

Abstract
Site-specific proteases are the most popular kind of enzymes for removing the fusion tags from fused target proteins. Nuclear inclusion protein a (NIa) proteases obtained from the family Potyviridae have become promising due to their high activities and stringencies of sequences recognition. NIa proteases from tobacco etch virus (TEV) and tomato vein mottling virus (TVMV) have been shown to process recombinant proteins successfully in vitro. In this report, recombinant PPV (plum pox virus) NIa protease was employed to process fusion proteins with artificial cleavage site in vitro. Characteristics such as catalytic ability and affecting factors (salt, temperature, protease inhibitors, detergents, and denaturing reagents) were investigated. Recombinant PPV NIa protease expressed and purified from Escherichia coli demonstrated efficient and specific processing of recombinant GFP and SARS-CoV nucleocapsid protein, with site F (N V V V H Q▾A) for PPV NIa protease artificially inserted between the fusion tags and the target proteins. Its catalytic capability is similar to those of TVMV and TEV NIa protease. Recombinant PPV NIa protease reached its maximal proteolytic activity at approximately 30 °C. Salt concentration and only one of the tested protease inhibitors had minor influences on the proteolytic activity of PPV NIa protease. Recombinant PPV NIa protease was resistant to self-lysis for at least five days.
Keywords: PPV NIa protease, Fusion protein, Fusion tag cleavage
The application of recombinant proteins is well-accepted approach for producing useful protein products such as industrial material and therapeutic proteins. Fusion tags have been very helpful, not only for the purpose of detection and purification of target proteins, but also for the purpose of improving the solubility of target proteins in different bioreactors (prokaryotic, yeast, animal, plant, and virus systems) [1], [2], [3], [4]. Examples of such tags are hexahistidine [5], streptavidin [6], glutathione-S-transferase (GST) 1 [7], and maltose-binding protein (MBP) [8], Flag [9], tandem affinity purification (TAP) [10], thioredoxin (Trx) [11]. However, the fusion tags may interfere with the structures and functions of the target proteins. It is preferable that fusion tags can be removed after purification and several strategies have been applied for this task [4]. Cleaving at a specifically recognized sequence that has been inserted between the tag and the target protein by a specific protease is a convenient way to eliminate the fusion tag. The commonly used proteases include factor Xa, thrombin, enterokinase, and tobacco etch virus (TEV) NIa protease, although each protease has its own advantages and disadvantages. Factor Xa can recognize and cleave at the sequence of Ile-Glu/Asp-Gly-Arg▾, which naturally converts prothrombin to thrombin [12]. However, under certain circumstances, factor Xa may cleave at non-specific sequences besides the predicted sites [13], [14]. Additionally, factor Xa may be disabled by adjacent hydrophobic sequences flanked on the C-terminal side of the potential site [15]. The optimal cleavage sites for thrombin have the following structures: (a) P4-P3-Pro-Arg▾P1′-P2′, where P3 and P4 are hydrophobic amino acid and P1′, P2′ are nonacidic amino acids; (b) P2-Arg▾P1′, where P2 or P1′ are Gly [16]. However, Chang et al. found out that thrombin can cleave at the lysine residues in the sequences Met-Lys▾Ser-Arg-Asn-Leu, Arg-Cys-Lys▾Pro-Val-Asn, and Ser-Ser-Lys▾Tyr-Pro-Asn in performic acid-oxidized ribonuclease when studied the action of thrombin on 30 polypeptide hormones [16]. Due to its high stringency of sequence recognition, TEV NIa protease has been frequently used in the recent time. The specificity of TEV NIa protease has been well investigated [17], [18], [19], [20]. The preferred site for TEV NIa protease is considered to be site F (P6)E (P5)N (P4)L (P3)Y (P2)F (P1)Q▾(P1′)G/S, in which P2, P4, and P5 positions may be non-conserved amino acids and P1′ can be any amino acid except Pro [21], [22]. Recombinant TEV NIa protease has been used for removing several fusion tags in vitro, processing on artificial site F efficiently [23], [24], [25], [26]. However, the wild-type TEV NIa protease may degrade into a truncated form with severely diminished enzymatic activity [27], which temporarily limited its range of application. Alternatively, TEV NIa protease mutants S219V or S219D, which are resistant against self-inactivation while maintaining enzymatic activity, have eliminated this limitation [20], [28].
TEV, TVMV, and PPV are members of the Potyviridae family and NIa proteases obtained from potyviruses have similar structures and functions [29], [30], [31]. The potyvirus genome is a (+) stranded RNA; it is approximately 10 kb in length with the VPg and poly(A) at 5′ and 3′ positions, respectively [32]. It is translated into a single polyprotein upon infection, which is processed by the virally encoded proteases P1, HC-pro, and NIa. Most of the cleavage events are performed by NIa (nuclear inclusion protein a) protease. NIa protease processes seven sites present in the potyvirus polyprotein, named as A, B, C, D, V, E, and F [33]. Different sites are cleaved at varied rates and efficiencies by NIa protease; therefore leading to the intermediate products with different functions [32], [34]. Analyses of TVMV and TEV NIa protease had shown that site F between NIb-Capsid protein in the potyvirus polyprotein was the best substrate for TVMV NIa protease [35] or highly preferred substrate for TEV NIa protease [17]. The potyvirus NIa protease has a His-Asp-Cys catalytic triad, which is homologous to the trypsin-like serine proteases except for Cys replacing Ser [36]. NIa proteases obtained from PPV, TVMV, and TEV share certain sequence identities (Fig. 1 ); however, they recognize distinct amino acid sequences at each recognition sites. Consequently, they cannot recognize the cleavage sites of each other efficiently [21], [31], [34]. This offers the possibility for adopting other NIa proteases when the fusion proteins happen to contain sequences similar to recognition sites of TEV NIa protease. Recombinant TVMV NIa protease was expressed in Escherichia coli and it can cleave at artificial site F between fusion tags and target proteins with high activity and sequence stringency, which is comparable to that of TEV NIa protease. Similar to the behavior of TEV NIa protease, the catalytic activity of TVMV NIa protease decreased at high salt concentration and low temperature. Nevertheless, TVMV NIa protease did not show self-degradation that was detected in wild-type TEV NIa protease [29]. It has been reported that PPV NIa protease recognized and processed natural and artificial site F efficiently within the E. coli expression system [37], [38]. Recombinant PPV NIa protease purified from E. coli can cleave at canonical cleavage site F (NIb-CP junction) in vitro [39]. These experiments suggested that PPV NIa protease has potential for removing fusion tags in vitro when site F (P6)N (P5)V (P4)V (P3)V (P2)H (P1)Q▾(P1′)A for PPV NIa protease is inserted between fusion tags and target proteins.
Fig. 1.

Amino acid sequences alignment for TEV, TVMV, and PPV NIa proteins (both the VPg and the NIa protease domains). The alignment was performed using the software DNAMAN, The catalytic triad His46–Asp81–Cys151 occurs at the same amino acid positions of the PPV, TEV, and TVMV NIa protease domain (indicated by  ).
).
This study mainly focused on the efficient and specific processing of fusion proteins by recombinant PPV NIa protease in vitro. A detailed evaluation of the efficiency, stability, and affecting factors of the purified protease was performed. We used tagged versions of GFP and SARS-coronavirus nucleocapsid protein as substrates, with the cleavage site F for PPV NIa protease inserted between fusion tags and target proteins. Both the protease and the fused target proteins were expressed in the E. coli cells and purified by Ni–NTA affinity chromatography.
Material and methods
Construction of expression vectors pET-P, pET-sN, and pET-sG
The potyvirus NIa protein contains the following two domains: the VPg domain at the N-terminus and the NIa protease domain at the C-terminus. The DNA sequence coding for the protease domain was amplified by PCR using plasmid pGGNIa as template. Plasmid pGGNIa contains the partial cDNA sequence of plum pox virus (Rankovic strain). Recognition site of the restriction endonuclease SacI was added to the 5′ end of primer 1 (5′-CAgagctcAGTAAATCACTGTTCAGAGG-3′) and HindIII site was added to the 5′ end of primer 2 (5′-CTaagcttGTGAGTGTAAACAAATTCCC-3′). This fragment was digested with SacI and HindIII, and subsequently inserted into the SacI–HindIII-treated pET-32a (+) (Novagen) vector. The consequential plasmid was referred to as pET-P, which contained the NIa protease domain coding sequence.
A BAC clone containing the cDNA of SARS-CoV (Urbani strain) was used to amplify the coding sequence for SARS-CoV nucleocapsid (N) protein by PCR. Primer 3 (5′-AAgcatgcgtcgacATGTCTGATAATGGACC-3′) and primer 4 (5′-ATacgcgtTTATGCCTGAGTTGAATC-3′) were used. Nucleotides gcatgc and gtcgac in primer 3 represent the recognition sequences for the restriction enzymes SphI and SalI, respectively, while the nucleotide acgcgt in primer 4 is an NcoI recognition site. The PCR product was subcloned into pGEM-T easy vector (Promega). This plasmid was referred to as pGEM-T-N. Oligodeoxynucleotides containing the site F for PPV NIa protease sequence (+) 5′-CGGTACCAACGTTGTTGTGCACCAAGCTGACGAACTGCAGCCATGGC-3′ and (−) 5′-TCGAGCCATGGCTGCAGTTCGTCAGCTTGGTGCACAACAACGTTGGTACCGCATG-3′ were synthesized and annealed, and subsequently inserted between the SphI–SalI digested pGEM-T-N vector, thereby resulting in a new plasmid pGEM-T-sN. The KpnI–SalI fragment obtained from pGEM-T-sN that contained the coding sequence for N protein was inserted into the KpnI–SalI-digested pET-32a (+) vector, thereby yielding the plasmid pET-sN. For the construction of pET-sG, primer 5 (5′-AGccatgggagctcATGGGTAAGGGAGAAGAACTT-3′) and primer 6 (5′-GGactgtgagctcTTATTTGTATAGTTCATCCAT-3′) were used to perform PCR amplification of the GFP gene obtained from pGFP-2 plasmid, which contains the GFP gene. This amplified fragment was digested with NcoI and SpeI and substituted in plasmid pET-sN for the NcoI–SpeI fragment that contained the coding sequence for N protein.
All these constructs were confirmed by restriction enzymes mapping and sequencing. The final constructs pET-P, pET-sG, and pET-sN were used to transform the competent cells of E. coli strain BL21AI. The recombinant clones were selected on LB agar plates supplemented with ampicillin (100 μg/mL) and confirmed by restriction enzymes mapping.
Expression in E. coli and purification of recombinant protease and substrates
The E. coli BL21AI cells carrying different constructs were grown overnight in LB medium supplemented with 100 μg/mL ampicillin at 37 °C. The overnight culture was diluted with LB to generate a start absorbance of 0.1 and was subsequently shaken at 18 °C. When the culture reached the mid-log phase (A 600 nm = 0.5), arabinose (final concentration of 0.1%) was added into the medium. An hour later, isopropyl-β-d-thiogalactopyranoside (IPTG) was added with a final concentration of 0.25 mM, and the incubation was prolonged for an additional 12–16 h. Under these conditions, high expression levels of protease and substrates were observed. The cells were harvested by centrifugation, washed extensively with PBS (137 mM NaCl, 2 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4, pH 7.4), and frozen at −20 °C. The cell pellets were resuspended and homogenized in ice-cold lysis buffer (50 mM NaH2PO4, 300 mM NaCl, pH 8.0) containing protease inhibitors (1 mM PMSF, 1 mg/mL benzamidine, 2 μg/mL aprotinin, and 1 μM pepstatin A), then disrupted by French Press. The homogenates were centrifuged at 12,000 rpm (CR 22G, Himac) at 4 °C for 45 min to remove the cell debris. The 6× His tag-fused proteins were subsequently purified with Ni–NTA agarose column (Cat. No. 30210, QIAGEN) according to the manufacturers instructions. The pure 6× His tag-fused proteins were eluted with a step-wise imidazole concentration gradient (100–500 mM). The eluted recombinant proteins were further concentrated and desalted by using an Amicon Ultra-15 membrane (Millipore). Purified PPV NIa protease was resuspended in 15% glycerol till a concentration of 1 mg/ml was achieved and then stored in aliquots at −80 °C. The substrates were resuspended in PBS and stored in aliquots at −80 °C. Protein concentrations were estimated by Bradford method and compared with the standard concentration of BSA (Bradford Protein Assay kit, Beyontime, China).
Assay of protease activity on NIb-CP protein synthesized in vitro
Behind the initiation codon ATG and the first 30 nucleotides of the PPV P3 cistron, the pGGP3ΔI plasmid contains the sequence coding for most part of the NIb protein and the complete capsid protein of PPV. This cDNA insert was downstream of a truncated T7 promoter. The TNT Coupled Reticulocyte Lysate System (Promega) was applied to perform in vitro transcription and translation of the PPV sequence in pGGP3ΔI in the presence of 35S-labeled Met and Cys. Pretreatment of the plasmid involved digestion with protease K, extraction with phenol–chloroform, and precipitation with ethanol; subsequently, it was resuspended in 20 μl nuclease-free water. Four microliters of translated products and 0.5 μg recombinant PPV NIa protease were incubated in 15 μl reaction buffer (20 mM HEPES, pH 7.5, 10 mM MgCl2, 10 mM KCl, and 1 mM DTT) based on the previous report [40]. The reaction was carried out at 20 or 30 °C for 5–10 min and terminated by adding 4× SDS–PAGE sample buffer. The samples were heated at 95 °C for 10 min, resolved by SDS–PAGE electrophoresis, and subsequently detected by 35S autoradiography.
PPV NIa protease processing of fusion proteins
One microgram of the recombinant PPV NIa protease was incubated with varied amounts of sG or sN at 30 °C in the reaction buffer. The reaction was terminated with 4× SDS–PAGE sample buffer and heated at 95 °C for 10 min. The samples were analyzed by SDS–PAGE and stained with Coomassie brilliant blue R-250 (CBB R-250).
Effects of temperature, salt concentration, protease inhibitors, and chemical reagents on recombinant PPV NIa proteases proteolytic activity
In order to analyze the effect of temperature on the enzymatic activity of recombinant PPV NIa protease, 50 μg of sG and 1 μg of recombinant PPV NIa protease were mixed in the reaction buffer, and incubated at 4, 20, 30, and 40 °C for up to 10 h. Additionally, the effect of salt concentration on the enzymatic activity of PPV NIa protease was examined. Fifty micrograms of sG and 1 μg recombinant PPV NIa protease were incubated in the reaction buffer mentioned above containing additional NaCl at increasing concentrations (50, 100, 200, 300, and 500 mM) at 30 °C for up to 6 h.
In order to assess the effect of protease inhibitors on the enzymatic activity of PPV NIa protease, 50 μg of sG substrate and 1 μg recombinant PPV NIa protease were incubated in the reaction buffer containing different commercial inhibitors of serine-, aspartic-, cysteine-, and metallo-proteases at 30 °C for 2 h. Those protease inhibitors are 1 mM PMSF (serine-, cysteine-, and metallo-protease inhibitors), 5 mM benzamidine (serine-proteases inhibitor), 0.1 mM pepstatin A (aspartic proteases inhibitor), 200 μg/ml aprotinin–dihydrochloride (serine proteases and esterase inhibitor), 1 mM leupeptin (cysteine- and serine-protease inhibitor), 1 mM antipain (cysteine- and serine-protease inhibitor), and 50 mM EDTA-Na2 (metallo proteases inhibitor). The effect of some denaturing reagents or detergents on the activity of PPV NIa protease was also analyzed. Eighty-five micrograms of sG substrate and 1 μg of recombinant PPV NIa protease were incubated at 30 °C for 3 h in the reaction mixture supplemented with different concentrations of urea, Triton X-100, and SDS. In all cases, the samples were analyzed by using SDS–PAGE and Coomassie blue staining as described above. Three independent experiments were conducted to obtain the numerical estimates of the fraction of the cleaved substrates at each time point. The Coomassie-stained gels were scanned with a Uniscan C1080 scanner (THUNIS), and the pixel densities of the bands were quantified by using the ImageQuant TL v2005 software (GE health). The average values and the standard errors were generated by Microsoft Excel. The kinetic parameters of recombinant PPV NIa protease were estimated by Origin 7.5 software (OriginLab).
Self-inactivation test of PPV NIa protease
One microgram of the recombinant PPV NIa proteases was incubated in the reaction buffer at 4 and 20 °C for up to 120 h. Aliquots were collected at different incubation times and analyzed by SDS–PAGE and Coomassie blue staining as explained above.
Results
Expression in E. coli and purification of recombinant PPV NIa protease and substrates
The NIa protease domain in the PPV NIa protein was selected for the purpose of evaluating its enzymatic activity. The cDNA coding region for the protease domain was cloned into pET-32a (+) expression vector, thereby resulting in the formation of pET-P vector (Fig. 2 a). pET-32a (+) expression vector provides Trx tag, 6× His tag, and S tag at the amino terminus and 6 × His tag at the carboxyl terminus for recombinant PPV NIa protease. The bacteria harboring this expression plasmid were induced with IPTG at a temperature of 18 °C and produced recombinant PPV NIa protease of 47 kDa (Fig. 2c, indicated as rProtease). No extra or degraded bands were observed for recombinant PPV NIa protease. Most of the recombinant PPV NIa protease was eluted by 250–500 mM imidazole. The recombinant PPV NIa protease was largely soluble and purified by using nickel affinity chromatography. Approximately 20 mg soluble recombinant PPV NIa protease was obtained from 1 liter of bacteria culture.
Fig. 2.
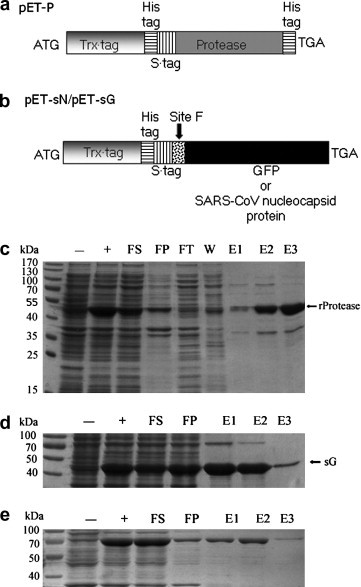
Expression and purification of recombinant PPV NIa protease and substrates. (a) Schematic representation of the expression vector pET-P for recombinant PPV NIa protease. (b) Schematic representation of the expression vectors pET-sN and pET-sG for recombinant substrates. (c) Expression and purification of recombinant PPV NIa protease. (d) Expression and purification of recombinant substrate sG. (e) Expression and purification of recombinant substrate sN. Each fraction corresponds to 0.2–0.5 ml cell culture (A600 nm = 0.5). The abbreviation are explained as following: −, before induction; +, after induction; FS, supernatant after French press lysis; FP, pellet after French press lysis; FT, flow through; W, Wash through; E1, elution with 100 mM imidazole; E2, elution with 250 mM imidazole; E3, elution with 500 mM imidazole.
Fused versions of GFP and SARS-CoV N (indicated as sG and sN) protein were expressed in E. coli as substrates for recombinant PPV NIa protease. A linker containing the most efficient recognition site F (N V V V H Q▾A) for PPV NIa protease was inserted between the N-terminal fusion tags and the C-terminal GFP or SARS-CoV N protein (Fig. 2b). Since the amino acids adjacent to the recognition site may interfere with the cleaving ability of the protease [34], the two original amino acids Asp, Glu behind Ala were incorporated in the linker to optimizing the cleavage efficiency. S tag, Trx tag, and 6× His tag in front of GFP or SARS-CoV N protein can be used to facilitate purification. Soluble 45 kDa sG protein and 64 kDa sN protein accumulated to high levels in the transformed bacteria and were easily purified by nickel affinity chromatography. The yields of purified sG and sN proteins from E. coli were 80 and 30 mg/L, respectively (Fig. 2d and e).
Primary test of NIa protease activity on a PPV NIb-CP substrate
In order to test whether the native NIb-CP junction (site F) could be recognized by the recombinant PPV NIa protease and estimate the proteolysis efficiency, NIb-CP protein was translated and 35S-labeled with a coupled in vitro transcription–translation system. Subsequently, the translated mixtures were treated with the purified PPV NIa protease. Most of the translated products were processed by 0.5 μg of recombinant PPV NIa protease at 20 or 30 °C within 5 min, resulting in two bands of 54.5 and 36.5 kDa. These two bands are corresponding to the sizes of PPV NIb and capsid protein translated from plasmid template, respectively (Fig. 3 ). This indicates that the recombinant PPV NIa protease produced and purified from E. coli functions well for the canonical PPV NIb-CP junction in vitro. Furthermore, NIb-CP protein of PPV expressed from E. coli was well-processed by purified recombinant PPV NIa protease (data not shown).
Fig. 3.
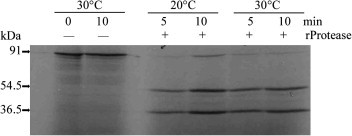
Processing of in vitro-synthesized NIb-CP substrate by recombinant PPV NIa protease. The in vitro-translated NIb-CP substrate was subjected to recombinant PPV NIa protease’s cleavage under the indicated reaction conditions. 35S-labeled proteins were resolved by SDS–PAGE and detected by autoradiography with X-ray films in dark for 48 h. −, in the absence of recombinant PPV NIa protease; +, in the presence of recombinant PPV NIa protease.
In vitro proteolytic assay for NIa protease processing specific fusion proteins
To investigate the capability of PPV NIa protease to process fusion proteins in vitro, we chose recombinant GFP (sG) and SARS-CoV Nucleocapsid protein (sN) as substrates. The GFP protein expressed and purified from E. coli is stable, well-characterized and widely used; however, therapeutic proteins are more often complex and vulnerable with regard to their structures and functions. In this test, substrate sG or sN (50–100 μg) was incubated in the reaction buffer in the presence of 1 μg of PPV NIa protease. More than 80% of the substrate (50 μg of sG or SN) could be cleaved efficiently and specifically in vitro by 1 μg recombinant PPV NIa protease at 30 °C within 2 h, yielding a free GFP protein (28 kDa) or a free SARS-CoV N protein (47 kDa) (Fig. 4 sG-lane 2, sN-lane 2). Furthermore, more than 95% of substrate (100 μg of sG or SN) could be cleaved at 30 °C after 10 h and no by-products occurred (Fig. 4 sG-lane 3, sN-lane 3). No significant difference of catalytic efficiency was observed in the processing of two substrates sG and sN.
Fig. 4.
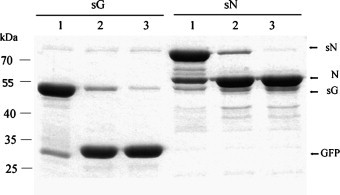
Specific processing of sG and sN by recombinant PPV NIa protease. Fifty micrograms of the substrate (sG or sN) and 1 μg protease were incubated for 2 h in a 50 μl reaction system at 30 °C. Aliquots of 5 μg substrate were withdrawn after 5 min (lane 1) and 2 h (lane 2). One hundred micrograms of the substrate (sG or sN) and 1 μg protease were incubated in a 100 μl reaction system at 30 °C for 10 h. Aliquots of 5 μg substrate were withdrawn after 10 h (lane 3).
Effects of temperature, salt concentration, protease inhibitors, and chemical reagents on the enzymatic activity of recombinant PPV NIa protease
Fifty micrograms of sG and 1 μg of recombinant PPV NIa protease were incubated at 4, 20, 30, and 40 °C to estimate the effect of temperature on the protease kinetics. The result showed the optimal temperature for recombinant PPV NIa protease is approximately 30 °C. However, 1 μg of recombinant PPV NIa protease exhibited adequate activity for processing 50 μg of the substrate at 20 °C in 10 h. Considerable activity was observed at 4 °C, though activity was severely diminished at 40 °C (Fig. 5 a). At 40 °C, degradation of GFP after cleavage was observed when the cleaved products were analyzed by SDS–PAGE; hence, high temperature is not recommended for the cleavage by PPV NIa protease. The initial velocity of recombinant PPV NIa protease at 30 °C were 1.21-fold and 1.79-fold of those at 4 and 40 °C, respectively.
Fig. 5.
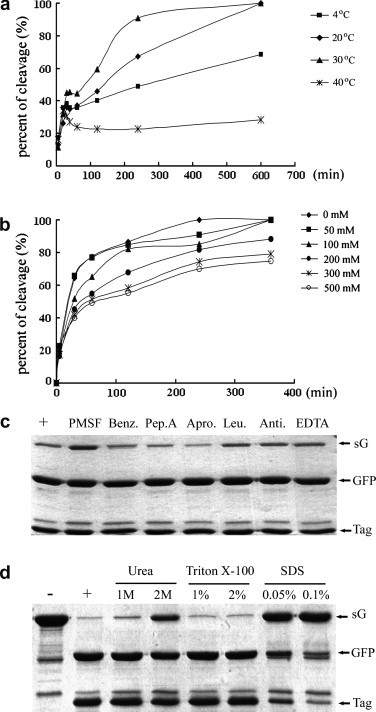
Impacts of temperature, ionic strength, and protease inhibitors on the enzymatic activity of recombinant PPV NIa protease. (a) Effect of temperature on the enzymatic activity of recombinant PPV NIa protease. Fifty micrograms of substrate sG and 1 μg of protease were incubated at different temperatures (4, 20, 30, and 40 °C) with a final substrate concentration of 1 μg/μl in the reaction system. The corresponding aliquots were collected and analyzed at different time points (5, 20, 30, 40, 60, 120, 240, and 600 min). (b) Effect of salt on the enzymatic activity of recombinant PPV NIa protease. Fifty micrograms of substrate sG and 1 μg of protease were incubated at 30 °C in the reaction buffer containing additional NaCl at increasing concentration (0, 50, 100, 200, 300, and 500 mM). The aliquots were collected and analyzed at each time point (5, 30, 60, 120, 240, and 360 min). (c) Effect of protease inhibitors on the enzymatic activity of recombinant PPV NIa protease. Fifty micrograms of substrate sG and 1 μg of protease were included in a 50 μl reaction system at 30 °C for 2 h in the presence of the indicated protease inhibitors. PMSF, phenylmethanesulfonyl fluoride; Benz, benzamidine; Pep. A, pepstatin A; Apro, aprotinin; Leu, leupeptin; Anti, antipain-dihydrochloride; EDTA, ethylene diamine tetraacetic acid. The reaction mixture in the absence of protease inhibitors was used as the positive control. (d) Effect of detergents and denaturing reagents on the enzymatic activity of recombinant PPV NIa protease. Eighty micrograms of substrate sG and 1 μg of protease were included in a 50 μl reaction system at 30 °C for 3 h in the presence of the indicated reagent. The concentration units of urea, Triton X-100, and SDS were mol/L, % (v/v), % (w/v), respectively. The reaction mixture without additional reagents was used as the positive control and uncleaved substrate sG was used as negative control.
Salt concentration was another common variable affecting the activity of the protease. The dependence of recombinant PPV NIa protease on salt is much less than that on temperature (Fig. 5b). Even in the presence of 500 mM NaCl, 40% of 50 μg sG was cleaved by 1 μg recombinant PPV NIa protease at 30 °C within 30 min; more than 60% substrate was specifically processed after 6 h. Monovalent ion strength, up to 200 mM, played a minor role on the performance of recombinant PPV NIa protease.
The naturally accruing proteases can be grouped into four classes according to the prominent functional residue at the active site: Ser, Cys, Asp, and metal [36], [41]. NIa proteases from TEV, TVMV, and PPV are similar to trypsin-like serine proteases but adopt unconventional catalytic triad in which Cys replaces Ser [42]. There are many reagents that can inhibit the protease function irreversibly or reversibly. To test whether commonly used protease inhibitors can affect the behavior of recombinant PPV NIa protease, recombinant PPV NIa protease (1 μg) and the substrate sG (50 μg) were incubated at 30 °C for 2 h, supplemented with different protease inhibitors. These protease inhibitors were used at high concentrations (even up to 100 times of their usual dosages). Only 1 mM PMSF had an effect, albeit slight, on the protease activity; other tested protease inhibitors (benzamidine, pepstatin A, aprotinin, leupeptin, antipain, and EDTA) had no effect on recombinant PPV NIa protease (Fig. 5c). Previous report showed human cystatin C affected PPV NIa protease activity in certain level when used at high concentration of 400 μg/ml [43]. In another way, the purified PPV NIa protease was not inhibited by other cystatins, such as oryzacystatins I and II (OCI and OCII), corn cystatin II (CCII), human stefin A (HSA), the domain 8 of tomato multicystatin (TMC-8) and a large 24 kDa tomato cystatin (LTCyst), when used at a molar ratio of 1:40 (NIa:cystatin) [39]. According to the detail manual of Devid S. Waugh laboratory (http://mcl1.ncifcrf.gov/waugh_research.html), TEV protease was not affected by PMSF (1 mM), AEBSF (1 mM), TLCK (1 mM), Bestatin (1 mg/ml), pepstatin A (1 mM), EDTA (1 mM) and E-64 (3 mg/ml), and a commercial “complete” protease inhibitor cocktail (Roche), which is frequently used to inhibit serine-, cysteine-, and metallo- proteases, as well as calpains, of bacterial, mammalian, yeast, and plant cell extracts. However, zwitterionic detergent (APO-10, DDMAB, DODMG, FC-12, HECAMEG, LDAO, OG, and ZW 3–12) notably inhibited the proteolytic activity of rTEV [44]. We chose two common detergents, SDS (also working as a denaturing reagent) and Triton X-100, to test their effects on the activity of PPV NIa protease. As we can see from Fig. 5d, recombinant PPV NIa protease was very sensitive to SDS, losing most of its proteolytic activity in the presence of 0.05% SDS, whereas recombinant PPV NIa protease was quite tolerant to Triton X-100 (up to 2%). Another denaturing reagent, urea, had almost no affect on the performance of recombinant PPV NIa protease when used at a final concentration of 1 M. About 30% activity of recombinant PPV NIa protease was blocked by 2 M urea.
Stability assay of PPV NIa protease
Wild-type TEV NIa protease is very prone to autolysis, thereby resulting in a truncated protein with poor cleavage activity [27], [28]. PPV NIa protease was homologous to TEV NIa protease; thus, the stability of recombinant PPV NIa protease was also analyzed. No self-degradation of recombinant PPV NIa proteases was observed during incubation for up to 120 h at 4 °C or 20 °C (Fig. 6 ). The autolysis of TEV NIa protease occurs between Met218 and Ser219 in the G H K V F M▾S sequence. This sequence can be aligned with the corresponding sequences S F T L V E D and S L Q L K R D in autolysis-resistant TVMV and PPV NIa proteases, respectively. TVMV NIa protease and PPV NIa protease both encode D219, while TEV NIa protease encodes S219 (Fig. 1, underlined region). Similarly, S219D mutant of TEV NIa protease was 10-fold more resistant to auto-inactivation than wild-type TEV NIa protease [28].
Fig. 6.
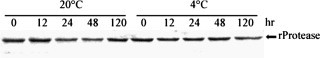
Self-lysis test of PPV NIa protease. Each of 1 μg recombinant PPV NIa protease was incubated in a 15 μl reaction system at 20 or 4 °C. Different incubation time points (0, 12, 24, 48, and 120 h) were selected for surveilling the possible self-lysis of recombinant PPV NIa protease.
Discussion
The large proportion of recombinant PPV NIa protease produced in E. coli BL21AI cells is soluble. This suggested the fusion tags (Trx tag, 6× His tag, and S tag) in front of PPV NIa protease favored the soluble status of recombinant PPV NIa protease. However, it was reported that only MBP, but not GST or Trx, can improve the solubility of recombinant TEV NIa protease [45]. For achieving soluble product, recombinant TVMV NIa protease was also expressed as MBP-TVMV NIa fusion protein [29]. On another aspect, recombinant PPV NIa protease was resistant to auto-degradation for at least 5 days at 20 °C, which has not been shown before and it is a crucial factor for prolonged cleavage activity. Recombinant TVMV NIa protease and mutant TEV NIa protease S219V were reported to remain stable during overnight incubation, but no prolonged assay was performed [29]. The recombinant PPV NIa proteases mentioned in this study was designed to be fused with 6× His tags, therefore, the protease can be easily removed from reaction mixture by passing through a nickle affinity column after cleavage. This property is similar as the commercial TEV NIa protease (AcTEVTM, Invitrogen).
Another concern is the purity of the protease. NIa proteases expressed as recombinant proteins in E. coli yield as highly pure product after purification through affinity chromatography, compared with animal-derived factor Xa and thrombin, which may have contamination of plasmin [46], [47], [48].
Many fusion tags are now available for optimizing the expression and purification of target proteins; therefore proteases are demanded to be highly specific towards the expected cleavage sites. We found that recombinant PPV NIa protease was extremely specific toward the cleavage sequence; no processing of Trx tag, hexahistidine tag, S tag, GST tag (data not shown) and MBP tag (data not shown) was observed, even during overnight cleavage. We chose the naturally preferred site F [32], [37], [38] as the recognition sequence in fused substrates for recombinant PPV NIa protease. Two different substrates (sG and sN) with insertion of site F were both cleaved efficiently by recombinant PPV NIa protease, indicating the interaction between protease and substrate was highly specific and efficient in both cases. The corresponding sites F for TEV, TVMV, and PPV were E N L Y F Q▾S, E T V R F Q▾S, and N V V V H Q▾A, all adopting a conserved residue Gln in the P1 position [21], [29], [37]. The specificity of PPV NIa protease has been studied, mainly illustrating with anomalous proteolytic processing caused by mutated proteases or mutated substrates [31], [38], [49], [50], thus a further analysis of the specificity and efficiency of PPV NIa protease–substrate complex by crystal structural is required. Although most studies about the cleavage determinant factors for NIa protease were performed with TEV and TVMV [20], [21], [33], [51], the high conserved amino acid sequence and function among PPV, TEV, and TVMV NIa protease indicate the recognition mechanism of TEV and TVMV NIa protease may also fit for PPV NIa protease [52]. The crystal structures of TEV and TVMV NIa protease suggested that the S4 and S3 pockets are primarily responsible for their different sequence specificities and that the residues in the P3 and P4 positions of the cleavage sites are the most important specificity discriminators [21].
The fact that recombinant PPV NIa protease maintained high activities in wide range of temperature (4–30 °C), high salt strength (even in 300 mM NaCl) and in the presence of protease inhibitors rendered it suitable for specific processing of labile proteins that demand such requirements. In addition, denaturing reagents and detergents were often required in the purification of insoluble or organellic proteins. The recombinant PPV NIa protease was sensitive to 0.05% SDS, but it was tolerant to both 2% Triton X-100 and 1 M urea. Comparatively, a commercial protease factor Xa (Cat. No. 69036-3, Novagen) is sensitive to urea, SDS, and high salt concentration (above 250 mM). The enzymatic ability of recombinant PPV NIa protease is similar to that reported by TVMV and TEV NIa protease with respect to molar ratio of protease and substrate [29], [53]. All the data indicates recombinant PPV NIa protease is a promising universal tool for fusion tags removal.
Proteins which are capable of in vivo-cleavage (for example, the intein protein from yeast) may be advanced in protein expression and purification [54]. Intracellular self-cleavage by TEV NIa protease on fusion protein MBP-TEVP-rsTEV-GFP-His6 (rsTEV: recognition site for TEV NIa protease) resulted in native and soluble EGFP protein [55]. When a fusion protein containing site F and the PPV NIa protease were driven by two compatible plasmids and expressed simultaneously within E. coli, the PPV NIa protease could process on site F correctly in vivo [56]. We can propose that two possible interacted-proteins and the PPV NIa protease may be fused together, separated by two cleavage sites, thereby the whole polyprotein can be expressed from one single vector. In this way, the protease component in the polyprotein is likely to perform self-cleavage in vivo, which will facilitate the analysis of the interaction between the two proteins in vivo.
Acknowledgments
This work was supported by Grants from the European Commission Sixth Framework Program (No. SP22-CT-2004-511060) and the National Natural Science Foundation of China (No. 30325030/30530400). We thank Dr. Luis Enjuanes from CNB-CSIC Spain for kindly providing the SARS-CoV cDNA clone.
Footnotes
Abbreviations used: NIa, nuclear inclusion protein a; TEV, tobacco etch virus; TVMV, tomato vein mottling virus; GST, glutathione-S-transferase; MBP, maltose-binding protein; TAP, tandem affinity purification; Trx, thioredoxin; IPTG, isopropyl-β-d-thiogalactopyranoside; OCI and OCII, oryzacystatins I and II; CCII, corn cystatin II; HSA, human stefin A; CP, capsid protein.
References
- 1.Chatterjee S., Schoepe J., Lohmer S., Schomburg D. High level expression and single-step purification of hexahistidine-tagged L-2-hydroxyisocaproate dehydrogenase making use of a versatile expression vector set. Protein Expr. Purif. 2005;39:137–143. doi: 10.1016/j.pep.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 2.Camprubi S., Bruguera M., Canalias F. Purification of recombinant histidine-tag streptolysin O using immobilized metal affinity expanded bed adsorption (IMA-EBA) Int. J. Biol. Macromol. 2006;38:134–139. doi: 10.1016/j.ijbiomac.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 3.Bernaudat F., Bulow L. Combined hydrophobic-metal binding fusion tags for applications in aqueous two-phase partitioning. Protein Expr. Purif. 2006;46:438–445. doi: 10.1016/j.pep.2005.09.026. [DOI] [PubMed] [Google Scholar]
- 4.Nilsson J., Stahl S., Lundeberg J., Uhlen M., Nygren P.A. Affinity fusion strategies for detection, purification, and immobilization of recombinant proteins. Protein Expr. Purif. 1997;11:1–16. doi: 10.1006/prep.1997.0767. [DOI] [PubMed] [Google Scholar]
- 5.Sharma S.K., Evans D.B., Vosters A.F., McQuade T.J., Tarpley W.G. Metal affinity chromatography of recombinant HIV-1 reverse transcriptase containing a human renin cleavable metal binding domain. Biotechnol. Appl. Biochem. 1991;14:69–81. [PubMed] [Google Scholar]
- 6.Ford C.F., Suominen I., Glatz C.E. Fusion tails for the recovery and purification of recombinant proteins. Protein Expr. Purif. 1991;2:95–107. doi: 10.1016/1046-5928(91)90057-p. [DOI] [PubMed] [Google Scholar]
- 7.Smith D.B., Johnson K.S. Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene. 1988;67:31–40. doi: 10.1016/0378-1119(88)90005-4. [DOI] [PubMed] [Google Scholar]
- 8.di Guan C., Li P., Riggs P.D., Inouye H. Vectors that facilitate the expression and purification of foreign peptides in Escherichia coli by fusion to maltose-binding protein. Gene. 1988;67:21–30. doi: 10.1016/0378-1119(88)90004-2. [DOI] [PubMed] [Google Scholar]
- 9.Einhauer A., Jungbauer A. The FLAG peptide a versatile fusion tag for the purification of recombinant proteins. J. Biochem. Biophys. Methods. 2001;49:455–465. doi: 10.1016/s0165-022x(01)00213-5. [DOI] [PubMed] [Google Scholar]
- 10.Puig O., Caspary F., Rigaut G., Rutz B., Bouveret E., Bragado-Nilsson E., Wilm M., Seraphin B. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods. 2001;24:218–229. doi: 10.1006/meth.2001.1183. [DOI] [PubMed] [Google Scholar]
- 11.Lockhart E., Griffin J.F., Chinn N., Lavallie E., Buchan G. The cloning expression and purification of cervine interleukin 2. Cytokine. 1996;8:603–612. doi: 10.1006/cyto.1996.0081. [DOI] [PubMed] [Google Scholar]
- 12.Radcliffe R., Nemerson Y. Mechanism of activation of bovine factor VII. Products of cleavage by factor Xa. J. Biol. Chem. 1976;251:4749–4802. [PubMed] [Google Scholar]
- 13.Kupke T., Stevanovic S., Ottenwalder B., Metzger J.W., Jung G., Gotz F. Purification and characterization of EpiA, the peptide substrate for post-translational modifications involved in epidermin biosynthesis. FEMS Microbiol. Lett. 1993;112:43–48. doi: 10.1111/j.1574-6968.1993.tb06421.x. [DOI] [PubMed] [Google Scholar]
- 14.Doskeland A.P., Martinez A., Knappskog P.M., Flatmark T. Phosphorylation of recombinant human phenylalanine hydroxylase: effect on catalytic activity substrate activation and protection against non-specific cleavage of the fusion protein by restriction protease. Biochem. J. 1996;313(Pt 2):409–414. doi: 10.1042/bj3130409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.He M., Jin L., Austen B. Specificity of factor Xa in the cleavage of fusion proteins. J. Protein Chem. 1993;12:1–5. doi: 10.1007/BF01024906. [DOI] [PubMed] [Google Scholar]
- 16.Chang J.Y. Thrombin specificity. Requirement for apolar amino acids adjacent to the thrombin cleavage site of polypeptide substrate. Eur. J. Biochem. 1985;151:217–224. doi: 10.1111/j.1432-1033.1985.tb09091.x. [DOI] [PubMed] [Google Scholar]
- 17.Dougherty W.G., Carrington J.C., Cary S.M., Parks T.D. Biochemical and mutational analysis of a plant virus polyprotein cleavage site. EMBO J. 1988;7:1281–1287. doi: 10.1002/j.1460-2075.1988.tb02942.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carrington J.C., Haldeman R., Dolja V.V., Restrepo-Hartwig M.A. Internal cleavage and trans-proteolytic activities of the VPg-proteinase (NIa) of tobacco etch potyvirus in vivo. J. Virol. 1993;67:6995–7000. doi: 10.1128/jvi.67.12.6995-7000.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schaad M.C., Haldeman-Cahill R., Cronin S., Carrington J.C. Analysis of the VPg-proteinase (NIa) encoded by tobacco etch potyvirus: effects of mutations on subcellular transport, proteolytic processing and genome amplification. J. Virol. 1996;70:7039–7048. doi: 10.1128/jvi.70.10.7039-7048.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Phan J., Zdanov A., Evdokimov A.G., Tropea J.E., Peters H.K., 3rd, Kapust R.B., Li M., Wlodawer A., Waugh D.S. Structural basis for the substrate specificity of tobacco etch virus protease. J. Biol. Chem. 2002;277:50564–50572. doi: 10.1074/jbc.M207224200. [DOI] [PubMed] [Google Scholar]
- 21.Tozser J., Tropea J.E., Cherry S., Bagossi P., Copeland T.D., Wlodawer A., Waugh D.S. Comparison of the substrate specificity of two potyvirus proteases. FEBS J. 2005;272:514–523. doi: 10.1111/j.1742-4658.2004.04493.x. [DOI] [PubMed] [Google Scholar]
- 22.Dougherty W.G., Cary S.M., Parks T.D. Molecular genetic analysis of a plant virus polyprotein cleavage site: a model. Virology. 1989;171:356–364. doi: 10.1016/0042-6822(89)90603-x. [DOI] [PubMed] [Google Scholar]
- 23.Cabrita L.D., Dai W., Bottomley S.P. A family of E. coli expression vectors for laboratory scale and high throughput soluble protein production. BMC Biotechnol. 2006;6:12. doi: 10.1186/1472-6750-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rubio V., Shen Y., Saijo Y., Liu Y., Gusmaroli G., Dinesh-Kumar S.P., Deng X.W. An alternative tandem affinity purification strategy applied to Arabidopsis protein complex isolation. Plant J. 2005;41:767–778. doi: 10.1111/j.1365-313X.2004.02328.x. [DOI] [PubMed] [Google Scholar]
- 25.Braud S., Moutiez M., Belin P., Abello N., Drevet P., Zinn-Justin S., Courcon M., Masson C., Dassa J., Charbonnier J.B., Boulain J.C., Menez A., Genet R., Gondry M. Dual expression system suitable for high-throughput fluorescence-based screening and production of soluble proteins. J. Proteome Res. 2005;4:2137–2147. doi: 10.1021/pr050230i. [DOI] [PubMed] [Google Scholar]
- 26.Parks T.D., Leuther K.K., Howard E.D., Johnston S.A., Dougherty W.G. Release of proteins and peptides from fusion proteins using a recombinant plant virus proteinase. Anal. Biochem. 1994;216:413–417. doi: 10.1006/abio.1994.1060. [DOI] [PubMed] [Google Scholar]
- 27.Parks T.D., Howard E.D., Wolpert T.J., ARP D.J., Dougherty W.G. Expression and purification of a recombinant Tobacco Etch Virus NIa protease: biochemical analysis of full-length and a naturally occurring truncated protease form. Virology. 1995;210:194–201. doi: 10.1006/viro.1995.1331. [DOI] [PubMed] [Google Scholar]
- 28.Kapust R.B., Tozser J., Fox J.D., Anderson D.E., Cherry S., Copeland T.D., Waugh D.S. Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Eng. 2001;14:993–1000. doi: 10.1093/protein/14.12.993. [DOI] [PubMed] [Google Scholar]
- 29.Nallamsetty S., Kapust R.B., Tozser J., Cherry S., Tropea J.E., Copeland T.D., Waugh D.S. Efficient site-specific processing of fusion proteins by tobacco vein mottling virus protease in vivo and in vitro. Protein Expr. Purif. 2004;38:108–115. doi: 10.1016/j.pep.2004.08.016. [DOI] [PubMed] [Google Scholar]
- 30.Kwon S.Y., Choi Y.J., Kang T.H., Lee K.H., Cha S.S., Kim G.H., Lee H.S., Kim K.T., Kim K.J. Highly efficient protein expression and purification using bacterial hemoglobin fusion vector. Plasmid. 2005;53:274–282. doi: 10.1016/j.plasmid.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 31.Garcia J.A., Lain S. Proteolytic activity of plum pox virus-tobacco etch virus chimeric NIa proteases. FEBS Lett. 1991;281:67–72. doi: 10.1016/0014-5793(91)80360-f. [DOI] [PubMed] [Google Scholar]
- 32.Urcuqui-Inchima S., Haenni A.L., Bernardi F. Potyvirus proteins: a wealth of functions. Virus Res. 2001;74:157–175. doi: 10.1016/s0168-1702(01)00220-9. [DOI] [PubMed] [Google Scholar]
- 33.Riechmann J.L., Lain S., Garcia J.A. Highlights and prospects of potyvirus molecular biology. J. Gen. Virol. 1992;73:1–16. doi: 10.1099/0022-1317-73-1-1. [DOI] [PubMed] [Google Scholar]
- 34.Garcia J.A., Martin M.T., Cervera M.T., Riechmann J.L. Proteolytic processing of the plum pox potyvirus polyprotein by the NIa protease at a novel cleavage site. Virology. 1992;188:697–703. doi: 10.1016/0042-6822(92)90524-s. [DOI] [PubMed] [Google Scholar]
- 35.Yoon H.Y., Hwang D.C., Choi K.Y., Song B.D. Proteolytic processing of oligopeptides containing the target sequences by the recombinant tobacco vein mottling virus NIa proteinase. Mol. Cells. 2000;10:213–219. doi: 10.1007/s10059-000-0213-3. [DOI] [PubMed] [Google Scholar]
- 36.Bazan J.F., Fletterick R.J. Viral cysteine proteases are homologous to the trypsin-like family of serine proteases: structural and functional implications. Proc. Natl. Acad. Sci. USA. 1988;85:7872–7876. doi: 10.1073/pnas.85.21.7872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garcia J.A., Riechmann J.L., Lain S. Artificial cleavage site recognized by plum pox potyvirus protease in Escherichia coli. J. Virol. 1989;63:2457–2460. doi: 10.1128/jvi.63.6.2457-2460.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garcia J.A., Riechmann J.L., Lain S. Proteolytic activity of the plum pox potyvirus NIa-like protein in Escherichia coli. Virology. 1989;170:362–369. doi: 10.1016/0042-6822(89)90426-1. [DOI] [PubMed] [Google Scholar]
- 39.Wen R., Zhang S.C., Michaud D., Sanfacon H. Inhibitory effects of cystatins on proteolytic activities of the Plum pox potyvirus cysteine proteinases. Virus Res. 2004;105:175–182. doi: 10.1016/j.virusres.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 40.Wyckoff E.E., Hershey J.W., Ehrenfeld E. Eukaryotic initiation factor 3 is required for poliovirus 2A protease-induced cleavage of the p220 component of eukaryotic initiation factor 4F. Proc. Natl. Acad. Sci. USA. 1990;87:9529–9533. doi: 10.1073/pnas.87.24.9529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scott B.L., Racz E., Lolley R.N., Bazan N.G. Developing rod photoreceptors from normal and mutant Rd mouse retinas: altered fatty acid composition early in development of the mutant. J. Neurosci. Res. 1988;20:202–211. doi: 10.1002/jnr.490200209. [DOI] [PubMed] [Google Scholar]
- 42.Hwang D.C., Kim D.H., Lee J.S., Kang B.H., Han J., Kim W., Song B.D., Choi K.Y. Characterization of active-site residues of the NIa protease from tobacco vein mottling virus. Mol. Cells. 2000;10:505–511. doi: 10.1007/s10059-000-0505-7. [DOI] [PubMed] [Google Scholar]
- 43.Garcia J.A., Cervera M.T., Riechmann J.L., Lopez-Otin C. Inhibitory effects of human cystatin C on plum pox potyvirus proteases. Plant Mol. Biol. 1993;22:697–701. doi: 10.1007/BF00047410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mohanty A.K., Simmons C.R., Wiener M.C. Inhibition of tobacco etch virus protease activity by detergents. Protein Expr. Purif. 2003;27:109–114. doi: 10.1016/s1046-5928(02)00589-2. [DOI] [PubMed] [Google Scholar]
- 45.Kapust R.B., Waugh D.S. Escherichia coli maltose-binding protein is uncommonly effective at promoting the solubility of polypeptides to which it is fused. Protein Sci. 1999;8:1668–1674. doi: 10.1110/ps.8.8.1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Donaldson V.H., Kleniewski J. The role of plasmin in kinin-release by preparations of human thrombin. Thromb Res. 1979;16:401–406. doi: 10.1016/0049-3848(79)90087-2. [DOI] [PubMed] [Google Scholar]
- 47.Suzuki S., Sakuragawa N. A study on the properties of commercial thrombin preparations. Thromb Res. 1989;53:271–277. doi: 10.1016/0049-3848(89)90102-3. [DOI] [PubMed] [Google Scholar]
- 48.Litwiller R.D., Jenny R.J., Mann K.G. Identification and isolation of vitamin K-dependent proteins by HPLC. Anal. Biochem. 1986;158:355–360. doi: 10.1016/0003-2697(86)90560-9. [DOI] [PubMed] [Google Scholar]
- 49.Garcia J.A., Riechmann J.L., Martin M.T., Lain S. Proteolytic activity of the plum pox potyvirus NIa-protein on excess of natural and artificial substrates in Escherichia coli. FEBS Lett. 1989;257:269–273. doi: 10.1016/0014-5793(89)81550-9. [DOI] [PubMed] [Google Scholar]
- 50.Garcia J.A., Lain S., Cervera M.T., Riechmann J.L., Martin M.T. Mutational analysis of plum pox potyvirus polyprotein processing by the NIa protease in Escherichia coli. J. Gen. Virol. 1990;71(Pt 12):2773–2779. doi: 10.1099/0022-1317-71-12-2773. [DOI] [PubMed] [Google Scholar]
- 51.Zhdanov A.S., Phan J., Evdokimov A.G., Tropea J.E., Kapust R.B., Li M., Wlodawer A., Waugh D.S. Tobacco etch virus proteinase: crystal structure of the active enzyme and its inactive mutant. Bioorg. Khim. 2003;29:457–460. [PubMed] [Google Scholar]
- 52.Jenny R.J., Mann K.G., Lundblad R.L. A critical review of the methods for cleavage of fusion proteins with thrombin and factor Xa. Protein Expr. Purif. 2003;31:1–11. doi: 10.1016/s1046-5928(03)00168-2. [DOI] [PubMed] [Google Scholar]
- 53.Marcos J.F., Beachy R.N. In vitro characterization of a cassette to accumulate multiple proteins through synthesis of a self-processing polypeptide. Plant Mol. Biol. 1994;24:495–503. doi: 10.1007/BF00024117. [DOI] [PubMed] [Google Scholar]
- 54.Hall M.C., Kunkel T.A. Purification of eucaryotic MutL homologs from Saccharomyces cerevisiae using self-cleaving. Protein Expr. Purif. 2001;22:25–30. doi: 10.1006/prep.2000.1379. [DOI] [PubMed] [Google Scholar]
- 55.Shih Y.P., Wu H.C., Hu S.M., Wang T.F., Wang A.H. Self-cleavage of fusion protein in vivo using TEV protease to yield native protein. Protein Sci. 2005;14:936–941. doi: 10.1110/ps.041129605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Perez-Martin J., Cases I., de Lorenzo V. Design of a solubilization pathway for recombinant polypeptides in vivo through processing of a bi-protein with a viral protease. Protein Eng. 1997;10:725–730. doi: 10.1093/protein/10.6.725. [DOI] [PubMed] [Google Scholar]