

Graphical abstract
Keywords: Epigenetic enzymes, Elastic network models, Protein structure networks, Domain motions, Allosteric sites
Highlights
-
•
Dynamics and allosteric potentials of the RFTS domain are proposed.
-
•
Hinge sites located at the RFTS-CD interface are key regulators for inter-domain interactions.
-
•
Network analysis reveals local allosteric networks and inter-domain communication pathways in DNMT1.
-
•
A potential allosteric site at the TRD interface for DNMT1 is identified.
Abstract
DNA methyltransferase 1 (DNMT1), a large multidomain enzyme, is believed to be involved in the passive transmission of genomic methylation patterns via methylation maintenance. Yet, the molecular mechanism of interaction networks underlying DNMT1 structures, dynamics, and its biological significance has yet to be fully characterized. In this work, we used an integrated computational strategy that combined coarse-grained and atomistic simulations with coevolution information and network modeling of the residue interactions for the systematic investigation of allosteric dynamics in DNMT1. The elastic network modeling has proposed that the high plasticity of RFTS has strengthened the correlated behaviors of DNMT1 structures through the hinge sites located at the RFTS-CD interface, which mediate the collective motions between domains. The perturbation response scanning (PRS) analysis combined with the enrichment analysis of disease mutations have further highlighted the allosteric potential of the RFTS domain. Furthermore, the long-range paths connect the intra-domain interactions through the TRD interface and catalytic interface, emphasizing some key inter-domain interactions as the bridges in the global allosteric regulation of DNMT1. The observed interplay between conserved intra-domain networks and dynamical plasticity encoded by inter-domain interactions provides insights into the intrinsic dynamics and functional evolution, as well as the design of allosteric modulators of DNMT1 based on the TRD interface.
1. Introduction
DNA methylation is a major epigenetic modification which plays an important role in a variety of biological processes, including regulation of transcription, genomic imprinting, genome stability, X-chromosome inactivation and transcriptional repression [1]. Aberrant DNA methylation has been observed in many human diseases, such as cancers and neurological disorders [2]. In mammals, DNA methylation mainly occurs at the C5 position of cytosines in a context of CpG dinucleotides [3]. Mammalian DNA methylation is established by three active DNMTs: the de novo DNA methyltransferases DNMT3A and DNMT3B, as well as the maintenance DNA methyltransferase DNMT1 [4], [5].
The crystal structures of DNMT1 suggest that DNMT1 is a large, multidomain protein containing a N-terminal platform, including a replication foci targeting sequence (RFTS) domain, a zinc-finger-like (CXXC) motif, two bromo-adjacent homology (BAH1 and BAH2) domains, a flexible linker composed of lysine-glycine (KG) repeats, and a catalytic C-terminal domain (CD) [6], [7], [8], [9] (Fig. 1A). The C-terminal domain can be further divided into two subdomains: the catalytic core and the target recognition domain (TRD). The early crystal structure of mDNMT1 fragment (residues 731–1602) covalently bound to a 12-mer hemimethylated DNA duplex provides insight into the productive state of DNMT1 [7]. Whereas the crystal structure of mDNMT1 fragment (residues 650–1602), spanning from the CXXC motif to CD, in complex with a 19-mer DNA duplex containing unmethylated CpG sites provides insight into an autoinhibitory state [6]. Structural comparison of the two states of mDNMT1 reveals that the largest conformational changes lie in two regions, one is the TRD region, and the other locates in the catalytic site (residues C1229-S1249) (as labeled in black boxes in Fig. 1B). In the active (designated as: helix-straight) state, the TRD region moves toward the catalytic site by about 2 to 3 Å compared with that in the autoinhibitory (designated as: helix-kinked) state. Meanwhile, the catalytic loop in the active state, which is followed by the straight catalytic helix, inserts into the DNA minor groove, making extensive protein-DNA contacts [7]. In comparison, the catalytic helix in autoinhibitory state adopts a kinked conformation, with the catalytic loop excluded from the DNA minor groove [6]. The disruption of this conformational transition leads to the impaired enzymatic activity of DNMT1, highlighting the importance of this conformational switch in DNMT1-mediated DNA methylation.
Fig. 1.

Structures of DNMT1s. (A) Color-coded domain architecture and numbering of hDNMT1 sequence, (B) Alignment of mDNMT1 (732–1600) in helix-straight (in color) and helix-kinked (in gray) states, (C) Alignment of the hDNMT1 (351–1600) (in color) and mDNMT1 (357–1608) (in gray) structures. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Recently, the crystal structures of DNMTs, including mDNMT1 (357–1608) and hDNMT1 (351–1600), containing the RFTS domain, have been reported (Fig. 1C) [8], [9]. Although DNMTs show different sequence lengths and structural folds in the N-terminal regions, they are all in the allosteric regulation by the variant N-terminal regions in specific molecular mechanism [10]. The RFTS domain is involved in the targeting of DNMT1 to DNA replication foci, thereby plays an important role in maintaining DNA methylation [11]. The RFTS domain directly interacts with the DNA-binding pocket of the catalytic domain, thereby exerting an autoinhibitory effect on DNMT1 activity [12]. Relieving the auto-inhibition can be triggered by UHRF1 (ubiquitin-like with PHD and ring finger domains 1) [13], which directly stimulate the catalytic activity of DNMT1 by interacting with the RFTS domain [14]. These findings suggest the RFTS domain is implicated in an autoinhibitory function, but how it regulates other domains in the context of the full-length DNMT1 has not been clarified. Furthermore, many reported disease mutations are enriched in the RFTS domain, which are corresponding to different phenotypes, such as hereditary sensory and autonomic neuropathy with dementia and hearing loss type IE (HSAN IE), and autosomal dominant cerebellar ataxia, deafness and narcolepsy (ADCA-DN) [15], [16]. It has become increasingly accepted that recent DNMT1 structures containing RFTS domain provide a probability to investigate the complex allosteric regulation of DNMT1 activity and specificity [10], [17]. In this manner, the allosteric regulation underlying DNMT1 structures at the increasing level of complexity remains to be systematically investigated.
Computational approaches including molecular dynamics (MD) simulations and normal mode analysis (NMA) play an important role in the investigation of allosteric mechanism of molecular machines [18]. In comparison with MD simulations, the coarse-grained approaches such as elastic network models (ENMs) provide more suitable methods for studying the conformational dynamics of large multidomain proteins and systematically dynamics of protein families [19]. Meantime, the physical based methods, including perturbation response scanning (PRS) approach and energetic-based method [20], are conducive to the identification of key residues and “hot spots” sites for protein dynamics and allosteric interactions [21], [22]. Furthermore, graph-based network models of protein structures yield a convenient description of residue interaction networks [23], providing a robust framework for understanding allosteric communications in protein molecular machines [24], [25], [26]. Following the “sequence-structures-dynamics” paradigm for protein function, protein sequence also provides noteworthy information from another dimension. Coevolution of protein residues are typically spatially close that can reflect a coordinated involvement of functional sites in mediating residue-residue contacts, thus also promoting allosteric signaling in terms of residue interaction networks. Using PDZ domains as an example, different coevolution methods have given the contradiction results for seeking allosteric networks [27]. Therefore, it argues that the combination of different methods from additional attributes to produce consistent results is necessary [28]. The effective combination of molecular dynamics methods, including MD and ENM, with protein structure network (PSN) and coevolutionary analysis provides suitable strategy to investigate the allosteric regulation in molecular machines [29]. Along this line, Verkhivkerand co-workers [30], [31], [32], [33], [34], [35] have significant contributions on the development of integrating computational approaches for understanding the allosteric mechanisms of molecular chaperone and protein kinases.
To date, multiple computational studies have been carried out for elucidating the conformational dynamics and protein–ligand interactions of DNMTs [36]. In our previous research [37], the MD study revealed that the dynamics switching between the two states (helix-straight and helix-kinked, as shown in Fig. 1B) was important for DNMT1 enzymatic activity, and key residues were validated by experiments in the conformational transition between both states. In addition, our recent studies have utilized integrated computational tools to study the allosteric properties of PRMT1 [38] and DNMT3A [39], which have opened a new way to probe the molecular mechanism in epigenetic enzymes. In this study, capitalizing on recent DNMT1 structures, we firstly examine the conformational dynamics of different DNMT1 structures using ENM and PCA methods, with the goal of elucidating and comparing collective mechanisms of motions and identifying key sites for inter-domain interactions. By combining functional dynamics with PRS analysis, a series of key residues in the RFTS domain that serve as effectors in transmitting perturbations in hDNMT1 are identified. Subsequently, the combination of MD simulations with PSN, as well as coevolutionary network provides valuable information for capturing the allosteric paths in modularity. Together with the molecular signature of pathogenic mutations and the identification of allosteric pockets, a potential allosteric site located at the RFTS-CD domain interface was suggested. The systematic study of intrinsic dynamics and allosteric network underlying DNMT1 structures proposed a novel allosteric model for DNMT1, shedding new lights for the design of allosteric modulator in future.
2. Materials and methods
2.1. DNMT1 structural models
Four DNMT1 structures were retrieved from the Protein Data Bank (https://www.rcsb.org), and the missing residues were added by modeler 9.20 [40]. Three DNMT1 crystal structures found in mouse were used in this study: mDNMT1 (731–1600) both in helix-straight (PDB ID: 4DA4) and helix-kinked (PDB ID: 3PT9) states contain three domains including BAH1, BAH2, and CD (BBC) domains, with sequence length from 732 to 1600, and mDNMT1 (357–1608) (PDB ID: 3AV5) is the structure with the further introduction of CXXC and RFTS domain, with sequence length from 357 to 1608. In addition, hDNMT1 (351–1600) (PDB ID: 4WXX) represent the DNMT1 structure found in human that contain BBC, CXXC, and RFTS domain, with sequence length from 351 to 1600.
2.2. Elastic network models
ENMs provide computationally efficient means for performing normal mode analysis on low-frequency modes of proteins and their complexes. In this work, the anisotropic network model (ANM) is used to analyze and visualize collective motions of DNMT1s. In ANM [41], each amino acid residue is represented by a node placed at its Cα coordinate. Residue pairs that fall within a cutoff distance rc = 15 Å are connected by harmonic springs of uniform force constant . For a network composed of N nodes, the total potential energy is a summation over all springs in the system and the (3N3N) Hessian matrix H is constructed based on the minimum energy structures that is the same as the crystal structures. Diagonalization of H yields (3N-6) nonzero eigenvalues () with corresponding eigenvectors (). Starting with the first non-zero eigenvalue, are sorted in ascending order, and also the corresponding . The modes with the lowest-frequencies (the smallest ) are called the softest modes and define the most cooperative/global motions intrinsically accessible to the structures. Gaussian network model (GNM) with isotropic residue fluctuations is used for evaluating the residue mean-square fluctuations (MSF) and mode shapes [42]. Briefly, the (N × N) Kirchhoff/connectivity matrix is constructed from the structural coordinates with a cutoff distance of 10 Å. Eigenvalue decomposition yields (N-1) non-zero modes for a folded structure. Both GNM and ANM analyses were performed by using the ProDy package [43].
2.3. Overlaps and structural changes
The similarity between the conformational space (ENM eigenvector sets) described by two different states/conformations of a system can be calculated according to the following equation [44]:
| (1) |
Here, the inner product ( quantifies the individual overlap between the ith and jth eigenvectors belonging to the different sets. Overlap (k) quantifies the overall correspondence between the first k modes of the sets. A structural change () between initial and final states of the same protein, such as an experimentally determined open-to-closed transition, may be described by a subset of ANM modes based on the initial structures. The agreement between an experimental transition and the kth ANM mode is quantified by (). The cumulative overlap measures how well a subset of low-frequency ANM modes (e.g., m of them) predicts the conformational changes as:
| (2) |
where m is the number of ANM modes.
2.4. Perturbation-Response Scanning analysis
Perturbation Response Scanning (PRS) approach is based on the linear response theory and allows evaluating residue displacements in response to external forces [20]. The PRS technique was combined with protein dynamics based on cross-correlations calculated from ANM by constructing the Hessian matrix H. The 3N-dimensional vector ΔR of node displacements in response to the application of a perturbation (a 3N-dimensional force vector F) obeys Hooke’s law . The idea in PRS is to exert a force of a given magnitude on the network, one residue at a time, and observe the response of the overall network. The force exerted on residue i is expressed as
| (3) |
and the resulting response is
| (4) |
ΔR(i) is a 3N-dimensional vector that describes the deformation of all the residues (in N blocks of dimension 3, each) in response to . A metric for the response of residue k is the magnitude of the kth block of ΔR(i) averaged over multiple F(i), expressed as the ikth element of the N × N PRS matrix, SPRS. The elements of SPRS refer to unit (or uniform) perturbing force. The response to unit deformation at each perturbation site is obtained by dividing each row by its diagonal value:
| (5) |
The average effect of the perturbed effector site i on all other residues is computed by averaging over all sensors (receivers) residues j and can be expressed as . The effector profile describes the average effect that local perturbation in the effector site i has on all other residues. The maxima along the effector and sensor profiles would correspond to functional mobile residues that undergo allosteric structural change.
2.5. Molecular dynamics analysis
All-atom molecular dynamics (MD) was performed for the hDNMT1 (351–1600). MD simulations were carried out with the AMBER03 force field (35) of Gromacs 4.5.3 [45]. Periodic boundary conditions were used to avoid edge effects in all calculations. In each system, the protein was solvated in a box with TIP3P water molecules to keep the boundary of the box at least 10 Å away from the protein on all sides (i.e., the starting structure had a 20 Å interval between periodic images). All the bonds with hydrogen atoms (e.g. C–H, O–H) were constrained using the linear constraint solver algorithm. Na+ and Cl− ions were subsequently added for charge neutralization under simulated physiological conditions. The final concentration of NaCl in the simulation system is 0.15 M. Long-range electrostatic interactions were treated using the particle-mesh Ewald method. Eventually, the systems that contain the water, ions, and protein were sequentially coupled to a temperature bath at 300 K with a coupling time of 1 ps using the Berendsen thermostat method. A cutoff distance of 10 Å was used for the Lennard-Jones interactions. The pressure was maintained by using the Berendsen pressure coupling for the equilibration of the systems. Before the conventional MD simulation run, energy minimization was then repeated on the whole system using the steepest descent algorithm. The systems were heated gradually from 0 to 300 K. Finally, conventional MD was performed, with coordinates saved every 10 ps throughout the entire process.
Principal component analysis (PCA) was performed based on MD ensembles to determine the essential dynamics of DNMT1. The calculation of the PCs involves the calculation of the covariance matrix, C, of the positional deviations, and the diagonalization of this matrix. The 3N dimensional covariance matrix is calculated based on an ensemble of protein structures, and the elements of C are defined as
| (6) |
where xi and xj are atomic coordinates and the brackets denote the ensemble average. The diagonalization of the symmetric matrix C is equivalent to solving the eigenvalue problem
| (7) |
where A represents the eigenvectors and λ the associated eigenvalues.
2.6. Protein structure network analysis
The Protein Structure Network (PSN) approach proposed by Vishveshwara and coworkers [46] was applied to unveil the allosteric communications in hDNMT1 (351–1600) from its MD ensembles. As a PSN for a protein structure, each amino acid is represented as a node, and these nodes are connected by edges based on the strength of noncovalent interactions between nodes. The so-called interaction strength value between two nodes is calculated as following,
| (8) |
where is the number of atom–atom pairs between residues i and j within a distance cutoff (4.5 Å); and and are normalization values for residues i and j, taking into account the differences in the side chains of different residue types and their propensity to make the maximum number of contacts with other residues in protein structures. The PSN analysis of hDNMT1 (351–1600) was implemented in the Wordom software [47]. Node inter-connectivity was used to calculate the shortest communication paths in hDNMT1 (351–1600). The search for the shortest path(s) by the Floyd-Warshall algorithm between pairs of nodes, as implemented in the PSNPath module of Wordom, combines information on node inter-connectivity from PSN analysis and cross-correlation of atomic fluctuations. The shortest path was identified as the path in which the two residues were non-covalently connected by the smallest number of intermediate nodes.
Firstly, the lowest accepted residue-residue interaction strength value (Imin) was set at 3.5, and the shortest paths in which at least one identified node featured a significant correlation value (0.5) with either one of the two extremities were retained. The recurrence analysis of path links can lead to the building of metapaths made of the most recurrent links in the whole pool of shortest paths. The metapaths obtained from the shortest paths form local allosteric networks in intra-domain. Subsequently, the lowest interaction strength value (Imin = 1.5) and correlation value (0.2) were adopted to probe the potential long-range paths across domain.
2.7. Sequence conservation and coevolution analyses
We evaluated the sequence conservation and co-evolution properties of the hDNMT1 (351–1600), starting from a multiple sequence alignment (MSA) of 500 sequences from the consurf server [48] in which MSA was generated using hidden Markov models (HMM). The MSA was refined in three steps: (i) a reference sequence was identified by screening hDNMT1 (351–1600) wild type sequence against all in the Pfam MSA using the Smith-Waterman algorithm; (ii) the MSA columns that correspond to the reference sequence residues were retained, and (iii) the resulting MSA was then subjected to further refinement, including removal of the redundant sequences using a threshold of 99%, and eliminating the sequences that had more than 20% gaps. Given the MSA, the Rate4Site algorithm is used to calculate position-specific evolutionary rates under an empirical Bayesian methodology. Coevolution analyses were performed by four different methods [49]: Direct Information (DI), and statistical coupling analysis (SCA), Observed-Minus-Expected-Squared (OMES) covariance algorithm, and Mutual Information with the APC correction (MIp). The latter two are particularly useful for removing indirect co-evolutionary effects. Given the high sensitivity of co-evolution results to the choice of methods as recently shown, we focus mostly on the residue pairs that are confirmed by the four methods. The coevolutionary networks were defined as graphs where nodes were defined as positions in the MSA of hDNMT1 and edges were defined between any pair of nodes of coevolution.
3. Results and discussion
3.1. Intrinsic dynamics of mDNMT1 (732–1600) in both the helix-straight and helix-kinked states
Collective motions are essential for protein functions, such as allosteric interactions and protein-substrate interactions [50]. Herein, the ANM method was used to compare and analyze the collective motions of the helix-straight (PDB ID: 4DA4) [7] and helix-kinked (PDB ID: 3PT9) [6] mDNMT1 (732–1600) structures. Fig. 2A shows the overlap map between the 20 softest modes of both mDNMT1s. In this map, each value describes the similarity of the pair of modes, in which assessed by overlaying the x, y, and z components of residue displacements. It has been shown that the three softest motions are extremely conserved in both states, just with reordering positions, underlying the DNMT1 fold-dependent dynamics. Specifically, mode 2 (u2helix-straight) in the helix-straight state resembles mode 4 (u4helix-kinked) in the helix-kinked state; both induce clamping motions between TRD region and BAH1 domain (Fig. 2B and Fig. S1(A)). In comparison between the two states, the formation of the productive mDNMT1-DNA complex involves the infiltration of DNA from both major (two TRD loops) and minor (catalytic loop (residues 1227–1243)) grooves, with the TRD region undergoing concerted movement toward the DNA major groove by about 2–3 Å upon complex formation [6], [7]. Besides, the structural alignment of different DNMT1 crystal structures showed that the TRD region and BAH1 domain fluctuates more or less in different states (Fig. S1(B)). Herein the shared clamping motions represent the conformational changes in the crystal structures, especially for the TRD region, which is important for substrate DNA binding and DNMT1 function.
Fig. 2.

The intrinsic dynamics of mDNMT1 (732–1600) in helix-straight and helix-kinked states. (A) Overlap of 20 ANM modes between the helix-straight and helix-kinked states. (B) The motions of ANM mode 2 of mDNMT1 in helix-straight, showing the clamping motion between TRD region and BAH1 domain. The overlap values between the 20 ANM modes and the deformation vector of the conformational changes for mDNMT1 (732–1600) in helix-straight (blue bars) (C) and helix-kinked (blue bars) (D), while red bars show the overlap values when only consider TRD regions in both states. (E) The cross-correlation map of the slowest ANM mode 1 for mDNMT1 (732–1600) in the helix-straight state. The color-coded domain organization is labeled for the X-Y scales for clarify. The values of the cross-correlation range from −1 to 1, where the positive value represents the residues moving in the same direction and the negative value means the residues moving in the opposite directions. Correspondingly, the regions in red color represent positive correlations and the blue regions denote negative correlations. (F) The cross-correlation map of the 20 slowest ANM modes for mDNMT1 (732–1600) in the helix-straight state. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Subsequently, we compared how well the structural changes observed between the helix-straight and helix-kinked states are reproduced by the low-energy modes. The experimental deformation vector has been calculated as the difference between the coordinates of both states after being structurally aligned. The blue bar plots show that the mode u2helix-straight yields an overlap (cosine correlation) of 0.25 (Fig. 2C), and u4helix-kinked yields an overlap of 0.35 (Fig. 2D), based on the whole structure of mDNMT1 (732–1660). This is consistent with the overlap table in Fig. 2A, in which the clamp motions between the TRD region and BAH1 in both states are important for substrate DNA binding. Whereas the conformational differences between the two states are mainly located in the TRD region and the catalytic helix as aforementioned, especially for the TRD region. The overlaps between ANM modes and structural changes were further calculated by focusing on TRD region in both states. As expected, the overlap values are much higher (red bars in Fig. 2C and D) with values of 0.83 and 0.86, respectively. It further indicates that the TRD clamp motion shared by two states is the functional mode for the interconversion. Compared with the coarse-grained ANMs, molecular dynamics (MD) simulations considering the detailed atomic interactions, can capture the local conformational changes, such as the straight or kinked catalytic helix. Through MD simulations and biochemical studies in our previous research, such conformational transition in the catalytic helix was observed, in which residues N1248 and R1279 have been proved to play crucial roles in biasing the catalytic helix to either the kinked or straight conformation [37].
In addition, we have evaluated the intra-/inter- modular cross-correlations between residue fluctuations for both mDNMT1 (732–1600) structures. Expect for the two functional modes, which facilitate the conversion between the two states, we have also investigated the lowest-frequency mode which is extremely conserved in both states. Fig. 2E shows the resulting correlation matrix based on the first ANM mode in the helix-straight state, which mainly shows the rigid-body motion defined by the strong coupling of amino acids through the intra-domain interactions. As the motion of this mode is shared in both states, the cross-correlation maps are accordingly identical in helix-kinked (data not shown). The coupled regions are in line with the shared modular architecture and quaternary arrangement of both states, e.g. strong (positive) correlations are observed within the BAH1, BAH2 and CD domains. For inter-domain couplings, the residues in BAH1 domain have high coupling motions with the residues in CD, and the BAH2-TRD loop also exhibits strong positive correlations with TRD region. Furthermore, the cross-correlation maps based on the first 20 modes (Fig. 2F) again emphasize the intra-domain coupling in the collective motions, and the inter-domain correlations are further represented. For inter-domain coupling, the regions (residues 800–820, 840–853 and 870–883) in BAH1, involved in BAH1 and CD core interactions, exhibit positive correlations with the residues (1100–1300) in CD core (blue box I). Besides, the BAH2-TRD loop also exhibits strong positive correlations with TRD region (blue box II). Taken together, the intrinsic dynamics of mDNMT1 (732–1600) are sequentially encoded by both the intra-domain interactions that is the main feature for correlated behaviors, and the inter-domain interactions, which are mainly involved in the domain interfaces, indicating the dynamical regulation encoded by the architectures of DNMT1 folds.
3.2. RFTS domain regulates the intrinsic dynamics of DNMT1 through hinge sites
Nowadays, DNMT1 structures encompassing the RFTS domain have been resolved in hDNMT1 (351–1600) (PDB ID: 4WXX) [9] and mDNMT1 (357–1608) (PDB ID: 3AV5) [8], in which the RFTS domain directly associates with the DNA-binding site in CD, acting as an autoinhibitory factor and inhibiting the binding of the substrate DNA. hDNMT1 and mDNMT1 share high sequence similarity (83%) and the root-mean-square deviation value is only 1.0 Å over their superimposed structures. Due to the high similarity in structural comparison, they share similar dynamics behaviors in collective coupling motions. In this section, ANM modes were used to describe the collective motions encoded by the architectures of DNMT1 with the introduction of RFTS, while principle component analysis (PCA) based on MD ensembles was also performed to detect the functional dynamics in consideration of the detailed atomic interactions.
Although there are weak overlaps between PCA and ANM modes (Fig. S2A, upper panel), both PC1 and ANM1 can detect the significant motions of RFTS domain. The first mode of PCA (Fig. 3A) consists of the predominant motions in which the N-lobe of RFTS and CXXC display the largest fluctuations, with BAH1 and BAH2 in relatively small coupling motions. Whereas in ANM mode 1 (Fig. 3B), the N-lobe of RFTS, together with CXXC region and TRD region, rotates in anti-clockwise mode; with BAH1 and BAH2 in relatively small coupling motions. The structural variations represented by PC1 show certain overlap (0.40) with ANM 1, in which the large motions of RFTS and CXXC indicate their potential allosteric regulation in DNMT1 activation. These motions of RFTS domain are considered to be functional relevant, as RFTS can inhibit the de novo methylation by occlusion of the catalytic site of DNMT1, which should be removed from the catalytic site for methylation to occur [6], [8], [9]. As expected, the overlaps between PCA modes and ANM modes become much higher when only consider RFTS domain motions (Fig. S2A, lower panel), reproducing the consistent large displacement for RFTS domain in both modes. Whereas in the mode 2, from both in PCA and ANM, the RFTS also displays largest fluctuations in the collective motions (Fig. S2B and C). Taken together, as the inhibitory element for occupying the binding site of DNA substrate, RFTS domain herein displays the largest fluctuations in the collective motions in both the coarse-grained ANM and sophisticated MD simulations, signifying their crucial roles in the allosteric regulation for DNMT1 accessible for DNA binding.
Fig. 3.

Intrinsic dynamics of hDNMT1 (351–1600). (A) The motion of PCA mode 1 and (B) ANM mode 1 of hDNMT1 (351–1600). (C) Distributions of the mode shapes in GNM mode 1, 2, 3 of hDNMT1 (351–1600), while the global hinge residues are labeled. (D) Structural mapping of global hinges predicted by GNM mode 1 (vertical plane), mode 2 (middle plane), and mode 3 (three triangles) in hDNMT1.
Towards further understanding the intrinsic dynamics of proteins, GNM was used to predict the hinge sites which are key residues just encoded in the molecular topology. Fig. 3C shows the mode shapes of the first three GNM modes of hDNMT1 (350–1600). In the GNM mode 1 (Upper panel), the RFTS together with CXXC, BAH2-TRD linker and TRD region, undergoes large displacement, while BAH1 and other residues in BAH2 and CD undergo relatively small but anti-correlated motion with respect to the RFTS. The hinge residues predicted in this mode (blue beads in Fig. 3D) are mainly located at the domain interface, representing the anchored sites for mediating coupling motions between the upper and lower parts. In the GNM mode 2 (Middle panel), BAH1 and BAH2 undergo the largest movement in anti-correlated motions, and the residues in the core of RFTS and CD serve as the anchors, which locate around the vertical plane of hDNMT1 (pink beads in Fig. 3D). Herein, E525, D526, D583, L587 and G589, involved in the association of RFTS with CD, play crucial roles in the coupling collective motions between domains. Whereas in the GNM mode 3 (Lower panel in Fig. 3D), the coupling motions mainly happen between the N-lobe and C-lobe of RFTS, and between BAH1 and BAH2 domains, with the hinge residues locating at the three triangle parts of hDNMT1 (gray beads in Fig. 3D). Specifically, E504-K505-I506-Y507 in the α-helix (αRS: residues 495–519 in hDNMT1 (350–1600)) are identified as the hinges for the coupling motions between the N-lobe and C-lobe of RFTS. This is consistent with the observation that the α-helix, connecting the two halves of the RFTS domain, adopts a straight conformation in hDNMT1 (350–1600) but is kinked in the structure of mDNMT1 (357–1608), resulting in the orientation of N-lobe by 19° [10].
The large fluctuations of RFTS domain and the location of hinge residues at the domain-domain interface, proposed that the RFTS domain may mediate the inter-domain interactions between RFTS and CD through hinge sites. To support this idea, we subsequently compared the modes for DNMT1 with and without RFTS, using the subsystem/environment coupling method. In this case, mDNMT1 (732–1600) without RFTS-CXXC is the subsystem, and RFTS-CXXC stands for the environment. Herein the overlaps between the 20 lowest-frequency modes accessible to mDNMT1 (732–1600) (y-axis) alone without RFTS-CXXC and the corresponding subsystem adding RFTS-CXXC (x-axis) are shown in Fig. 4A (Upper panel). All the modes accessible to mDNMT1 (357–1608) encompassing RFTS are maintained with an overlap of 0.7 or above in the isolated mDNMT1 (732–1600). Whereas almost half global modes (13th–20th) are only favored by the isolated mDNMT1 (732–1600), which are not observed in the whole mDNMT1 (357–1608). As the global modes (13th–20th) in mDNMT1 (732–1600) that induce high fluctuations at particularly in TRD and catalytic helix, are practically restrained in mDNMT1 (357–1608) with the introduction of RFTS. As shown in the structural representation in Fig. 4A (Lower panel), the TRD region and catalytic helix are the key elements at the inter-domain interface for RFTS insertion, and this binding imposes high constrains quenching their motions through inter-domain interactions. The modes (13th–20th) accessible in mDNMT1 (351–1600) show high fluctuations in RFTS and CXXC regions, indicating their regulation roles in the catalytic methylation of DNMT1.
Fig. 4.

Difference of intrinsic dynamics between mDNMT1 (357–1608) and mDNMT1 (732–1600). (A) The overlap values represented by the correlations between the top 20 modes accessible to mDNMT1 (732–1600) alone and in the whole mDNMT1 (357–1608) (upper panel). The mDNMT1 (732–1600) stands for the subsystem represented in blue and the RFTS and CXX C region stand for the environment in gray (lower panel). (B) The square fluctuations of 20 ANM modes in mDNMT1 (732–1600) and mDNMT1 (357–1608) (Upper panel). The square fluctuations for the trajectories of MD simulations in mDNMT1 (732–1600) and hDNMT1 (351–1600) (Lower panel). (C) The cross-correlation map of the 100 slowest modes for mDNMT1 (357–1608). (D) The difference map of cross-correlations between mDNMT1 (732–1600) and mDNMT1 (357–1608). In particular, the larger positive correlations between BAH1 and CD, between BAH2-TRD loop and CD, as well as between the TRD region and the residues in CD core have been highlighted (blue boxes). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
We further calculated and compared the square fluctuations for mDNMT1 structures with RFTS and without RFTS, based on the first 20 ANM modes and MD trajectories retried from our previous study [37], as shown in Fig. 4B. Both results clarify that the main effect by the introduction of RFTS in DNMT1 structures is to suppress the mobility of the catalytic site and TRD region in CD (as shown in the black boxes in Fig. 4B), which are involved in the RFTS-CD interface. The reduced dynamics of both regions are much remarkable in MD simulations, in the consideration of atomic interactions. These inter-domain interactions limit the movement of CD and make it accessible to the helix-kinked state (also named autoinhibitory state). This observation is consistent with the overlap value of the global modes between DNMT1 structures with and without RFTS, in which the introduction of RFTS quenched certain motions observed in the one without RFTS. Subsequently, we have also investigated the cross-correlations of dynamical behaviors encoded by mDNMT1 (357–1608) (Fig. 4C), in which the N-lobe of RFTS exhibits obvious negative coupling correlations with CD and small positive correlations with BAH1 and BAH2 domains, whereas the C-lobe of RFTS has positive coupling motions with CD (blue box). The further investigation of the difference of cross-correlations between mDNMT1 (357–1608) and mDNMT1 (732–1600) was performed. It is worth noting that with the introduction of the RFTS domain, the cross-correlations between residues in intra-domains have obviously been strengthened, especially for CD (as shown in the difference map in Fig. 4D). On the other hand, the cross-correlations between residues in inter-domains, including between BAH1 and CD and between BAH2-TRD loop and CD (blue boxes), have also been strengthened. Thus, the strengthened cross-correlations within intra- or inter- domains introduced by RFTS limits the dynamical modes of the overall topology to some extent. This observation indicated that the introduction of RFTS, which possesses large flexibility itself in collective motions, strengthened the inter-domain regulation mainly through the enhanced modularity in intra-domain.
3.3. PRS reveals the allosteric potential of the RFTS domain in hDNMT1 (351–1600)
Based on ANM calculation, the perturbation-response scanning (PRS) approach was employed to quantify the allosteric effect of each residue in the protein structures on all other residues in response to external perturbation. First, the PRS map provides information on the sensitivity and effect of a given residue in transmitting signals (Fig. 5A). Then, two groups of residues including sensors and effectors can be predicted from this map, which are potentially involved in allosteric signal sensing and transmission. In Fig. 5A, the PRS map highlights some key residues that can be served as the strongest sensors and effectors in hDNMT1 (351–1600) structure. The sensor profile shows some peaks at E393, S394, G742, K743, N1006, G1007, K1115, and G1116, which are located at the top of RFTS domain, the bottom of BAH1 and BAH2domains, and the KG linker (Fig. 5B). Whereas the effector profile shows some peaks at H405, K406, L407, F410, S436, G437, S438, and Y486 in the core region of RFTS, F792 and L825 in BAH1, E971 in BAH2-TRD linker, K1015 and I1016 in BAH2, and P1501 in TRD region, which are clustered into four regions as labeled in Fig. 5C.
Fig. 5.

PRS of hDNMT1 (351–1600) highlights residues acting as effectors and sensors for signal transduction. (A) Normalized PRS map, where the strongest signals are shown in red (as scale on the right). The plot along the above ordinate shows the average propensity to transmit perturbation (each point therein is the average over all elements of the PRS matrix in the corresponding column); and that along the left is the average propensity to sense perturbations (average over all elements in each row). The corresponding peaks are effectors and sensors, respectively. (B) The hDNMT1 (351–1600) structure color-coded by ability to sense perturbations, where red regions are strongest sensors. (C) The hDNMT1 (351–1600) structure is color-coded by ability to propagate perturbations, in which the red regions correspond to the strongest effectors, which are grouped into several clusters. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Through structural observation, the sensor residues are mostly involved in the protein–protein interactions, including RFTS-ubiquitin complex and hDNMT1-USP7 complex. In the crystal structures of hDNMT1 RFTS domain in complex with two ubiquitins (PDB ID: 5YDR) and H3-ubiquitins (PDB ID: 5WVO), the long loop (L386-Q404) protruding RFTS domain, which is predicted to be enriched as sensors in the PRS calculation, is sandwiched by both ubiquitins (Fig. S3A). In detail, Glu384, Asn392, Phe396, Glu397 and Try399 in the protruding loop of RFTS form salt bridge or H-bonds with the residues in ubiquitin 1. On the other hand, Glu384, Ile388, Asp390 and Glu397 form salt bridges or H-bonds with the residues in ubiquitin 2. At the same time, a large number of hydrophobic interactions are also formed between the interfaces of RFTS and both ubiquitins. The mutants, including Y399G and E384A/E397A, displayed impaired binding to ubiquitin, and were unable to restore global DNA methylation in DNMT1-/- ES cells [51]. The disruption of the interactions between DNMT1 and ubiquitinated histone H3 could also affect the nuclear localization of DNMT1. The molecular mechanism underlying the binding of ubiquitinated H3 results in the spatial rearrangement of the two lobes in RFTS, indicating the displacement of RFTS and the subsequent opening of the active site in DNMT1 [17]. Whereas in the complex structure of hDNMT1 with USP7 (PDB ID: 4YOC) [52], lysine residues within the KG linker loop (K1109–K1119), also acting as sensors in PRS calculation, form a network of H-bonds and salt bridge contacts with acidic residues in USP7 (Fig. S3B). The acetylation mimic mutant (K1111Q/K1113Q/K1115Q/K1117Q) or acetylation of the four lysine residues on the KG linker lead to a decrease in its binding affinity to USP7 and promote the degradation of DNMT1, suggesting these intermolecular interactions are required for USP7-mediated stabilization of DNMT1.
The effectors are grouped into several clusters through mapping into the structure (Fig. 5C), including the core regions of RFTS domain, BAH1 and BAH2 domains, and TRD region. Most importantly, the residues distributed in the N-lobe of RFTS domain represented as the strongest effectors, including H405, K406, L407, F410, S436, G437, S438, A439, and Y486, near the H3-ubiquitin binding site. In BAH domains, F792 and L825 in BAH1, and K1015 and I1016 in BAH2 serve as the strong effectors. The ablation of BAH domains causes DNMT1 to be excluded from replication foci even in the presence of RFTS in vivo, suggesting the essential roles of BAH domains in the methylation maintenance [53]. Besides, the effectors include residues P1501 and H1502 in TRD and E971 in BAH2-TRD linker. P1501 is adjacent to the TRD loop 1, which inserts into DNA from the major groove. The side-chains of H1502 and T1503 form H-bonds with RFTS domain, acting as hinge sites in GNM mode 2 and thus acting as crucial effectors in allosteric signal transduction. Furthermore, the effectors, enriched in the RFTS domain, presumably affect its folding and stability, which are efficient in the allosteric signal transduction and in turn lead to the dysregulation of DNMT1-mediated methylation. The PRS analysis of hDNMT1 (351–1600) again emphasized the allosteric roles of RFTS in sensing perturbations and effectively transmitting the allosteric signals in the regulation of DNMT1 methylation.
3.4. Network indicates that RFTS-CD interactions facilitate the communication between the local allosteric networks
Allosteric regulation, as well as the structural stability of proteins, relies on its complex network of inter-amino acid interactions. Understanding of these interactions and their networks provides for a systems-based powerful analysis to quantify the allosteric modulation of protein machines. Hence, the dynamics-based network analysis [46] was used to detect the allosteric networks within hDNMT1 (351–1600). In this method, the protein structure networks (PSNs) of hDNMT1 (351–1600) was constructed based on 1131 frames from the trajectory of 200 ns MD simulation. The shortest paths of long-range paths were then calculated, which are likely to transmit a “signal” over long distances within the protein structures more efficiently.
With the restricted condition in PSNPath module, the shortest paths with occurrence probability >30%, cluster in intra-domains. The recurrence analysis of path links led to the building of metapaths made of the most recurrent links in the whole pool of paths. The cross talks among these metapaths form several local allosteric networks with effective signal transmitting in intra-domains. As depicted in Fig. 6A, the local allosteric networks based on the metapaths were clustered within each domain, with the residues in the catalytic site forming the largest one (Fig. 6B). The residue pairs, including T621/L623-D1256, T619-L1195/N1192, and F631-L1282/R1285, form H-bonds or hydrophobic contacts for docking the RFTS-CXXC linker region against CD. Besides, of particular note is the salt-bridge forming pair D1256-R1259 and the electrostatic-π stacking interaction between R1259-W843 at the BAH1-CD interface. Herein, T619 and W843, captured near the hinge sites in GNM, combined with other dense hydrophobic contacts, facilitate the perturbations from the CXXC linker and BAH1 transmitting to the catalytic site. In the N-lobe of RFTS (Fig. 6C, upper panel), it is noteworthy that Q379 was captured as sensor, and F410, F435 and Y486 serve as effectors in PRS calculation, supporting the signal transmission from external perturbations, such as H3-ubiquitin binding. Whereas in the C-lobe of RFTS (Fig. 6C, lower panel), the links involved in the α-helix (residues 495–519) mediate the orientations between the N-lobe and C-lobe of RFTS. The appearance of hinge residues H553 and F556 in the network further emphasized their crucial roles in mediating the coupling motions and allosteric signal transduction. In the adjacent TRD region (Fig. 6D, upper panel), W1498 and H1502 in the TRD interface and adjacent to hinge sites, most likely play roles in mediating the coupling motions and allosteric regulation. H1502 was also captured as strong effector in PRS, and several residues were captured in the long range paths, contributing to the signal transmission from RFTS to the TRD local allosteric network. Whereas in the TRD region (Fig. 6D, lower panel), the salt-bridge forming contacts form the dense allosteric network for signal transmission in this region.
Fig. 6.

Intra- and inter- domain allosteric interactions in hDNMT1 (351–1600) structure. (A) The local allosteric networks formed by metapaths are represented, with residues shown in small sphere and the links in red, while the long-range paths across RFTS-CD are also represented, with residues shown in big sphere and the links in black. The detailed representation of the local allosteric networks in the catalytic site (B), in the N-lobe and C-lobe of RFTS (C), and in the TRD region (D). The representative long paths across the TRD interface (E) and catalytic interface (F). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
PSN combined with MD not only unveiled the hierarchical modularity of local allosteric networks, but also proposed the inter-domain interactions served as the allosteric functional hotspots responsible for the signaling regulation. The RFTS domain has be suggested as a mediator of interdomain allostery in DNMT1, we thus focus on the intra-domain interactions between RFTS and CD domains. Although there are a multitude of energy transfer pathways across domains, these paths share a few common nodes that represent effective “chokepoints” for communication. These “chokepoint” residues were identified as the strongest and best conserved contacts involved in inter-domain interactions. As shown in Fig. 6A, two clusters of paths were identified from RFTS to CD, including through the TRD interface and catalytic interface, in agreement with the collective dynamics as aforementioned.
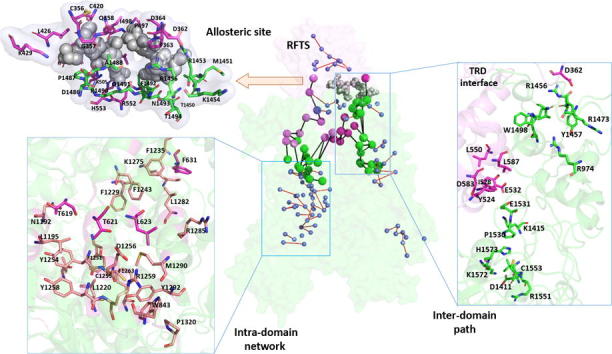
Through TRD interface (Fig. 6E), the high-concurrency paths, including L550/D583/L587 → Y524 → I528 → E1531/E532 → K1415 → P1530 → H1573 → C1553/V1550 → K1572/D1411 → R1551 starting from L550, which is located in the kinked helix, and D362 → R1456 → Q1491 → W1498 → V1475 → Y1457 → R974 → R1473, transmit signals between the local allosteric networks in the C-lobe of RFTS and TRD respectively. High occurrence of residue pairs revealed a strong flow of allosteric information between RFTS and TRD region. A closer inspection reveals that these pairs are mainly involved in electrostatic or H-bond interactions, including I528-E1531, E532-K1415, D362-R1456, D548-R1424, L587-W1510, L551-M1533, and Q555-R1490. Again medium/long-chain charged or polar residues appear to be fundamental in conveying allosteric information through the TRD interface. In particular, TRD loop1 (residue 1501–1516) and TRD loop2 (residue 1530–1537), which penetrate in DNA binding, appear to form an important allosteric center for mediating communication between RFTS and TRD. Whereas through the catalytic interface (Fig. 6F), paths W464 → Q598 → Q560 → V513 → Y564 → E610 → S570 → K611 → D571 → R1238, E572 → K1242 → P615 → S1246 → F1229 → F1274 → N1270 and L550 → Y524 → D583 → K586 → E703 → R582 → D702 → N1233 → E698 transmit signals from RFTS, through CXXC and the linker region, to the largest local allosteric network surrounding the catalytic site in CD. Specifically, residue pairs D571-R1238, P615-S1246, D702-N1233, D702-K1275, E703-S1230, and E572-K1242, represent the “chokepoints” for the inter-domain interactions. Consistent with the collective motions, these “chokepoints” were mostly captured as hinge residues, indicating their crucial roles in coupling motions and information flow. This is consistent with the fact that the hinge residues appear as the most frustrated region in the complex. Such chokepoints displaying high occurrence are likely to play important roles in allosteric mechanisms and should be good candidates for mutagenesis studies. The biochemical experiments identified that mutants, including R582E, and with removing residues 694–701 (denoted as Δ694-701), obtain higher methylation activity, demonstrating their allosteric inhibitory roles for the enzymatic activity [9]. In comparison, the connections are strong between the C-lobe of RFTS and local allosteric network in TRD, whereas the strength of communications between RFTS, CXXC, the linker and active site in CD appears to be quite weak.
Besides the dynamics information, the sequence evolution adds another dimension for probing the communication within proteins. By using four methods to explore the coevolutionary information within DNMTs sequences, the maps of the coevolutionary matrixes are presented in Fig. S4. The coevolutionary network with the residue pairs captured by the four methods further supported the local networks within each domain (Fig. S5). We have found that coevolutionary networks can recapitulate key regulatory sites and interactions responsible for allosteric signaling and can form local allosteric networks as the dynamical networks in DNMT1. This is consistent with the evolutionary remark that the domain is evolutionary conserved for their functions. Taken together, the network analysis has recapitulated the long-range paths across RFTS and CD, mainly through the TRD interface and catalytic interface. The long-distance communication paths between RFTS and CD domains are a reminder of its dynamical plasticity and the biological role of its activation segment. The “chokepoint” residues involved in the inter-domain interactions are also captured as hinge sites in collective motions. These long paths connect the local allosteric networks within each domain that are highly efficient in transmit signals within domains, especially for the catalytic site. The allosteric regulation of the plastic RFTS are mediated by the long-range paths across domains and dense local allosteric networks simultaneously.
3.5. Mutational analysis validates the functionality of the RFTS domain
Recent advances in whole genome association studies have identified that the RFTS domain has been found to be enriched with a lot of mutation hotspots associated with several loss-of-function phenotypes [54] (Fig. S6 and Table S1). The pathogenesis mutations in RFTS cause both hereditary sensory and autonomic neuropathy with dementia and hearing loss type IE (HSAN IE), and autosomal dominant cerebellar ataxia, deafness and narcolepsy (ADCA-DN). Amongst, mutation hotspots K505, Y524, N529, I531, H553, A554, C580, G589 and V590 are identified as hinge sites in GNM low-frequency modes. Specifically, mutations D490E&P491Y and Y495C (amino acids 351–600), located near the hinge sites, form inclusion bodies which are insoluble attributed to the misfolded structures [55]. In addition, three mutation hotspots Y495, K505, and H553 are also involved in the local allosteric network, further consolidate that the disruption of these atomic interactions would transform the structure of RFTS and affect the recognition and binding procedure of hemimethylated DNA, creating abnormal methylation and gene silencing. The mutations, captured as the hinge sites, would affect the coupling motions and rearrangement of DNMT1 domains. It is thus interesting to investigate the conformational dynamics of all disease mutations. Herein, we have examined these loss-of-function mutations in RFTS domain, using dynamics-, sequence-, energetic- based computations and identify their shared molecular features.
Firstly, mean square fluctuation (MSF) based on all GNM modes were calculated to describe the conformational dynamics (Fig. 7A). As expected, the MSF profile displayed quite small values (MSF < 0.01) for all reported disease mutations, showing a significant structural stability as observed in the collective motions. Next, the effective profiles of the RFTS domain (Fig. 7B) calculated by PRS analysis were used to measures the average impact of the perturbed site on all other residues. Overall, disease mutations displayed moderate effectiveness. The PRS analysis revealed that some disease mutations locate at the RFTS-CD domain interface, including Y524, I531, A554, V590, were distributed at the peaks of thee effector profiles. The RFTS-H3-ubiquitin interfacial residue C353 also emerged as the peak in the effector profile. This residue is of special interest, since it may be involved in long-range allosteric signaling through the protein–protein interface. The observed effectiveness of disease-associated mutations also verifies their involvement in functional motions and potential regulatory role in the allosteric mechanism. We suggested that mutations located at interfaces may cause not only local destabilizing effects but also lead to the increased flexibility across the interface and impair the long-range allosteric cooperativity.
Fig. 7.

Results of molecular signatures of disease mutations on the RFTS domain. Mapping disease mutations to (A) the MSF, (B) the effectors, (C) the conservation score and (D) the stability changes ΔΔG profiles. The positions of clinical disease mutations are annotated as pink dots. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Other two relevant parameters were used to describe the molecular signatures of disease mutations are the evolutionary conservation and energetic stability patterns. The conservation score of the RFTS domain was calculated using MSA profiles of the HATPase_c family (PF02518) to evaluate the sequence conservation of the RFTS domain, where 1 indicates the most rapidly evolving positions, and 9 indicates the most evolutionarily conserved positions. As shown in Fig. 7(C), expect for P491 and I531withmoderate conservation grade, other disease-associated mutations are corresponding to highly conserved node (i.e., with a conservation grade ≥8). Finally, the free energy change ΔΔG of RFTS domain was calculated to explore the thermodynamic determinants of mutational hotspots. Using MAESTRO [56] and alanine scanning methods, each residue in RFTS domain was mutated to alanine to compare the stability changes brought by the specific residue, so as to obtain the results of mutation hotspots and residual sensitivity (Fig. 7D). It was observed that alanine mutations cause most sites to be unstable (ΔΔG > 0), while the mutation sites known to cause disease have relatively large ΔΔG values. Amongst, hereditary sensory and autonomic neuropathy with dementia and hearing loss type IE (HSAN IE) involves sites (C353, T481, Y495, Y524, I531, H553) corresponding to the peaks of the mutation hotspots. Interestingly, the energetic stability profile showed some significant peaks of C353, Y524, A554, V590, which correspond to the effectors. It may suggest that allosteric sites process the large destabilization effect upon alanine mutations.
In summary, we have employed four matrixes to describe the molecular signatures of mutational hotspots in the RFTS domain. Our findings have suggested that mutations with severe phenotype have high structural stability and evolutional conservation. Some variations act as major effectors of allosteric interactions with other domains or protein partners. Thus the variation could cause adverse effects on the structural stability, disrupting the recognition and binding procedure of hemimethylated DNA and leading to abnormal methylation as well as gene silencing. The result suggests that these mutations in RFTS domain are not only implicated in severe phenotypes, but also consist of central receivers of allosteric signals and hinge sites in collective dynamics. Similar results have been obtained for several tumor suppressor proteins [57].
3.6. Allosteric modes of hDNMT1 (351–1600) and its implication in drug design
Allostery is a universal phenomenon in complex protein systems. Two classical models of allostery have been proposed: MWC (Monod–Wyman–Changeux) model assumes that a concerted, all-or-nonechange in all subunits [58], and KNF (Koshland–Nemethy–Filmer) model assumes that a sequential transition of individual subunits [59]. Allostery is not always accompanied with conformational change, but also based on the vibrational modes of the proteins without any major structural changes [60]. Despite much progresses having been made in the repertoire of allostery since the turn of the millennium, the identification of allosteric drugs against therapeutic targets and the elucidation of allosteric mechanisms still present substantial challenges [61]. These challenges are derived from the difficulties in the identification of allosteric sites, the assessment of allosteric protein–modulator interactions, the screening of allosteric modulators, and the elucidation of allosteric mechanisms in biological systems. Amongst, the identification of allosteric sites is the first step for structure-based allosteric modulator discovery. Despite some successes in understanding allostery of DNMT1 have been achieved [62], the physical principle of allosteric regulations is still unclear due to its complexity, and diversity of multi-domains.
In our study, both dynamics and coevolutionary network models were used to reveal the role of key amino acids in allosteric mechanisms and their evolutionary conservation among homologous proteins. Although there is no one-to-one correspondence, both allosteric and co-evolution network analysis have divided DNMT1 into local allosteric networks, with each corresponding to evolutionarily allosteric residues within different domains. The result also means that different domains have different biological functions, while functional dynamics of each domain is conserved during the evolution [63]. For example, the residues in CD constituting evolutionary network contribute to the functions towards modulating the catalytic activity. On the other hand, the hierarchical organization of DNMT1 endows the RFTS domain with inherent regulatory plasticity, while the PRS analysis indicates the allosteric potential of RFTS domain that plays regulatory interactions with other domains through the inter-domain interactions. The RFTS domain has long been implicated in subcellular localization, protein association, and catalytic function. The kinetic analysis with a fluorogenic DNA substrate indicates the RFTS domain as a 600-fold inhibitor of DNMT1 enzymatic activity [12]. The DNMT1 E572R/D575R variant with the removal of RFTS domain has a higher DNA methylation activity in human cells, further supporting the autoinhibitory role of the RFTS domain [64]. In the movement of RFTS domain from the catalytic site, both ubiquitin and UHRF binding with RFTS could contribute to its translocation and DNMT1 activation [51]. Besides, in the research of the cell-cycle dependent dynamics of DNMT1 by fluorescence recovery after photobleaching (FRAP) and diffusion-coupled modeling [65], the RFTS domain could bind to both PCNA-enriched replication sites and nearby pericentromeric heterochromatin subregions for DNMT1 localization. The RFTS domain-dependent binding in the late S phase is substantially stronger (residence time, Tres ~ 22 s) than PBD-dependent interaction (Tres ≤ 10 s) in the early S phase. Here in our research, according to the observed interplay between conserved allosteric, coevolution network and structural plasticity regulated by the RFTS domain (Fig. 8A), our proposed allostery model contains two properties: (i) the domain based-allostery model featured with local allosteric networks with high signal transmission, fitting with dynamics-driven allosteric model [66], regulated by (ii) the structural rearrangement of the RFTS domain and long-range paths across RFTS and CD, which allosterically regulate their catalytic activity. Taken together, our proposed model not only gives insights into the allosteric mechanism of DNMT1, but also suggests a novel strategy for drug discovery.
Fig. 8.

(A) The proposed allosteric model for hDNMT1 (351–1600), including both local allosteric networks within each domain and long-term communication pathways induced by the RFTS domain. (B) The allosteric sites 1–5 are represented in hDNMT1 (351–1600) structure, which were predicted by both Allosite and AlloPred web servers. (C) The binding properties, scoring functions, and constituting residues for the pocket 4 located at the DFTS-CD domain interface.
DNMT1 has emerges as an important drug target for various human diseases. Current structure-based drug design against DNMT1 mainly targets its highly conserved orthosteric pocket of the catalytic domain [67], [68]. As allosteric modulators bound to structurally diverse allosteric sites can achieve better pharmacological advantages than orthosteric ligands. Herein, the web servers of Allosite [69] and AlloPred [70] were used to detect the allosteric pockets in hDNMT1 (351–1600). Both methods were applied to predict allosteric sites using SVM based on topological and physiochemical pocket features from Fpocket algorithm. As shown in Fig. 8B, five potential allosteric pockets were identified in hDNMT1 (351–1600), and the allosteric binding properties, scoring functions and constituting residues of these pockets are denoted in Table S2. Of particular interesting is the pocket 4, which is located at the RFTS-CD domain interface, with the detail information also shown in Fig. 8C. Among pocket residues, K505, R552, H553, P1487, and A1488 were identified as crucial hinge residues in the GNM collective motions. Meanwhile, residues D362, R1456, and Q1491 were captured in the long-range path for inter-domain communications. Moreover, most residues in pocket 4 are involved in the local allosteric networks in the C-lobe of RFTS and TRD region, indicating the allosteric perturbation introduced by binding modulators in this site. The dynamics properties of this allosteric pocket are thus well defined by locally correlated motions of these residues, which are involved in the local allosteric networks and connected by long-range paths. Moreover, K505 and H553 were found overlap with disease mutations. Taking these residues together as allo-targeting site would impact the coupling between RFTS-CD interface and the catalytic site, providing new avenues for the rational design of allosteric modulators of DNMT1.
4. Conclusions
DNMT1 is responsible for preserving DNA methylation patterns that play important regulatory roles in cell differentiation and development. Recently, a series of multidomain crystal structures of DNMT1 were determined, which provide us an opportunity to understand its additional allosteric regulation in the full-length DNMT1 structures. The study of the structural organization of allosteric residues and their detailed interactions in DNMT1s is crucial for studying biological regulation and allosteric drug design. In this work, we have employed an integrated computational method for the systematic modeling the intrinsic dynamics and allosteric regulations in DNMT1s. A summary of the computational approaches and related results, as well as the agreement with known experimental data, are shown in Fig. 9. Major findings are listed as following:
-
1)
The ANM analysis was performed to investigate the intrinsic dynamics of mDNMT1 (732–1600) with two different conformations, including their collective motions and cross-correlations. Both the helix-straight and helix-kinked mDNMT1 (732–1600) captured the shared global modes of clamping motions between TRD region and BAH1 domain, which are consistent with the conformational changes involved in different crystal structures, especially for the TRD region [6], [7] (red box in Fig. 9). This clamp motion is proposed as the functional modes for interconversion, and important for substrate DNA binding. Through MD simulations and biochemical studies in our previous work [37], the conformational transition in the catalytic helix was observed, in which residues N1248 and R1279 have been proved to play crucial roles in biasing the catalytic helix to either the kinked or straight conformation. Herein, the shared TRD clamp motion agrees with the conformational change and is the complement for our previous MD study. The combination of ANM and MD results could better decipher the conformational space of DNMT1, underlying the dynamics for substrate binding and methylation activity.
-
2)
The ENM and PCA study has given the first insight of the intrinsic dynamics of DNMT1 structures with the RFTS domain. Both the slow modes in ANM and PCA analysis have captured the large domain motions of RFTS, which restrains the fluctuations of CD and strengthens the positive correlations between intra- and inter- domains. The large domain motions of RFTS are consistent with its autoinhibitory roles for occupying the catalytic site and being released for substrate DNA binding and methylation activity. The research focusing on the crystal structure of RFTS complexed with H3-K18Ub/23Ub and MD simulations has proved that the RFTS domain can be dissociated from the catalytic site upon ubiquitin binding, leading to the activation of DNMT1 and supporting the large domain motions of RFTS herein [17] (red box in Fig. 9). Therefore, our computational results have demonstrated the dynamical potential of the RFTS domain, showing an agreement with the NMR [51] and fluorescence [65] experimental data.
-
3)
The GNM calculation of hDNMT1 (351–1600) has predicted the hinges, serving as the mediators of interdomain correlations by the introduction of RFTS domain. In addition, the PRS analysis further gave two kinds of key residues in allostery, namely effectors and sensors. The strongest effectors locate at the N-lobe of RFTS domain, and the overlap with the hinge sites further confirmed the functional roles of these residues. To date, many experimental reported mutational hotspots shared the same region of RFTS domain with these hinge sites and effectors (purple box in Fig. 9). Recently, two mutants of G589A and V590F significantly decrease the thermal stability of DNMT1 and partially relieve the autoinhibition by RFTS domain [71]. The two residues G589 and V590 in the RFTS domain are captured as hinge sites in GNM mode 2, emphasizing their crucial roles in domain dynamics. In addition, the predicted sensor residues are mostly involved in RFTS-ubiquitin and DNMT1-USP7 interactions. Alterations of some sensor residues, including Y399G, E384A/E397A, and K1111Q/K1113Q/K1115Q/ K1117Q, led to reduced binding to its cofactors and resulted in impaired methylation activity [52] (purple box in Fig. 9).
-
4)
The integration of PSN and MD analysis revealed both intra and inter-domain allosteric communications in hDNMT1 (351–1600), while the coevolutionary network further emphasized the key functional residues and complemented the correlation from the evolutionary pressure. In the meantime, many hinge sites and mutational hotspots were captured as the mediators of allosteric communications. During the two clusters of paths predicted by the dynamics-based network analysis, several residues were validated to be important for methylation activity of DNMT1. For example, mutations in mDNMT, including W1512A (corresponding to W1510 in hDNMT), M1535A (M1533 in hDNMT), S1233A (S1230 in hDNMT), N1236A (S1233 in hDNMT), and S1249A (S1246 in hDNMT), showed reduced methylation activities in different degrees [7], [37], confirming the functional roles of the predicted pathways (blue box in Fig. 9). In the end, the pocket proposed in the TRD interface, consisting of the residues captured by the hinge sites, the mediators in the pathways, and gain-of-function mutations, represents the high potential allosteric site for targeting DNMT1.
Fig. 9.

A summary of the computational approaches and results of DNMT1s, the comparisons with experimental data are also briefly given (three boxes in the right panel).
In summary, our computational study has shown that the allosteric potential of RFTS domain in DNMT1 structures from different aspects, including intrinsic dynamics and communication pathways. The results of the computational investigation are compared with different experimental studies, including crystal structures, known disease mutations and enzymatic kinetic assays, to reveal the role of RFTS domain in the allosteric regulation of DNMT1. The potential allosteric model was finally proposed based on the combination of computational and experimental results, and the role of TRD interface in allosteric drug design for DNMT1 was highlighted (orange pocket in the last box of Fig. 9). Of course, there are still gaps between the understanding of allosteric properties and real drug discovery, and more experimental verifications are needed to advance the allosteric drug design targeting DNMT1.
Funding
This work was supported by the National Natural Science Foundation of China (31872723), a Project Funded by the Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions, the China Postdoctoral Science Foundation (2016M590495), and the Jiangsu Planned Projects for Postdoctoral Research Funds (1601168C).
Conflict of interest
Authors declare no conflict of interest.
CRediT authorship contribution statement
Zhongjie Liang: Conceptualization, Methodology, Formal analysis, Writing - original draft. Yu Zhu: Methodology, Formal analysis. Jie Long: Data curation, Visualization. Fei Ye: Software, Resources, Writing - review & editing. Guang Hu: Conceptualization, Supervision, Writing - review & editing.
Acknowledgement
We thank the reviewers for their valuable comments.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2020.03.016.
Contributor Information
Fei Ye, Email: yefei@zstu.edu.cn.
Guang Hu, Email: huguang@suda.edu.cn.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 2.Baylin S.B., Jones P.A. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer. 2011;11:726–734. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Law J.A., Jacobsen S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet. 2010;11:204–220. doi: 10.1038/nrg2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tajima S., Suetake I., Takeshita K., Nakagawa A., Kimura H. Domain structure of the Dnmt1, Dnmt3a, and Dnmt3b DNA methyltransferases. Adv Ecp Med Biol. 2016;945:63–86. doi: 10.1007/978-3-319-43624-1_4. [DOI] [PubMed] [Google Scholar]
- 5.Ren W., Gao L., Song J. Structural basis of DNMT1 and DNMT3A-mediated DNA methylation. Genes (Basel) 2018;9 doi: 10.3390/genes9120620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Song J., Rechkoblit O., Bestor T.H., Patel D.J. Structure of DNMT1-DNA complex reveals a role for autoinhibition in maintenance DNA methylation. Science. 2011;331:1036–1040. doi: 10.1126/science.1195380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Song J., Teplova M., Ishibe-Murakami S., Patel D.J. Structure-based mechanistic insights into DNMT1-mediated maintenance DNA methylation. Science. 2012;335:709–712. doi: 10.1126/science.1214453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takeshita K., Suetake I., Yamashita E., Suga M., Narita H., Nakagawa A. Structural insight into maintenance methylation by mouse DNA methyltransferase 1 (Dnmt1) Proc Natl Acad Sci U S A. 2011;108:9055–9059. doi: 10.1073/pnas.1019629108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Z.M., Liu S., Lin K., Luo Y., Perry J.J., Wang Y. Crystal structure of human DNA methyltransferase 1. J Mol Biol. 2015;427:2520–2531. doi: 10.1016/j.jmb.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jeltsch A., Jurkowska R.Z. Allosteric control of mammalian DNA methyltransferases - a new regulatory paradigm. Nucleic Acids Res. 2016;44:8556–8575. doi: 10.1093/nar/gkw723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leonhardt H., Page A.W., Weier H.U., Bestor T.H. A targeting sequence directs DNA methyltransferase to sites of DNA replication in mammalian nuclei. Cell. 1992;71:865–873. doi: 10.1016/0092-8674(92)90561-p. [DOI] [PubMed] [Google Scholar]
- 12.Syeda F., Fagan R.L., Wean M., Avvakumov G.V., Walker J.R., Xue S. The replication focus targeting sequence (RFTS) domain is a DNA-competitive inhibitor of Dnmt1. J Biol Chem. 2011;286:15344–15351. doi: 10.1074/jbc.M110.209882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berkyurek A.C., Suetake I., Arita K., Takeshita K., Nakagawa A., Shirakawa M. The DNA methyltransferase Dnmt1 directly interacts with the SET and RING finger-associated (SRA) domain of the multifunctional protein Uhrf1 to facilitate accession of the catalytic center to hemi-methylated DNA. J Biol Chem. 2014;289:379–386. doi: 10.1074/jbc.M113.523209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arita K., Ariyoshi M., Tochio H., Nakamura Y., Shirakawa M. Recognition of hemi-methylated DNA by the SRA protein UHRF1 by a base-flipping mechanism. Nature. 2008;455:818–821. doi: 10.1038/nature07249. [DOI] [PubMed] [Google Scholar]
- 15.Hamidi T., Singh A.K., Chen T. Genetic alterations of DNA methylation machinery in human diseases. Epigenomics-UK. 2015;7:247–265. doi: 10.2217/epi.14.80. [DOI] [PubMed] [Google Scholar]
- 16.Maresca A., Zaffagnini M., Caporali L., Carelli V., Zanna C. DNA methyltransferase 1 mutations and mitochondrial pathology: is mtDNA methylated? Front Genet. 2015;6:90. doi: 10.3389/fgene.2015.00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ishiyama S., Nishiyama A., Saeki Y., Moritsugu K., Morimoto D., Yamaguchi L. Structure of the Dnmt1 reader module complexed with a unique two-mono-ubiquitin mark on histone H3 reveals the basis for DNA methylation maintenance. Mol Cell. 2017;68:350–360. doi: 10.1016/j.molcel.2017.09.037. [DOI] [PubMed] [Google Scholar]
- 18.Schueler-Furman O., Wodak S.J. Computational approaches to investigating allostery. Curr Opin Struct Biol. 2016;41:159–171. doi: 10.1016/j.sbi.2016.06.017. [DOI] [PubMed] [Google Scholar]
- 19.Bahar I., Lezon T.R., Yang L.W., Eyal E. Global dynamics of proteins: bridging between structure and function. Annu Rev Biophys. 2010;39:23–42. doi: 10.1146/annurev.biophys.093008.131258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Atilgan C., Gerek Z.N., Ozkan S.B., Atilgan A.R. Manipulation of conformational change in proteins by single-residue perturbations. Biophys J. 2010;99:933–943. doi: 10.1016/j.bpj.2010.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Penkler D., Sensoy O., Atilgan C., Tastan Bishop O. Perturbation-response scanning reveals key residues for allosteric control in Hsp70. J Chem Inf Model. 2017;57:1359–1374. doi: 10.1021/acs.jcim.6b00775. [DOI] [PubMed] [Google Scholar]
- 22.Penkler D.L., Atilgan C., Tastan Bishop O. Allosteric modulation of human Hsp90alpha conformational dynamics. J Chem Inf Model. 2018;58:383–404. doi: 10.1021/acs.jcim.7b00630. [DOI] [PubMed] [Google Scholar]
- 23.O'Rourke K.F., Gorman S.D., Boehr D.D. Biophysical and computational methods to analyze amino acid interaction networks in proteins. Comput Struct Biotechnol J. 2016;14:245–251. doi: 10.1016/j.csbj.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blacklock K., Verkhivker G.M. Allosteric regulation of the Hsp90 dynamics and stability by client recruiter cochaperones: protein structure network modeling. PLoS ONE. 2014;9 doi: 10.1371/journal.pone.0086547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Felline A., Ghitti M., Musco G., Fanelli F. Dissecting intrinsic and ligand-induced structural communication in the beta3 headpiece of integrins. Biochim Biophys Acta Gen Subj. 1861;2017:2367–2381. doi: 10.1016/j.bbagen.2017.05.018. [DOI] [PubMed] [Google Scholar]
- 26.Johnson L.E., Ginovska B., Fenton A.W., Raugei S. Chokepoints in mechanical coupling associated with allosteric proteins: the pyruvate kinase example. Biophys J. 2019;116:1598–1608. doi: 10.1016/j.bpj.2019.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gautier C., Laursen L., Jemth P., Gianni S. Seeking allosteric networks in PDZ domains. Protein Eng Des Sel. 2018;31:367–373. doi: 10.1093/protein/gzy033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Amamuddy O.S., Veldman W., Manyumwa C., Khairallah A., Agajanian S., Oluyemi O. Integrated computational approaches and tools for allosteric drug discovery. Int J Mol Sci. 2020;21:847. doi: 10.3390/ijms21030847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liang Z., Verkhivker G.M., Hu G. Integration of network models and evolutionary analysis into high-throughput modeling of protein dynamics and allosteric regulation: theory, tools and applications. Brief Bioinform. 2019 doi: 10.1093/bib/bbz029. [DOI] [PubMed] [Google Scholar]
- 30.Astl L., Verkhivker G.M. Atomistic modeling of the ABL kinase regulation by allosteric modulators using structural perturbation analysis and community-based network reconstruction of allosteric communications. J Chem Theory Comput. 2019;15:3362–3380. doi: 10.1021/acs.jctc.9b00119. [DOI] [PubMed] [Google Scholar]
- 31.Verkhivker G.M. Dynamics-based community analysis and perturbation response scanning of allosteric interaction networks in the TRAP1 chaperone structures dissect molecular linkage between conformational asymmetry and sequential ATP hydrolysis. Biochim Biophys Acta Proteins Proteom. 1866;2018:899–912. doi: 10.1016/j.bbapap.2018.04.008. [DOI] [PubMed] [Google Scholar]
- 32.Astl L., Verkhivker G.M. Data-driven computational analysis of allosteric proteins by exploring protein dynamics, residue coevolution and residue interaction networks. Biochim Biophys Acta Gen Subj. 2019 doi: 10.1016/j.bbagen.2019.07.008. [DOI] [PubMed] [Google Scholar]
- 33.Stetz G., Verkhivker G.M. Functional role and hierarchy of the intermolecular interactions in binding of protein kinase clients to the Hsp90-Cdc37 chaperone: structure-based network modeling of allosteric regulation. J Chem Inf Model. 2018;58:405–421. doi: 10.1021/acs.jcim.7b00638. [DOI] [PubMed] [Google Scholar]
- 34.Stetz G., Verkhivker G.M. Computational analysis of residue interaction networks and coevolutionary relationships in the Hsp70 chaperones: a community-hopping model of allosteric regulation and communication. Plos Comput Biol. 2017;13 doi: 10.1371/journal.pcbi.1005299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tse A., Verkhivker G.M. Molecular dynamics simulations and structural network analysis of c-Abl and c-Src kinase core proteins: capturing allosteric mechanisms and communication pathways from residue centrality. J Chem Inf Model. 2015;55:1645–1662. doi: 10.1021/acs.jcim.5b00240. [DOI] [PubMed] [Google Scholar]
- 36.Caulfield T., Medina-Franco J.L. Molecular dynamics simulations of human DNA methyltransferase 3B with selective inhibitor nanaomycin A. J Struct Biol. 2011;176:185–191. doi: 10.1016/j.jsb.2011.07.015. [DOI] [PubMed] [Google Scholar]
- 37.Ye F., Kong X., Zhang H., Liu Y., Shao Z., Jin J. Biochemical studies and molecular dynamic simulations reveal the molecular basis of conformational changes in DNA methyltransferase-1. ACS Chem Biol. 2018;13:772–781. doi: 10.1021/acschembio.7b00890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou R., Xie Y., Hu H., Hu G., Patel V.S., Zhang J. Molecular mechanism underlying PRMT1 dimerization for SAM binding and methylase activity. J Chem Inf Model. 2015;55:2623–2632. doi: 10.1021/acs.jcim.5b00454. [DOI] [PubMed] [Google Scholar]
- 39.Liang Z., Hu J., Yan W., Jiang H., Hu G., Luo C. Deciphering the role of dimer interface in intrinsic dynamics and allosteric pathways underlying the functional transformation of DNMT3A. Biochim Biophys Acta Gen Subj. 1862;2018:1667–1679. doi: 10.1016/j.bbagen.2018.04.015. [DOI] [PubMed] [Google Scholar]
- 40.Webb B., Sali A. Comparative protein structure modeling using MODELLER. Curr Protoc Protein Sci. 2016;86:2–9. doi: 10.1002/cpps.20. [DOI] [PubMed] [Google Scholar]
- 41.Atilgan A.R., Durell S.R., Jernigan R.L., Demirel M.C., Keskin O., Bahar I. Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys J. 2001;80:505–515. doi: 10.1016/S0006-3495(01)76033-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bahar I., Atilgan A.R., Erman B. Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential. Fold Des. 1997;2:173–181. doi: 10.1016/S1359-0278(97)00024-2. [DOI] [PubMed] [Google Scholar]
- 43.Bakan A., Meireles L.M., Bahar I. ProDy: protein dynamics inferred from theory and experiments. Bioinformatics. 2011;27:1575–1577. doi: 10.1093/bioinformatics/btr168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marcos E., Crehuet R., Bahar I. Changes in dynamics upon oligomerization regulate substrate binding and allostery in amino acid kinase family members. Plos Comput Biol. 2011;7 doi: 10.1371/journal.pcbi.1002201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pronk S., Pall S., Schulz R., Larsson P., Bjelkmar P., Apostolov R. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29:845–854. doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ghosh A., Vishveshwara S. A study of communication pathways in methionyl-tRNA synthetase by molecular dynamics simulations and structure network analysis. Proc Natl Acad Sci U S A. 2007;104:15711–15716. doi: 10.1073/pnas.0704459104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Seeber M., Felline A., Raimondi F., Muff S., Friedman R., Rao F. Wordom: a user-friendly program for the analysis of molecular structures, trajectories, and free energy surfaces. J Comput Chem. 2011;32:1183–1194. doi: 10.1002/jcc.21688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ashkenazy H., Abadi S., Martz E., Chay O., Mayrose I., Pupko T. an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016;44(2016):W344–W350. doi: 10.1093/nar/gkw408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bakan A., Dutta A., Mao W., Liu Y., Chennubhotla C., Lezon T.R. Evol and ProDy for bridging protein sequence evolution and structural dynamics. Bioinformatics. 2014;30:2681–2683. doi: 10.1093/bioinformatics/btu336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bahar I., Cheng M.H., Lee J.Y., Kaya C., Zhang S. Structure-encoded global motions and their role in mediating protein-substrate interactions. Biophys J. 2015;109:1101–1109. doi: 10.1016/j.bpj.2015.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li T., Wang L., Du Y., Xie S., Yang X., Lian F. Structural and mechanistic insights into UHRF1-mediated DNMT1 activation in the maintenance DNA methylation. Nucleic Acids Res. 2018;46:3218–3231. doi: 10.1093/nar/gky104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cheng J., Yang H., Fang J., Ma L., Gong R., Wang P. Molecular mechanism for USP7-mediated DNMT1 stabilization by acetylation. Nat Commun. 2015;6:7023. doi: 10.1038/ncomms8023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yarychkivska O., Shahabuddin Z., Comfort N., Boulard M., Bestor T.H. BAH domains and a histone-like motif in DNA methyltransferase 1 (DNMT1) regulate de novo and maintenance methylation in vivo. J Biol Chem. 2018;293:19466–19475. doi: 10.1074/jbc.RA118.004612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Norvil A.B., Saha D., Saleem D.M., Gowher H. Effect of disease-associated germline mutations on structure function relationship of DNA methyltransferases. Genes (Basel) 2019;10 doi: 10.3390/genes10050369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Klein C.J., Botuyan M.V., Wu Y., Ward C.J., Nicholson G.A., Hammans S. Mutations in DNMT1 cause hereditary sensory neuropathy with dementia and hearing loss. Nat Genet. 2011;43:595–600. doi: 10.1038/ng.830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Laimer J., Hofer H., Fritz M., Wegenkittl S., Lackner P. MAESTRO–multi agent stability prediction upon point mutations. BMC Bioinf. 2015;16:116. doi: 10.1186/s12859-015-0548-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Verkhivker G.M. Biophysical simulations and structure-based modeling of residue interaction networks in the tumor suppressor proteins reveal functional role of cancer mutation hotspots in molecular communication. Biochim Biophys Acta Gen Subj. 1863;2019:210–225. doi: 10.1016/j.bbagen.2018.10.009. [DOI] [PubMed] [Google Scholar]
- 58.Monod J., Wyman J., Changeux J.P. On the nature of allosteric transitions: a plausibale model. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 59.Koshland D.J., Nemethy G., Filmer D. Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochemistry-US. 1966;5:365–385. doi: 10.1021/bi00865a047. [DOI] [PubMed] [Google Scholar]
- 60.Del S.A., Tsai C.J., Ma B., Nussinov R. The origin of allosteric functional modulation: multiple pre-existing pathways. Structure. 2009;17:1042–1050. doi: 10.1016/j.str.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wagner J.R., Lee C.T., Durrant J.D., Malmstrom R.D., Feher V.A., Amaro R.E. Emerging computational methods for the rational discovery of allosteric drugs. Chem Rev. 2016;116:6370–6390. doi: 10.1021/acs.chemrev.5b00631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ye F., Huang J., Wang H., Luo C., Zhao K. Targeting epigenetic machinery: emerging novel allosteric inhibitors. Pharmacol Ther. 2019;107406 doi: 10.1016/j.pharmthera.2019.107406. [DOI] [PubMed] [Google Scholar]
- 63.Creixell P., Pandey J.P., Palmeri A., Bhattacharyya M., Creixell M., Ranganathan R. Hierarchical organization endows the kinase domain with regulatory plasticity. Cell Syst. 2018;7:371–383. doi: 10.1016/j.cels.2018.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bashtrykov P., Rajavelu A., Hackner B., Ragozin S., Carell T., Jeltsch A. Targeted mutagenesis results in an activation of DNA methyltransferase 1 and confirms an autoinhibitory role of its RFTS domain. ChemBioChem. 2014;15:743–748. doi: 10.1002/cbic.201300740. [DOI] [PubMed] [Google Scholar]
- 65.Schneider K., Fuchs C., Dobay A., Rottach A., Qin W., Wolf P. Dissection of cell cycle-dependent dynamics of Dnmt1 by FRAP and diffusion-coupled modeling. Nucleic Acids Res. 2013;41:4860–4876. doi: 10.1093/nar/gkt191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ahuja L.G., Taylor S.S., Kornev A.P. Tuning the “violin” of protein kinases: the role of dynamics-based allostery. IUBMB Life. 2019;71:685–696. doi: 10.1002/iub.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xie T., Yu J., Fu W., Wang Z., Xu L., Chang S. Insight into the selective binding mechanism of DNMT1 and DNMT3A inhibitors: a molecular simulation study. Phys Chem Chem Phys. 2019;21:12931–12947. doi: 10.1039/c9cp02024a. [DOI] [PubMed] [Google Scholar]
- 68.Yu J., Xie T., Wang Z., Wang X., Zeng S., Kang Y. DNA methyltransferases: emerging targets for the discovery of inhibitors as potent anticancer drugs. Drug Discov Today. 2019 doi: 10.1016/j.drudis.2019.08.006. [DOI] [PubMed] [Google Scholar]
- 69.Huang W., Lu S., Huang Z., Liu X., Mou L., Luo Y. Allosite: a method for predicting allosteric sites. Bioinformatics. 2013;29:2357–2359. doi: 10.1093/bioinformatics/btt399. [DOI] [PubMed] [Google Scholar]
- 70.Greener J.G., Sternberg M.J. AlloPred: prediction of allosteric pockets on proteins using normal mode perturbation analysis. BmcBioinformaics. 2015;16:335. doi: 10.1186/s12859-015-0771-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dolen E.K., McGinnis J.H., Tavory R.N., Weiss J.A., Switzer R.L. Disease-associated mutations G589A and V590F relieve replication focus targeting sequence-mediated autoinhibition of DNA methyltransferase 1. Biochemistry. 2019;51:5151–5159. doi: 10.1021/acs.biochem.9b00749. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.