.2893. [DOI] [PubMed] [Google Scholar]
- 28.Uhal B.D., Joshi I., Ramos C., Pardo A., Selman M. Alveolar epithelial cell death adjacent to underlying myofibroblasts in advanced fibrotic human lung. Am J Physiol Lung Cell Mol Physiol. 1998;275:1192–1199. doi: 10.1152/ajplung.1998.275.6.L1192. [DOI] [PubMed] [Google Scholar]