

Abstract
The spike (S) glycoprotein is one of the major structure proteins of SARS-associated coronavirus (CoV). Fragment 450–650 (S450-650) of the S protein contains receptor-binding domain and neutralizing epitopes. In this study, S450-650 was expressed with a histidine tag in Escherichia coli BL21. Bacterial inclusion bodies containing the recombinant S450-650 were solubilized with 8 M urea and then applied onto a Ni–nitrilotriacetic acid column. On-column refolding and purification was performed. Reduced glutathione and oxidized glutathione were included in the refolding buffer. In the wash and elution buffers, glycerol and glucose were necessary additives to prevent protein aggregation during purification. This refolding and purification procedure allowed production of S450-650 at up to 500 μg/ml in soluble form, which maintained appropriate antigenicity and immunogenicity. It was able to induce strong IgG responses in BALB/c mice. In Western blot assays, the recombinant S450-650 was recognized by monoclonal Ab against the His-tag and also sera from a convalescent SARS patient. S450-650-based ELISA system was able to detect anti-SARS-CoV IgG Abs in patient sera.
Keywords: SARS-coronavirus, Spike protein, On-column refolding
Severe acute respiratory syndrome (SARS) is caused by SARS-associated coronavirus (SARS-CoV), an enveloped, positive-stranded RNA virus of the Coronaviridae family [1], [2], [3]. The genome of SARS-CoV encodes several structural proteins including the spike (S) glycoprotein, nucleocapsid protein, membrane protein, and envelope protein [3]. Membrane fusion between SARS-CoV and the host cell is mediated by its S glycoprotein [4], [5], [6], [7], [8], [9], which is also the main target for neutralization Abs against SARS-CoV [5], [8]. The S protein consists of 1255 amino acid residues with approximately 25% homology to those of the other human CoVs [10], [11]. Expression of full-length, or fragments of, S protein in soluble form and relatively large quantity is desirable for study of viral invasion, construction of diagnostic kits, and potential application as subunit vaccines. Computational modeling and sequence analysis indicates that the S protein of SARS-CoV contains 14 disulfide bonds [12] and is highly hydrophobic (446 out of its 1255 amino acid residues are hydrophobic ones). Further studies (EMBOSS: antigenic program) revealed that residues 450–650 of the S glycoprotein (S450-650) of SARS-CoV are largely solvent accessible and contain dominant B cell epitopes. There is also evidence suggesting that fragment 485–625 might contain the receptor-binding domain and also neutralization epitopes [13]. In this study, we expressed histidine-tagged recombinant S450-650 in E. coli and purified the polypeptide using a rapid on-column refolding procedure.
Materials and methods
Reagents
High fidelity Taq DNA polymerase was purchased from TaKaRa Biotech (Japan). Restriction enzymes and T4 ligase were from Invitrogen (USA). The DNA extraction and purification kit was from Qiagen (Germany). pET-28a expression vector was from Novagen (Germany). The Ni–nitrilotriacetic acid agarose was from Novagen (Germany). Mouse anti-His monoclonal antibody (mAb) was purchased from BD Clontech (USA). Horseradish peroxidase (HRP) labeled goat anti-human IgG and anti-mouse IgG were obtained from Zhongshan Biotech (China). Complementary DNA encoding full-length S proteins of SARS-CoV was obtained from China CDC. Sera from convalescent SARS patients were provided by Beijing Red Cross Blood Center.
Construction of the pET28a-S450-650 expression vector
DNA coding for S450-650 was amplified using PCR with SARS-CoV S protein cDNA as template. The sequences of the primers employed in the PCR were 5′CGC GGA TCC ATG CCC TTT GAG AGA GAC ATA TCT 3′ (forward sequence containing a BamHI site) and 5′CCC GAA TTC TTA AAT GTC GCA CTC ATA AGA AGT G 3′ (reverse sequence containing an EcoRI site).
PCR was performed under the following conditions: 5 min at 95 °C for full denaturation, 30 s at 95 °C, 1 min at 50 °C, 1 min at 72 °C for 35 circles of amplification, and 10 min at 72 °C for an additional extension. The amplified product was digested with BamHI and EcoRI, and then ligated with cut vector, pET28a. The resulting plasmid was named pET28a-S450-650.
Expression, on-column refolding, and purification of S450-650
Freshly transformed Escherichia coli BL21 (DE3) cells harboring plasmid pET28a-S450-650 were cultured in 1 L of 2YT medium containing kanamycin (25 μg/ml) at 37 °C. When the cell density reached 0.8–1.0 (OD600), isopropyl-β-d-thiogalactopyranoside (IPTG, Sigma) was added to a final concentration of 0.1 mM, and the bacteria were cultured for a further 3.5 h at 37 °C. The culture was then harvested by centrifugation at 5,000g for 15 min at 4 °C. The cell pellet was suspended in 100 ml buffer A (Table 1 ). After sonication (4 s pulse, 4 s pause, 200 W for 50 times), the lysed cells were centrifuged at 5000g for 15 min at 4 °C and pellet was resuspended in 100 ml buffer B.
Table 1.
Buffers used for the refolding purification procedure
| Buffer A | 20 mM Tris–HCl, 500 mM NaCl, 5 mM imidazole, 5 mM β-mercaptoethanol, pH 7.9 |
| Buffer B | Buffer A + 0.5% Triton X-100 |
| Buffer C | Buffer A + 8 M urea |
| Buffer D | 20 mM Tris–HCl, 500 mM NaCl, 5 mM imidazole, 1 mM GSH, 0.1 mM GSSG, 20% glycerol, pH 7.9 |
| Buffer E | 20 mM Tris–HCl, 500 mM NaCl, 100 mM imidazole, 20% glycerol, 5% glucose, pH 7.9 |
| Buffer F | 20 mM Tris–HCl, 500 mM NaCl, 800 mM imidazole, 20% glycerol, 5% glucose, pH 7.9 |
The purified inclusion bodies were solubilized in 40 ml buffer C by incubating at 4 °C overnight. After centrifugation at 16,000g for 30 min, the supernatant was filtered through a 0.45 μm membrane and incubated with 4 ml Ni–nitrilotriacetic acid (NTA) agarose at 4 °C for 1 h. Then, the agarose was poured into a column and washed with 30 ml buffer C at a flow rate of 2 ml/min. A linear gradient urea buffer D between 8 and 0 M was applied. Finally, the column was washed with 10 ml buffer E, and the recombinant S450-650 was eluted with 15 ml buffer F. The flow rate was maintained at 1 ml/min throughout. The recombinant S450-650 was adjusted to proper concentration and dialyzed with PBS (pH 7.2) containing 20% glycerol, 5% glucose.
Immunization of animals
Female BALB/c mice of 6–8 weeks of age were obtained from Institute of Genetics and Developmental Biology Chinese Academy of Sciences, Beijing, China. The mice were immunized S.C. with S480-650 (50 μg/mouse) or PBS emulsified in complete Freund’s adjuvant (CFA, Sigma) at the base of the tail. Two more booster immunization was given on days 15 and 29 with Ag mixed with incomplete Freund’s adjuvant (IFA). Mouse blood was collected by tail bleeding on day 0, 7, 14, 21, 28, 35, 42, and the sera were kept at −80 °C until use.
Western blot assays
The nitro-cellulose membranes, on which the recombinant S450-650 protein bands had been transferred, were blocked at room temperature for 2 h with blocking solution (5% non-fat dried milk) and then incubated for 2 h at room temperature with the first antibody. After washes in Tris-buffered saline (TBS, pH 8.0) containing 0.05% Tween 20 (TBS-T), the membranes were incubated with HRP-labeled goat anti-human IgG or goat anti-mouse IgG. 3,3′-diaminobenzidine tetrahydrochloride (DAB, Sigma) was used to visualize the reaction.
ELISAs
ELISA plates were coated at 4 °C overnight with recombinant protein (2.5 pmol/well) resuspended in carbonate buffer (pH 9.6). The wells were then incubated with blocking solution (2% BSA in PBS) for 2 h at 37 °C. After four washes with PBS containing 0.05% Tween 20 (PBS-T), one hundred microliters of diluted sera was added and incubated for 90 min at 37 °C. After washes with PBS-T, the plates were then incubated with HRP-labeled goat anti-human IgG antibody for 1 h at 37 °C. OPD (ortho-phenylenediamine) substrate (100 μl/well) was added after five washes with PBS-T and incubated for 2 min at room temperature. Fifty microliters of 2 M H2SO4 solution was added to each well to stop the reaction, and the optical density (OD) was immediately read at 492 nm.
Results
Cloning and expression of S450-650 in E. coli
As illustrated in Fig. 1 , complementary DNA encoding S450-650 of the S protein of SARS-CoV was inserted into pET28a, a vector for His-tag fusion protein expression under the control of T7 RNA polymerase and lac operator [14], [15]. Following IPTG induction (0.1 mM, 37 °C), the recombinant protein was overexpressed in inclusion bodies of E. coli BL21 cells (Fig. 2 ), reaching approximate 40% of total bacterial proteins, as estimated by scanning imaging analysis of the SDS–PAGE gels (data not shown).
Fig. 1.
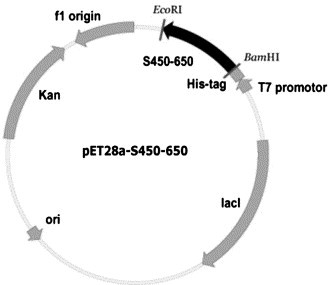
Recombinant plasmid pET28a-S450-650. S450-650 was introduced into pET28a, a T7 driven prokaryotic expression vector, in between BamHI and EcoRI sites.
Fig. 2.
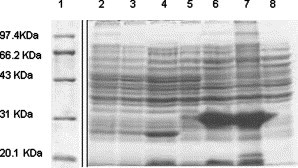
Expression of recombinant S450-650 in E. coli. Samples of different fractions of E. coli were run in a Coomassie blue-stained SDS–PAGE 12% gel, with protein molecular weight markers (M) in lane 1 and total cell lysate of E. coli BL21 in lane 2. Total lysates of cells harboring pET28a before and after IPTG induction were run in lanes 3 and 4, respectively; total lysates of cells harboring pET28a-S450-650 before and after IPTG induction were run in lanes 5 and 6; pellet and supernatant of the lysate of cells harboring pET28a-S450-650 after IPTG induction were in lanes 7 and 8.
Purification of the recombinant S450-650
Escherichia coli BL21 cells expressing S450-650 were harvested, washed with wash buffer containing β-mercaptoethanol and Triton X-100 (Table 1), and then subjected to several cycles of sonication and washes to obtain inclusion bodies, which were then lysed in lysis buffer containing 8 M urea and 5 mM β-mercaptoethanol (Table 1) overnight at 4 °C. After centrifugation, the supernatant was harvested and incubated with Ni resin for 1 h at 4 °C with continuous rotation. Most of the recombinant S450-650 in the supernatant bound to the column, as only a small amount of S450-650 was detected in the flow-through (Fig. 3 ).
Fig. 3.
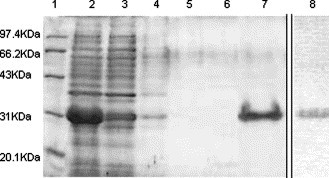
Purification of S450-650. Inclusion bodies from bacteria harboring pET28a-S450-650 were purified and lysed. A sample of the lysate of purified was run in lane 2. The supernatant was then mixed with Ni–nitrilotriacetic acid (NTA) resin for an hour at 4 °C with continuous rotation before packed into a column. A sample of the flow-through was run in lane 3. Lanes 4, 5, and 6 represent fractions of wash-through using buffers C, D, and E, respectively. Lane 7 represents eluate by buffer F and lane 8 represents the protein eluted by the conventional used buffer (20 mM Tris–HCl, 500 mM NaCl, and 800 mM imidazole). Standard protein marker was in lane 1.
Our on-column refolding and purification procedure was based on that reported by previous investigators [16], [17], [18], [19], [20], [21], [22], [23]. After the protein-bound column was pre-cleared with low concentration imidazole (Fig. 3), a linear gradient of urea in refolding buffer containing glycerol, reduced glutathione (GSH), and oxidized glutathione (GSSG) (Table 1) was applied, with flow rate held at 1 ml/min. The column was then washed with buffer E containing imidazole at 100 mM, eluting contaminant bacterial proteins but not the target protein (Fig. 3). Finally, the S450-650 protein was eluted using 800 mM imidazole containing glycerol and glucose (Table 1). The purity of the recombinant S450-650 protein thus obtained was approximately 95% as judged by SDS–PAGE analysis (Fig. 3). Inclusion of glycerol and glucose in the refolding buffer and elute buffers appeared to be crucial, as the yield and the concentration of S450-650 significantly decreased when buffers without glycerol and glucose were used (Fig. 3, Table 2 ). The purification and yield of S450-650 from E. coli are summarized in Table 2. The eluted protein was dialyzed in PBS (pH 7.2) containing glycerol (20%) and glucose (5%), and stored at 300 μg–500 μg/ml at −80 °C.
Table 2.
Purification of recombinant S450-650 protein from E. coli in 1 L culture
| Purification steps | Total proteins (mg)c | S450-650 (mg)d | Recovery rate (%)e |
|---|---|---|---|
| Cell lysate | 132 | 54 | 100 |
| Inclusion bodies | 106 | 47 | 87 |
| Eluatea | 17.9 | 17 | 31.5 |
| Eluateb | 1.4 | 1.3 | 2.4 |
With glycerol and glucose in refolding and elute buffers.
Without glycerol and glucose in refolding and elute buffers.
The amount of total proteins was determined with a Coomassie protein assay reagent (Pierce) using bovine γ-globulin as the standard protein.
The amount of recombinant S450-650 protein was estimated from the protein band intensity in SDS–PAGE gels by scanning imaging analysis.
Recovery rate = (recombinant S450-650 protein in each purification step)/(recombinant S450-650 protein in cell lysate) × 100%.
Characterization of the recombinant S450-650
In Western blot assays, the recombinant S450-650 was recognized by anti-His-tag mAb as well as convalescent serum from one of the two SARS patients (Fig. 4 ). The S450-650 polypeptide was also used as coating Ag in an ELISA system, which was able to detect S protein-specific IgG Abs in sera from convalescent SARS patients (Figs. 5 A and B). Subcutaneous immunization of BALB/c mice with S450-650 induced very strong IgG response against the immunizing Ag, as determined in ELISAs (Fig. 6).
Fig. 4.
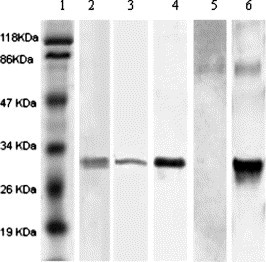
Western blot assay of the recombinant S450-650. Affinity purified S450-650 was run in two identical 12% SDS–PAGE gels with pre-stained protein molecular weight markers (M) in lane 1. One of the gels was stained with Coomassie blue (lane 2), and the other transferred onto cellulose nitrite membrane. WB was performed using mouse anti-His mAb (lane 3), serum from PT31 (lane 4) or PT32 (lane 5) or mice immunized with S450-650 (lane 6) was used as the first Ab. The secondary Ab was HRP-conjugated goat-anti-human IgG (lanes 4 and 5) or goat-anti-mouse IgG (lanes 3 and 6).
Fig. 5.
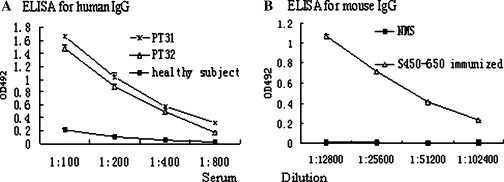
Sensitivity of the S450-650-based ELISA. (A) Serum samples from two convalescent SARS patients (PT31 × and PT32 ▵) and a healthy individual (Cont. ■) were serial diluted and assayed for IgG Abs in S450-650-based ELISA. (B) Serial diluted sera from mice immunized with S450-650 (▴) checked and PBS were used as a control (■).
Fig. 6.
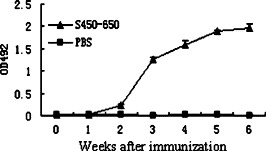
Time course of specific Ab production in S450-650-immunized mice. Groups of BALB/c mice were s.c. immunized with recombinant S450-650 (▴) or PBS (■) emulsified in CFA and bled at different time points thereafter. Abs in mouse sera (diluted 1:800) were determined using S450-650-based ELISA with HRP-labeled goat anti-mouse IgG as secondary Ab. The results are expressed as absorbance reading at 492 nm wavelength.
Discussion
The predicted molecular weight of the S450-650 recombinant protein is 25.8 kDa (total 201 + 38 amino acid residues). However, it appears to be 31 kDa in SDS–PAGE gels (Fig. 2, Fig. 3). We reckon this is because so the S450-650 protein contains 20 acidic residues (D, E) with a predicted iso-electric point of 4.3. It has been demonstrated that strongly acidic proteins behave abnormally in SDS-gels since the acidic amino acids do not bind SDS [24].
Solubility of the recombinant S450-650 was very poor, it was mainly expressed in inclusion bodies of the bacterial cells even at 20 °C with different IPTG concentrations (data not shown). This is probably due to the fact that S450-650 contains 64 hydrophobic amino acid residues (31.8% of the sequence). To obtain, reasonably high concentration S450-650 in soluble form, we adopted an on-column refolding and purification procedure. β-Mercaptoethanol and Triton X-100 were included in the inclusion body wash buffer and lysis buffer (Table 1), which help to solubilize hydrophobic proteins, reduce inter-chain disulfide bonds, and minimize non-specific binding of proteins to Ni column [16]. Glycerol and glucose were included in the refolding and elution buffers (Table 1). As powerful cosolvents, they are able to enhance native protein stability [25], [26], [27], [28], [29], [30], [31]. GSH/GSSG (1 mM GSH, 0.1 mM GSSG) were also helpful additives, because of the fact that the oxidizing conditions maintained by GSH/GSSG promote correct formation of disulfide bonds and dramatically increase the recombinant protein yield [20]. Yin et al. [16] showed that flow rate of the refolding buffer is a factor affecting protein refolding, whick was maintained at 1 ml/min in our experiments.
Apparently, our recombinant S450-650 maintained appropriate antigenicity and immunogenicity. When BALB/c mice were immunized with S450-650, a very strong IgG response was observed (Fig. 6 ). In ELISAs and Western blot assays, the S450-650 recombinant protein was recognized by serum Abs from convalescent SARS patients (Fig. 4, Fig. 5). It is of interest to note that sera from patient PT32 recognized the S450-650 polypeptide in ELISAs but not Western blot assays (Fig. 4, Fig. 5A). It is highly likely that Abs in PT32 serum were specific mainly for conformational rather than linear epitope(s) in the S450-650 fragment. In ELISAs, Abs can recognize native (undenatured and non-reduced) Ags, whilst in Western blot assays they can only react with Ags in denatured and reduced form. It can therefore be concluded that a certain proportion of the molecules of our recombinant S450-650 polypeptide refolded correctly following our on-column refolding procedure.
Acknowledgments
This study was supported by grants from the National Key Basic Research Programs (2001CB510007, 2003CB514109), National High Technology Research & Development Program (2003AA208412A) and “211” Program of China. Patient’s consent was obtained before blood collection.
Contributor Information
Jin-Cun Zhao, Email: zhaojincun@hotmail.com.
Zhen-Dong Zhao, Email: zhaoz@bjmu.edu.cn.
References
- 1.Rota P.A., Oberste M.S., Monroe S.S. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 2.Peiris J.S., Lai S.T., Poon L.L. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet. 2003;361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marra M., Jones S., Astell C.R. The genome sequence of the SARS-associated coronavirus. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 4.Li W., Moore M.J., Vasilieva N., Sui J.H., Wong S.K., Berne M.A., Somasundaran M., Sullivan J.L., Luzuriaga K., Greenough T.C., Choe H., Farzan M. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xiao X., Chakraborti S., Dimitrov A.S., Gramatikoff K., Dimitrov D.S. The SARS-CoV S glycoprotein: expression and functional characterization. Biochem. Biophys. Res. Commun. 2003;312(4):1159–1164. doi: 10.1016/j.bbrc.2003.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dimitrov D.S. The secret life of ACE2 as a receptor for the SARS virus. Cell. 2003;115(6):652–653. doi: 10.1016/S0092-8674(03)00976-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wong S.K., Li W., Moore M.J., Choe H., Farzan M. A 193-amino acid fragment of the SARS coronavirus S protein efficiently binds angiotensin-converting enzyme 2. J. Biol. Chem. 2004;279(5):3197–3201. doi: 10.1074/jbc.C300520200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang P., Chen J., Zheng A. Expression cloning of functional receptor used by SARS coronavirus. Biochem. Biophys. Res. Commun. 2003;312:1159–1164. doi: 10.1016/j.bbrc.2004.01.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sui J., Li W., Murakami A., Tamin A., Matthews L.J. Potent neutralization of severe acute respiratory syndrome (SARS) coronavirus by a human mAb to S1 protein that blocks receptor association. Proc. Natl. Acad. Sci. USA. 2004;101(8):2536–2541. doi: 10.1073/pnas.0307140101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ho T.Y., Wu S.L., Cheng S.E., Wei Y.C., Huang S.P., Hsiang C.Y. Antigenicity and receptor-binding ability of recombinant SARS coronavirus spike protein. Biochem. Biophys. Res. Commun. 2004;313(4):938–947. doi: 10.1016/j.bbrc.2003.11.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krokhin O., Li Y., Andonov A., Feldmann H., Flick R. Standing KG. Mass spectrometric characterization of proteins from the SARS virus: a preliminary report. Mol. Cell Proteomics. 2003;2(5):346–356. doi: 10.1074/mcp.M300048-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spiga O., Bernini A., Ciutti A. Molecular modeling of S1 and S2 subunits of SARS coronavirus spike glycoprotein. Biochem. Biophys. Res. Commun. 2003;310:78–83. doi: 10.1016/j.bbrc.2003.08.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou T., Wang H., Luo D. An exposed domain in the severe acute respiratory syndrome coronavirus spike protein induces neutralizing antibodies. J. Virol. 2004;78:7217–7226. doi: 10.1128/JVI.78.13.7217-7226.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Studier F.W., Moffatt B.A. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 1986;189:113–130. doi: 10.1016/0022-2836(86)90385-2. [DOI] [PubMed] [Google Scholar]
- 15.Lee M.H., Park T.I., Park Y.B., Kwak J.W. Bacterial expression and in vitro refolding of a single-chain Fv antibody specific for human plasma apolipoprotein B-100. Protein Expr. Purif. 2002;25:166–173. doi: 10.1006/prep.2002.1623. [DOI] [PubMed] [Google Scholar]
- 16.Yin S.M., Zheng Y., Tien P. On-column purification and refolding of recombinant bovine prion protein: using its octarepeat sequences as a natural affinity tag. Protein Expr. Purif. 2003;31:104–109. doi: 10.1016/S1046-5928(03)00195-5. [DOI] [PubMed] [Google Scholar]
- 17.Matsumoto M., Misawa S., Tsumoto K. On-column refolding and characterization of soluble human interleukin-15 receptor α-chain produced in Escherichia coli. Protein Expr. Purif. 2003;31:64–71. doi: 10.1016/s1046-5928(03)00143-8. [DOI] [PubMed] [Google Scholar]
- 18.Lemercier G., Bakalara N., Santarelli X. On-column refolding of an insoluble tag recombinant exopolyphosphatase from Trypanosoma brucei overexpressed in Escherichia coli. J. Chromatogr. B. 2003;786:305–309. doi: 10.1016/s1570-0232(02)00745-6. [DOI] [PubMed] [Google Scholar]
- 19.Shi Y., Jiang C., Chen Q., Tang H. One-step on-column affinity refolding purification and functional analysis of recombinant human VDAC1. Biochem. Biophys. Res. Commun. 2003;303:475–482. doi: 10.1016/s0006-291x(03)00359-0. [DOI] [PubMed] [Google Scholar]
- 20.Guo J.Q., You S.Y., Li Le. Construction and high-level expression of a single-chain Fv antibody fragment specific for acidic isoferritin in Escherichia coli. J. Biotechnol. 2003;102:177–189. doi: 10.1016/s0168-1656(03)00020-8. [DOI] [PubMed] [Google Scholar]
- 21.Li M., SU Zhi-Guo, Janson Jan-Christer. In vitro protein refolding by chromatographic procedures. Protein Expr. Purif. 2004;33:1–10. doi: 10.1016/j.pep.2003.08.023. [DOI] [PubMed] [Google Scholar]
- 22.Schauer S., Lüer C., Moser J. Large scale production of biologically active Escherichia coli glutamyl-tRNA reductase from inclusion bodies. Protein Expr. Purif. 2003;31:271–275. doi: 10.1016/s1046-5928(03)00184-0. [DOI] [PubMed] [Google Scholar]
- 23.Gulnik S.V., Afonina E.I., Gustchina E. Utility of (His)6 Tag for purification and refolding of proplasmepsin-2 and mutants with altered activation properties. Protein Expr. Purif. 2002;24:412–419. doi: 10.1006/prep.2001.1590. [DOI] [PubMed] [Google Scholar]
- 24.R. Westermeier, Section 1, 1, Electrophoresis, Electrophoresis in Practice, second ed., Weinhein: VCH 1997, pp. 35
- 25.Bondos S.E., Bicknell A. Detection and prevention of protein aggregation before, during, and after purification. Anal. Biochem. 2003;316:223–231. doi: 10.1016/s0003-2697(03)00059-9. [DOI] [PubMed] [Google Scholar]
- 26.Clark E.D.B. Refolding of recombinant proteins. Curr. Opin. Biotechnol. 1998;9:157–163. doi: 10.1016/s0958-1669(98)80109-2. [DOI] [PubMed] [Google Scholar]
- 27.Arakawa T., Timasheff S.N. Stabilization of protein structure by sugars. Biochemistry. 1982;21:6536–6544. doi: 10.1021/bi00268a033. [DOI] [PubMed] [Google Scholar]
- 28.Timasheff S.N. Control of protein stability and reactions by weakly interacting cosolvents: the simplicity of the complicated. Adv. Protein Chem. 1998;51:355–432. doi: 10.1016/s0065-3233(08)60656-7. [DOI] [PubMed] [Google Scholar]
- 29.Collins K., Washabaugh M.W. The Hofmeister effect and the behavior of water at interafaces. Q. Rev. Biophys. 1985;18:323–422. doi: 10.1017/s0033583500005369. [DOI] [PubMed] [Google Scholar]
- 30.Xie G., Timasheff S.N. Mechanisms of the stabilization of ribonuclease A by sorbitol: preferential hydration is great for the denatured than for the native protein. Protein Sci. 1997;6:211–221. doi: 10.1002/pro.5560060123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xie G., Timasheff S.N. Temperature dependence of the preferential interactions of ribonuclease A in aqueous cosolvent systems: thermodynamic analysis. Protein Sci. 1997;6:222–232. doi: 10.1002/pro.5560060124. [DOI] [PMC free article] [PubMed] [Google Scholar]