

Abstract
The sudden appearance and potential lethality of severe acute respiratory syndrome associated coronavirus (SARS-CoV) in humans has focused attention on understanding its origins. Here, we assess phylogenetic relationships for the SARS-CoV lineage as well as the history of host-species shifts for SARS-CoV and other coronaviruses. We used a Bayesian phylogenetic inference approach with sliding window analyses of three SARS-CoV proteins: RNA dependent RNA polymerase (RDRP), nucleocapsid (N) and spike (S). Conservation of RDRP allowed us to use a set of Arteriviridae taxa to root the Coronaviridae phylogeny. We found strong evidence for a recombination breakpoint within SARS-CoV RDRP, based on different, well supported trees for a 5′ fragment (supporting SARS-CoV as sister to a clade including all other coronaviruses) and a 3′ fragment (supporting SARS-CoV as sister to group three avian coronaviruses). These different topologies are statistically significant: the optimal 5′ tree could be rejected for the 3′ region, and the optimal 3′ tree could be rejected for the 5′ region. We did not find statistical evidence for recombination in analyses of N and S, as there is little signal to differentiate among alternative trees. Comparison of phylogenetic trees for 11 known host-species and 36 coronaviruses, representing coronavirus groups 1–3 and SARS-CoV, based on N showed statistical incongruence indicating multiple host-species shifts for coronaviruses. Inference of host-species associations is highly sensitive to sampling and must be considered cautiously. However, current sampling suggests host-species shifts between mouse and rat, chicken and turkey, mammals and manx shearwater, and humans and other mammals. The sister relationship between avian coronaviruses and the 3′ RDRP fragment of SARS-CoV suggests an additional host-species shift. Demonstration of recombination in the SARS-CoV lineage indicates its potential for rapid unpredictable change, a potentially important challenge for public health management and for drug and vaccine development.
Keywords: Severe acute respiratory syndrome, Coronavirus, SARS-CoV Nidovirales, Phylogeny, Recombination, Host-shift, RNA-dependent RNA polymerase
1. Introduction
The sudden appearance and potential lethality of severe acute respiratory syndrome associated coronavirus (SARS-CoV) in humans has focused attention on understanding its origins. The host reservoir from which humans were infected remains to be determined. However, molecular phylogenetics can be used to assess SARS-CoV’s evolutionary origin and history of change by analyzing genes from SARS-CoV with homologous genes from other coronaviruses. Though surveys and sampling of coronaviruses from both wild and domestic host-species are limiting, comparative phylogenetic analyses for viruses and hosts is important in elucidating the history of host associations as well.
Coronaviruses have been divided into three groups based on serological and genetic criteria (Siddell, 1995). To date, group 3 coronaviruses have been found only in birds, group 1 coronaviruses have been found in carnivores, cetartiodactyls and primates, and group 2 coronaviruses have been found in cetartiodactyls, perissodactyls, rodents, and birds. Previous phylogenetic analyses, all of which were unrooted, suggested that SARS-CoV represents a relatively early diverging coronavirus lineage equally distantly related to the three groups of coronaviruses noted above. On this basis, SARS-CoV was proposed as representing a fourth, distinct group within the genus Coronavirus (Marra et al., 2003, Rota et al., 2003). These previous studies, which focused on characterizing and sequencing SARS-CoV, did not yield evidence for recombination within the SARS-CoV genome, although Marra et al. (2003) commented that the s2m motif within the SARS-CoV UTR may be the product of horizontal transfer, given the disjunct presence of s2m in many if not all astroviruses, a picornavirus (ERBV) and only one group 3 coronavirus (avian infectious bronchitis viruses (IBV)).
Here, we use phylogenetic analyses of SARS-CoV and other coronaviruses, rooted with diverse viruses from the family Artiviridae, to show that the RNA dependent RNA polymerase (RDRP) of SARS-CoV is a recombinant. We also compare phylogenetic trees for known coronaviruses and their hosts to assess the history of host associations for SARS-CoV and other coronaviruses.
2. Materials and methods
2.1. Identification and alignment of proteins
To identify and align three SARS-CoV proteins with homologs in the non-redundant GenBank CDS translations (also includes PDB, SwissProt and PIR; 20 April 2003), we used PFAM hidden Markov models (HMM) (Bateman et al., 2002) with the software HMMER (Eddy, 1998). The HMMs we used are: PF05183 for RNA dependent RNA polymerase, PF00937 for nucleocapsid (N) and PF01601 for spike (S). For identical RDRP sequences, we only retained a single representative. For N and S we used BLASTCLUST to retain a single representative from 95% identity groups to reduce the abundance of these sequences for computational efficiency. Envelope and membrane proteins were not analyzed because of their short size and lack of conservation across coronaviruses.
2.2. Detection of recombination within genes and phylogenetic analyses
To detect recombination within RDRP, S and N genes, we used sliding window phylogenetic analyses where windows of 100 amino acids in 25 amino acid intervals were analyzed using Bayesian inference (BI) (Mau et al., 1999, Yang and Rannala, 1997). This approach is analogous to bootscanning (e.g. Salminen et al., 1995), however, we use Bayesian inference rather than neighbor joining (NJ) and amino acid sequences rather than nucleotides. For BI, four chains were run for 200 K generations with a 100 K generation burn-in using a γ distribution of rates and the transition matrices WAG for S and N, and rtREV for RDRP (Dimmic et al., 2002) in MrBayes v.3b4 (Huelsenbeck and Ronquist, 2001) and summarized as a 50% majority rule consensus tree. We used the differential phylogenetic position of different SARS-CoV gene fragments (‘windows’) with respect to other coronavirus groups to identify potential recombination breakpoints and to divide the alignment into segments for additional phylogenetic analyses (Fig. 2A for RDRP) with BI and NJ bootstrap. For the analysis of these segments, BI parameters were as above, except each chain was run for 1 million generations. For NJ bootstrap, we used 1000 bootstrap replicates and NJ searches under default parameters in PAUP*, summarized as a 50% majority rule consensus tree. We used the approximately unbiased (AU) test (Shimodaira, 2002) to assess the validity of these breakpoints by determining whether alternative phylogenetic placements for different SARS-CoV gene regions can be statistically rejected using the program CONSEL (Shimodaira and Hasegawa, 2001) with branch lengths and model parameters estimated in PAML (Yang, 1997).
Fig. 2.
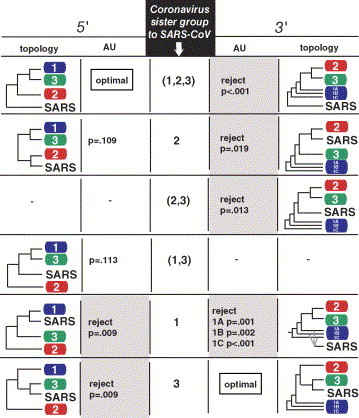
Results of the AU topological test (Shimodaira, 2002) for alternative trees based on the 5′ and 3′ RDRP putative recombinant fragments. Compatible topologies for the 5′ and 3′ fragments are located in each row. Putative recombinant fragments were inferred from the results of the sliding window analysis shown in Fig. 2B. The topologies shown are summaries, with each group represented by a single terminal taxa; see Fig. 1 for details.
2.3. Host association
In order to evaluate the types of evolutionary events (codivergence, duplication, sorting, host switching) that explain the fit between coronavirus evolution and host evolution, we considered the nucleocapsid coronavirus phylogeny and its host phylogeny, where mammalian relationships in the host tree follow Murphy et al. (2001). We used TreeFitter v.1 (Ronquist, 2000) which incorporates differential costs to the four types of potential events of a host–parasite association: codivergence (C), duplication (D), sorting (S) and host switching (H). We used various event costs to test a variety of situations (see Desdevises et al., 2002, Ronquist and Liljeblad, 2001). Significance of fit was determined by comparing the cost of the observed tree with 10,000 random permutations of the coronavirus tree terminals.
3. Results
3.1. Recombination within RDRP
The RDRP HMM detected 27 unique sequences in GenBank related to SARS-CoV from Arteriviridae and Coronaviridae. The relationship between SARS-CoV and these other coronaviruses for each 100 amino acid window in RDRP, as indicated by BI phylogeny, is shown in Fig. 1A . Three contiguous, overlapping windows spanning 150 amino acids in the 5′ region of the SARS-CoV RDRP are sister to a clade including groups 1–3 (all other known coronaviruses). Alternatively, seven contiguous windows, spanning 259 amino acids in the 3′ region, are sister to group 3 coronaviruses. Using this diagram (Fig. 1A), we split RDRP into two fragments, 5′ and 3′.
Fig. 1.
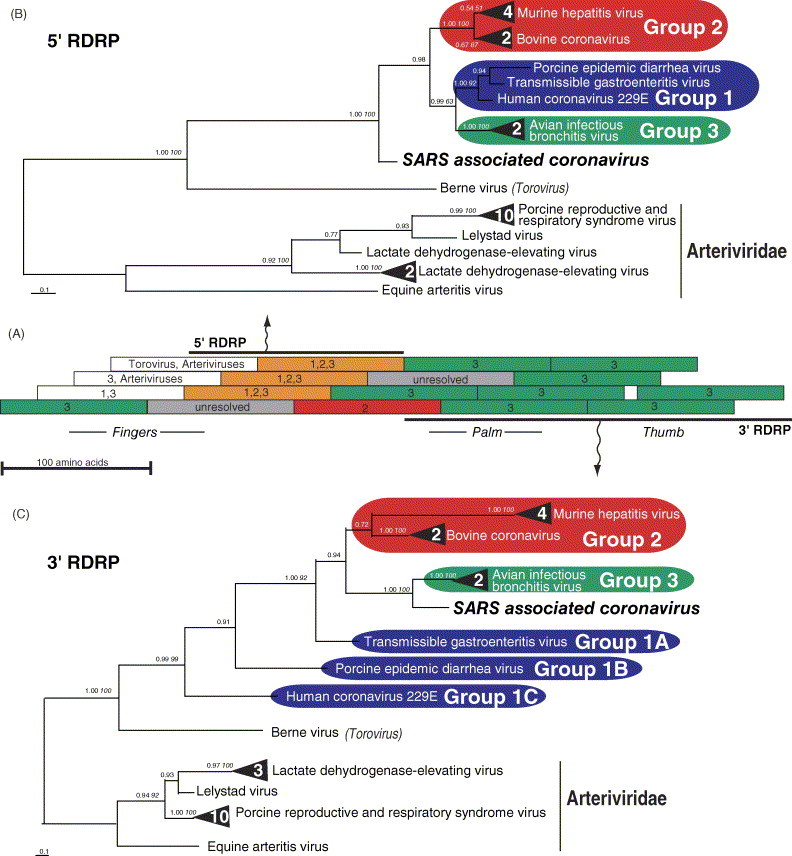
Recombinant nature of SARS-CoV RNA dependent RNA polymerase, as indicated by different sister relationships with other coronaviruses for different gene regions. (A) Schematic diagram showing Bayesian inference (BI) sliding window (100 amino acids long in 25 amino acid intervals) analyses used to assess recombination breakpoints within RDRP. The sister relationship of SARS-CoV RDRP with other coronavirus groups (1, 2 and/or 3) for each fragment is indicated by color code and numbers. BI phylogenies are shown for the entire 5′ (B) and 3′ (C) regions of RDRP. Numbers by each node are posterior probabilities, and when applicable, are followed by neighbor joining bootstrap percentages in italics. Multiple terminal nodes from a single virus species are represented by a black triangle, with the number of terminals indicated in white numerals. GenInfo identifiers for the proteins used in this analysis: 482297, 564004, 7769353, 93916, 233625, 14917044, 6625761, 13752450, 10242469, 94017, 12744851, 10179430, 25121660, 20271248, 11878197, 7650194, 17529672, 11878201, 25361011, 26008080, 12082740, 133455, 29293454, 14250963, 12240326, 10181074, 9635157, 29837504.
To assess the significance of this inference, we performed extensive phylogenetic analyses on each fragment. The optimal tree for the 5′ region (Fig. 1B) and the 3′ region (Fig. 1C) mirrored the results of the sliding window analysis (Fig. 1A). To assess the potential impact of the outgroups on these results we also analyzed both regions for the 12 coronavirus taxa alone. These unrooted topologies (not shown) are compatible with the rooted topologies, indicating that the results for SARS-CoV in Fig. 1 are not due to long branch attraction involving the outgroup. We then used the approximately unbiased (AU) tree selection test (Shimodaira, 2002) to see if the alternative, competing trees for each gene fragment can be statistically rejected in favor of the optimal tree, or if the conflicting results between the 5′ and 3′ regions are not statistically significant. As shown in Fig. 2 , the optimal sister relationship for the 5′ region, with SARS-CoV as sister to a clade including groups 1–3 combined, is rejectable for the 3′ region, and the optimal sister relationship for the 3′ region, with SARS-CoV sister to group 3, is rejectable for the 5′ region. Additionally, SARS-CoV RDRP as sister to group 1 RDRP is rejectable in both regions, and SARS-CoV as sister to group 2 is rejectable in the 3′ region. To assess the impact of the outgroups on these results, we repeated the AU tests with only the 12 coronavirus taxa. All alternative topologies for both regions were rejectable in favor of the optimal topology according to the AU test.
For S, the HMM detected 120 unique relatives of SARS-CoV, which we reduced to 24 by 95% identity clustering. For N, the HMM detected 93 unique relatives of SARS-CoV, which we reduced to 32 by identity clustering. For S and N, we were not able to reject alternative topologies for segments when following the above procedure (results not shown) and thus considered each gene as a historical unit for further analysis. According to their HMMs, both S and N are too variable to allow inclusion of an outgroup in alignment and phylogenetic analyses, therefore their phylogenies are unrooted. For N (Fig. 3 ) and S (not shown) coronavirus groups 1–3 are each monophyletic with respect to SARS-CoV, however, we cannot say with statistical confidence (according to the AU test) which group (1, 2 or 3) is most closely related to SARS-CoV.
Fig. 3.
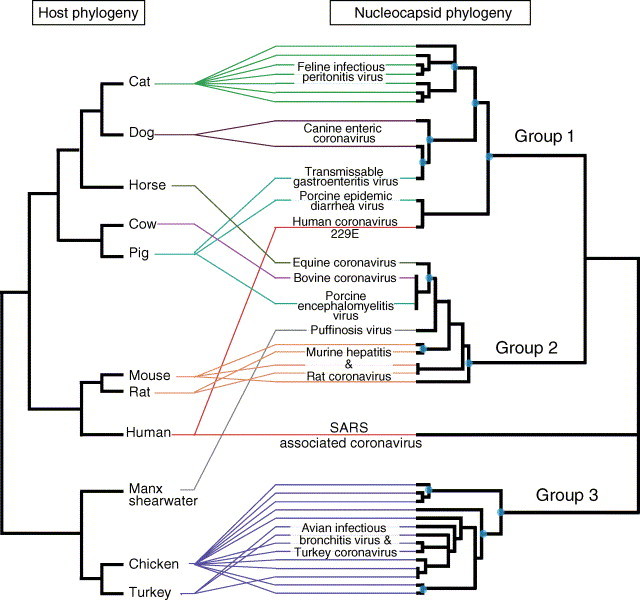
Bayesian inference phylogeny of the nucleocapsid protein of coronaviruses in comparison with the phylogeny of their hosts (after Murphy et al., 2001 for mammals). Lines drawn between the two phylogenies indicate the host status of each coronavirus. For the nucleocapsid phylogeny, all nodes are supported by >50% Bayesian posterior probability. Nodes overlaid with circles are also supported by >75% of neighbor joining bootstraps. GenInfo identifiers for the proteins used in this analysis: 1220375, 395178, 127872, 11096193, 543643, 3132999, 1515361, 21624372, 222585, 29840828, 13448682, 28916465, 281107, 74863, 28460530, 11640712, 29836503, 14253137, 1515365, 1515367, 6689852, 6689856, 320020, 1515375, 1515373, 1515371, 331869, 547999, 21624295, 21624366, 21624369, 28932648, 28932650.
3.2. Host-shifts
Examination of the fit of the N virus tree to the host tree was performed in TreeFitter v.1 (Ronquist, 2000). Under default settings (H=2, S=1, D=0, C=0), nine host switches (P⪡0.001) describe a significant fit (P⪡0.001) of the virus and host trees, while codivergences, duplications and sorting events are rare (P⪢0.05). As further evidence of this, when the program settings are changed to maximize codivergence events (H=0; or H=0 and C=−1) the global fit between the two trees is no longer significant (P⪢0.05). Together, these results indicate that, given current sampling, host switches have been extremely important in the evolution of coronaviruses and their hosts.
4. Discussion
4.1. Phylogeny and recombination
The difference in the phylogenies inferred from the 5′ (Fig. 1B) and 3′ (Fig. 1C) RDRP regions, and the significant differences in support and rejectability of alternative trees for each gene region, all strongly support the hypothesis of an ancient recombination event between two co-infecting viruses. We say ‘ancient’ to denote that both regions are sister to clades of other sequences, rather than to any single recently diverged sequence. These results indicate that the two SARS-CoV RDRP regions do indeed represent two unique histories. Thus, it is preferable not to analyze them together, as has been done in previous analyses (Marra et al., 2003, Rota et al., 2003) because their history cannot be represented by a single tree. These previous analyses used distance methods (NJ) less able to accommodate heterogeneity in rates of sequence character change, and found RDRP as a whole to be closest to group 2 coronaviruses. This may result from conflicts within the data stemming from recombination and/or from effects of rate heterogeneity. The authors do not report whether alternative trees could be rejected based on their analyses of multiple genes, though it seems unlikely, as when we performed similar analyses for the S and N proteins we were not able to differentiate between alternative SARS-CoV sister relationships using the AU test. We note that the approach for detecting recombination implemented here is rigorous in comparison to traditional bootscanning, in that it analyzes more conserved amino acids using Bayesian inference rather than NJ, and explicitly tests the significance of alternative topologies using the AU test. It is possible and likely that more recombination events have happened within RDRP, N, S or other SARS-CoV genes, than we have detected here, although the evidence for recombination generally becomes more difficult to discern over time.
Inclusion of an outgroup, as we have done with RDRP from 15 Arteriviridae taxa (Fig. 1), allows inference of the sister relationships and the relative age and timing of Coronaviridae (Coronavirus and Torovirus) divergence events, missing from the previous unrooted analyses which only could assess distance between clades. Our rooted analyses indicate that the 5′ RDRP fragment diverged from other Coronavirus taxa prior to divergences between and within groups 1–3. Fig. 1C indicates that the 3′ RDRP fragment diverged from other coronavirus homologs more recently, after divergences between and within groups 1–3. Interestingly, Fig. 1C also shows non-monophyly for group 1 coronaviruses. This is not surprising, given that groups 1–3 were initially distinguished based on serological tests rather than phylogenetic analyses and given the capacity for recombination.
The sister relationship between the more recently diverged SARS-CoV 3′ RDRP fragment and group 3 avian infectious bronchitis viruses (Fig. 1C), suggests that potential horizontal transmissions of s2m to SARS-CoV (Marra et al., 2003) and the 3′ region of RDRP are correlated. They may have even been incorporated concomitantly, perhaps on transmission from an ancestor of IBV, the only coronavirus with s2m (Jonassen et al., 1998). As the 5′ region of RDRP and the s2m motif are disjunct in the SARS-CoV genome, putative replication-dependent recombination would have involved several consecutive template switches, as was inferred for the transfer of s2m to IBV (Jonassen et al., 1998). Alternatively, horizontal transfer from astroviruses or a picornavirus cannot be ruled out, as horizontal transfer of s2m accounts for its presence in three different virus families (Jonassen et al., 1998).
Phylogenetic indication of recombination for SARS-CoV makes sense, as coronaviruses are unique among single-stranded, non-segmented RNA viruses in their propensity for recombination, a mechanism purportedly useful in eliminating frequent deleterious mutations in large RNA viruses (Lai, 1996). Coronavirus genomes generally appear resilient and able to tolerate deletions, insertions and rearrangements (de Haan et al., 2002). Similar to our findings, Decimo et al. (1993) suggested that N genes from murine hepatitis viruses were the result of double recombination, and several authors have reported evidence for recombination among natural isolates of IBV (e.g. Jia et al., 1995, Wang et al., 1993). Recombination can be important in gain of novel functions. For example, in HIV recombination is considered to be a powerful adaptive mechanism for antiviral agent resistance and cytotoxic T-cell escape (e.g. Morris et al., 1999). The combination of horizontal transfer and recombination results in complex phylogenies that may blur the evolutionary history of genes (e.g. Keeling and Palmer, 2001, Rest and Mindell, 2003), especially since horizontal transfer and recombination are often associated processes.
4.2. Host association
Despite the limitations imposed on inference of historical host association by the restricted sampling of hosts and coronaviruses to date, some preliminary observations can be made. Coronaviruses have been shown to be particularly host specific (Lai, 1990, Sturman and Holmes, 1983) and it had been assumed by some that they coevolved (diversified in tandem) with their hosts (Decimo et al., 1993). However, based on current sampling and analyses summarized in Fig. 3, some host switching events are implicated in accounting for incongruence between the host and coronavirus phylogenies. For example, chicken and turkey are sister taxa within the host phylogeny, yet isolates from each do not form host-specific monophyletic groups. Rather, some isolates from chicken are most closely related to turkey isolates, suggesting host-shifts for coronaviruses between these two bird species (Fig. 3). A third avian species, the manx shearwater (Puffinus puffinus) is the host to a group 2 coronavirus (Kirkwood et al., 1995), which are otherwise known only from mammals. The phylogenetically nested position of this avian coronavirus within mammalian-host isolates suggests a possible bird–mammal host-shift. Coronavirus host-shifts between mouse and rat are implicated in the same manner as for chicken and turkey; isolates from the two rodent species are not reciprocally monophyletic. Similarly, isolates from pig are not monophyletic, including two group 1 and one group 2 coronavirus in Fig. 3. With the outbreak of SARS, coronavirus isolates from humans are also non-monophyletic.
Using an earlier and more limited sampling of host species (n=6) and coronaviruses (n=9), Decimo et al. (1993) suggested a host-shift between cats and pigs. Inclusion of additional host and virus sampling in Fig. 3, including two isolates from dogs, implicates potential host-shift between dogs and pigs, rather than cats and pigs. This demonstrates sensitivity to sampling, although the implication of host-shifting as a phenomenon remains. The emerging picture of coronavirus host associations is increasingly indicative of host-shifts. This is supported in Fig. 3 by the observations mentioned above as well as (1) the sister relationship between human coronavirus 229E and porcine (pig) epidemic diarrhea virus and (2) non-monophyly for human coronavirus 229E and SARS-CoV. Further, according to statistical tests in TreeFitter, allowing host-shifts to occur results in significant fit while duplication, codivergence and sorting play no detectable role. Depending on the assigned costs, TreeFitter estimates between 9 and 15 host-shifts in reconciling the host and coronavirus phylogenies shown in Fig. 3, though the actual number is unknown. In light of the genomic disparity among diverse coronaviruses from some individual host-species (e.g. humans, pigs), it seems unlikely that increased sampling of coronaviruses will yield monophyly for all isolates from each of the individual host-species in Fig. 3.
The finding of recombination for SARS-CoV RDRP, the relatively early phylogenetic divergence for RDRP fragments (prior to the most recent divergences within coronavirus groups 1–3), as well as the inference of multiple coronavirus hosts switches, suggests that SARS-CoV belongs to an old, potentially diverse, and changeable coronavirus lineage that remains to be discovered in its natural hosts. Demonstration of recombination in the SARS associated coronavirus lineage indicates its potential for rapid unpredictable change, a potentially important challenge for public health management and for drug and vaccine development. The known non-human coronaviruses come from only nine, mostly domestic, mammal or bird species, and searches for the zoonotic reservoir might reasonably focus on other species, including non-domesticated animals, that are used as food for humans in the geographic region of the SARS outbreak.
Acknowledgments
Support was provided by Museum of Zoology Hinsdale-Walker and Department of Ecology and Evolutionary Biology, University of Michigan block grants to JSR and by National Science Foundation Grant DBI-9974525 to DPM and R. A. Goldstein.
References
- Bateman A, Birney E, Cerruti L, Durbin R, Etwiller L, Eddy S.R, Griffiths-Jones S, Howe K.L, Marshall M, Sonnhammer E.L. The Pfam protein families database. Nucleic Acids Res. 2002;30:276–280. doi: 10.1093/nar/30.1.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan C.A, Volders H, Koetzner C.A, Masters P.S, Rottier P.J. Coronaviruses maintain viability despite dramatic rearrangements of the strictly conserved genome organization. J. Virol. 2002;76:12491–12502. doi: 10.1128/JVI.76.24.12491-12502.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decimo D, Philippe H, Hadchouel M, Tardieu M, Meunier-Rotival M. The gene encoding the nucleocapsid protein: sequence analysis in murine hepatitis virus type 3 and evolution in Coronaviridae. Arch. Virol. 1993;130:279–288. doi: 10.1007/BF01309660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desdevises Y, Morand S, Jousson O, Legendre P. Coevolution between Lamellodiscus (Monogenea: Diplectanidae) and Sparidae (Teleostei): the study of a complex host-parasite system. Evolution. 2002;56:2459–2471. doi: 10.1111/j.0014-3820.2002.tb00171.x. [DOI] [PubMed] [Google Scholar]
- Dimmic M.W, Rest J.S, Mindell D.P, Goldstein R.A. rtREV: an amino acid substitution matrix for inference of retrovirus and reverse transcriptase phylogeny. J. Mol. E. 2002;55:65–73. doi: 10.1007/s00239-001-2304-y. [DOI] [PubMed] [Google Scholar]
- Eddy S.R. Profile hidden Markov models. Bioinformatics. 1998;14:755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- Huelsenbeck J.P, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- Jia W, Karaca K, Parrish C.R, Naqi S.A. A novel variant of avian infectious bronchitis virus resulting from recombination among three different strains. Arch. Virol. 1995;140:259–271. doi: 10.1007/BF01309861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonassen C.M, Jonassen T.O, Grinde B. A common RNA motif in the 3′ end of the genomes of astroviruses, avian infectious bronchitis virus and an equine rhinovirus. J. Gen. Virol. 1998;79:715–718. doi: 10.1099/0022-1317-79-4-715. [DOI] [PubMed] [Google Scholar]
- Keeling P.J, Palmer J.D. Lateral transfer at the gene and subgenic levels in the evolution of eukaryotic enolase. Proc. Natl. Acad. Sci. U.S.A. 2001;98:10745–10750. doi: 10.1073/pnas.191337098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood J.K, Cunningham A.A, Hawkey C, Howlett J, Perrins C.M. Hematology of fledgling Manx shearwaters (Puffinus puffinus) with and without ‘puffinosis’. J. Wildl. Dis. 1995;31:96–98. doi: 10.7589/0090-3558-31.1.96. [DOI] [PubMed] [Google Scholar]
- Lai M.M. Coronavirus: organization, replication and expression of genome. Annu. Rev. Microbiol. 1990;44:303–333. doi: 10.1146/annurev.mi.44.100190.001511. [DOI] [PubMed] [Google Scholar]
- Lai M.M.C. Recombination in large RNA viruses: coronaviruses. Semin. Virol. 1996;7:381–388. doi: 10.1006/smvy.1996.0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marra M.A, Jones S.J, Astell C.R, Holt R.A, Brooks-Wilson A, Butterfield Y.S, Khattra J, Asano J.K, Barber S.A, Chan S.Y, Cloutier A, Coughlin S.M, Freeman D, Girn N, Griffith O.L, Leach S.R, Mayo M, McDonald H, Montgomery S.B, Pandoh P.K, Petrescu A.S, Robertson A.G, Schein J.E, Siddiqui A, Smailus D.E, Stott J.M, Yang G.S, Plummer F, Andonov A, Artsob H, Bastien N, Bernard K, Booth T.F, Bowness D, Czub M, Drebot M, Fernando L, Flick R, Garbutt M, Gray M, Grolla A, Jones S, Feldmann H, Meyers A, Kabani A, Li Y, Normand S, Stroher U, Tipples G.A, Tyler S, Vogrig R, Ward D, Watson B, Brunham R.C, Krajden M, Petric M, Skowronski D.M, Upton C, Roper R.L. The Genome sequence of the SARS-associated coronavirus. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- Mau B, Newton M.A, Larget B. Bayesian phylogenetic inference via Markov chain Monte Carlo methods. Biometrics. 1999;55:1–12. doi: 10.1111/j.0006-341x.1999.00001.x. [DOI] [PubMed] [Google Scholar]
- Morris A, Marsden M, Halcrow K, Hughes E.S, Brettle R.P, Bell J.E, Simmonds P. Mosaic structure of the human immunodeficiency virus type 1 genome infecting lymphoid cells and the brain: evidence for frequent in vivo recombination events in the evolution of regional populations. J. Virol. 1999;73:8720–8731. doi: 10.1128/jvi.73.10.8720-8731.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy W.J, Eizirik E, O’Brien S.J, Madsen O, Scally M, Douady C.J, Teeling E, Ryder O.A, Stanhope M.J, de Jong W.W, Springer M.S. Resolution of the early placental mammal radiation using Bayesian phylogenetics. Science. 2001;294:2348–2351. doi: 10.1126/science.1067179. [DOI] [PubMed] [Google Scholar]
- Rest J.S, Mindell D.P. Retroids in archaea: phylogeny and lateral origins. Mol. Biol. E. 2003;20:1134–1142. doi: 10.1093/molbev/msg135. [DOI] [PubMed] [Google Scholar]
- Ronquist, F., 2000. TreeFitter. Ver. 1.0. Software avialable via http://www.ebc.uu.se/systzoo/research/treefitter/treefitter.html. Uppsala University, Uppsala, Sweden.
- Ronquist F, Liljeblad J. Evolution of the gall wasp-host plant association. Evolution. 2001;55:2503–2522. doi: 10.1111/j.0014-3820.2001.tb00765.x. [DOI] [PubMed] [Google Scholar]
- Rota P.A, Oberste M.S, Monroe S.S, Nix W.A, Campagnoli R, Icenogle J.P, Penaranda S, Bankamp B, Maher K, Chen M.H, Tong S, Tamin A, Lowe L, Frace M, DeRisi J.L, Chen Q, Wang D, Erdman D.D, Peret T.C, Burns C, Ksiazek T.G, Rollin P.E, Sanchez A, Liffick S, Holloway B, Limor J, McCaustland K, Olsen-Rasmussen M, Fouchier R, Gunther S, Osterhaus A.D, Drosten C, Pallansch M.A, Anderson L.J, Bellini W.J. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- Salminen M.O, Carr J.K, Burke D.S, McCutchan F.E. Identification of breakpoints in intergenotypic recombinants of HIV type 1 by bootscanning. AIDS Res. Hum. Retroviruses. 1995;11:1423–1425. doi: 10.1089/aid.1995.11.1423. [DOI] [PubMed] [Google Scholar]
- Shimodaira H. An approximately unbiased test of phylogenetic tree selection. Syst. Biol. 2002;51:492–508. doi: 10.1080/10635150290069913. [DOI] [PubMed] [Google Scholar]
- Shimodaira H, Hasegawa M. CONSEL: for assessing the confidence of phylogenetic tree selection. Bioinformatics. 2001;17:1246–1247. doi: 10.1093/bioinformatics/17.12.1246. [DOI] [PubMed] [Google Scholar]
- Siddell, S.G., 1995. The Coronaviridae: an introduction. In: Siddell, S.G. (Ed.), The Coronaviridae: An Introduction. Plenum Press, New York, NY, pp. 1–10.
- Sturman L.S, Holmes K.V. The molecular biology of coronaviruses. Adv. Virus Res. 1983;28:35–112. doi: 10.1016/S0065-3527(08)60721-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Junker D, Collisson E.W. Evidence of natural recombination with the S1 gene of infectious-bronchitis virus. Virology. 1993;192:710–716. doi: 10.1006/viro.1993.1093. [DOI] [PubMed] [Google Scholar]
- Yang Z.H. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 1997;13:555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- Yang Z.H, Rannala B. Bayesian phylogenetic inference using DNA sequences: a Markov Chain Monte Carlo method. Mol. Biol. E. 1997;14:717–724. doi: 10.1093/oxfordjournals.molbev.a025811. [DOI] [PubMed] [Google Scholar]