

Highlights
-
•
Review of hierarchical virtual screening approaches is presented.
-
•
Hierarchical combination of ligand and structure-based methods is preferred over parallel combination.
-
•
Molecular docking is a key component of many hierarchical virtual screening schemes.
-
•
Hierarchical virtual screening may be useful in plucking high-hanging fruits.
Keywords: Hierarchical virtual screening, Molecular docking, Similarity search, Pharmacophore modeling
Abstract
Virtual screening has played a significant role in the discovery of small molecule inhibitors of therapeutic targets in last two decades. Various ligand and structure-based virtual screening approaches are employed to identify small molecule ligands for proteins of interest. These approaches are often combined in either hierarchical or parallel manner to take advantage of the strength and avoid the limitations associated with individual methods. Hierarchical combination of ligand and structure-based virtual screening approaches has received noteworthy success in numerous drug discovery campaigns. In hierarchical virtual screening, several filters using ligand and structure-based approaches are sequentially applied to reduce a large screening library to a number small enough for experimental testing. In this review, we focus on different hierarchical virtual screening strategies and their application in the discovery of small molecule modulators of important drug targets. Several virtual screening studies are discussed to demonstrate the successful application of hierarchical virtual screening in small molecule drug discovery.
1. Introduction
Modern drug discovery process starts with the identification of initial hits that are further optimized to improve the potency, selectivity, metabolic stability and oral bioavailability. Among many ways of identifying initial hits in drug discovery, high-throughput screening (HTS) and virtual screening (VS) are most common. The VS was originally developed to bring down the cost of discovering new molecules using HTS. In the last two decades, advances in computational programs and processing power have made VS an important tool to identify starting points, inhibitors and chemical probes in various drug discovery campaigns [1], [2], [3], [4].
In light of the immense potential of VS methodologies in the identification of initial hits, various structure and ligand-based approaches were developed [1], [5], [6], [7], [8]. Structure-based methods rely on the structural information of the protein target and typically include methods such as molecular docking [9], [10], structure-based pharmacophores [11], [12] and de novo design [13], [14]. Ligand-based methods can work in the absence of structural information for the protein target. These methods include two or three-dimensional (2D or 3D) similarity searches [15], [16], ligand-based pharmacophore screenings [5], [17], machine learning approaches [18], [19], quantitative structure activity relationships (QSAR) [5], [20] among others. Ligand-based methods, however, require the availability of at least one known active molecule. Although utilizing these structure and ligand-based methods individually have demonstrated immense potential in retrieving initial hits, these methods are unable to fulfill all the practical requirements of drug discovery alone. Furthermore, with ever-increasing screening library size [21] and computational cost associated with some VS approaches especially flexible molecular docking [22], [23], [24], [25], it is indispensable to integrate different VS approaches to filter compounds. Ligand and structure-based methods can be combined in a sequential or parallel manner (Fig. 1 ). The most common way of combining these methods is to use them in a sequential funnel like manner commonly known as hierarchical VS (HLVS). In HLVS, a large small molecule library is reduced to a number of compounds that is small enough for biological assay by applying a series of filters (generally two or three) sequentially (Fig. 1A). In contrast to HLVS, there is parallel virtual screening (PVS) where several complementary methods are run in parallel and the best hits ranked according to each method are selected for biological testing (Fig. 1B). Although the retrospective analysis of literature data has shown the successful application of PVS [26], [27], [28], [29], [30], only a few applications in real world scenario could be found [27], [31], [32].
Fig. 1.
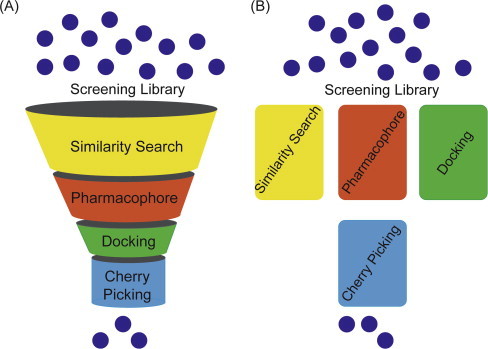
Integration of ligand and structure-based approaches. (A) Hierarchical virtual screening (HLVS): series of filters (here similarity search, pharmacophore and molecular docking) are sequentially applied to bring down the number of compounds to be cherry-picked for biological assay. (B) Parallel virtual screening (PVS): ligand and structure-based filters are performed independently on the same or similar number of compounds.
In this paper, we review the current status of commonly used HLVS approaches and try to understand their utility in drug discovery campaigns of important therapeutic targets. Although the scientific literature is inundated by hierarchical computational approaches and protocols, we have restricted our review on only those studies that were validated by experimental assays. In the following sections we will outline the types of HLVS approaches utilized in various small molecule discovery campaigns. Later, we will discuss recent cases of successful hit identification utilizing HLVS protocols. Finally, we will describe the usefulness of HLVS in discovering inhibitors of important drug targets.
2. Hierarchical combination of VS methods
Hierarchical combination of ligand and structure-based VS approaches generally involves sequential execution of dissimilar VS methods. Mostly, computationally inexpensive ligand based approaches such as similarity search and pharmacophore screening are used during initial steps of an HLVS protocol. Methods demanding comparatively high computational resources such as molecular docking and molecular dynamics (MD) simulation are used once the number of compounds to be screened decreases to a reasonable number. The final step in a majority of HLVS campaigns incorporates the visual selection of compounds by expert researcher commonly known as “cherry picking”. In this step, ranking from VS methods is combined with expert chemical intuition and with literature-based knowledge. The HLVS can be classified in three categories based on the combination of VS methods: ligand-based HLVS, structure-based HLVS and hybrid HLVS, which will be described in detail below. A few successful cases of small molecule discovery using these three classes of HLVS are summarized in Table 1 .
Table 1.
A few cases of successful hit identification utilizing HLVS protocols.
| Drug target | Role | Structure of best compound | Activity of best compound | HLVS methods used | Reference |
|---|---|---|---|---|---|
| LB-HLVS | |||||
| Liver X Receptor | Cholesterol metabolism | 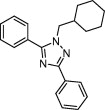 |
Three fold activation at 10 μM | Shape similarity, Pharmacophore modeling | Temml et al. [42] |
| NAADP receptor | NAADP signaling | 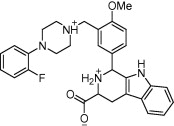 |
IC50 = 2 μM | Shape similarity and electrostatic potential matching | Naylor et al. [45] |
| Enoyl reductase | Antibacterials | 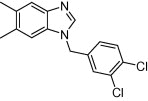 |
IC50 = 0.3 μM | Shape similarity, electrostatic potential matching, 2D fingerprints | Hevener et al. [44] |
| Melanin-concentrating hormone receptor 1 (MCHr1) | Obesity | 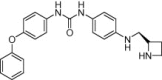 |
MCHr1 binding IC50 = 2.2 nM | Shape similarity, electrostatic potential matching, 2D fingerprints | Muchmore et al. [43] |
| Dihydrofolate reductase and thymidylate synthase | Ovarian cancer | 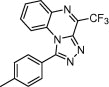 |
IC50 = 42.5 and 26.7 μM against cDDP-sensitive and IC50 = 33 and 35.7 μM against resistant ovarian cancer cell line | Physiochemical properties and pharmacophore fingerprint similarity | Carosati et al. [46] |
| Human bitter taste receptors (TAS2Rs) | Asthma and infection | 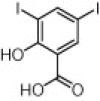 |
0.5 μM effective concentration | 1D molecular descriptors, fingerprint similarity, pharmacophore modeling and shape similarity | Levit et al. [48] |
| Cytohesin | Vesicle trafficking and insulin signaling | 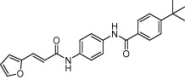 |
IC50 = 3.1 μM, Binding Kd = 7.5 μM | 2D fingerprints and support vector machine | Stumpfe et al. [50] |
| SB-HLVS | |||||
| SUMO activating enzyme 1 | Sumoylation pathway | IC50 = 11.1 μM | Molecular docking, molecular dynamics and MM-PBSA binding free energy | Kumar et al. [51] | |
| Tubulin | Cancer | 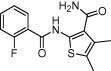 |
IC50 = 30 μM | Molecular docking, molecular dynamics and MM-PBSA and MM-GBSA binding free energy | Cao et al. [55] |
| HIV-1 protease | AIDS | 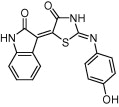 |
IC50 = 14 μM | Molecular docking and molecular dynamics simulation | Kunze et al. [56] |
| Cytochrome P450 aromatase | Estrogen-dependent breast cancer | 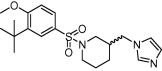 |
IC50 = 9.4 nM | Multistep hierarchical docking | Caporuscio et al. [65] |
| FabI | Bacterial fatty acid biosynthesis | 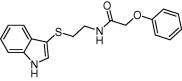 |
IC50 = 3.4 μM | Molecular docking, molecular dynamics and MM-PBSA binding free energy | Hu et al. [152] |
| LBSB-HLVS | |||||
| Serotonin transporter | Neurotransmission | 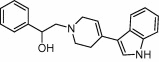 |
Ki = 1.5 nM | 2D fingerprints, ADMET filtering, 3D pharmacophore modeling and molecular docking | Gabrielsen et al. [66] |
| B-RafV600E | Ras/Raf/MEK/ERK signaling pathway | 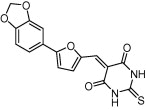 |
IC50 = 0.3 μM | SHAFT 3D ligand similarity and molecular docking | Kong et al. [67] |
| Protein kinase CK2 | Cancer | 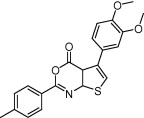 |
85% inhibition at 10 μM | Bayesian modeling, pharmacophore modeling and molecular docking | Di-wu et al. [69] |
| Coagulation factor VIII | Anticoagulants | 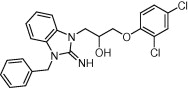 |
IC50 = 3.5 μM | Pharmacophore modeling and molecular docking | Nicolaes et al. [70] |
| SUMO specific protease 2 | Sumoylation pathway | 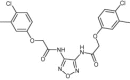 |
IC50 = 3.7 μM | Shape similarity, electrostatic potential matching and molecular docking | Kumar et al. [108] |
| Insulin-like growth factor-1 receptor (IGF-1R) | Cell growth, proliferation and apoptosis | 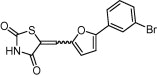 |
IC50 = 57 nM | Pharmacophore modeling and molecular docking | Liu et al. [72] |
| DNA G-quadruplex | Cellular aging and cancer | 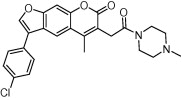 |
Ability to bind and stabilize telomeric G-quadruplex shown using fluorescence and CD methods. | 2D fingerprints, shape similarity and molecular docking | Alcaro et al. [89] |
| Cruzain | Cysteine protease, Chagas disease | 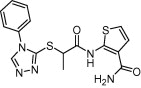 |
IC50 = 48.8 μM | Shape similarity, molecular docking, molecular hologram QSAR | Wiggers et al. [111] |
2.1. Ligand based HLVS (LB-HLVS)
LB-HLVS sequentially combines methods based on similarity search or compound classification. Similarity based techniques include methods accessing the similarity of one or a few experimentally identified hits with molecules in a large library in terms of their physicochemical properties [33], structural fingerprints [34], 3D-shape [35], electrostatic potential [36] and pharmacophore features [37] etc. Compound classification techniques include clustering [38], [39] or machine learning based methods such as Bayesian methods and support vector machines [18], [40]. Although most of the ligand-based VS methods are used either standalone or in combination with structure-based VS methods, only ligand-based methods were reported to have been effectively combined in prospective applications. Yao et al. [41] reported an efficient multi-step ligand-based VS protocol that included physicochemical property filtering, pharmacophore-based screening, protein–ligand interaction fingerprint similarity analysis and 2D-fingerprint structural similarity search. Their protocol significantly improved the hit rate when compared with individual methods. Among the prospective applications, LB-HLVS that included a combination of shape-based VS and pharmacophore modeling has been used by Temml et al. [42] to identify two agonist of liver X receptor. The reported agonist activated both subtypes of liver X receptor (LXR α and β). Shape similarity has also been combined with electrostatic potential matching in the discovery of melanin-concentrating hormone receptor 1 antagonist [43], Francisella tularensis enoyl-reductase inhibitors [44] and chemical probe for nicotinic acid adenine dinucleotide phosphate (NAADP) [45]. In another application [46], ZINC database [47] subset enriched with quinoxaline scaffold was filtered based on pharmacokinetic properties. The resulting molecules were again investigated for pharmacophore fingerprint similarity with known quinoxaline based inhibitors of folate cycle proteins. Associated biological assay resulted in three compounds, which interfered with dihydrofolate reductase and thymidylate synthase and reduced their levels. Levit et al. [48] integrated 1D molecular descriptors, 2D fingerprint-based molecular similarity, ligand-based pharmacophore models and a shape-based VS method to identify activators of human bitter taste receptor TAS2R14. Bayesian analysis has been used with pharmacophore modeling to identify inhibitors of breast cancer resistance protein (BCRP) [49]. VS was carried out against 2000 FDA-approved drugs and 19 drugs were found to exhibit significant effect on BCRP transport function. Another machine learning technique, support vector machine (SVM) in combination with fingerprint similarity search was also used in the identification of molecular probes for the study of cytohesins [50]. Ligand-based methods have the advantage of being computationally inexpensive and have the ability to retrieve more potent hits than the structure-based methods. However, most of these methods suffer from the limitation of being biased towards active chemical scaffolds and thereby generate less diverse hits. Moreover it is difficult to decide the right input query as it affects the quality of output results. In this scenario, hierarchical combination of ligand-based methods such as pharmacophore modeling and similarity-based methods assure chemical diversity with potency.
2.2. Structure-based HLVS (SB-HLVS)
Structure-based methods are also occasionally combined in hierarchical style. Structure-based methods are used standalone or in combination with ligand-based methods. Hierarchical combination of structure-based methods suffered from the limitation of being computationally expensive. However, recent computational developments that enabled VS to be executed on massively parallel computing architectures allowed successful implementation of CPU intensive approaches such as flexible molecular docking and MD simulation in SB-HLVS protocols. Using structure-based methods in hierarchical manner, our group identified compounds with biaryl urea scaffold as inhibitors of small ubiquitin-like modifier (SUMO) activating protein 1 (SUMO E1) [51], [52]. We docked a small molecule library to the ATP binding site in SUMO E1 using a two-step molecular docking strategy. Initially, a fast rigid protein docking was used to dock Maybridge small molecule library. The top ranking compounds were then re-docked using a method that incorporates both ligand and protein flexibilities. Top ranking hits from docking were subsequently prioritized for biological assay using the MD simulation-based binding free energy calculation. Similar hierarchical docking and molecular-mechanics Poisson–Boltzmann surface area (MM-PBSA) scoring [53], [54] procedure has been also used in the identification of novel tubulin inhibitors [55]. Recently, Kunze et al. [56] reported low molecular weight inhibitors of human immunodeficiency virus 1 protease subtype B utilizing SB-HLVS. MD simulation was used to identify transient surface pockets, one of which was translated into a structure-based pharmacophore query and used to screen compound databases. Molecular docking was then used to prioritize compounds for biological assay that resulted in the identification of two active low molecular weight inhibitors. Apart from sequentially combining multiple structure-based approaches, molecular docking based VS was sometimes performed in multi-step hierarchical fashion [52], [57], [58]. One such approach is implemented in Glide program [59], [60], [61], [62] which starts with coarse-grained docking followed by finer levels of sampling with more strict description of scoring functions. This hierarchical docking approach has been successfully used to identify highly potent inhibitors [57], [58], [63], [64], [65]. The main advantage of hierarchical docking is that it can rapidly screen large small molecule libraries without requiring high performance computing infrastructures. Fast and simple docking methods reduce a big library to a focused set with comparatively fewer molecules that are later docked using more computationally expensive methods.
2.3. Hybrid HLVS (LBSB-HLVS)
A hybrid approach that combines the ligand and structure-based HLVS (LBSB-HLVS) is the most commonly used among the three types of HLVS. LBSB-HLVS holds significant potential as it takes advantage of available structural and ligand information in hit identification. In general, fast computational approaches are used initially followed by computationally expensive approaches. Historically, LBSB-HLVS has enjoyed more successes than either LB-HLVS or SB-HLVS. Recent identification of serotonin transporter ligands [66] is an excellent example of LBSB-HLVS where five small molecule databases were screened using a VS workflow that combines 2D fingerprint based screening, ADMET filtering, 3D pharmacophore-based screening and molecular docking. In silico screened compounds were tested using in vitro screening and full binding assays, and 74 active compounds belonging to 16 structural classes were obtained. In another study [67], a hybrid 3D molecular similarity approach, SHAFTS [68] was combined with docking-based VS to identify nine low μM inhibitors of B-RafV600E. Analog search and subsequent chemical optimization of hits resulted in compounds with nM potency. Di-wu et al. [69] also employed a LBSB-HLVS comprising of Bayesian modeling, pharmacophore modeling and molecular docking to identify casein kinase 2 inhibitors. Nicolaes et al. [70] prefiltered a Chembridge compound collection for drug-likeness and subsequently used it to screen four pharmacophore models developed from known actives. Molecular docking was then used to prioritize a list of 1000 compounds for surface plasmon resonance based biological assay. Extensive biological characterization resulted in the identification of 9 compounds targeting the C2 domain of coagulation factor VIII with activities ranging from 2.1 to 19.9 μM. We have earlier developed an LBSB-HLVS workflow that utilized protein ligand interaction information to prioritize hits from ligand and structure-based VS. This workflow was employed to identify compounds with potent anti-tubercular activities [71]. In another excellent LBSB-HLVS study, Liu et al. [72] reported potent inhibitors of insulin-like growth factor-1 receptor (IGF-1R). Structure-based pharmacophore modeling combined with molecular docking resulted in two hits with IGF-1R inhibitory activity. These initial hits were subjected to substructure search to retrieve structural analogs that were prioritized using pharmacophore constrained molecular docking. Subsequent biological assays identified 15 more hits with good IGF-1R inhibitory activity. The implementation of LBSB-HLVS is particularly advantageous in cases where both ligand and structural information are available, as it takes into account all possible information to prioritize hits for biological assay. It has been already established that no single structure or ligand-based method performs well on all targets. The hierarchical fusion of ligand and structure based methods improve the VS performance by decreasing the number of false positives while increasing true positive hits.
3. Molecular docking is a key component in most HLVS
Molecular docking is one of the commonly used VS method and a central component in the majority of HLVS schemes. Molecular docking algorithms predict the orientation and conformation of ligands with respect to target receptor binding site. Hundreds of binding poses are generated that are evaluated by scoring functions. Last decade has witnessed tremendous growth in the development of molecular docking methodologies, algorithms and programs [9], [10]. These methods adequately use protein structural information to prioritize hits for biological assay. Some of these methodologies were effectively utilized to identify active hits from screening libraries, though there were reports that molecular docking failed to outperform ligand-based virtual screening methods even in the presence of protein structural information [73], [74], [75]. Most molecular docking programs are quite fast and are able to dock a small library in reasonable amount of time. However, docking a library of a few million compounds still requires huge computational resources and can be effectively performed only on massively parallel computer clusters. To circumvent this issue, HLVS approaches combining molecular docking with different levels of filtering have been proposed to speed-up the time consuming procedure of structure-based VS. Several of these approaches use structural (2D or 3D) similarity, shape or electrostatic similarity with the ligand to filter a large screening library. Others utilize ligand or structure-based pharmacophore modeling to reduce a large small molecule library to a reasonable number (a few hundred to tens of thousands of compounds) for molecular docking. In the following section, we will outline these fast VS methods that were effectively combined with molecular docking.
3.1. Similarity search – molecular docking
Similarity search is a simple and computationally inexpensive ligand-based method that evaluates the similarity of a library of small molecules with known ligands in terms of their 2D or 3D properties [15], [16]. Similarity analysis can be performed using topological indices [76], substructure search [77], [78], molecular fingerprints [79], [80], [81], molecular descriptors [82], shape and electrostatic properties [73], [83], [84], [85], [86]. Among all these similarity measures, substructure search is sometimes performed to screen large ligand libraries for the molecules that contain defined molecular fragment [87], [88]. 2D and 3D molecular fingerprints are routinely used to filter compounds for molecular docking in HLVS campaigns. The molecular access system (MACCS) fingerprint [81] is one of the most popular molecular fingerprint [15], [16]. MACCS fingerprint has been successfully integrated with molecular docking in several virtual screening studies [89]. Additionally, MACCS fingerprints are routinely used to improve the VS hit rate by identifying structural analogs of hit compounds [52], [90], [91], [92]. The extended connectivity fingerprints [80] from Accelrys and 2-point (TGD) and two 3-point (TGT and GpiDAPH3) pharmacophore-based fingerprints [93] from molecular operating environment (MOE) are other fingerprints that are commonly combined with molecular docking in several VS studies [88], [94], [95]. Steric complementarity between ligand and receptor is one of the key factors determining the strength of binding. This forms the basis of a hypothesis that two molecules with similar shape should have similar biological activities. Although the shape similarity alone was sufficient to retrieve highly potent binders [44], [45], [96], [97], [98], [99], the true potential of shape similarity was realized when it was combined with molecular docking in hierarchical manner. In one study, Acharya et al. [100] screened ZINC database [47] by coupling ROCS [73], [85] shape based approach with GOLD docking [101] method to identify small molecule inhibitors of chemokine receptor 5 (CCR5)-N terminus binding to gp120 protein. Xia et al. [102] employed HLVS protocol combining shape-based approach with molecular docking to discover selective 11β-hydroxysteroid dehydrogenase 1 (11β-HSD1) inhibitors. By performing VS against the Maybridge database, they identified seven hits showing IC50 values lower than 100 nM. Salo et al. [103] performed VS against the Chembridge library using shape-based filtering and flexible docking to identify two novel SIRT3 scaffolds. Similar VS approaches have also been used to identify novel non-steroidal FXR ligands, PPARγ partial agonists, Grb7-based antitumor agents and fungal trihydroxynaphthalene reductase inhibitors [104], [105], [106], [107]. In some studies, instead of using shape-based similarity alone with molecular docking, it has been complemented with approaches such as 2D similarity search, electrostatic potential matching and pharmacophore modeling. Alcaro et al. [89] merged compounds retrieved by 2D MACCS fingerprint and ROCS similarity search using seven known actives. Drug-likeness filter and molecular docking were used to further prioritize these compounds. Biological testing of 28 selected compounds resulted in a few compounds that interact with the human telomeric G-quadruplex. Recently, our group has identified 1,2,5-oxadiazoles as novel inhibitors of SUMO specific protease 2 (SENP2) utilizing a VS protocol that combines shape and electrostatic potential based similarity searches with molecular docking [108]. This inhibitor discovery process utilizing LBSB-HLVS is outlined in Fig. 2 . A similar HLVS approach was also used to identify inhibitors of biotin carboxylase [109] and F. tularensis enoyl-reductase [44]. In another study, Hamza et al. [110] employed a VS protocol that integrated a pharmacophore model, consensus molecular shape patterns of active ligands and molecular docking to identify inhibitors with μM potency against mycobacterial mycosin protease-1. Furthermore, Wiggers et al. [111] developed a multi-step cascade strategy that integrated shape based approaches with molecular docking and molecular hologram QSAR. This protocol successfully identified non-peptidic cruzain inhibitors with trypanocidal activity.
Fig. 2.
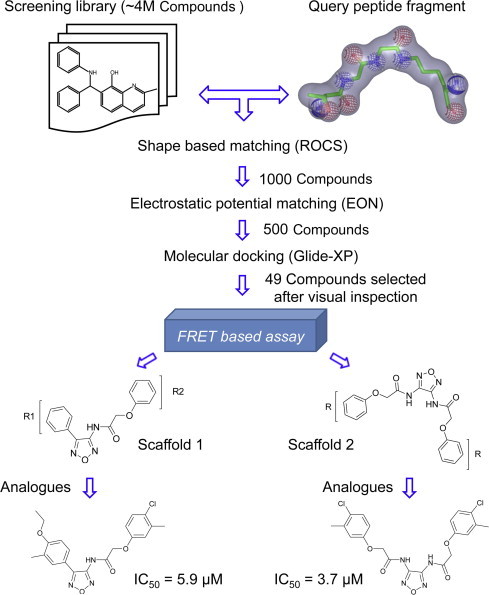
Outline of the discovery process of novel SENP2 inhibitors utilizing LBSB-HLVS. A four million compound small molecule library was filtered based on shape and electrostatic similarity with a fragment query prepared from the conjugate of SUMO1 C-terminal residues and substrate protein lysine. Molecular docking further prioritized the hits that were tested using a FRET based assay. Biological testing revealed two scaffolds that were later optimized for potency by identifying analogs.
3.2. Pharmacophore – molecular docking
As recommended by International Union of Pure and Applied Chemistry (IUPAC), the term pharmacophore represents an “ensemble of steric and electronic features that is necessary to ensure the optimal supra-molecular interactions with a specific biological target structure and to trigger (or to block) its biological response” [112]. In recent years, several pharmacophore modeling approaches and programs were developed [113], [114], [115]. Pharmacophore modeling has been extensively used in drug discovery either as a standalone VS method [116], [117], [118] or in combination with other VS approaches such as molecular docking. With a goal to identify inhibitors of matrix metalloproteinase 2 (MMP2), Pizio et al. [119] created and validated the seven-feature pharmacophore query based on the co-crystal structure of MMP8. This query was used to screen a library of ∼300,000 drug-like compounds. Molecular docking was then used to rank-order hits from pharmacophore-based screenings. Enzyme inhibition assay revealed nine active compounds. In another study, novel PKR-like endoplasmic reticulum kinase (PERK) inhibitors were discovered using hierarchical combination of pharmacophore screening and molecular docking [120]. Recently, similar VS protocols have been used to identify inhibitors of SARS-CoV 3-chymotrypsin-like protease [121], carbonic anhydrase VII [122], acetohydroxyacid synthase [123], malarial cysteine protease [124] and 5-lipoxygenase-activating protein [125]. Occasionally, studies combining structure and ligand based pharmacophore modeling have been published [126], [127], [128]. Using p53-MDM2 complex co-crystal structures, Xue et al. [126] built a structure-based pharmacophore model that was refined using a ligand-based pharmacophore model. The consensus model was used to screen a library of ∼239,735 compounds. Cascade docking further prioritized the hits and six active compounds were discovered. Ligand and receptor based pharmacophore screening in combination with molecular docking has been also used to discover CXC chemokine receptor 4 (CXCR4) antagonist [127] and p38α mitogen-activated protein kinase inhibitors [128]. Pharmacophore modeling has been also used as a post-filtering tool for molecular docking results [129], [130], [131]. In order to discover inhibitors of Met tyrosine kinase, Peach et al. [129] utilized a series of pharmacophore queries to filter out the docking poses that did not form critical interactions. Voet et al. [130] utilized pharmacophore modeling both as pre- and post-filtering tool for molecular docking. In a VS study to identify novel human androgen receptor (hAR) antagonist, they first created two pharmacophore queries, one for antagonist and the other for agonist, based on all hAR ligands co-crystal structures and homology modeling. The pharmacophore queries were then used to filter a library of commercially available small molecules to select compounds that fulfill the antagonist query and then to remove all those that match the agonist query. Molecular docking, pharmacophore post-filtering and visual selection were performed to select 31 compounds for evaluation of biological activity. The study resulted in the identification of two novel chemotypes with antagonist activities in low μM range while devoid of any agonist activity. As described above, the hierarchical combination of pharmacophore modeling and molecular docking has been extensively exploited in drug discovery. However, the performance of this HLVS scheme highly depends upon the accuracy of pharmacophore modeling. There are number of free and commercial pharmacophore modeling programs including Phase [132], [133], MOE-pharmacophore [93], LigandScout [134], Gasp [135], Catalyst [136], PharmID [137], etc. As different pharmacophore modeling programs vary in their performance, so the strength and weakness of each approach should be considered before applying them in HLVS protocols. The advantages and shortcomings of utilizing these pharmacophore modeling methods in drug discovery have been reported by Sanders et al. [138].
3.3. Molecular docking – molecular dynamics simulation – free energy
Recent advances in high performance computing has allowed the implementation of computationally demanding but more precise methods such flexible molecular docking and molecular dynamics simulation as VS tools. The success of a VS protocol depends on the accurate modeling of protein–ligand interactions, which require the consideration of protein and ligand in a dynamic environment. Several methodologies have been developed to account for protein flexibilities [139] that include soft docking [140], [141], sampling side-chain rotamer libraries and docking to an ensemble of receptor conformations [142], [143]. Molecular docking in combination with MD simulation has been especially recommended [144], [145]. MD simulation can be used to generate an ensemble of protein conformations for molecular docking [146], [147]. Moreover, MD simulation and free energy based ranking can be utilized in prioritizing molecular docking hits [148]. Both of these strategies have been effectively used in HLVS schemes for the identification of potent inhibitors. Wang et al. [149] carried out a docking-based VS using MD simulation to generate an ensemble of protein conformations. MD simulations optimized the docked complexes and led to the selection of compounds to be tested for human aldose reductase inhibition. Finally, in vitro assay identified two sub-micromolar compounds with novel chemotypes. Apart from generating receptor ensembles for molecular docking, MD simulation has also been used to identify functionally relevant receptor conformations, e.g. inactive DFG-out conformations in kinases to achieve selectivity against various kinases or the active state of GPCRs for the identification of agonists. These receptor conformations were then employed in corresponding HLVS campaigns [150], [151]. Hu et al. [152] reported F. tularensis FabI inhibitors identified from a HLVS protocol that integrated molecular docking with MD simulations. In their study, around 1000 hits from docking-based VS were subjected to a short MD simulation in implicit solvent. MM-PBSA binding free energy [53], [54] was used to rank-order compounds and 75 hits were singled out for biological assay that revealed 3 active hits. Using a similar approach, our group also successfully identified inhibitors of SUMO E1 protein [52]. In another study, Cao et al. [55] discovered novel tubulin inhibitors by taking advantage of HLVS protocol. They selected nine compounds for biological assay after screening an in-house library of around 100,000 compounds sequentially using molecular docking and MD simulation based MM-PBSA binding free energy calculation. Five out of 9 compounds demonstrated tubulin inhibitory activity. To efficiently take advantage of MM-PBSA binding free energy based ranking, computationally inexpensive VS methodologies have been developed [153], [154], [155]. Some of these studies reduce the time required by calculating MM-PBSA binding free energy from energy minimized single structure. These strategies were successfully integrated with molecular docking in HLVS schemes and resulted in the identification of potent inhibitors of plasmepsin II and Myt1 kinase [156], [157]. Although the integration of molecular docking with molecular dynamics simulation and binding free energy calculations seems to be an attractive option due to precise handling of protein–ligand-solvent interactions, it is still limited by some challenges. One obvious need is the requirement of huge computational resources to facilitate adequate sampling for accurate free energy calculations. Apart from computational requirements, the force fields used in molecular dynamics simulations are approximations and need further refinement [158]. Further, the calculation of binding free energy using relatively fast methods such as MM-PBSA rescoring [148], [153] relies on the ligand’s starting pose predicted by molecular docking and a wrong starting pose may lead to erroneous results. Rescoring of 10 docking poses has been suggested as a possible solution to achieve good accuracy [159].
4. HLVS for the identification of SMPPIIs
Recent advances in the discovery of small molecule protein–protein interaction inhibitors (SMPPIIs) have made the modulation of protein–protein interactions (PPIs) achievable [160], [161], [162], [163], [164]. VS approaches have played a significant role in changing the notion about the undruggability of PPIs. Several examples of VS studies have been reported in literature that targeted PPIs, including for HIV-1 IN–LEDGF/p75 [165], [166], [167], [168], XIAP–Caspase9 [169], Nrf2–Keap1 [64], [170], BCL2–BAK BH3 domain [171], uPAR–uPA [147], ΔF508–CFTR [172], RUNX1/ETO tetramerization [173], ERCC1–XPF [174], Annexin A2–S100A10 [175], SUMO–SIM [176], IFN-α–IFNAR [177], HIV Nef–SH3 [178] and p53–HDM2 [179]. Some of these VS studies took advantage of the hierarchical approach to identify initial hits that were later optimized into highly potent SMPPIIs [147], [165], [167], [168], [175], [176], [177], [178]. LBSB-HLVS was employed in some of these studies while other studies reported SB-HLVS approach. Substructure search in combination with molecular docking has been used to identify compounds disrupting the interaction of 14-3-3σ with aminopeptidase N [180]. Christ et al. [165] presented an excellent example of the employment of LBSB-HLVS for the identification of SMPPIIs for HIV-1 IN–LEDGF/p75 interaction. They have utilized pharmacophore-based screening, molecular docking and pharmacophore post-filtering to select 25 diverse molecules for biological assay. Optimization of initial hits first by testing analogs and then by medicinal chemistry led to the identification of compounds with potency in low μM range. Using a similar LBSB-HLVS approach comprising of pharmacophore screening and molecular docking, Reddy et al. [175] identified substituted 1,2,4-triazoles as inhibitors of the Annexin A2–S100A10 PPI. Voet et al. [176] translated the PPI between SUMO and SUMO interaction motif (SIM) into a pharmacophore query. This query was then used to screen a small molecule library and the resulting compounds were further prioritized using molecular docking and pharmacophore post-filtering. Multiple biological assays and HSQC-NMR revealed low μM inhibitors of SUMO-SIM PPI. Betzi et al. [178] applied drug-like filter, docking and pharmacophoric filter to identify inhibitors of HIV Nef–SH3 interaction with potency in μM range. Furthermore, novel SMPPIIs of Nrf2–Keap1 interaction were identified using HLVS scheme comprising of pharmacophore modeling and molecular docking. The most promising compound reported in this study disrupted the Nrf2–Keap1 interaction in vitro EC50 of 9.80 μM [170]. The Nrf2–Keap1 interaction was again targeted for SMPPIIs discovery, but this time utilizing an SB-HLVS approach [64]. Glide hierarchical docking protocol was used to identify nine hits with three novel scaffolds. In another study, Lavecchia et al. [57] built a theoretical model of frataxin/ubiquitin complex to reveal potential ubiquitin-binding site in frataxin. This interface was used to perform hierarchical docking-based VS that resulted in the identification of one compound that prevented frataxin ubiquitination and degradation. Subsequent scaffold searches identified a series of compounds able to disrupt frataxin/ubiquitin PPI with μM affinity. A similar hierarchical docking approach has been also used to identify inhibitors of PA–PB1 [181], p53–MDM2 [126], Cdc42–intersectin [182] and TWEAK–Fn14 [183] PPI. Using SB-HLVS, Khanna et al. [147] reported the discovery of SMPPIIs inhibiting the uPAR–uPA PPI. The compounds for biological assay were selected utilizing multi-step docking-based VS performed on a conformational ensemble of uPAR sampled from MD simulation. Extensive biochemical characterization revealed a compound inhibiting uPAR–uPA PPI with an IC50 of 10 μM. In an SB-HLVS study, Hu et al. [168] employed induced fit docking and MM-GBSA [184], [185] rescoring to identify 18 compounds targeting HIV-1 IN–LEDGF/p75 PPI. The two most potent compounds displayed IC50 values at 0.32 and 0.26 μM.
5. HLVS for the identification of GPCR modulators
G protein-coupled receptors (GPCRs) constitute the largest family of membrane proteins [186] and are the most important targets for the treatment of various diseases. GPCRs were the subject of many VS studies [94], [127], [187], [188], [189], [190], [191], [192], [193]. Several of these VS studies reported GPCR modulators following a hierarchical multi-step protocol where a large small molecule library was filtered to a smaller set to be tested by various assays. Due to the unavailability of crystal structures for most GPCRs, a majority of these HLVS were carried out using homology models [94], [151], [190], [192], [194]. Tikhonova et al. [94] used a structural model of free fatty acid receptor 1 (FFAR1) to perform VS employing 2D similarity search, 3D pharmacophore search and molecular docking. The biological assay of 70 compounds revealed 15 compounds acting as either antagonists or agonists of FFAR1. Engel et al. [190] created a structure-based pharmacophore that described interaction potential within the thyrotropin-releasing hormone (TRH) receptor binding pocket. ZINC small molecule library [47] was screened and the resulting compounds were prioritized for assay using flexible molecular docking and molecular descriptors. Five structurally diverse antagonists of the TRH receptors were identified. Recently using multiple crystal structure templates, Yoshikawa et al. [192] developed a homology model of CXCR7. The validated homology model was utilized for VS employing molecular docking and pharmacophore post-filtering. The study resulted in 21 CXCR7 ligands with novel scaffolds and IC50 ranging from 1.29 to 11.4 μM. In another recent report [151], dynamic pharmacophore modeling and molecular docking were used to discover agonists of serotonin receptor subtype 1A (5-HT1AR). Homology model of 5-HT1AR based on the active state of beta-2 adrenergic receptor was used for VS. Furthermore, de Graaf et al. [194] applied LBSB-HLVS to identify modulators of glucagon receptor (GLR) and glucagon-like peptide 1 receptor (GLP-1R). Both GLR and GLP-1R are challenging targets belonging to class B GPCRs. A library of 1.9 million drug-like compounds was filtered based on structural similarity with known antagonists. Subsequent hits were then docked to the trans-membrane cavity in a GLR homology model and finally selected for biological assay utilizing protein–ligand interaction information. Two of the 23 evaluated compounds displayed in vitro binding to GLR and inhibited glucagon effect. Interestingly, one compound inactive against GLR showed in vitro binding to GLP-1R. Lin et al. [195] combined homology modeling, molecular docking and MD simulations to establish kinase inhibitor Sorafenib as a nanomolar antagonist of 5-HT receptor. Other successful studies discovering GPCR modulators by performing HLVS virtual screening on homology models include reports for alpha1A adrenergic receptor [191], neurokinin-1 receptor [196] and nonpeptide CCR5 receptor [197]. Recent advances in the crystallography of GPCRs [198], [199] opened excellent opportunities for structure-based VS studies [200], [201], [202], [203], [204], [205], [206], [207]. A majority of these studies utilized only molecular docking as VS tool [201], [202], [203], [204], [206], [207]. Some of these studies applying HLVS approach on crystal structures include the discovery of histamine H1 receptor [200], β-2 adrenoreceptor [208], adenosine A2A receptor [208] and sphingosine 1-phosphate receptor [208] ligands. Among these studies, de Graaf et al. [200] developed and validated a HLVS method that combined molecular docking with interaction fingerprint (IFP) based scoring. The method was successfully applied to the identification of 19 human histamine H1 receptor ligands with potency ranging from 10 μM to 6 nM. In another study, Sanders et al. [208] combined ligand and structure-based methods to carry out prospective VS against three GPCRs (β-2 adrenoreceptor, adenosine A2A receptor and sphingosine 1-phosphate receptor). Biological assay revealed 6, 18 and 3 active ligands for β-2 adrenoreceptor, adenosine A2A receptor and sphingosine 1-phosphate receptor respectively. As described above, the majority of HLVS efforts were focused on the identification of small molecule modulators of class A or rhodopsin-like GPCRs. The reason may be the availability of plentiful ligand information and ever increasing crystal structures for this class of receptors. Barring a few studies [194], [209], [210], HLVS has not been exploited in the discovery of small molecule modulators for class B and C GPCRs, which may be due to the unavailability of sufficient structural information about receptor’s trans-membrane domain. However, the recent availability of trans-membrane domain crystal structures of class B [211], [212] and class C [213] GPCRs should augment drug discovery efforts. Specially, these developments should fuel efforts exploiting the power of LBSB-HLVS in the discovery of small molecular modulators of class B and C GPCRs.
6. Conclusion
We have witnessed tremendous growth in successful applications of VS in various drug discovery campaigns in recent years. VS methods relying on ligand and structure-based approaches have been developed and successfully employed in prospective applications. In order to enhance VS efficiency and improve hit rate, integration of several ligand and structure-based approaches has been suggested. A survey of recently published VS studies revealed hierarchical combination as the most common way of integrating ligand and structure-based methods. Mostly, computationally inexpensive ligand-based methods are used at the beginning of a multi-step hierarchical procedure while computationally demanding methods are used towards the end. Pharmacophore modeling and molecular docking are two extensively employed VS methods in HLVS schemes. The hierarchical combination of these two methods has been proven effective in the discovery of inhibitors of difficult drug targets such as PPIs. GPCRs are another class of important drug targets where HLVS has shown potential in retrieving promising hits, especially, considering the recent advances in GPCR crystallography.
Acknowledgments
We thank members of our laboratory for help and discussions. We acknowledge RIKEN, Japan for funding.
References
- 1.Tanrikulu Y., Krüger B., Proschak E. Drug Discov. Today. 2013;18:358–364. doi: 10.1016/j.drudis.2013.01.007. [DOI] [PubMed] [Google Scholar]
- 2.Walters W.P., Stahl M.T., Murcko M.A. Drug Discov. Today. 1998;3:160–178. [Google Scholar]
- 3.Muegge I. Mini Rev. Med. Chem. 2008;8:927–933. doi: 10.2174/138955708785132792. [DOI] [PubMed] [Google Scholar]
- 4.Muegge I., Oloff S. Drug Discov. Today Technol. 2006;3:405–411. doi: 10.1016/j.ddtec.2006.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sliwoski G., Kothiwale S., Meiler J., Lowe E.W. Pharmacol. Rev. 2014;66:334–395. doi: 10.1124/pr.112.007336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Drwal M.N., Griffith R. Drug Discov. Today Technol. 2013;10:e395–e401. doi: 10.1016/j.ddtec.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 7.Lavecchia A., Di Giovanni C. Curr. Med. Chem. 2013;20:2839–2860. doi: 10.2174/09298673113209990001. [DOI] [PubMed] [Google Scholar]
- 8.M. Lill, in: S. Kortagere (Ed.), In Silico Models for Drug Discovery, Humana Press 2013, pp. 1–12.
- 9.Yuriev E., Agostino M., Ramsland P.A. J. Mol. Recognit. 2011;24:149–164. doi: 10.1002/jmr.1077. [DOI] [PubMed] [Google Scholar]
- 10.Yuriev E., Ramsland P.A. J. Mol. Recognit. 2013;26:215–239. doi: 10.1002/jmr.2266. [DOI] [PubMed] [Google Scholar]
- 11.Pirhadi S., Shiri F., Ghasemi J.B. Curr. Top. Med. Chem. 2013;13:1036–1047. doi: 10.2174/1568026611313090006. [DOI] [PubMed] [Google Scholar]
- 12.Sanders M.P.A., McGuire R., Roumen L., de Esch I.J.P., de Vlieg J., Klomp J.P.G., de Graaf C. MedChemComm. 2012;3:28–38. [Google Scholar]
- 13.Kutchukian P.S., Shakhnovich E.I. Expert Opin. Drug Discov. 2010;5:789–812. doi: 10.1517/17460441.2010.497534. [DOI] [PubMed] [Google Scholar]
- 14.Loving K., Alberts I., Sherman W. Curr. Top. Med. Chem. 2010;10:14–32. doi: 10.2174/156802610790232305. [DOI] [PubMed] [Google Scholar]
- 15.Maggiora G., Vogt M., Stumpfe D., Bajorath J. J. Med. Chem. 2013;57:3186–3204. doi: 10.1021/jm401411z. [DOI] [PubMed] [Google Scholar]
- 16.Stumpfe D., Bajorath J. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2011;1:260–282. [Google Scholar]
- 17.Caporuscio F., Tafi A. Curr. Med. Chem. 2011;18:2543–2553. doi: 10.2174/092986711795933669. [DOI] [PubMed] [Google Scholar]
- 18.Melville J.L., Burke E.K., Hirst J.D. Comb. Chem. High Throughput Screen. 2009;12:332–343. doi: 10.2174/138620709788167980. [DOI] [PubMed] [Google Scholar]
- 19.Gertrudes J.C., Maltarollo V.G., Silva R.A., Oliveira P.R., Honorio K.M., da Silva A.B. Curr. Med. Chem. 2012;19:4289–4297. doi: 10.2174/092986712802884259. [DOI] [PubMed] [Google Scholar]
- 20.Guha R. Methods Mol. Biol. 2013;993:81–94. doi: 10.1007/978-1-62703-342-8_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ruddigkeit L., van Deursen R., Blum L.C., Reymond J.-L. J. Chem. Inf. Model. 2012;52:2864–2875. doi: 10.1021/ci300415d. [DOI] [PubMed] [Google Scholar]
- 22.Davis I.W., Baker D. J. Mol. Biol. 2009;385:381–392. doi: 10.1016/j.jmb.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 23.Davis I.W., Raha K., Head M.S., Baker D. Protein Sci. 2009;18:1998–2002. doi: 10.1002/pro.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meiler J., Baker D. Proteins. 2006;65:538–548. doi: 10.1002/prot.21086. [DOI] [PubMed] [Google Scholar]
- 25.Kaufmann K.W., Meiler J. PLoS One. 2012;7:e50769. doi: 10.1371/journal.pone.0050769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Svensson F., Karlén A., Sköld C. J. Chem. Inf. Model. 2011;52:225–232. doi: 10.1021/ci2004835. [DOI] [PubMed] [Google Scholar]
- 27.Swann S.L., Brown S.P., Muchmore S.W., Patel H., Merta P., Locklear J., Hajduk P.J. J. Med. Chem. 2011;54:1223–1232. doi: 10.1021/jm1013677. [DOI] [PubMed] [Google Scholar]
- 28.Tan L., Geppert H., Sisay M.T., Gütschow M., Bajorath J. ChemMedChem. 2008;3:1566–1571. doi: 10.1002/cmdc.200800129. [DOI] [PubMed] [Google Scholar]
- 29.Holliday J., Kanoulas E., Malim N., Willett P. J. Cheminformatics. 2011;3:29. doi: 10.1186/1758-2946-3-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kang H., Sheng Z., Zhu R., Huang Q., Liu Q., Cao Z. J. Chem. Inf. Model. 2012;52:834–843. doi: 10.1021/ci200481c. [DOI] [PubMed] [Google Scholar]
- 31.Sharma R., Lawrenson A.S., Fisher N.E., Warman A.J., Shone A.E., Hill A., Mbekeani A., Pidathala C., Amewu R.K., Leung S., Gibbons P., Hong D.W., Stocks P., Nixon G.L., Chadwick J., Shearer J., Gowers I., Cronk D., Parel S.P., O’Neill P.M., Ward S.A., Biagini G.A., Berry N.G. J. Med. Chem. 2012;55:3144–3154. doi: 10.1021/jm3001482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Langdon S.R., Westwood I.M., van Montfort R.L.M., Brown N., Blagg J. J. Chem. Inf. Model. 2013;53:1100–1112. doi: 10.1021/ci400100c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pozzan A. Curr. Pharm. Des. 2006;12:2099–2110. doi: 10.2174/138161206777585247. [DOI] [PubMed] [Google Scholar]
- 34.P. Willett, in: J. Bajorath (Ed.) Chemoinformatics and Computational Chemical Biology, Humana Press, 2011, pp. 133–158.
- 35.Ebalunode J.O., Zheng W. Curr. Top. Med. Chem. 2010;10:669–679. doi: 10.2174/156802610791111489. [DOI] [PubMed] [Google Scholar]
- 36.Gane P.J., Chan A.W. Methods Mol. Biol. 2013;1008:479–499. doi: 10.1007/978-1-62703-398-5_18. [DOI] [PubMed] [Google Scholar]
- 37.Yang S.Y. Drug Discov. Today. 2010;15:444–450. doi: 10.1016/j.drudis.2010.03.013. [DOI] [PubMed] [Google Scholar]
- 38.Olah M.M., Bologa C.G., Oprea T.I. Curr. Drug Discov. Technol. 2004;1:211–220. doi: 10.2174/1570163043334965. [DOI] [PubMed] [Google Scholar]
- 39.Stahl M., Mauser H., Tsui M., Taylor N.R. J. Med. Chem. 2005;48:4358–4366. doi: 10.1021/jm040213p. [DOI] [PubMed] [Google Scholar]
- 40.Geppert H., Vogt M., Bajorath J.r. J. Chem. Inf. Model. 2010;50:205–216. doi: 10.1021/ci900419k. [DOI] [PubMed] [Google Scholar]
- 41.Yao S., Lu T., Zhou Z., Liu H., Yuan H., Ran T., Lu S., Zhang Y., Ke Z., Xu J., Xiong X., Chen Y. Mol. Divers. 2014;18:183–193. doi: 10.1007/s11030-013-9493-3. [DOI] [PubMed] [Google Scholar]
- 42.Temml V., Voss C.V., Dirsch V.M., Schuster D. J. Chem. Inf. Model. 2014;54:367–371. doi: 10.1021/ci400682b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Muchmore S.W., Souers A.J., Akritopoulou-Zanze I. Chem. Biol. Drug Des. 2006;67:174–176. doi: 10.1111/j.1747-0285.2006.00341.x. [DOI] [PubMed] [Google Scholar]
- 44.Hevener K.E., Mehboob S., Su P.-C., Truong K., Boci T., Deng J., Ghassemi M., Cook J.L., Johnson M.E. J. Med. Chem. 2012;55:268–279. doi: 10.1021/jm201168g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Naylor E., Arredouani A., Vasudevan S.R., Lewis A.M., Parkesh R., Mizote A., Rosen D., Thomas J.M., Izumi M., Ganesan A., Galione A., Churchill G.C. Nat. Chem. Biol. 2009;5:220–226. doi: 10.1038/nchembio.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carosati E., Sforna G., Pippi M., Marverti G., Ligabue A., Guerrieri D., Piras S., Guaitoli G., Luciani R., Costi M.P., Cruciani G. Biorg. Med. Chem. 2010;18:7773–7785. doi: 10.1016/j.bmc.2010.09.065. [DOI] [PubMed] [Google Scholar]
- 47.Irwin J.J., Sterling T., Mysinger M.M., Bolstad E.S., Coleman R.G. J. Chem. Inf. Model. 2012;52:1757–1768. doi: 10.1021/ci3001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Levit A., Nowak S., Peters M., Wiener A., Meyerhof W., Behrens M., Niv M.Y. FASEB J. 2014;28:1181–1197. doi: 10.1096/fj.13-242594. [DOI] [PubMed] [Google Scholar]
- 49.Pan Y., Chothe P.P., Swaan P.W. Mol. Pharm. 2013;10:1236–1248. doi: 10.1021/mp300547h. [DOI] [PubMed] [Google Scholar]
- 50.Stumpfe D., Bill A., Novak N., Loch G., Blockus H., Geppert H., Becker T., Schmitz A., Hoch M., Kolanus W., Famulok M., Bajorath J.r. ACS Chem. Biol. 2010;5:839–849. doi: 10.1021/cb100171c. [DOI] [PubMed] [Google Scholar]
- 51.Kumar A., Ito A., Hirohama M., Yoshida M., Zhang K.Y.J. Bioorg. Med. Chem. Lett. 2013;23:5145–5149. doi: 10.1016/j.bmcl.2013.07.022. [DOI] [PubMed] [Google Scholar]
- 52.Kumar A., Ito A., Hirohama M., Yoshida M., Zhang K.Y.J. J. Chem. Inf. Model. 2013;53:809–820. doi: 10.1021/ci300618e. [DOI] [PubMed] [Google Scholar]
- 53.Kollman P.A., Massova I., Reyes C., Kuhn B., Huo S., Chong L., Lee M., Lee T., Duan Y., Wang W., Donini O., Cieplak P., Srinivasan J., Case D.A., Cheatham T.E. Acc. Chem. Res. 2000;33:889–897. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- 54.Srinivasan J., Cheatham T.E., Cieplak P., Kollman P.A., Case D.A. J. Am. Chem. Soc. 1998;120:9401–9409. [Google Scholar]
- 55.Cao R., Liu M., Yin M., Liu Q., Wang Y., Huang N. J. Chem. Inf. Model. 2012;52:2730–2740. doi: 10.1021/ci300302c. [DOI] [PubMed] [Google Scholar]
- 56.Kunze J., Todoroff N., Schneider P., Rodrigues T., Geppert T., Reisen F., Schreuder H., Saas J., Hessler G., Baringhaus K.-H., Schneider G. J. Chem. Inf. Model. 2014;54:987–991. doi: 10.1021/ci400712h. [DOI] [PubMed] [Google Scholar]
- 57.Lavecchia A., Di Giovanni C., Cerchia C., Russo A., Russo G., Novellino E. J. Med. Chem. 2013;56:2861–2873. doi: 10.1021/jm3017199. [DOI] [PubMed] [Google Scholar]
- 58.Xu L., Zhang Y., Zheng L., Qiao C., Li Y., Li D., Zhen X., Hou T. J. Med. Chem. 2014;57:3737–3745. doi: 10.1021/jm401908w. [DOI] [PubMed] [Google Scholar]
- 59.Glide, version 5.7, Schrödinger, LLC, New York, NY, 2011.
- 60.Friesner R.A., Banks J.L., Murphy R.B., Halgren T.A., Klicic J.J., Mainz D.T., Repasky M.P., Knoll E.H., Shelley M., Perry J.K., Shaw D.E., Francis P., Shenkin P.S. J. Med. Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 61.Friesner R.A., Murphy R.B., Repasky M.P., Frye L.L., Greenwood J.R., Halgren T.A., Sanschagrin P.C., Mainz D.T. J. Med. Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 62.Halgren T.A., Murphy R.B., Friesner R.A., Beard H.S., Frye L.L., Pollard W.T., Banks J.L. J. Med. Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 63.Zhou Z., Khaliq M., Suk J.-E., Patkar C., Li L., Kuhn R.J., Post C.B. ACS Chem. Biol. 2008;3:765–775. doi: 10.1021/cb800176t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhuang C., Narayanapillai S., Zhang W., Sham Y.Y., Xing C. J. Med. Chem. 2014;57:1121–1126. doi: 10.1021/jm4017174. [DOI] [PubMed] [Google Scholar]
- 65.Caporuscio F., Rastelli G., Imbriano C., Del Rio A. J. Med. Chem. 2011;54:4006–4017. doi: 10.1021/jm2000689. [DOI] [PubMed] [Google Scholar]
- 66.Gabrielsen M., Kurczab R., Siwek A., Wolak M., Ravna A.W., Kristiansen K., Kufareva I., Abagyan R., Nowak G., Chilmonczyk Z., Sylte I., Bojarski A.J. J. Chem. Inf. Model. 2014;54:933–943. doi: 10.1021/ci400742s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kong X., Qin J., Li Z., Vultur A., Tong L., Feng E., Rajan G., Liu S., Lu J., Liang Z., Zheng M., Zhu W., Jiang H., Herlyn M., Liu H., Marmorstein R., Luo C. Org. Biomol. Chem. 2012;10:7402–7417. doi: 10.1039/c2ob26081f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu X., Jiang H., Li H. J. Chem. Inf. Model. 2011;51:2372–2385. doi: 10.1021/ci200060s. [DOI] [PubMed] [Google Scholar]
- 69.Di-wu L., Li L.-L., Wang W.-J., Xie H.-Z., Yang J., Zhang C.-H., Huang Q., Zhong L., Feng S., Yang S.-Y. J. Mol. Graph. Model. 2012;36:42–47. doi: 10.1016/j.jmgm.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 70.Nicolaes G.A.F., Kulharia M., Voorberg J., Kaijen P.H., Wroblewska A., Wielders S., Schrijver R., Sperandio O., Villoutreix B.O. Blood. 2014;123:113–120. doi: 10.1182/blood-2013-05-503227. [DOI] [PubMed] [Google Scholar]
- 71.Kumar A., Chaturvedi V., Bhatnagar S., Sinha S., Siddiqi M.I. J. Chem. Inf. Model. 2008;49:35–42. doi: 10.1021/ci8003607. [DOI] [PubMed] [Google Scholar]
- 72.Liu X., Xie H., Luo C., Tong L., Wang Y., Peng T., Ding J., Jiang H., Li H. J. Med. Chem. 2010;53:2661–2665. doi: 10.1021/jm901798e. [DOI] [PubMed] [Google Scholar]
- 73.Hawkins P.C.D., Skillman A.G., Nicholls A. J. Med. Chem. 2006;50:74–82. doi: 10.1021/jm0603365. [DOI] [PubMed] [Google Scholar]
- 74.McGaughey G.B., Sheridan R.P., Bayly C.I., Culberson J.C., Kreatsoulas C., Lindsley S., Maiorov V., Truchon J.-F., Cornell W.D. J. Chem. Inf. Model. 2007;47:1504–1519. doi: 10.1021/ci700052x. [DOI] [PubMed] [Google Scholar]
- 75.Zhang Q., Muegge I. J. Med. Chem. 2006;49:1536–1548. doi: 10.1021/jm050468i. [DOI] [PubMed] [Google Scholar]
- 76.Ivanciuc O. Curr. Comput. Aided Drug Des. 2013;9:153–163. doi: 10.2174/1573409911309020002. [DOI] [PubMed] [Google Scholar]
- 77.Golovin A., Henrick K. J. Chem. Inf. Model. 2009;49:22–27. doi: 10.1021/ci8003013. [DOI] [PubMed] [Google Scholar]
- 78.Ehrlich H.C., Henzler A.M., Rarey M. J. Chem. Inf. Model. 2013;53:1676–1688. doi: 10.1021/ci400107k. [DOI] [PubMed] [Google Scholar]
- 79.Bender A., Mussa H.Y., Glen R.C., Reiling S. J. Chem. Inf. Comput. Sci. 2004;44:1708–1718. doi: 10.1021/ci0498719. [DOI] [PubMed] [Google Scholar]
- 80.Rogers D., Hahn M. J. Chem. Inf. Model. 2010;50:742–754. doi: 10.1021/ci100050t. [DOI] [PubMed] [Google Scholar]
- 81.MACCS Keys, MDL Information Systems Inc., San Leandro, CA, 1979.
- 82.Todeschini R., Consonni V. In: Methods and Principles in Medicinal Chemistry. Mannhold R., Kubinyi H., Folkers G., editors. Wiley-VCH; Weinheim: 2009. [Google Scholar]
- 83.Armstrong M.S., Morris G., Finn P., Sharma R., Moretti L., Cooper R., Richards W.G. J. Comput.-Aided Mol. Des. 2010;24:789–801. doi: 10.1007/s10822-010-9374-0. [DOI] [PubMed] [Google Scholar]
- 84.Vainio M.J., Puranen J.S., Johnson M.S. J. Chem. Inf. Model. 2009;49:492–502. doi: 10.1021/ci800315d. [DOI] [PubMed] [Google Scholar]
- 85.ROCS 3.2.0.4: OpenEye Scientific Software, Santa Fe, NM. http://www.eyesopen.com.
- 86.Berenger F., Voet A., Lee X., Zhang K. J. Cheminformatics. 2014;6:23. doi: 10.1186/1758-2946-6-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jansen C., Wang H., Kooistra A.J., de Graaf C., Orrling K.M., Tenor H., Seebeck T., Bailey D., de Esch I.J.P., Ke H., Leurs R. J. Med. Chem. 2013;56:2087–2096. doi: 10.1021/jm3017877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vidler L.R., Filippakopoulos P., Fedorov O., Picaud S., Martin S., Tomsett M., Woodward H., Brown N., Knapp S., Hoelder S. J. Med. Chem. 2013;56:8073–8088. doi: 10.1021/jm4011302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Alcaro S., Musetti C., Distinto S., Casatti M., Zagotto G., Artese A., Parrotta L., Moraca F., Costa G., Ortuso F., Maccioni E., Sissi C. J. Med. Chem. 2013;56:843–855. doi: 10.1021/jm3013486. [DOI] [PubMed] [Google Scholar]
- 90.Daidone F., Montioli R., Paiardini A., Cellini B., Macchiarulo A., Giardina G., Bossa F., Borri Voltattorni C. PLoS One. 2012;7:e31610. doi: 10.1371/journal.pone.0031610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Broos K., Trekels M., Jose R.A., Demeulemeester J., Vandenbulcke A., Vandeputte N., Venken T., Egle B., De Borggraeve W.M., Deckmyn H., De Maeyer M. J. Biol. Chem. 2012;287:9461–9472. doi: 10.1074/jbc.M111.311431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Okamoto M., Takayama K., Shimizu T., Ishida K., Takahashi O., Furuya T. J. Med. Chem. 2009;52:7323–7327. doi: 10.1021/jm901191q. [DOI] [PubMed] [Google Scholar]
- 93.Chemical Computing Group Inc.; Montreal, Quebec, Canada: 2010. Molecular Operating Environment (MOE), Version 2011.10. [Google Scholar]
- 94.Tikhonova I.G., Sum C.S., Neumann S., Engel S., Raaka B.M., Costanzi S., Gershengorn M.C. J. Med. Chem. 2008;51:625–633. doi: 10.1021/jm7012425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Muddassar M., Jang J.W., Gon H.S., Cho Y.S., Kim E.E., Keum K.C., Oh T., Cho S.-N., Pae A.N. Biorg. Med. Chem. 2010;18:6914–6921. doi: 10.1016/j.bmc.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 96.Sun H., Xu X., Wu X., Zhang X., Liu F., Jia J., Guo X., Huang J., Jiang Z., Feng T., Chu H., Zhou Y., Zhang S., Liu Z., You Q. J. Chem. Inf. Model. 2013;53:2093–2102. doi: 10.1021/ci400114f. [DOI] [PubMed] [Google Scholar]
- 97.Kaoud T.S., Yan C., Mitra S., Tseng C.-C., Jose J., Taliaferro J.M., Tuohetahuntila M., Devkota A., Sammons R., Park J., Park H., Shi Y., Hong J., Ren P., Dalby K.N. ACS Med. Chem. Lett. 2012;3:721–725. doi: 10.1021/ml300129b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Vasudevan S.R., Moore J.B., Schymura Y., Churchill G.C. J. Med. Chem. 2012;55:7054–7060. doi: 10.1021/jm300671m. [DOI] [PubMed] [Google Scholar]
- 99.Kumar A., Parkesh R., Sznajder L.J., Childs-Disney J.L., Sobczak K., Disney M.D. ACS Chem. Biol. 2012;7:496–505. doi: 10.1021/cb200413a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Acharya P., Dogo-Isonagie C., LaLonde J.M., Lam S.N., Leslie G.J., Louder M.K., Frye L.L., Debnath A.K., Greenwood J.R., Luongo T.S., Martin L., Watts K.S., Hoxie J.A., Mascola J.R., Bewley C.A., Kwong P.D. ACS Chem. Biol. 2011;6:1069–1077. doi: 10.1021/cb200068b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jones G., Willett P., Glen R.C., Leach A.R., Taylor R. J. Mol. Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 102.Xia G., Xue M., Liu L., Yu J., Liu H., Li P., Wang J., Li Y., Xiong B., Shen J. Bioorg. Med. Chem. Lett. 2011;21:5739–5744. doi: 10.1016/j.bmcl.2011.08.019. [DOI] [PubMed] [Google Scholar]
- 103.Salo H.S., Laitinen T., Poso A., Jarho E., Lahtela-Kakkonen M. Bioorg. Med. Chem. Lett. 2013;23:2990–2995. doi: 10.1016/j.bmcl.2013.03.033. [DOI] [PubMed] [Google Scholar]
- 104.Fu J., Si P., Zheng M., Chen L., Shen X., Tang Y., Li W. Bioorg. Med. Chem. Lett. 2012;22:6848–6853. doi: 10.1016/j.bmcl.2012.09.045. [DOI] [PubMed] [Google Scholar]
- 105.Vidović D., Busby S.A., Griffin P.R., Schürer S.C. ChemMedChem. 2011;6:94–103. doi: 10.1002/cmdc.201000428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ambaye N.D., Gunzburg M.J., Lim R.C.C., Price J.T., Wilce M.C.J., Wilce J.A. ChemMedChem. 2013;8:280–288. doi: 10.1002/cmdc.201200400. [DOI] [PubMed] [Google Scholar]
- 107.Brunskole Švegelj M., Turk S., Brus B., Lanišnik Rižner T., Stojan J., Gobec S. J. Chem. Inf. Model. 2011;51:1716–1724. doi: 10.1021/ci2001499. [DOI] [PubMed] [Google Scholar]
- 108.Kumar A., Ito A., Takemoto M., Yoshida M., Zhang K.Y.J. J. Chem. Inf. Model. 2014;54:870–880. doi: 10.1021/ci4007134. [DOI] [PubMed] [Google Scholar]
- 109.Mochalkin I., Miller J.R., Narasimhan L., Thanabal V., Erdman P., Cox P.B., Prasad J.V.N.V., Lightle S., Huband M.D., Stover C.K. ACS Chem. Biol. 2009;4:473–483. doi: 10.1021/cb9000102. [DOI] [PubMed] [Google Scholar]
- 110.Hamza A., Wagner J.M., Evans T.J., Frasinyuk M.S., Kwiatkowski S., Zhan C.-G., Watt D.S., Korotkov K.V. J. Chem. Inf. Model. 2014;54:1166–1173. doi: 10.1021/ci500025r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wiggers H.J., Rocha J.R., Fernandes W.B., Sesti-Costa R., Carneiro Z.A., Cheleski J., da Silva A.B., Juliano L., Cezari M.H., Silva J.S., McKerrow J.H., Montanari C.A. PLoS Negl. Trop. Dis. 2013;7:e2370. doi: 10.1371/journal.pntd.0002370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Güner O.F., Bowen J.P. J. Chem. Inf. Model. 2014;54:1269–1283. doi: 10.1021/ci5000533. [DOI] [PubMed] [Google Scholar]
- 113.Horvath D. Methods Mol. Biol. 2011;672:261–298. doi: 10.1007/978-1-60761-839-3_11. [DOI] [PubMed] [Google Scholar]
- 114.Sun H. Curr. Med. Chem. 2008;15:1018–1024. doi: 10.2174/092986708784049630. [DOI] [PubMed] [Google Scholar]
- 115.Kim K.H., Kim N.D., Seong B.L. Expert Opin. Drug Discov. 2010;5:205–222. doi: 10.1517/17460441003592072. [DOI] [PubMed] [Google Scholar]
- 116.Hinsberger S., Hüsecken K., Groh M., Negri M., Haupenthal J., Hartmann R.W. J. Med. Chem. 2013;56:8332–8338. doi: 10.1021/jm400485e. [DOI] [PubMed] [Google Scholar]
- 117.Pauli I., dos Santos R.N., Rostirolla D.C., Martinelli L.K., Ducati R.G., Timmers L.F.S.M., Basso L.A., Santos D.S., Guido R.V.C., Andricopulo A.D., Norberto de Souza O. J. Chem. Inf. Model. 2013;53:2390–2401. doi: 10.1021/ci400202t. [DOI] [PubMed] [Google Scholar]
- 118.Rechfeld F., Gruber P., Kirchmair J., Boehler M., Hauser N., Hechenberger G., Garczarczyk D., Lapa G.B., Preobrazhenskaya M.N., Goekjian P., Langer T., Hofmann J. J. Med. Chem. 2014;57:3235–3246. doi: 10.1021/jm401605c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Di Pizio A., Laghezza A., Tortorella P., Agamennone M. ChemMedChem. 2013;8:1475–1482. doi: 10.1002/cmdc.201300186. [DOI] [PubMed] [Google Scholar]
- 120.Wang Q., Park J., Devkota A.K., Cho E.J., Dalby K.N., Ren P. J. Chem. Inf. Model. 2014;54:1467–1475. doi: 10.1021/ci500114r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lee H., Mittal A., Patel K., Gatuz J.L., Truong L., Torres J., Mulhearn D.C., Johnson M.E. Biorg. Med. Chem. 2014;22:167–177. doi: 10.1016/j.bmc.2013.11.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.De Luca L., Ferro S., Damiano F.M., Supuran C.T., Vullo D., Chimirri A., Gitto R. Eur. J. Med. Chem. 2014;71:105–111. doi: 10.1016/j.ejmech.2013.10.071. [DOI] [PubMed] [Google Scholar]
- 123.Wang D., Zhu X., Cui C., Dong M., Jiang H., Li Z., Liu Z., Zhu W., Wang J.-G. J. Chem. Inf. Model. 2013;53:343–353. doi: 10.1021/ci3004545. [DOI] [PubMed] [Google Scholar]
- 124.Shah F., Mukherjee P., Gut J., Legac J., Rosenthal P.J., Tekwani B.L., Avery M.A. J. Chem. Inf. Model. 2011;51:852–864. doi: 10.1021/ci200029y. [DOI] [PubMed] [Google Scholar]
- 125.Banoglu E., Çalışkan B., Luderer S., Eren G., Özkan Y., Altenhofen W., Weinigel C., Barz D., Gerstmeier J., Pergola C., Werz O. Biorg. Med. Chem. 2012;20:3728–3741. doi: 10.1016/j.bmc.2012.04.048. [DOI] [PubMed] [Google Scholar]
- 126.Xue X., Wei J.-L., Xu L.-L., Xi M.-Y., Xu X.-L., Liu F., Guo X.-K., Wang L., Zhang X.-J., Zhang M.-Y., Lu M.-C., Sun H.-P., You Q.-D. J. Chem. Inf. Model. 2013;53:2715–2729. doi: 10.1021/ci400348f. [DOI] [PubMed] [Google Scholar]
- 127.Vitale R.M., Gatti M., Carbone M., Barbieri F., Felicità V., Gavagnin M., Florio T., Amodeo P. ACS Chem. Biol. 2013;8:2762–2770. doi: 10.1021/cb400521b. [DOI] [PubMed] [Google Scholar]
- 128.Gangwal R.P., Das N.R., Thanki K., Damre M.V., Dhoke G.V., Sharma S.S., Jain S., Sangamwar A.T. J. Mol. Graph. Model. 2014;49:18–24. doi: 10.1016/j.jmgm.2014.01.002. [DOI] [PubMed] [Google Scholar]
- 129.Peach M.L., Tan N., Choyke S.J., Giubellino A., Athauda G., Burke T.R., Nicklaus M.C., Bottaro D.P. J. Med. Chem. 2009;52:943–951. doi: 10.1021/jm800791f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Voet A., Helsen C., Zhang K.Y.J., Claessens F. ChemMedChem. 2013;8:644–651. doi: 10.1002/cmdc.201200549. [DOI] [PubMed] [Google Scholar]
- 131.He S., Li C., Liu Y., Lai L. J. Med. Chem. 2013;56:3296–3309. doi: 10.1021/jm301900x. [DOI] [PubMed] [Google Scholar]
- 132.Dixon S., Smondyrev A., Knoll E., Rao S., Shaw D., Friesner R. J. Comput.-Aided Mol. Des. 2006;20:647–671. doi: 10.1007/s10822-006-9087-6. [DOI] [PubMed] [Google Scholar]
- 133.Dixon S.L., Smondyrev A.M., Rao S.N. Chem. Biol. Drug Des. 2006;67:370–372. doi: 10.1111/j.1747-0285.2006.00384.x. [DOI] [PubMed] [Google Scholar]
- 134.Wolber G., Langer T. J. Chem. Inf. Model. 2004;45:160–169. doi: 10.1021/ci049885e. [DOI] [PubMed] [Google Scholar]
- 135.Jones G., Willett P., Glen R. J. Comput.-Aided Mol. Des. 1995;9:532–549. doi: 10.1007/BF00124324. [DOI] [PubMed] [Google Scholar]
- 136.Barnum D., Greene J., Smellie A., Sprague P. J. Chem. Inf. Comput. Sci. 1996;36:563–571. doi: 10.1021/ci950273r. [DOI] [PubMed] [Google Scholar]
- 137.Feng J., Sanil A., Young S.S. J. Chem. Inf. Model. 2006;46:1352–1359. doi: 10.1021/ci050427v. [DOI] [PubMed] [Google Scholar]
- 138.Sanders M.P.A., Barbosa A.J.M., Zarzycka B., Nicolaes G.A.F., Klomp J.P.G., de Vlieg J., Del Rio A. J. Chem. Inf. Model. 2012;52:1607–1620. doi: 10.1021/ci2005274. [DOI] [PubMed] [Google Scholar]
- 139.B-Rao C., Subramanian J., Sharma S.D. Drug Discov. Today. 2009;14:394–400. doi: 10.1016/j.drudis.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 140.Jiang F., Kim S.H. J. Mol. Biol. 1991;219:79–102. doi: 10.1016/0022-2836(91)90859-5. [DOI] [PubMed] [Google Scholar]
- 141.Claussen H., Buning C., Rarey M., Lengauer T. J. Mol. Biol. 2001;308:377–395. doi: 10.1006/jmbi.2001.4551. [DOI] [PubMed] [Google Scholar]
- 142.Knegtel R.M., Kuntz I.D., Oshiro C.M. J. Mol. Biol. 1997;266:424–440. doi: 10.1006/jmbi.1996.0776. [DOI] [PubMed] [Google Scholar]
- 143.Osterberg F., Morris G.M., Sanner M.F., Olson A.J., Goodsell D.S. Proteins. 2002;46:34–40. doi: 10.1002/prot.10028. [DOI] [PubMed] [Google Scholar]
- 144.Okimoto N., Futatsugi N., Fuji H., Suenaga A., Morimoto G., Yanai R., Ohno Y., Narumi T., Taiji M. PLoS Comput. Biol. 2009;5:e1000528. doi: 10.1371/journal.pcbi.1000528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Rastelli G. Pharm. Res. 2013;30:1458–1463. doi: 10.1007/s11095-013-1012-9. [DOI] [PubMed] [Google Scholar]
- 146.Amaro R., Baron R., McCammon J.A. J. Comput.-Aided Mol. Des. 2008;22:693–705. doi: 10.1007/s10822-007-9159-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Khanna M., Wang F., Jo I., Knabe W.E., Wilson S.M., Li L., Bum-Erdene K., Li J., Sledge G.W., Khanna R., Meroueh S.O. ACS Chem. Biol. 2011;6:1232–1243. doi: 10.1021/cb200180m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Homeyer N., Gohlke H. Mol. Inform. 2012;31:114–122. doi: 10.1002/minf.201100135. [DOI] [PubMed] [Google Scholar]
- 149.Wang L., Gu Q., Zheng X., Ye J., Liu Z., Li J., Hu X., Hagler A., Xu J. J. Chem. Inf. Model. 2013;53:2409–2422. doi: 10.1021/ci400322j. [DOI] [PubMed] [Google Scholar]
- 150.Zhao H., Huang D., Caflisch A. ChemMedChem. 2012;7:1983–1990. doi: 10.1002/cmdc.201200331. [DOI] [PubMed] [Google Scholar]
- 151.Xu L., Zhou S., Yu K., Gao B., Jiang H., Zhen X., Fu W. J. Chem. Inf. Model. 2013;53:3202–3211. doi: 10.1021/ci400481p. [DOI] [PubMed] [Google Scholar]
- 152.Hu X., Compton J.R., AbdulHameed M.D.M., Marchand C.L., Robertson K.L., Leary D.H., Jadhav A., Hershfield J.R., Wallqvist A., Friedlander A.M., Legler P.M. J. Med. Chem. 2013;56:5275–5287. doi: 10.1021/jm4001242. [DOI] [PubMed] [Google Scholar]
- 153.Rastelli G., Degliesposti G., Del Rio A., Sgobba M. Chem. Biol. Drug Des. 2009;73:283–286. doi: 10.1111/j.1747-0285.2009.00780.x. [DOI] [PubMed] [Google Scholar]
- 154.Wichapong K., Lawson M., Pianwanit S., Kokpol S., Sippl W. J. Chem. Inf. Model. 2010;50:1574–1588. doi: 10.1021/ci1002153. [DOI] [PubMed] [Google Scholar]
- 155.Lindström A., Edvinsson L., Johansson A., Andersson C.D., Andersson I.E., Raubacher F., Linusson A. J. Chem. Inf. Model. 2011;51:267–282. doi: 10.1021/ci100354x. [DOI] [PubMed] [Google Scholar]
- 156.Degliesposti G., Kasam V., Da Costa A., Kang H.-K., Kim N., Kim D.-W., Breton V., Kim D., Rastelli G. ChemMedChem. 2009;4:1164–1173. doi: 10.1002/cmdc.200900111. [DOI] [PubMed] [Google Scholar]
- 157.Wichapong K., Rohe A., Platzer C., Slynko I., Erdmann F., Schmidt M., Sippl W. J. Chem. Inf. Model. 2014;54:881–893. doi: 10.1021/ci4007326. [DOI] [PubMed] [Google Scholar]
- 158.Durrant J.D., McCammon J.A. BMC Biol. 2011;9:71. doi: 10.1186/1741-7007-9-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zhang X., Wong S.E., Lightstone F.C. J. Chem. Inf. Model. 2013;54:324–337. doi: 10.1021/ci4005145. [DOI] [PubMed] [Google Scholar]
- 160.Voet A., Zhang K.Y. Curr. Pharm. Des. 2012;18:4586–4598. doi: 10.2174/138161212802651616. [DOI] [PubMed] [Google Scholar]
- 161.Berg T. Curr. Opin. Drug Discov. Devel. 2008;11:666–674. [PubMed] [Google Scholar]
- 162.Fry D.C. Curr. Pharm. Des. 2012;18:4679–4684. doi: 10.2174/138161212802651634. [DOI] [PubMed] [Google Scholar]
- 163.Arkin M.R., Wells J.A. Nat. Rev. Drug Discov. 2004;3:301–317. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- 164.Voet A., Banwell E.F., Sahu K.K., Heddle J.G., Zhang K.Y. Curr. Top. Med. Chem. 2013;13:989–1001. doi: 10.2174/1568026611313090003. [DOI] [PubMed] [Google Scholar]
- 165.Christ F., Voet A., Marchand A., Nicolet S., Desimmie B.A., Marchand D., Bardiot D., Van der Veken N.J., Van Remoortel B., Strelkov S.V., De Maeyer M., Chaltin P., Debyser Z. Nat. Chem. Biol. 2010;6:442–448. doi: 10.1038/nchembio.370. [DOI] [PubMed] [Google Scholar]
- 166.De Luca L., Morreale F., Christ F., Debyser Z., Ferro S., Gitto R. Eur. J. Med. Chem. 2013;68:405–411. doi: 10.1016/j.ejmech.2013.07.025. [DOI] [PubMed] [Google Scholar]
- 167.De Luca L., Barreca M.L., Ferro S., Christ F., Iraci N., Gitto R., Monforte A.M., Debyser Z., Chimirri A. ChemMedChem. 2009;4:1311–1316. doi: 10.1002/cmdc.200900070. [DOI] [PubMed] [Google Scholar]
- 168.Hu G., Li X., Zhang X., Li Y., Ma L., Yang L.-M., Liu G., Li W., Huang J., Shen X., Hu L., Zheng Y.-T., Tang Y. J. Med. Chem. 2012;55:10108–10117. doi: 10.1021/jm301226a. [DOI] [PubMed] [Google Scholar]
- 169.Nikolovska-Coleska Z., Xu L., Hu Z., Tomita Y., Li P., Roller P.P., Wang R., Fang X., Guo R., Zhang M., Lippman M.E., Yang D., Wang S. J. Med. Chem. 2004;47:2430–2440. doi: 10.1021/jm030420+. [DOI] [PubMed] [Google Scholar]
- 170.Sun H.-P., Jiang Z.-Y., Zhang M.-Y., Lu M.-C., Yang T.-T., Pan Y., Huang H.-Z., Zhang X.-J., You Q.-d. MedChemComm. 2014;5:93–98. [Google Scholar]
- 171.Enyedy I.J., Ling Y., Nacro K., Tomita Y., Wu X., Cao Y., Guo R., Li B., Zhu X., Huang Y., Long Y.-Q., Roller P.P., Yang D., Wang S. J. Med. Chem. 2001;44:4313–4324. doi: 10.1021/jm010016f. [DOI] [PubMed] [Google Scholar]
- 172.Odolczyk N., Fritsch J., Norez C., Servel N., da Cunha M.F., Bitam S., Kupniewska A., Wiszniewski L., Colas J., Tarnowski K., Tondelier D., Roldan A., Saussereau E.L., Melin-Heschel P., Wieczorek G., Lukacs G.L., Dadlez M., Faure G., Herrmann H., Ollero M., Becq F., Zielenkiewicz P., Edelman A. EMBO Mol. Med. 2013;5:1484–1501. doi: 10.1002/emmm.201302699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Metz A., Schanda J., Grez M., Wichmann C., Gohlke H. J. Chem. Inf. Model. 2013;53:2197–2202. doi: 10.1021/ci400332e. [DOI] [PubMed] [Google Scholar]
- 174.Jordheim L.P., Barakat K.H., Heinrich-Balard L., Matera E.-L., Cros-Perrial E., Bouledrak K., El Sabeh R., Perez-Pineiro R., Wishart D.S., Cohen R., Tuszynski J., Dumontet C. Mol. Pharmacol. 2013;84:12–24. doi: 10.1124/mol.112.082347. [DOI] [PubMed] [Google Scholar]
- 175.Reddy T.R.K., Li C., Guo X., Myrvang H.K., Fischer P.M., Dekker L.V. J. Med. Chem. 2011;54:2080–2094. doi: 10.1021/jm101212e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Voet A.R.D., Ito A., Hirohama M., Matsuoka S., Tochio N., Kigawa T., Yoshida M., Zhang K.Y.J. MedChemComm, 2014;5:783–786. [Google Scholar]
- 177.Geppert T., Bauer S., Hiss J.A., Conrad E., Reutlinger M., Schneider P., Weisel M., Pfeiffer B., Altmann K.-H., Waibler Z., Schneider G. Angew. Chem. Int. Ed. 2012;51:258–261. doi: 10.1002/anie.201105901. [DOI] [PubMed] [Google Scholar]
- 178.Betzi S., Restouin A., Opi S., Arold S.T., Parrot I., Guerlesquin F., Morelli X., Collette Y. Proc. Natl. Acad. Sci. U.S.A. 2007;104:19256–19261. doi: 10.1073/pnas.0707130104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Czarna A., Beck B., Srivastava S., Popowicz G.M., Wolf S., Huang Y., Bista M., Holak T.A., Dömling A. Angew. Chem. Int. Ed. 2010;49:5352–5356. doi: 10.1002/anie.201001343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Thiel P., Roglin L., Meissner N., Hennig S., Kohlbacher O., Ottmann C. Chem. Commun. 2013;49:8468–8470. doi: 10.1039/c3cc44612c. [DOI] [PubMed] [Google Scholar]
- 181.Tintori C., Laurenzana I., Fallacara A.L., Kessler U., Pilger B., Stergiou L., Botta M. Bioorg. Med. Chem. Lett. 2014;24:280–282. doi: 10.1016/j.bmcl.2013.11.019. [DOI] [PubMed] [Google Scholar]
- 182.Friesland A., Zhao Y., Chen Y.-H., Wang L., Zhou H., Lu Q. Proc. Natl. Acad. Sci. U.S.A. 2013;110:1261–1266. doi: 10.1073/pnas.1116051110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Dhruv H., Loftus J.C., Narang P., Petit J.L., Fameree M., Burton J., Tchegho G., Chow D., Yin H., Al-Abed Y., Berens M.E., Tran N.L., Meurice N. J. Biol. Chem. 2013;288:32261–32276. doi: 10.1074/jbc.M113.493536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Bashford D., Case D.A. Annu. Rev. Phys. Chem. 2000;51:129–152. doi: 10.1146/annurev.physchem.51.1.129. [DOI] [PubMed] [Google Scholar]
- 185.Tsui V., Case D.A. Biopolymers. 2000;56:275–291. doi: 10.1002/1097-0282(2000)56:4<275::AID-BIP10024>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 186.Fredriksson R., Lagerström M.C., Lundin L.-G., Schiöth H.B. Mol. Pharmacol. 2003;63:1256–1272. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- 187.Kiss R., Kiss B., Könczöl Á., Szalai F., Jelinek I., László V., Noszál B., Falus A., Keserű G.M. J. Med. Chem. 2008;51:3145–3153. doi: 10.1021/jm7014777. [DOI] [PubMed] [Google Scholar]
- 188.Varady J., Wu X., Fang X., Min J., Hu Z., Levant B., Wang S. J. Med. Chem. 2003;46:4377–4392. doi: 10.1021/jm030085p. [DOI] [PubMed] [Google Scholar]
- 189.Becker O.M., Marantz Y., Shacham S., Inbal B., Heifetz A., Kalid O., Bar-Haim S., Warshaviak D., Fichman M., Noiman S. Proc. Natl. Acad. Sci. U.S.A. 2004;101:11304–11309. doi: 10.1073/pnas.0401862101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Engel S., Skoumbourdis A.P., Childress J., Neumann S., Deschamps J.R., Thomas C.J., Colson A.-O., Costanzi S., Gershengorn M.C. J. Am. Chem. Soc. 2008;130:5115–5123. doi: 10.1021/ja077620l. [DOI] [PubMed] [Google Scholar]
- 191.Evers A., Klabunde T. J. Med. Chem. 2005;48:1088–1097. doi: 10.1021/jm0491804. [DOI] [PubMed] [Google Scholar]
- 192.Yoshikawa Y., Oishi S., Kubo T., Tanahara N., Fujii N., Furuya T. J. Med. Chem. 2013;56:4236–4251. doi: 10.1021/jm400307y. [DOI] [PubMed] [Google Scholar]
- 193.van der Horst E., van der Pijl R., Mulder-Krieger T., Bender A., Ijzerman A.P. ChemMedChem. 2011;6:2302–2311. doi: 10.1002/cmdc.201100369. [DOI] [PubMed] [Google Scholar]
- 194.de Graaf C., Rein C., Piwnica D., Giordanetto F., Rognan D. ChemMedChem. 2011;6:2159–2169. doi: 10.1002/cmdc.201100317. [DOI] [PubMed] [Google Scholar]
- 195.Lin X., Huang X.-P., Chen G., Whaley R., Peng S., Wang Y., Zhang G., Wang S.X., Wang S., Roth B.L., Huang N. J. Med. Chem. 2012;55:5749–5759. doi: 10.1021/jm300338m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Evers A., Klebe G. J. Med. Chem. 2004;47:5381–5392. doi: 10.1021/jm0311487. [DOI] [PubMed] [Google Scholar]
- 197.Kellenberger E., Springael J.-Y., Parmentier M., Hachet-Haas M., Galzi J.-L., Rognan D. J. Med. Chem. 2007;50:1294–1303. doi: 10.1021/jm061389p. [DOI] [PubMed] [Google Scholar]
- 198.Katritch V., Cherezov V., Stevens R.C. Trends Pharmacol. Sci. 2012;33:17–27. doi: 10.1016/j.tips.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Venkatakrishnan A.J., Deupi X., Lebon G., Tate C.G., Schertler G.F., Babu M.M. Nature. 2013;494:185–194. doi: 10.1038/nature11896. [DOI] [PubMed] [Google Scholar]
- 200.de Graaf C., Kooistra A.J., Vischer H.F., Katritch V., Kuijer M., Shiroishi M., Iwata S., Shimamura T., Stevens R.C., de Esch I.J.P., Leurs R. J. Med. Chem. 2011;54:8195–8206. doi: 10.1021/jm2011589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Carlsson J., Yoo L., Gao Z.-G., Irwin J.J., Shoichet B.K., Jacobson K.A. J. Med. Chem. 2010;53:3748–3755. doi: 10.1021/jm100240h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Kolb P., Rosenbaum D.M., Irwin J.J., Fung J.J., Kobilka B.K., Shoichet B.K. Proc. Natl. Acad. Sci. U.S.A. 2009;106:6843–6848. doi: 10.1073/pnas.0812657106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Carlsson J., Coleman R.G., Setola V., Irwin J.J., Fan H., Schlessinger A., Sali A., Roth B.L., Shoichet B.K. Nat. Chem. Biol. 2011;7:769–778. doi: 10.1038/nchembio.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Sabio M., Jones K., Topiol S. Bioorg. Med. Chem. Lett. 2008;18:5391–5395. doi: 10.1016/j.bmcl.2008.09.046. [DOI] [PubMed] [Google Scholar]
- 205.Katritch V., Jaakola V.-P., Lane J.R., Lin J., Ijzerman A.P., Yeager M., Kufareva I., Stevens R.C., Abagyan R. J. Med. Chem. 2010;53:1799–1809. doi: 10.1021/jm901647p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Weiss D.R., Ahn S., Sassano M.F., Kleist A., Zhu X., Strachan R., Roth B.L., Lefkowitz R.J., Shoichet B.K. ACS Chem. Biol. 2013;8:1018–1026. doi: 10.1021/cb400103f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Mysinger M.M., Weiss D.R., Ziarek J.J., Gravel S., Doak A.K., Karpiak J., Heveker N., Shoichet B.K., Volkman B.F. Proc. Natl. Acad. Sci. U.S.A. 2012;109:5517–5522. doi: 10.1073/pnas.1120431109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Sanders M.P.A., Roumen L., van der Horst E., Lane J.R., Vischer H.F., van Offenbeek J., de Vries H., Verhoeven S., Chow K.Y., Verkaar F., Beukers M.W., McGuire R., Leurs R., Ijzerman A.P., de Vlieg J., de Esch I.J.P., Zaman G.J.R., Klomp J.P.G., Bender A., de Graaf C. J. Med. Chem. 2012;55:5311–5325. doi: 10.1021/jm300280e. [DOI] [PubMed] [Google Scholar]
- 209.Tresadern G., Bemporad D., Howe T. J. Mol. Graph. Model. 2009;27:860–870. doi: 10.1016/j.jmgm.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 210.Li G.-B., Yang L.-L., Feng S., Zhou J.-P., Huang Q., Xie H.-Z., Li L.-L., Yang S.-Y. Bioorg. Med. Chem. Lett. 2011;21:1736–1740. doi: 10.1016/j.bmcl.2011.01.087. [DOI] [PubMed] [Google Scholar]
- 211.Hollenstein K., Kean J., Bortolato A., Cheng R.K.Y., Dore A.S., Jazayeri A., Cooke R.M., Weir M., Marshall F.H. Nature. 2013;499:438–443. doi: 10.1038/nature12357. [DOI] [PubMed] [Google Scholar]
- 212.Siu F.Y., He M., de Graaf C., Han G.W., Yang D., Zhang Z., Zhou C., Xu Q., Wacker D., Joseph J.S., Liu W., Lau J., Cherezov V., Katritch V., Wang M.W., Stevens R.C. Nature. 2013;499:444–449. doi: 10.1038/nature12393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Wu H., Wang C., Gregory K.J., Han G.W., Cho H.P., Xia Y., Niswender C.M., Katritch V., Meiler J., Cherezov V., Conn P.J., Stevens R.C. Science. 2014;344:58–64. doi: 10.1126/science.1249489. [DOI] [PMC free article] [PubMed] [Google Scholar]