

Highlights
► Effects of CXCL10 in human T lymphocytes in terms of apoptosis and survival. ► The signaling pathways that are involved in the process were studied. ► New perspective about the role of CXCL10 during viral infections.
Keywords: CXCL10, CXCR3, Apoptosis, Lymphocytes
Abstract
CXCL10 is part of the group of interferon-stimulated genes and it plays an important role during different viral infections by inducing cell activation, chemotaxis and lymphocyte priming toward the Th1 phenotype. In this study, we investigated in vitro the effects of CXCL10 in activated human primary T lymphocytes in terms of apoptosis or survival, and delineated the signaling pathways that are involved. CXCL10, in combination with IL-2 and/or IFNα, induces apoptosis in T lymphocytes. Moreover, CXCL10-induced activation of CXCR3 also triggers pro-survival signals that can be blocked by pertussis toxin. The analysis of the downstream signaling kinases shows that apoptosis is p38 MAPK-dependent and the pro-survival signals rely on the sustained activation of PI3K and the transient activation of Akt. On the other hand, the transient activation of p44/p42 ERK did not have an impact on T lymphocyte survival. We propose an immunological model in which CXCL10, together with other co-stimulating cytokines, participates in the activation of T lymphocytes, promotes survival and expansion of certain lymphocyte subsets, and induces chemotaxis toward the infected tissues. On the other hand, CXCL10 might contribute to the triggering of apoptosis in other subsets of T lymphocytes, including those lymphocytes that were transiently activated but later lacked the appropriate sets of specific co-stimulating signals to ensure their survival.
1. Introduction
CXCL10 (IFNγ-inducible protein of 10 kDa or IP10) plays an important role during viral infections such as influenza [1], SARS coronavirus [2], HIV [3] and West Nile virus [4]. Although initially identified to be an IFNγ-responsive gene, a number of different cytokines including IFNα/β are capable of inducing CXCL10 expression in diverse cell types. The chemokines CXCL4, CXCL9 (Mig), CXCL10 (IP-10), and CXCL11 (I-TAC) bind the receptor CXCR3 and exert a potent chemotactic effect on activated T lymphocytes [5]; however, CXCL10 is the one that plays the most prominent role during the antiviral responses. CXCR3 is expressed on 35–40% of normal blood T cells and its expression is enhanced by treatment [6] with IL-2. Additionally, CXCR3 is expressed on other cell types such as natural killer cells (NKs), eosinophils, B lymphocytes, and different epithelial and endothelial cell types. CXCR3 is a receptor that is coupled to a G protein-signaling pathway. Binding of chemokines to CXCR3 activates G protein-mediated signaling and results in downstream activation of phospholipase C (PLC), kinase A and C, formation of phosphatidyl inositol-1,4,5-triphophate, and also the increase of intracellular calcium ions [7]. Despite the functional diversity of the cells that can express CXCR3, the downstream activation seems to be quite homogeneous, involving p44/p42 ERK, Akt, PI3K and p38 [5], [8], [9]. However, the effects of CXCR3 activation are very diverse, ranging from chemotaxis in T lymphocytes [8], [10], [11] and airway epithelial cells [9] to anti-proliferation in human embryonic kidney cells [10] and induction of apoptosis in acinar cells [12].
Programmed cell death (apoptosis) is an active process morphologically characterized by shrinkage of the cell, nuclear fragmentation, chromatin condensation and chromosomal DNA fragmentation [13]. Apoptosis plays an essential role in tissue development and is required for maintaining tissue homeostasis [14]. Although apoptosis can be triggered by many different stimuli, executioner caspases always participate in the organized degradation of the cellular organelles; when activated, executioner caspases cleave a variety of cytoplasmic and nuclear proteins including caspases themselves and their downstream substrates [15] through controlled proteolysis [16]. Induction of apoptosis is a tightly regulated process and it is the balance between pro-survival and pro-apoptosis signals that eventually determines the fate of the cells [17]. Vlahakis et al. have shown that G protein-coupled chemokine receptors can activate both the survival and the apoptotic signaling pathways [18]. It has been reported that CXCL10 can trigger apoptosis in several cellular models, such as acinar cells [12], neurons [19], HeLa cells [20] and microvascular endothelial cells [21]; however, the role of CXCL10 in determining survival or death in blood T lymphocytes remains largely unknown.
In this study, we have delineated critical signaling pathways activated by CXCL10 and its receptor CXCR3. We demonstrated that CXCL10 is able to transiently activate the Akt and p44/42 ERK signaling cascades and sustainably activate p38 MAPK signaling in primary human T lymphocytes. Moreover, we determined the role that these pathways have in the induction of either apoptosis or cell survival.
2. Materials and methods
2.1. Antibodies and reagents
The monoclonal antibodies (MAb) mouse anti-human Akt, mouse anti-human phospho-Akt, mouse anti-human CXCR3, mouse anti-human CD4, CD8, CD20, and mouse anti-human actin, were obtained from BD Biosciences (San Diego, CA, USA). Mouse anti-human p38, rabbit anti-human phospho-p38, rabbit anti-human p44/42 ERK, rabbit anti-human phospho-p44/42 ERK, rabbit anti-human phospho-Bcl2, rabbit anti-human Bcl2, rabbit anti-human caspase 3 (CASP3), and cleaved CASP3 were obtained from Cell Signaling Technology (Boston, MA, USA). SYBR®Green PCR Master Mix was obtained from Applied Biosystems (Foster City, CA, USA). Goat anti-mouse IgG-HRP and donkey anti-rabbit IgG-HRP were purchased from Santa Cruz Biotechnology (Santa Cruz, CA. USA). Recombinant CXCL10, IL-2, and IFNα protein were purchased from R&D Systems (Minneapolis, MN). RPMI-1640 supplemented with 2 mM L-glutamine HEPES Buffer solution, and 0.005% (w/v) gentamycin was obtained from GIBCO BRL (Burlington, Ontario, Canada). Fetal calf serum (FCS) was purchased from Cansera (Rexdale, Ontario, Canada) and heat-inactivated at 56 °C for 30 min upon arrival. The PI3K inhibitor (wortmannin), Phosphate-buffered saline (PBS) pH 7.4 and Pertussis toxin were obtained from Sigma, Aldrich Inc. (Oakville, Ontario, Canada). The MEK inhibitor (PD98059) and the p38 inhibitor (SB202190) were purchased from Biosource International Inc. (Camarillo, CA, USA).
2.2. Isolation of peripheral blood mononuclear cells (PBMCs)
Blood was obtained from healthy donors with ages ranging from 23 to 40 years old. Density gradient centrifugation was used to separate PBMCs from human blood. First, approximately 60 ml of blood were collected from the brachial vein of a healthy human volunteer using a 28-gauge needle and a sterile 60 ml syringe containing 50–100 units sodium heparin (Becton–Dickinson, Mississauga, Ontario) per ml blood. Next 20 ml sterile PBS containing penicillin and streptomycin (Pen/Strep) (GIBCO BRL) (2 ml/50 ml PBS) was added to 20 ml blood in a sterile 50 mL centrifuge tube. The tube was underlayed with 7 ml of sterile ficoll-Hypaque 1077 (Sigma–Aldrich Inc.) and centrifuged at 400 g for 45 min at room temperature. Using a transfer pipette, approximately 5 ml of plasma was removed and discarded. Using a fresh sterile transfer pipette, the PBMC layer was transferred to a fresh 50 ml tube. The tube was tilted at a 45-degree angle to facilitate removal of the PBMC layer and 45 ml PBS-pen/strep was added to the isolated PBMCs. The PBMC sample was centrifuged at 400 g for 10 min and the supernatant was discarded. The resulting cell pellet was gently resuspended in approximately 3 ml of PBS-pen/strep. 5–10 ml of sterile distilled water was added before swirling for a maximum of 10–15 s to lyse the RBCs. Finally, 30 ml of PBS were immediately added and the resuspended cells were centrifuged again for 10 min at 300–400 g to remove lysed RBCs and any remaining ficoll.
2.3. Isolation and culture of human T lymphocytes
For the study of CXCR3 expression, lymphocytes were separated from PBMCs by magnetic beads positive selection using the Monocyte Isolation Kit II (Miltenyi Biotech, Auburn, CA). In all the other experiments, untouched T lymphocytes were separated from PBMCs using the Pan T Cell Isolation Kit II (Miltenyi). Cells were resuspended in PBS and centrifuged for 10 min at 300–400 g. The supernatant was discarded and the cells resuspended in 5–10 ml complete RPMI-1640 medium supplemented with 10% of heat inactivated FCS, 2 mM L-glutamine (Fisher Scientific, Nepean, Ontario, Canada), 0.1 mM β-mercaptoethanol (Fisher Scientific) and pen/strep. The cells were counted using Unopettes (Becton Dickinson and company, Franklin Lake, NJ, USA) and a hemocytometer and the desired cell concentration was prepared by dilution with complete RPMI medium. To induce expression of CXCR3, lymphocytes were cultured for 10 days in complete RPMI 1640 medium with different combinations of IL-2 (400 U/ml), IFNα (5000 U/ml) and CXCL10 (50 nM); cells received cytokine re-treatment every 3 days. Selected T cell cultures were preincubated for 1 hour at 37 °C with inhibitors such as pertussis toxin (1 μg/ml), wortmannin (100 nM), PD9809 (10 μM), SB203580 (10 μM) before the addition of CXCL10 + IL-2 + IFNα. All experiments were performed in triplicate.
2.4. Flow cytometry
Lymphocyte cells were pelleted by centrifugation, resuspended in 0.1% BSA in PBS, aliquotted into FACS tubes, and incubated with antibody for 30 min in the dark. To detect apoptotic cells, the samples were incubated with Annexin V-PE in a buffer containing 7-Amino-actinomycin (7-AAD) (BD Biosciences, San Diego, CA) and analyzed by FACS. Stained cells were washed in 2 ml PBS fixed in 2% paraformaldehyde in PBS, and analyzed using a dual-laser flow cytometer (FACS Calibur, Becton Dickinson, San Jose, CA) and CellQuest Software (BD Biosciences). Post-acquisition cytometry data was analyzed using FlowJo v. 5.7.2 (TreeStar, Inc., Ashland, OR).
2.5. RNA extraction and quantitative real-time PCR
After 10 days of culture, cells were pelleted by centrifugation at 300 g, lysed by addition of 1 ml TRIZOL® reagent (Invitrogen Canada) per 5–10 × 106 cells and the RNA was extracted according to the manufacture’s protocol. The concentration of RNA was measured as optical density (OD) at 260 nm using an Eppendorf Biophotometer (Mississauga, Ontario, Canada). The integrity and quality of total RNA was confirmed by RNase-free agarose gel electrophoresis using DEPC-treated TAE buffer. Quantitative real timePCR (QRT-PCR) was performed using an ABI-PRISM 7900HT Sequence Detection System and SYBR green PCR Master Mix (Applied Biosystems, Foster City, California). Total RNA (250–1000 ng) was reverse transcribed in a 20 μl reaction under the following conditions: 6.25 μM random hexanucleotide primer (Applied Biosystems), 50 mM TrisHCl pH 8.3, 3 mM MgCl2, 75 mM KCl, 500 μM dATP, dGTP, dTTP, and dCTP, 10 mM DTT and 200U SuperScript II RNAse H-reverse transcriptase, at 42 °C for 60 min using a thermal cycler (PTC-150 Minicycler, MJ Research-Bio-Rad Laboratories Canada Ltd., Mississauga, Ontario). The resulting cDNA was subjected either to conventional PCR amplification or QRT-PCR. Each QRT-PCR reaction was performed in a volume of 10 μl with 0.125 μl cDNA, primer pair and 5 μl of SYBR green PCR Master Mix in ABI-PRISM optical 384 well plates. Primer pairs were designed to generate intron-spanning products of approximately 100 bp using Primer Express v2.0 software (Applied Biosystems). Each primer pair was tested with a logarithmic dilution of cDNA to generate a standard curve, which was used to calculate the starting quantity of target RNA. Primers specific for 18S rRNA were used as an endogenous standard to normalize samples. Fold-changes were calculated by dividing the normalized post-treated sample quantity with the normalized pre-treated control quantity. 5′–3′ sequences of primer pairs were as follows: CXCL10-forward-TCC ACG TGT TGA GAT CAT TGC, reverse-TCT TGA TGG CCT TCG ATT CTG, 18S-rRNA-forward-CGG CTA CCA CAT CCA AGG AA, reverse-GCT GGA ATT ACC GCG GCT.
2.6. Cell lysis and protein extraction
Lymphocytes were cultured in 24 well plates at 37 °C and, following the treatments indicated in their respective figure legends (Fig. 3, Fig. 4, Fig. 5 and Supp. Fig. 3), reactions were terminated by centrifugation, followed by supernatant aspiration and cell resuspension in 50 μl of ice-cold lysis buffer (20 mM Tris (pH 7.4), 137 mM NaCl, 1 mM MgCl2, 1 mM CaCl2, 10% glycerol (w/v), 1% Nonidet P-40 (w/v), and protease inhibitors, 1 mM PMSF, 2 μg/ml aprotinin, 2 μg/ml leupeptin, 2 μg/ml pepstatin A, and tyrosine phosphotase inhibitor, 1 mM sodium orthovanadate). Lysates were incubated on a rotator for 20 min, and non-soluble material was removed by centrifugation at 14,000 rpm for 10 min. Protein levels in the supernatants were determined by Bradford protein assay (Bio-Rad) and adjusted to equal protein concentration between samples by dilution with lysis buffer.
Fig. 3.
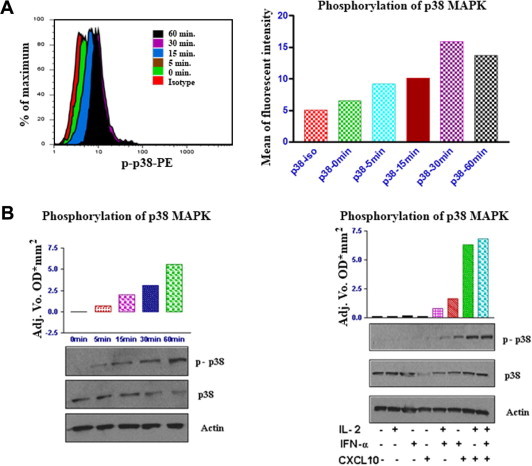
CXCL10 induces sustainable phosphorylation of p38 MAPK in CXCR3-positive T cells. (A) Histogram analysis of intracellular staining of p-p38 in CXCR3 positive lymphocytes (left) and mean fluorescent intensity of p-p38 in T cells (right). Freshly isolated human T cells were cultured in medium alone, or treated with IL-2, IFNα, or CXCL10 or a combination of the three, for the times indicated. Cells were then washed and analyzed by FACS. FACS was performed using an antibody specific for the activated p38 MAPK (phospho-p38 [p-p38]). The results are shown for events in the live cell gate and are representative of at least three independent experiments. (B) Western blots using a phospho-p38-specific antibody (left) were performed on protein derived from human T cells cultured either in medium alone or treated with CXCL10 + IL-2 + IFNα, for the times indicated. The phosphorylation was quantified by chemiluminescence and standardized against the same blots stripped and re-probed with antibodies that recognize total p38 protein. Actin was used as a loading control. Western blots and band quantification (right) in T cells that were untreated or cultured with IL-2, IFNα, CXCL10, or a combination of the three, as indicated, for 16 h. Cells were then lysed and phosphorylation of p38 was determined by western blot analysis using specific anti-phospho-p38 antibodies.
Fig. 4.
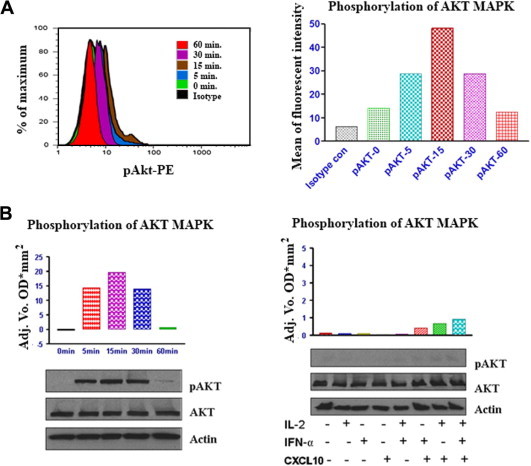
CXCL10 induces transient activation (phosphorylation) of AKT MAPK in CXCR3-positive T lymphocytes. A) Freshly isolated human T cells were cultured in medium alone, or treated with IL-2 or IFNα or CXCL10, or with a combination of all three for the times indicated. Cells were then washed and analyzed by FACS. FACS was performed for activated AKT MAPK (phospho-AKT [p-AKT]). FACS results are shown for events in the live cell gate and are representative of at least three independent experiments. Left: histogram analysis of intracellular staining of p-AKT in CXCR3+ cells. Right: mean fluorescent intensity of p-AKT in T cells. B) Left: human T cells were cultured in medium alone or treated with IL-2, IFNα, and CXCL10 for the times indicated. Cells were then lysed and phosphorylation levels of AKT were determined by western blot analysis using specific anti-phospho-p38 antibodies. Phosphorylation was quantified by chemiluminescence and corrected for total AKT MAPK expression on the strip blot. Total AKT and actin were used as loading controls. Right, T cells were untreated or treated with IL-2 or IFNα or CXCL10, or in combination, as indicated for16 h; phosphorylation of p38 was determined by western blot analysis using specific anti-phospho-AKT antibodies.
Fig. 5.
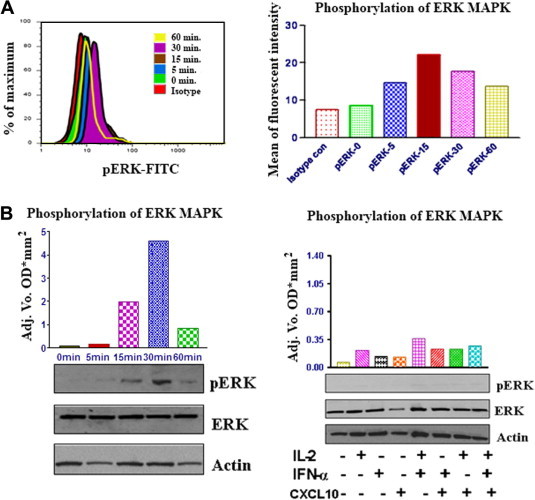
CXCL10 induces activation (phosphorylation) of p44/42 ERK in CXCR3-positive T lymphocytes. (A) Freshly isolated human T cells were cultured in medium alone, or treated with IL-2 or IFNα or CXCL10 or with a combination of all three for the times indicated. Cells were then washed and analyzed by FACS. FACS was performed for activated p44/p42 ERK (phospho-ERK [p-ERK]). FACS results are shown for events in the live cell gate and are representative of at least three independent experiments. Left: histogram analysis of intra-cellular staining of p-ERK in CXCR3 + lymphocytes. Right: mean fluorescent intensity of p-ERK in T cells. (B) CXCL10 induces phosphorylation of p44/p42 ERK in T lymphocytes: (left) human T cells were cultured in medium alone or treated with IL-2, IFNα, and CXCL10 for the times indicated. Cells were then lysed and phosphorylation levels of ERK were determined by western blot analysis using specific anti-phospho-ERK antibodies. Phosphorylation was quantified by chemiluminescence and corrected for total p44/p42 ERK expression on strip blot; total ERK and actin were used as loading controls (right). T cells were untreated or treated with IL-2 or IFNα or CXCL10, or a combination of the three, as indicated, for16 h; phosphorylation of ERK was determined by western blot analysis using specific anti-phospho-ERK antibodies.
2.7. Immunoblotting
Cell lysates were boiled for 5 min in Laemmli buffer and samples were loaded onto 4–14% SDS–PAGE gels (Bio-Rad) and the protein was separated by electrophoresis. The protein was transferred onto nitrocellulose membranes by semi-dry electroblotting. Immunoblotting was performed as previously described (42). Briefly, membranes were incubated for 1 hour with blocking solution (5% nonfat milk/0.05% sodium azide in TBS (10 mM Tris (pH 7.5) and 100 mM NaCl)) and then incubated overnight in the required dilution of the appropriate primary antibody in TBS/0.1% Tween-20. After washing twice, membranes were incubated with goat anti-mouse or donkey anti-rabbit horseradish peroxidase-conjugated secondary antibodies. Protein bands were detected with enhanced chemiluminescence (ECL) (Amersham-Pharmacia Biotech) and directly quantified using an image station (Bio-Rad, Hercules, CA). Where necessary, membranes were stripped for re-probing by incubation in stripping buffer (62.5 mM Tris (pH 6.8), 2% SDS, and 100 mM 2-ME) at room temperature for 30 min.
3. Results
3.1. IL-2, but neither CXCL10 nor IFNα, increases the expression of CXCR3 on human T lymphocytes
We examined the effects of IL-2 and CXCL10 on the expression of CXCR3 in human primary lymphocytes using flow cytometry. CXCR3 was found to be expressed on untreated lymphocytes, however, we cannot rule out the possibility that the expression levels of CXCR3 were altered, up to some extent, during the initial process of positive cell selection. Upon treatment with IL-2, primary lymphocytes showed greatly enhanced expression of CXCR3. The increase in CXCR3 expression occurred in the CD8+ population, but not in CD4+ or CD20+ cells (Fig. 1 A, B and E). However, incubation with CXCL10 did not alter the expression levels of CXCR3 (Fig. 1C–E). We also showed that the expression of CXCR3 is not induced by concentrations of IFNα up to 15,000 U/ml in neither CD4+ nor CD8+ cells (Supp. Fig. 1). Using real time PCR, we showed that CXCL10 is induced by IFNα both in primary lymphocytes and human embryonic kidney cells (HEK239) (Supp. Fig. 2), ensuring that T lymphocytes were responsive to IFNα under our experimental conditions.
Fig. 1.
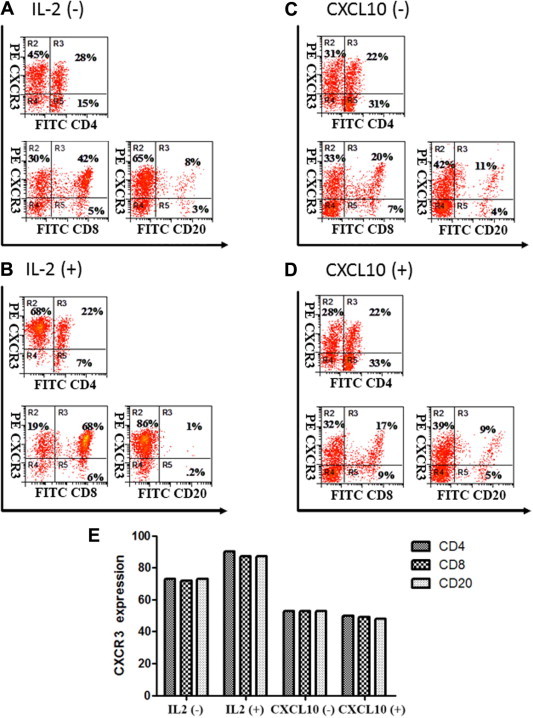
CXCR3 expression on human T cells is enhanced by IL-2 but not induced by CXCL10. Lymphocytes were purified from healthy donor PBMCs using magnetic bead positive selection (Monocyte Isolation Kit II, Miltenyi Biotech) and cultured in the absence of IL-2 (A), the presence of IL-2 (B), the absence of CXCL10 (C) and the presence of CXCL10 (D). Phenotypic flow cytometry analysis was performed to assess CXCR3, CD8, CD84, and CD20 expression at the protein level. Percentages of CXCR3+ cells on gated viable cell populations are indicated within the panels, where the cutoff is determined by negative controls obtained by isotype-matched control antibody conjugated with PE. The results are representative of three independent experiments. Gates were based on the isotype controls.
3.2. CXCL10 in combination with IL-2 and/or IFNα, induces caspase 3-mediated apoptosis in primary T lymphocytes
We examined the rate of cell death induced in primary T lymphocytes after incubation with CXCL10 alone (50 nM) after 16 h. FACS analysis of cells stained with Annexin V and 7-AAD demonstrated that treatment of CXCR3-positive T cells with recombinant CXCL10 induces early apoptosis (Annexin V positive, 7-AAD negative), although to a very limited extent (Fig. 2 A). Furthermore, pre-incubation of T cells with IL-2 (400 U/ml), which increases CXCR3 expression but not apoptosis rates (Fig. 2B and C), enhanced CXCL10-induced early apoptosis dramatically (Fig. 4C). Cell incubation with IFNα alone (5000 U/ml) also leads to T cell early apoptosis. Moreover, the highest rates of early apoptosis were observed with simultaneous treatment with CXCL10 + IL-2 + IFNα (Fig. 2C). The cytokine treatments that were tested also produced increased necrosis rates (Annexin V positive, 7-AAD positive) in parallel with the observed rates of early apoptosis (Fig. 2A and B). To determine whether the cell death was CASP3-mediated, we analyzed whole cell lysates from T lymphocytes treated with CXCL10 + IL-2 + IFNα; the presence of active cleaved fragments of CASP3 was assessed by western blotting using antibodies that recognize both procaspase 3 (p32) and cleaved CASP3 (p19, p17 and p12). As shown in Fig. 2D, independent treatments with CXCL10 or IL-2 or IFNα did not result in appreciable CASP3 overexpression or cleavage. It was the combined treatment with CXCL10 + IL-2 + IFNα that caused the highest levels of CASP3 activation, closely followed by CXCL10 + IL-2. Interestingly, treatment with IL-2 + IFNα also resulted in CASP3 activation.
Fig. 2.
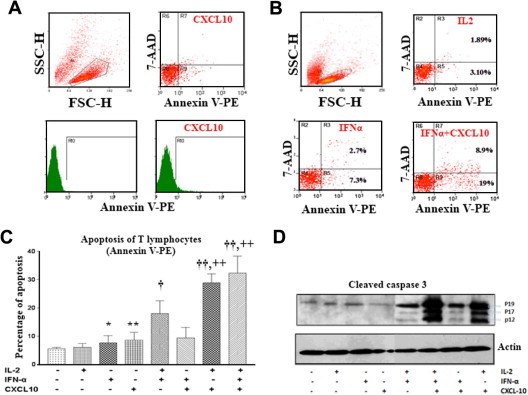
Recombinant CXCL10-induced apoptosis in T cells. (A) Primary T lymphocytes were treated with CXCL10 and analyzed by FACS for apoptosis. (B) Primary lymphocytes were treated with IL2, IFNα and CXCL10 and analyzed by FACS for apoptosis. (C) Primary lymphocytes were treated with IL-2, IFNα and CXCL10 and analyzed by FACS for apoptosis using BD Annexin-V Apoptosis Detection Kit. Statistical significance, respect to isotype control: *p < 0.05 and **p < 0.01; respect to IL-2 only: †p < 0.05 and ††p < 0.01; respect to IL-2 + IFNα: ++p < 0.01. Gates were based on the isotype controls. (D) Activation of p38 MAPK in T lymphocytes induces activation of caspase 3. Protein extracts derived from lysates from T cells treated with different combinations of IL-2, IFNα, and CXCL10, were subjected to western blot analysis using antibodies that recognize full-length (p32) and cleaved (p19, p17, p12) caspase 3. The protein level of actin was examined as a loading control.
3.3. CXCL10 induces sustained activation of p38 MAPK pathways in T cells
To elucidate the signaling pathways responsible for CXCL10-mediated apoptosis, we studied the phosphorylation of different MAPK kinases, which are essential signaling mediators that lead cells toward apoptosis or survival. First we examined the putative role of CXCL10 as an activator of p38 MAPK in activated T lymphocytes by flow cytometric analysis using phospho-specific antibodies and western blotting, (Fig. 3A). We found that CXCL10 treatment resulted in rapid and sustained phosphorylation of p38. The maximal response was observed within 60 min following CXCL10 stimulation and continued for 24 h (Fig. 3B). We also observed that CXCL10 alone, but not IFNα or IL-2, was able to induce modest p38 phosphorylation. It was the combined treatment with CXCL10 and IL-2 that caused a dramatic increase in the phosphorylation levels of p38, which is in accordance with the induction of the abovementioned apoptosis.
3.4. CXCL10 induces transient phosphorylation of Akt and p44/p42 ERK in T lymphocytes
Later, we investigated Akt phosphorylation after stimulation of T lymphocytes with CXCL10 + IL2 + IFNα by FACS analysis. CXCL10 (50 nM) induced a transient, time-dependent increase in Akt phosphorylation, reaching the maximum levels in 15 min and returning close to the basal levels in the 60-min time-point (Fig. 4A). When using a lower concentration of CXCL10 (10 nM), the phosphorylation peak was detected after 45 min of incubation, and the levels were still high 60 min after the beginning of the stimulation (data not shown). These findings were confirmed using western blot analysis (Fig. 4B). Our results also suggest that CXCL10-mediated Akt phosphorylation is stronger when IL-2 and/or IFNα are present (Fig. 4B). Next, we examined the activation of the p44/p42 ERK kinases following exposure of peripheral T cells to CXCL10 + IL2 + IFNα. Exposure of CXCR3-expressing lymphocyte cells to CXCL10 (50 nM) resulted in a transient increase in p44/p42 ERK phosphorylation which peaked after 30 min of exposure to CXCL10 (Fig. 5A and B). However, in the assay where different combinations of cytokines were tested, the incubation time was too long (16 h) and the ERK phosphorylation was indistinguishable from the basal activation (Fig. 5B), indicating that the transient ERK activation had already extinguished. However, considering the other assays within this study, it is reasonable to conclude that ERK activation is stronger when CXCL10 incubation occurs in combination with IL-2 and/or IFNα.
3.5. Evaluation of the implication of different signaling mediators in apoptosis and survival by using specific inhibitors
Human primary T lymphocytes were incubated with CXCL10 + IL-2 + IFNα together with different pathway-specific inhibitors, and after 24 h, the percentage of cell death was determined by FACS analysis (Fig. 6 ). As expected, cells treated with CXCL10 + IL-2 + IFNα, but not inhibitors, showed a notable increase in cell death as compared with the untreated cells (medium only). Both pertussis toxin (inhibitor of G-coupled protein signaling) and wortmannin (PI3K inhibitor) induced a significant increase in the percentage of cell death as compared with the no-inhibitors group. On the other hand, cells treated with SB203580 (p38 MAPK inhibitor) showed similar levels of cell death as the untreated group (medium only), indicating that the inhibitor was able to revert the apoptosis induced by CXCL10 + IL-2 + IFNα. Finally, the inhibitor PD98509 (MEK inhibitor, upstream of ERK) did not modify the levels of cell death as compared with the no-inhibitor group, suggesting that the activation of p44/p42 ERK is not connected with the induction of either cell death or survival. Additionally, T lymphocytes were treated separately with all the inhibitors and no increase in the levels of apoptosis was observed for any of them (data not shown).
Fig. 6.
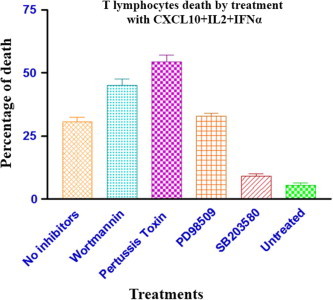
CXCL10 mediated death of T cells via initiation of proapoptotic signals. T lymphocytes were untreated or treated with a combination of IL-2, IFNα, and CXCL10, following separate pre-incubations with the following inhibitors: pertussis toxin (inhibits G-protein coupled signaling), wortmannin (PI3K inhibitor), SB203580 (p38 MAPK inhibitor), and PD98509 (MEK inhibitor). 24 h later, the percentage of T cell death was determined using an Annexin V-PE Apoptosis Detection Kit (BD Biosciences).
3.6. CXCL10 blocks Bcl2 anti-apoptotic activity by inducing its phosphorylation in T cells via p38 MAPK
To investigate the anti-apoptotic activity of Bcl2 upon treatment with CXCL10 + IL-2 + IFNα, we performed western blots on whole cell lysates of T cells, using antibodies that recognized both phosphorylated and total Bcl2. As shown in Supp. Fig. 3A, we were unable to detect changes in the expression levels of the Bcl2 protein, whose upregulation is normally linked with the presence of stronger anti-apoptotic signals. However, phosphorylated Bcl2 was present in CXCL10-treated T cells, but not in medium-only cells. This suggests that the treatment with CXCL10 in activated T lymphocytes blocks, at least in part, the anti-apoptotic activity of Bcl2 by phosphorylation, which can be exerted by active p38 MAPK.
4. Discussion
The expression of CXCL10 is part of the interferon-stimulated genes (ISGs) response and its presence is one of the hallmarks of the immune responses during viral infections [2]. During a self-limited influenza infection process, high levels of CXCL10 can be detected during the early stages of the disease, and shortly after the virus has been cleared CXCL10 returns to its basal levels, even though other markers of inflammation and repair may remain elevated beyond that point [1]. On the other hand, in situations where the host fails to control the viral infection the levels of CXCL10, together with other ISGs and inflammatory mediators, remain highly elevated throughout the process and possibly cause detrimental effects by means of excessive immune activation and cell death [22]. We found that CXCL10 alone is a very modest inducer of apoptosis (according to the Annexin V results). Consequently, we needed to consider the addition of other lymphocyte activators that are present during antiviral responses in vivo to unleash the full potential of CXCL10. Since CXCL10 shows its true biological effects in combination with other cytokines, such as IL2 and IFNα, the signaling status upon treatment with this combination of cytokines was the focus of our study. It can be argued that the observed downstream signaling might not correspond to a pure CXCL10 response and some of the observed effects might be overlapping with those from IL2 or IFNα. On the other hand, it should be noted that the presence of these cytokines during viral infections is a proven fact that merits scientific attention, while an in vivo response mediated by CXCL10 alone is an unlikely scenario.
CXCR3 is a seven-transmembrane-spanning domain glycoprotein receptor that is coupled to a G-protein signaling pathway. Binding of different chemokines to CXCR3, such as CXCL4, CXCL9, CXCL10 and CXCL11, causes downstream activation of phospholipase C (PLC), kinase A and C, formation of phosphatidyl inositol-1,4,5-triphophate, and increases intracellular calcium ions [7], [23]. Here we have confirmed that CXCR3 is expressed on blood T lymphocytes and its expression is greatly enhanced by IL-2 treatment [6] (Fig. 1). Moreover, we showed that IFNα treatment significantly increases the expression of CXCL10 in T cells and HEK239 cells (Supp. Fig. 2); however, CXCL10 does not induce changes in the expression of its receptor, CXCR3 (Fig. 1C and D). Our observations are in accordance with previous studies which revealed that CXCR3 expression in T lymphocytes is fine-tuned and it is determined by the presence of certain cytokines [24]. It would appear that both CXCR3 + and CXCR3- populations of cells are undergoing apoptosis. While IL2 did not cause a detectable increase in the level of apoptosis (Annexin V) or CASP3 activation (Fig. 2C and D), CXCL10 and INFα each alone caused a modest increase in apoptosis. The dual combination of Il-2 plus IFNα induced substantially greater levels of apoptosis, and the combination of CXCL10 and IL-2 and the triple combination of IL-2, IFNα and CXCL10 induced the highest levels of apoptosis. Therefore, it is likely that CXCL10 responsive CXCR3+ and CXCR3- cells are undergoing apoptosis.
Chemotaxis is the best-known CXCR3-mediated effect in leukocytes, and the activation of this signaling pathway can be inhibited by pertussis toxin [8]. Our results and also previous studies have shown that CXCL10 and other CXCR3 ligands induce activation of p38 MAPK [11] (Fig. 3), PI3K/Akt [5], [8] (Fig. 4) and p44/42 ERK (Fig. 5). However, studies with small molecule kinase inhibitors were unable to link conclusively these kinases with the induction of chemotaxis in T lymphocytes [8], although some evidence of the involvement of kinase-mediated signaling has been provided [11]. Smit et al. proposed that chemotaxis is not mediated by ERK or Akt but rather by phospholipase C-dependent pathways and phosphatidylinositol kinases other than PI3Kγ [8].
To better characterize the biological functions of CXCL10, we decided to explore its implication in the induction of apoptosis and to delineate the signaling pathways that are involved in the process. Our results show that CXCL10 contributes to the triggering of apoptosis in activated human T lymphocytes, as shown by Annexin V-PE staining (Fig. 2A–C). To determine whether cell death was CASP3-mediated, we analyzed whole cell lysates from T lymphocytes treated with CXCL10 + IL-2 + IFNα for the presence of active and cleaved fragments of CASP3 using western blotting probed with antibodies that recognize both procaspase 3 (p32) and cleaved CASP3 (p19, p17 and p12). As shown in Fig. 2D, independent treatments with CXCL10 or IL-2 or IFNα did not result in appreciable CASP3 overexpression or cleavage. The highest levels of CASP3 activation resulted only with the combined treatment with CXCL10 + IL-2 + IFNα, closely followed by CXCL10 + IL-2 treatment. Interestingly, treatment with IL-2 + IFNα also resulted in CASP3 activation but at much lower levels.
We found that CXCL10 induces sustained phosphorylation of p38 MAPK in T lymphocytes (Fig. 3), a critical mediator of apoptosis that also participates in other cellular processes, including inflammation, cell differentiation, and cell growth [25]. Moreover, in our study we have shown that CXCL10-induced apoptosis was abrogated by the p38 inhibitor SB203580 (Fig. 6), which confirms the role of p38 in the pro-apoptotic signaling during incubation with CXCL10. The anti-apoptotic Bcl2 has been identified as a p38 MAPK substrate [26], and by direct phosphorylation of Bcl2, p38 MAPK prevents Bcl2 from exerting its anti-apoptotic function and allows the apoptotic process to occur [27]. Our results did not clearly show any changes in the expression levels of Bcl2 upon incubation of T lymphocytes with CXCL10 + IL2 + IFNα; however, the phosphorylated form of Bcl2 was more prominent in the chemokine-treated group (Supp. Fig. 3) which confirms the activity of p38 MAPK.
Induction of cell survival is also a critical role of CXCL10 and other CXCR3 ligands. We found that pertussis toxin dramatically increased the rate of apoptotic T cells treated with CXCL10 + IL2 + IFNα, indicating that anti-apoptotic signals are G-protein dependent (Fig. 6); also, pre-treatment of T cells with wortmannin (PI3K inhibitor) led to a significant increase in cell death (Fig. 6). The controls treated only with either pertussis toxin or wormannin did not show increased levels of apoptosis (data not shown), ruling out the possibility that these inhibitors triggered pro-apoptotic signals directly. These results show that CXCR3-mediated signaling plays a critical role in the induction of pro-survival signals in activated T lymphocytes; however, the exact mechanism requires further investigation. Later, we studied the activation profile of Akt, which is a downstream signaling mediator of PI3K. Akt was found to be phosphorylated in T lymphocytes treated with CXCL10 together with IL2 and/or IFNα, and its activation was found to be transient; strong Akt phosphorylation can be detected as early as 5 min after stimulation and it is barely detectable after 60 min (Fig. 4). Akt is crucial for cell survival, proliferation, gene expression and cell migration [8], and the pro-survival effects are explained, at least in part, by the fact that Akt inactivates the apoptotic factor BAD by direct phosphorylation [28]. PI3K and downstream activation of Akt are closely related; however, additional studies are required to elucidate some aspects of their association, such as the involvement of other PI3K downstream signaling mediators, the mechanism through which Akt phosphorylation is rapidly ended, or the conditions that would be necessary for sustained Akt activation. As previously demonstrated for CXCL12 and CXCR4, the loss of Akt signaling after transient activation may be the triggering event for chemokine-induced apoptosis [8], [18]. This process might be required not only to promote cell migration, but also to induce programmed cell death in T cells when CXCR3 ligands are highly expressed. As mentioned above, we found that CXCL10 induced transient phosphorylation of ERK in activated T lymphocytes; however, pretreatment of T cells with the MAPK kinase (MEK) inhibitor PD98059 (MEK is an upstream regulator of p44/p42 ERK) does not alter the percentage of apoptotic cells (Fig. 6), suggesting that CXCL10-induced activation of p44/p42 ERK does not play a relevant role in the survival/apoptosis signaling. Taken together, these results highlight the relevance of G-coupled chemokine receptor signaling to promote survival during lymphocyte activation, a mechanism in which the downstream signaling is mediated by PI3K and Akt.
In this study, In vitro experiments have shown that CXCL10 induces p38-mediated apoptosis in a relevant proportion of T lymphocytes (Fig. 2), and at the same time, CXCL10 + IL2 + IFNα treatment promotes cell survival through PI3K and Akt activation. In vivo, CXCL10 participates in the activation of T lymphocytes during viral infections by priming toward the Th1 phenotype and inducing chemotaxis toward the infected tissues, however, the biological role of CXCL10 as modulator of apoptosis and survival in T lymphocytes is still unclear. We speculate that CXCL10 may help to focus the adaptive immune responses by inducing apoptosis of low-affinity T lymphocyte subsets [29], [30], including those lymphocytes that initially were activated but later lacked the appropriate sets of specific co-stimulating signals to ensure their survival (Fig. 7 ). Additional studies in terms of cell maturity, antigen selectivity, and Th1/Th2 phenotype are required to delineate the features of the T lymphocytes that are positively selected by CXCL10 from those which are depleted by apoptosis in the context of a viral infection.
Fig. 7.
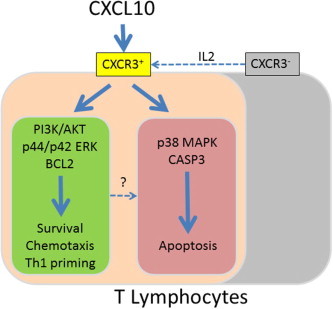
Proposed model of CXCL10 functions during lymphocyte activation. During viral infections and other insults, CXCL10 is produced in response to interferon signaling. IL-2, which is also part of the early immune response, augments the proportion of CXCR3-expressing T lymphocytes and increases their responsiveness. CXCL10 binds its receptor CXCR3, and through transient phosphorylation of Akt and p44/p42 ERK, CXCL10 participates in T lymphocyte activation, survival, proliferation, differentiation and chemotaxis toward the infection sites. However, additional co-signaling is required to maintain long-standing activation, and without it, CXCL10 induces p38-mediated apoptosis, which causes the elimination of low-affinity T lymphocyte subsets.
Acknowledgements
This project was supported by Immune Diagnostics and Research (Toronto, Canada), and Grants from the NIH/NIAID and the Canadian Institute of Health Research.
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cyto.2012.05.002.
Appendix A. Supplementary material
References
- 1.Rowe T., Leon A.J., Crevar C.J., Carter D.M., Xu L., Ran L. Modeling host responses in ferrets during A/California/07/2009 influenza infection. Virology. 2010;401:257–265. doi: 10.1016/j.virol.2010.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cameron M.J., Ran L., Xu L., Danesh A., Bermejo-Martin J.F., Cameron C.M. Interferon-mediated immunopathological events are associated with atypical innate and adaptive immune responses in patients with severe acute respiratory syndrome. J Virol. 2007;81:8692–8706. doi: 10.1128/JVI.00527-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bosinger S.E., Hosiawa K.A., Cameron M.J., Persad D., Ran L., Xu L. Gene expression profiling of host response in models of acute HIV infection. J Immunol. 2004;173:6858–6863. doi: 10.4049/jimmunol.173.11.6858. [DOI] [PubMed] [Google Scholar]
- 4.Klein R.S., Lin E., Zhang B., Luster A.D., Tollett J., Samuel M.A. Neuronal CXCL10 directs CD8+ T-cell recruitment and control of West Nile virus encephalitis. J Virol. 2005;79:11457–11466. doi: 10.1128/JVI.79.17.11457-11466.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Korniejewska A., McKnight A.J., Johnson Z., Watson M.L., Ward S.G. Expression and agonist responsiveness of CXCR3 variants in human T lymphocytes. Immunology. 2011;132:503–515. doi: 10.1111/j.1365-2567.2010.03384.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Loetscher M., Loetscher P., Brass N., Meese E., Moser B. Lymphocyte-specific chemokine receptor CXCR3: regulation, chemokine binding and gene localization. Eur J Immunol. 1998;28:3696–3705. doi: 10.1002/(SICI)1521-4141(199811)28:11<3696::AID-IMMU3696>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 7.Romagnani P., Annunziato F., Lazzeri E., Cosmi L., Beltrame C., Lasagni L. Interferon-inducible protein 10, monokine induced by interferon gamma, and interferon-inducible T-cell alpha chemoattractant are produced by thymic epithelial cells and attract T-cell receptor (TCR) alphabeta+ CD8+ single-positive T cells, TCR gammadelta+ T cells, and natural killer-type cells in human thymus. Blood. 2001;97:601–607. doi: 10.1182/blood.v97.3.601. [DOI] [PubMed] [Google Scholar]
- 8.Smit M.J., Verdijk P., van der Raaij-Helmer E.M., Navis M., Hensbergen P.J., Leurs R. CXCR3-mediated chemotaxis of human T cells is regulated by a Gi- and phospholipase C-dependent pathway and not via activation of MEK/p44/p42 MAPK nor Akt/PI-3 kinase. Blood. 2003;102:1959–1965. doi: 10.1182/blood-2002-12-3945. [DOI] [PubMed] [Google Scholar]
- 9.Shahabuddin S., Ji R., Wang P., Brailoiu E., Dun N., Yang Y. CXCR3 chemokine receptor-induced chemotaxis in human airway epithelial cells: role of p38 MAPK and PI3K signaling pathways. Am J Physiol Cell Physiol. 2006;291:C34–C39. doi: 10.1152/ajpcell.00441.2005. [DOI] [PubMed] [Google Scholar]
- 10.Petrai I., Rombouts K., Lasagni L., Annunziato F., Cosmi L., Romanelli R.G. Activation of p38(MAPK) mediates the angiostatic effect of the chemokine receptor CXCR3-B. Int J Biochem Cell Biol. 2008;40:1764–1774. doi: 10.1016/j.biocel.2008.01.008. [DOI] [PubMed] [Google Scholar]
- 11.Kukhtina N.B., Arefieva T.I., Krasnikova T.L. Intracellular signal cascade in CD4+ T-lymphocyte migration stimulated by interferon-gamma-inducible protein-10. Biochemistry (Mosc) 2005;70:652–656. doi: 10.1007/s10541-005-0165-5. [DOI] [PubMed] [Google Scholar]
- 12.Singh L., Arora S.K., Bakshi D.K., Majumdar S., Wig J.D. Potential role of CXCL10 in the induction of cell injury and mitochondrial dysfunction. Int J Exp Pathol. 2010;91:210–223. doi: 10.1111/j.1365-2613.2009.00697.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kerr J.F., Wyllie A.H., Currie A.R. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jacobson M.D., Weil M., Raff M.C. Programmed cell death in animal development. Cell. 1997;88:347–354. doi: 10.1016/s0092-8674(00)81873-5. [DOI] [PubMed] [Google Scholar]
- 15.Lazebnik Y.A., Kaufmann S.H., Desnoyers S., Poirier G.G., Earnshaw W.C. Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature. 1994;371:346–347. doi: 10.1038/371346a0. [DOI] [PubMed] [Google Scholar]
- 16.Cardone M.H., Salvesen G.S., Widmann C., Johnson G., Frisch S.M. The regulation of anoikis: MEKK-1 activation requires cleavage by caspases. Cell. 1997;90:315–323. doi: 10.1016/s0092-8674(00)80339-6. [DOI] [PubMed] [Google Scholar]
- 17.Youle R.J., Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 18.Vlahakis S.R., Villasis-Keever A., Gomez T., Vanegas M., Vlahakis N., Paya C.V. G protein-coupled chemokine receptors induce both survival and apoptotic signaling pathways. J Immunol. 2002;169:5546–5554. doi: 10.4049/jimmunol.169.10.5546. [DOI] [PubMed] [Google Scholar]
- 19.Sui Y., Potula R., Dhillon N., Pinson D., Li S., Nath A. Neuronal apoptosis is mediated by CXCL10 overexpression in simian human immunodeficiency virus encephalitis. Am J Pathol. 2004;164:1557–1566. doi: 10.1016/S0002-9440(10)63714-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang H.M., Yuan J., Cheung P., Chau D., Wong B.W., McManus B.M. Gamma interferon-inducible protein 10 induces HeLa cell apoptosis through a p53-dependent pathway initiated by suppression of human papillomavirus type 18 E6 and E7 expression. Mol Cell Biol. 2005;25:6247–6258. doi: 10.1128/MCB.25.14.6247-6258.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feldman E.D., Weinreich D.M., Carroll N.M., Burness M.L., Feldman A.L., Turner E. Interferon gamma-inducible protein 10 selectively inhibits proliferation and induces apoptosis in endothelial cells. Ann Surg Oncol. 2006;13:125–133. doi: 10.1245/ASO.2006.03.038. [DOI] [PubMed] [Google Scholar]
- 22.Cameron C.M., Cameron M.J., Bermejo-Martin J.F., Ran L., Xu L., Turner P.V. Gene expression analysis of host innate immune responses during Lethal H5N1 infection in ferrets. J Virol. 2008;82:11308–11317. doi: 10.1128/JVI.00691-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Datta D., Flaxenburg J.A., Laxmanan S., Geehan C., Grimm M., Waaga-Gasser A.M. Ras-induced modulation of CXCL10 and its receptor splice variant CXCR3-B in MDA-MB-435 and MCF-7 cells: relevance for the development of human breast cancer. Cancer Res. 2006;66:9509–9518. doi: 10.1158/0008-5472.CAN-05-4345. [DOI] [PubMed] [Google Scholar]
- 24.Chen J., Vistica B.P., Takase H., Ham D.I., Fariss R.N., Wawrousek E.F. A unique pattern of up- and down-regulation of chemokine receptor CXCR3 on inflammation-inducing Th1 cells. Eur J Immunol. 2004;34:2885–2894. doi: 10.1002/eji.200425318. [DOI] [PubMed] [Google Scholar]
- 25.Ono K., Han J. The p38 signal transduction pathway: activation and function. Cell Signal. 2000;12:1–13. doi: 10.1016/s0898-6568(99)00071-6. [DOI] [PubMed] [Google Scholar]
- 26.De Chiara G., Marcocci M.E., Torcia M., Lucibello M., Rosini P., Bonini P. Bcl-2 phosphorylation by p38 MAPK: identification of target sites and biologic consequences. J Biol Chem. 2006;281:21353–21361. doi: 10.1074/jbc.M511052200. [DOI] [PubMed] [Google Scholar]
- 27.Merritt C., Enslen H., Diehl N., Conze D., Davis R.J., Rincon M. Activation of p38 mitogen-activated protein kinase in vivo selectively induces apoptosis of CD8(+) but not CD4(+) T cells. Mol Cell Biol. 2000;20:936–946. doi: 10.1128/mcb.20.3.936-946.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Datta S.R., Dudek H., Tao X., Masters S., Fu H., Gotoh Y. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 29.Jiang J., Lau L.L., Shen H. Selective depletion of nonspecific T cells during the early stage of immune responses to infection. J Immunol. 2003;171:4352–4358. doi: 10.4049/jimmunol.171.8.4352. [DOI] [PubMed] [Google Scholar]
- 30.McNally J.M., Zarozinski C.C., Lin M.Y., Brehm M.A., Chen H.D., Welsh R.M. Attrition of bystander CD8 T cells during virus-induced T-cell and interferon responses. J Virol. 2001;75:5965–5976. doi: 10.1128/JVI.75.13.5965-5976.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.