

Abstract
Objective
Peroxisome proliferator-activated receptors (PPARs) are key transcription factors that regulate adipose development and function, and the conversion of white into brown-like adipocytes. Here we investigated whether PPARα and PPARγ activation synergize to induce the browning of white fat.
Methods
A selection of PPAR activators was tested for their ability to induce the browning of both mouse and human white adipocytes in vitro, and in vivo in lean and obese mice.
Results
All dual PPARα/γ activators tested robustly increased uncoupling protein 1 (Ucp1) expression in both mouse and human adipocytes in vitro, with tesaglitazar leading to the largest Ucp1 induction. Importantly, dual PPARα/γ activator tesaglitazar strongly induced browning of white fat in vivo in both lean and obese male mice at thermoneutrality, greatly exceeding the increase in Ucp1 observed with the selective PPARγ activator rosiglitazone. While selective PPARγ activation was sufficient for the conversion of white into brown-like adipocytes in vitro, dual PPARα/γ activation was superior to selective PPARγ activation at inducing white fat browning in vivo. Mechanistically, the superiority of dual PPARα/γ activators is mediated at least in part via a PPARα-driven increase in fibroblast growth factor 21 (FGF21). Combined treatment with rosiglitazone and FGF21 resulted in a synergistic increase in Ucp1 mRNA levels both in vitro and in vivo. Tesaglitazar-induced browning was associated with increased energy expenditure, enhanced insulin sensitivity, reduced liver steatosis, and an overall improved metabolic profile compared to rosiglitazone and vehicle control groups.
Conclusions
PPARγ and PPARα synergize to induce robust browning of white fat in vivo, via PPARγ activation in adipose, and PPARα-mediated increase in FGF21.
Keywords: Brown adipocytes, Beige adipocytes, Thermogenesis, UCP1, FGF21, PPAR
Abbreviations: BAT, brown adipose tissue; DIO, diet-induced obese; EE, energy expenditure; eWAT, epididymal WAT; FGF21, fibroblast growth factor 21; iWAT, inguinal WAT; iBAT, interscapular BAT; PPAR, peroxisome proliferator-activated receptors; QUICKI, quantitative insulin-sensitivity check index; RT, room temperature; TN, thermoneutrality; TZD, thiazolidinedione; TG, triglycerides; UCP1, uncoupling protein 1; WAT, white adipose tissue
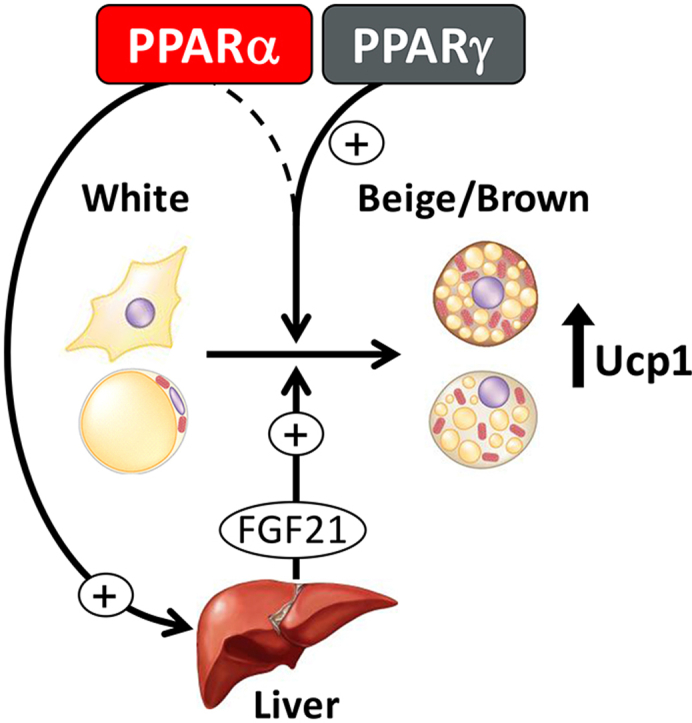
Graphical abstract
Dual PPARα/γ activation is superior to selective PPARγ activation at inducing browning of white fat in vivo: via PPARγ action in the adipose and a PPARα-mediated action in the liver resulting in increased circulating fibroblast growth factor 21 (FGF21).
Highlights
-
•
Dual PPARα/γ activators robustly induce browning of white fat in vitro and in vivo.
-
•
PPARγ action alone is sufficient to convert white into brown-like adipocytes in vitro.
-
•
Dual PPARα/γ activators are superior to PPARγ activators for browning of white fat in vivo.
-
•
PPARγ action in adipose and PPARα-mediated increase in FGF21 synergize to induce browning of white fat in vivo.
1. Introduction
The primary function of white adipose tissue (WAT) is to store energy for future systemic use [1]. By contrast, brown adipose tissue (BAT) consumes energy and plays a central role in thermogenesis and overall energy expenditure [2]. This unique energy-wasting capacity of BAT is due to the expression of uncoupling protein 1 (UCP1) in specialized brown adipocytes. UCP1 is expressed in the mitochondria of brown adipocytes and functions by leaking protons across the mitochondrial membrane, dissipating chemical energy in the form of heat [2]. Adult humans possess significant BAT depots which can be activated by cold or β-adrenergic receptor agonists, and BAT mass and activity inversely correlate with obesity [[3], [4], [5], [6], [7]]. Upon cold or β-adrenergic stimuli, a distinct type of thermogenic brown-like adipocytes (termed beige or brite) can also arise within WAT [8]. These adipocytes share several key characteristics with brown adipocytes, including the expression of UCP1 [9,10]. In animal models, the browning of WAT is associated with increased energy expenditure, decreased body weight, and improved glucose tolerance [11]. Thus, increased browning of WAT has therapeutic potential for treating obesity and type 2 diabetes [12,13].
Peroxisome proliferator-activated receptors (PPARs) are ligand-gated transcription factors. Three PPAR subtypes have been identified (α, δ and γ). PPARγ expression is predominately restricted to adipose and is the master regulator of adipogenesis. Upon its activation by a suitable ligand, PPARγ promotes differentiation of preadipocytes into adipocytes [14]. Some selective PPARγ activators, including the thiazolidinediones (TZDs) such as rosiglitazone, have been shown to induce a brown fat gene transcription program in white adipocytes both in vitro [[15], [16], [17], [18], [19]] and in vivo [16,[20], [21], [22], [23]], via SIRT1-, PRDM16-, C/EBPα-, and PGC1α-mediated mechanisms [16,[24], [25], [26]]. However, despite leading to an increased capacity for UCP1-mediated uncoupled respiration in adipocytes, browning induced by TZD treatment is not associated with increased energy expenditure or weight loss in vivo [21,27]. PPARα is predominantly expressed in tissues with high oxidative capacity, such as BAT, liver, heart and kidney, where it promotes fatty acid oxidation, ketogenesis and glucose sparing [28,29]. Selective PPARα activators, such as fibrates, can also induce the browning of WAT via PGC1α- and PRDM16-mediated mechanisms [[30], [31], [32], [33]].
In the present study, we investigated whether PPARα and PPARγ synergize to induce the browning of white fat. We found that selective PPARγ activation is sufficient for the conversion of white adipocytes into brown-like adipocytes in vitro. However, dual PPARα/γ activation is superior to selective PPARγ activation at inducing browning of WAT in vivo due to a concomitant PPARα-mediated increase in fibroblast growth factor 21 (FGF21). Furthermore, in contrast to the previously reported browning of WAT induced by the selective PPARγ activator rosiglitazone, the browning of WAT induced by the dual PPARα/γ activator tesaglitazar is associated with increased energy expenditure, improved metabolism and decreased hepatic steatosis in diet-induced obese insulin resistant mice.
2. Materials and methods
2.1. Mouse preadipocyte and adipocyte cultures
Primary preadipocytes were obtained by digesting inguinal subcutaneous white adipose tissue from 6 to 8 week old lean C57BL/6J male mice with collagenase as previously described [34]. Immortalized preadipocytes were obtained by immortalizing primary inguinal subcutaneous preadipocytes from C57BL/6J male mice via retroviral expression of SV40 large-T antigen. Both mouse primary (Figure 1, Figure 5, Figure 6g, Figures S1, S6a and S9d) and immortalized (Figure 1A, Figures S1 and S2a) preadipocytes were grown in DMEM/F12, 10% FBS, 1% penicillin-streptomycin. Upon confluence (day 0), preadipocytes were differentiated in DMEM/F12 containing 20 nM insulin, 1 nM T3, 125 nM indomethacin (Sigma, I7378), 1 μM dexamethasone (Sigma, D2915) and 0.5 mM isobutylmethylxanthine (IBMX, Sigma, I5879). Indomethacin, dexamethasone, and IBMX were omitted from the medium from day 2. Mouse adipocytes (Figure 1, Figure 5b, Figures S2b and S6b) were obtained by differentiating immortalized preadipocytes for 6 days. For all protocols, the medium was changed every other day.
Figure 1.

Dual PPARα/γ activators induce UCP1 in both mouse and human white adipocytes in vitro. (a)Ucp1 and Fabp4 mRNA expression in immortalized mouse white preadipocytes following 8 days of treatment with various dual PPARα/γ activators during differentiation (n = 3/group). (b)Ucp1 mRNA expression in mouse white adipocytes (pre-differentiated from immortalized preadipocytes for 6 days) following 2 days of treatment (n = 3/group). (c)UCP1 and FABP4 mRNA expression in primary human white preadipocytes following 12 days of treatment during differentiation (n = 4–8/group). (d)UCP1 mRNA expression in freshly isolated mature human adipocytes following 7 days of treatment (n = 4/group). All compounds in a-d were added at a final concentration of 10 μM. (e) qPCR analysis (n = 3/group) of BAT-enriched genes (left panel) and lipid/glucose metabolism genes (right panel), (f) Western blot analysis of Pparγ, mitochondrial subunits and Ucp1 protein levels and (g) mitochondrial/nuclear DNA ratio (n = 6/group), measured in primary mouse white preadipocytes after 8 days of treatment during differentiation with either vehicle or tesaglitazar (10 μM). Data presented as mean ± SEM: ∗P < 0.05; ∗∗P < 0.01; ∗∗∗P < 0.001 by two-tailed, unpaired Student's t-test.
Figure 5.

PPARγ alone is sufficient to convert white into brown-like adipocytes in vitro, but PPARα and PPARγ have synergistic effects in vivo. (a–d) qPCR analysis of Ucp1 and Cidea mRNA levels, following treatment with either vehicle, WY14643, rosiglitazone, rosiglitazone + WY14643 or tesaglitazar (10 μM for all except rosiglitazone 1 μM) in (a) primary preadipocytes isolated from the iWAT depot of lean mice (8-day treatment during differentiation, n = 3/group), (b) mouse white adipocytes (pre-differentiated from immortalized preadipocytes for 6 days followed by a 2-day treatment after differentiation, n = 3/group), (c) primary human white preadipocytes (12-day treatment during differentiation, n = 4–8/group) and (d) freshly isolated mature human adipocytes (7-day treatment, n = 4/group). (e) qPCR analysis of Ucp1 mRNA in iWAT, eWAT and iBAT of lean male mice, acclimated for 3 weeks at thermoneutrality (TN, 30 °C) and treated for 2 weeks at TN (q.d., oral gavage, n = 4–6/group) with either vehicle, fenofibrate (isopropyl 2-[4-(4-chlorobenzoyl)phenoxy]-2-methylpropanoate, 416 μmol/kg), rosiglitazone (28 μmol/kg), rosiglitazone + fenofibrate, tesaglitazar (1.0 μmol/kg) or saroglitazar (2.3 μmol/kg). Data presented as mean ± SEM: ∗P < 0.05; ∗∗P < 0.01; ∗∗∗P < 0.001 by one-way ANOVA with Tukey's multiple comparisons test.
Figure 6.

Tesaglitazar induces browning of white fat in vivo, partly via FGF21. Plasma Fgf21, Fgf21 mRNA in liver and iBAT and Fabp3 and Ehhadh mRNA in liver of (a) TN and TN→RT DIO male mice (conditions and treatments as described in Figure 3A, n = 4–5/group) and (b) lean male mice (conditions and treatments as described in Figure 5E). Linear regression analysis of individual Ucp1 gene expression in iWAT, eWAT and iBAT vs. plasma Fgf21 concentrations following various dual PPARα/γ treatments in (c) DIO and (d and e) lean mice; squares and dashed lines represent 2-week treatment at TN whereas circles and solid lines represent animals treated for 2 weeks at TN followed by 2 weeks treatment at RT (TN→RT). (f) qPCR analysis of Ucp1, Cidea and Pdk4 iWAT mRNA levels in lean male mice acclimated to TN (30 °C) and treated with either vehicle, FGF21 (0.15 mg/kg), rosiglitazone (28 μmol/kg), FGF21 + rosiglitazone or tesaglitazar (1.0 μmol/kg) for two weeks (n = 6/group). (g) qPCR analysis of Ucp1, Fabp4, Cidea and Pdk4 following treatment with either vehicle, Fgf21 (100 nM), rosiglitazone (0.01 μM), rosiglitazone + Fgf21 or tesaglitazar (10 μM) in primary preadipocytes isolated from the iWAT depot of lean mice (8-day treatment during differentiation, n = 5/group). Data in a, b, f and g presented as mean ± SEM: ∗P < 0.05; ∗∗P < 0.01; ∗∗∗P < 0.001 by one-way ANOVA and Tukey's multiple comparisons test.
2.2. Patient consent
Anonymous adipose tissue samples were collected from the subcutaneous abdominal region of female patients undergoing elective surgery at Sahlgrenska University Hospital in Gothenburg, Sweden. All study subjects received written and oral information before giving written informed consent for the use of the tissue. The studies were approved by The Regional Ethical Review Board in Gothenburg, Sweden.
2.3. Human preadipocyte and adipocyte cultures
Human subcutaneous preadipocytes (Figure 1, Figure 5c, Figures S1, S2c, S6c and S9e) were isolated and differentiated as previously described [35]. Briefly, isolated cells were seeded and cultured in proliferation medium (Zenbio, PM-1) containing 3% FBS Gold (PAA, A15-104), 1% penicillin/streptomycin and 1 nM bFGF (Sigma, F0291). After becoming confluent (day 0), cells were differentiated in basal medium (Zenbio, BM-1) containing 3% FBS Gold (PAA, A15-104), 1% penicillin/streptomycin, 0.5 mM IBMX (Sigma, I5879), 100 nM dexamethasone (Sigma, D2915), 20 nM insulin, 1 nM T3, and 10 ng/mL BMP4 (R&D Systems, 314-BP-010/CF). After 7 days of differentiation, the medium was replaced but IBMX was omitted, and the cells were harvested on day 12 of differentiation. Freshly isolated human mature subcutaneous adipocytes were cultured as MAAC (membrane mature adipocyte aggregate cultures, Figure 1, Figure 5d, Figures S1, S2d, S6d and S9f) based on a previously described method [35]. Briefly, following isolation, adipocytes were placed under a membrane and into 1 mL of medium (DMEM/F12) containing 10% FBS (Gibco, 10270), 1% penicillin/streptomycin and 20 nM insulin (day 0).
2.4. In vitro compound treatments
All PPAR activators were used at a final concentration of 10 μM (except rosiglitazone, used at 1 μM, or 0.01 μM, specifically in Figure 6G) and were obtained from AstraZeneca compound management. Recombinant mouse Fgf21 (R&D Systems, 8409-FG-025) and human Fc-FGF21 (AstraZeneca compound management) were used at a final concentration of 100 nM.
2.5. RNA isolation, cDNA synthesis, and real-time qPCR
Total RNA was extracted from adipocyte cultures using RNeasy Mini Kits (Qiagen, 74106). Adipose and liver tissue pieces were placed in a plastic tube containing a stainless-steel bead (Qiagen, 69989) and 500 μL of TRIzol, and lysed for 2 min using a tissue lyser. Following chloroform phase separation, RNA from the aqueous phase was subsequently purified using RNeasy Mini Kits (Qiagen, 74106). Isolated RNAs from cells and tissues were reverse transcribed using the High-Capacity cDNA Synthesis kit (Applied Biosystems, 4368814) and gene expression was measured using real-time PCR with Power SYBR Green master mix (Applied Biosystems) on a Quantstudio 7 Flex Real-Time PCR machine (Applied Biosystems). Tata-binding protein (TBP) was used as an internal normalization control. Primer sequences are included in Table S1.
2.6. Immunoblotting and mitochondrial DNA isolation
Protein extracts were prepared from adipocytes by aspirating the medium, washing two times with PBS, and lysing the cells in RIPA buffer (50 mM Tris (pH 8.0), 150 mM NaCl, 5 mM EDTA, 1% NP40, 0.5% sodium deoxycholate, 0.1% SDS) containing protease inhibitors (Roche, 04693159001). Whole tissue pieces were placed in a plastic tube containing a stainless-steel bead (Qiagen, 69989) and RIPA buffer containing protease inhibitors (Roche, 04693159001) and lysed for 2 min using a tissue lyser. Protein content was determined using a BCA Protein Assay kit (Pierce). Protein samples were denatured with Laemmli buffer 2 × (Merck, S3401) by heating at 95 °C for 5 min. Proteins were separated on 4%–12% Bis-Tris NuPAGE gels (Invitrogen) and transferred to PVDF membranes. Primary antibodies were against PPARγ (SantaCruz, SC-7196), mitochondrial subunits (Mitoscience, MS601/F1208), UCP1 [36], β-actin (Sigma, A5441), ERK (SantaCruz, SC-93), and GAPDH (Cell signaling, 2118). Bands were detected using either standard ECL substrate (Pierce, 32106) or SuperSignal West Femto (Thermo, 34096). Images were obtained and quantified using Image Lab (version 5.2, Bio-Rad Laboratories).
2.7. Mitochondrial DNA isolation
DNA was isolated by digesting adipocytes overnight in a buffer containing 100 mM Tris (pH 8), 5 mM EDTA, 200 mM NaCl, 0.5% SDS, and 100 mg/mL Proteinase K. DNA was ethanol precipitated and resuspended in Tris–EDTA. The ratio mitochondria/genomic DNA was quantified by measuring MTCO1 and Ndufv1 genes by real-time PCR. Primer sequences are given in Table S1.
2.8. Animal studies
All experimental procedures were approved by the Gothenburg ethics review committee on animal experiments. Animals were kept in an Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) accredited facility and attended daily. Lean C57BL/6J male mice were purchased at 20–25 g from Charles River (Germany). Diet-induced obese (DIO) C57BL/6J male mice, pre-conditioned from 6 weeks of age with 60% kcal high fat diet (D12492, Research Diets, USA), were purchased at 18 weeks of age from Charles River (Germany). Upon arrival, all animals were acclimated to thermoneutrality (TN, 30 °C) for 3 weeks before randomization and the start of interventions at 21 weeks of age. Throughout, animals were housed individually, supplied with wood chips, a cardboard house, paper-wool, a wooden stick, and maintained on a 12:12 h light: dark cycle (lights off at 6 p.m.) with free access to food and water. Lean mice received standard rodent chow diet (R70, Lactamin AB, Sweden) and DIO mice were maintained on a D12492 diet.
Animals were randomized into groups based on body weight and treated with compounds solubilized in 0.1% Tween 80 and 0.5% hydroxypropyl methylcellulose by oral dosing (gavage, 5 mL/kg); Fc-FGF21 was dissolved in PBS containing 0.1% BSA, given s.c. (5 mL/kg). Compounds were supplied by AstraZeneca compound management. Doses and treatment paradigms are given in corresponding figure legends. Energy expenditure was assessed by indirect calorimetry using an Oxymax open-circuit indirect calorimeter, Comprehensive Lab Animal Monitoring System (Columbus Instruments, Columbus, OH, USA). The mice were acclimated to the metabolic chambers for 24 h before measurements and housed individually with free access to food and water with a 12-hour light/dark cycle and ambient temperatures maintained at 30 °C or 22 °C.
Termination was performed under isoflurane anesthesia in animals fasted for 4 h after the last compound administration. Orbital blood was collected in EDTA-coated tubes, immediately centrifuged and plasma samples were stored at −80 °C pending analysis. Tissues were dissected, weighed, and placed in 4% buffered formaldehyde for histology or snap frozen in liquid nitrogen and stored at −80 °C pending analysis.
Insulin tolerance test (ITT) was performed in 4 h fasted animals. Insulin (0.75 U/kg, Novo Nordisk, Actrapid) was administered via i.p. injection and circulating glucose levels (tail vein sampling) were monitored for 90 min using a portable glucose analyzer (ACCU-CHEK, Roche Diagnostics). Immediately following the 90 min blood sample, glucose (oral gavage, 1 g/kg) was administered to the tesaglitazar-treated mice, due to severe hypoglycemia.
2.9. Blood chemistry
An ABX Pentra 400 (Horiba Medical) was used to analyze plasma free fatty acids (Wako Chemicals), glucose (Horiba ABX, A11A01667), triglycerides (Roche Diagnostics, 11877771216), total cholesterol (Horiba ABX, A11A01634), and β-hydroxybutyrate (ketone bodies, Randox, RB1007). MultiCal (Horiba ABX, A110A01652) was used as a calibrator for plasma glucose, triglycerides and total cholesterol analyses. Plasma insulin was analyzed using a MESO SECTOR S 600 (mouse/rat insulin kit, Meso Scale Diagnostics, K152BZC). Plasma FGF21 was analyzed using an ELISA kit (R&D Systems, MF2100). Plasma drug exposures were analyzed by LC-MSMS.
2.10. Liver triglyceride content
Liver triglyceride content (Horiba ABX, A11A01640) with MultiCal (Horiba ABX, A110A01652) was measured with an ABX Pentra 400 (Horiba Medical) following isopropanol extraction.
2.11. Statistics
Statistical significance was evaluated by two-tailed Student's t test, Mann Whitney test or where appropriate (as indicated) one-way or two-way ANOVA with Tukey's multiple comparisons test, and linear regression was used to test association between two variables, performed using GraphPad Prism 7.04 (GraphPad Software Inc., La Jolla, CA, USA). Statistical evaluation of the regression plots of energy expenditure vs. body mass was performed by ANCOVA (R version 3.4.0) with body mass as covariate. P < 0.05 was considered statistically significant, and results are reported as mean ± SEM throughout.
3. Results
3.1. Tesaglitazar robustly induces browning of white adipocytes in vitro
A selection of dual PPARα/γ activators (aleglitazar, imiglitazar, reglitazar, chiglitazar, saroglitazar, muraglitazar, ragaglitazar and tesaglitazar), all of which reached late stage clinical development, were tested for their ability to induce browning of white adipocytes. Upon treating differentiating mouse preadipocytes for 8 days, the compounds significantly increased Ucp1 mRNA levels. Tesaglitazar, ragaglitazar and muraglitazar were identified as the strongest Ucp1 inducers, resulting in ∼1700-, 1600- and 1300-fold inductions, respectively (Figure 1A). Aleglitazar and imiglitazar both resulted in a 50-fold Ucp1 induction, whereas reglitazar, chiglitazar and saroglitazar produced ∼200-, 300- and 400-fold Ucp1 inductions, respectively. The adipocyte differentiation marker and PPARγ responsive gene Fabp4 was increased 4-fold in all compound treated cells compared to vehicle controls, but there was no significant difference in Fabp4 levels across treatments, indicating a similar level of adipocyte differentiation (Figure 1A). Importantly, the browning effect of the dual PPARα/γ activators was also observed in differentiating human preadipocytes. Tesaglitazar and ragaglitazar were found to be the strongest UCP1 inducers, resulting in >10-fold induction, but all dual PPARα/γ activators, except imiglitazar, significantly induced UCP1 in human preadipocytes (Figure 1C). The dual PPARα/γ activators also induced Ucp1 robustly in adipocytes: treatment of mouse adipocytes (pre-differentiated for 6 days) with saroglitazar, ragaglitazar and tesaglitazar for 2 days resulted in a ∼80-fold increase in Ucp1 (Figure 1B). We have recently developed a method for long-term culture of freshly isolated mature human adipocytes [35]. Saroglitazar, ragaglitazar, and tesaglitazar treatment of freshly isolated mature human adipocytes for 7 days led to a ∼200-fold increase in UCP1 expression. (Figure 1D). To quantitatively compare the effect of the dual PPARα/γ activators across the different mouse and human cell models, we generated UCP1 cDNA standard curves, allowing the determination of absolute UCP1 mRNA levels. We found that UCP1 mRNA levels were higher in tesaglitazar-treated mouse cells compared to human cells, but there was no obvious difference in UCP1 levels when comparing treatment with tesaglitazar during differentiation or of mature adipocytes directly (Figure S1). It is unclear, however, if the differences in UCP1 levels between mouse and human cells reflect differences in the intrinsic capacity of these cells to brown, differences in the function of PPARγ, PPARα and/or their associated cofactors, or if it merely represents differences in culture conditions. Expression of other brown fat markers such as Cidea, Cpt1b and Pdk4 were also strongly upregulated following dual PPARα/γ activator treatment (Figure S2). In addition to Ucp1, tesaglitazar caused significant increases in gene expression of several lipid and glucose metabolism genes and mitochondrial proteins (Figure 1E). Protein levels of mitochondrial proteins Atp5a, Uqcrc2, Sdhb and Ndufb8 (Figure 1F), and mitochondrial/nuclear DNA ratio (Figure 1G) were also increased, indicating increased mitochondria biogenesis. Importantly, tesaglitazar also increased Ucp1 protein levels (Figure1F). Together, these results indicate that tesaglitazar and other dual PPARα/γ activators induce browning of both mouse and human white adipocytes in vitro.
3.2. Tesaglitazar robustly induces browning of white fat in vivo in lean mice
Standard housing conditions (22 °C) is a cold stimulus for mice [37], which in itself induces some degree of white fat browning. The ability of compounds to induce browning of WAT in mice was therefore assessed at mouse thermoneutrality (TN, ∼30 °C), to better mimic standard human living thermoneutral conditions. At TN, lean mice were treated for two or five weeks with tesaglitazar and benchmarked against the known browning agent, the selective PPARγ activator rosiglitazone. A two-week tesaglitazar treatment resulted in a ∼120-fold increase in Ucp1 mRNA levels in both the inguinal WAT (iWAT) and epididymal WAT (eWAT), relative to vehicle controls. In contrast, rosiglitazone treatment had modest effects, resulting in only 3- and 6-fold induction of Ucp1 in the iWAT and eWAT, respectively (Figure 2A). The marked induction of Ucp1 in iWAT upon tesaglitazar treatment was also observed at the protein level (Figure 2B). Ucp1 mRNA was also increased 4-fold in the interscapular BAT (iBAT) following tesaglitazar treatment, and 2-fold following rosiglitazone treatment (Figure 2A). Maximal induction of Ucp1 was seen after two weeks of tesaglitazar treatment, with no further increase with a five-week treatment (Figure 2A). Pgc1α and Cidea were also robustly increased in iWAT and eWAT, and to a lesser extent in iBAT, following tesaglitazar treatment (Figure 2C, Figure S3). Importantly, tesaglitazar, but not rosiglitazone treatment, was associated with increased energy expenditure (EE) (Figure 2D–E) with no difference in locomotor activity (Figure S3d).
Figure 2.

Tesaglitazar robustly induces browning of white fat and increases energy expenditure in lean mice housed at thermoneutrality. Lean male mice were acclimated to thermoneutrality (30 °C) for 3 weeks and treated (b.i.d. oral gavage) with either vehicle, rosiglitazone (15 μmol/kg) or tesaglitazar (1.0 μmol/kg) for a period of 2 weeks (2w) or 5 weeks (5w). (a) qPCR analysis of Ucp1 mRNA in iWAT, eWAT and iBAT. (b) Western blot analysis and quantification of Ucp1 protein levels in iWAT of mice treated for 2 weeks. (c) qPCR analysis of Pgc1a and Cidea mRNA in iWAT, eWAT and iBAT after 2 weeks treatment. (d) Average energy expenditure for the full day (24 h), dark and light periods and (e) regression plot of average energy expenditure vs. body mass. Data presented as mean ± SEM (n = 4–5/group): ∗P < 0.05; ∗∗P < 0.01; ∗∗∗P < 0.001 by one-way ANOVA (a–d) or ANCOVA with body mass as covariate (e), with Tukey's multiple comparisons test.
3.3. Tesaglitazar robustly induces browning of white fat in vivo in obese mice
To test whether tesaglitazar could induce the browning of WAT in obese mice and alter body weight and metabolism, diet-induced obese (DIO) mice were acclimated to TN and treated with either vehicle, rosiglitazone or tesaglitazar. Tesaglitazar was administered at three different doses, to titrate the degree of Ucp1 induction required to elicit metabolic improvements. Animals were terminated either after two weeks of treatment at TN (TN group), or after treatment for an additional two weeks while the temperature was reduced to room temperature (TN→RT group) to achieve a mild cold stimulus in order to activate Ucp1 in the newly formed beige adipocytes (Figure 3A). Observed plasma drug concentrations following rosiglitazone and high dose tesaglitazar treatment indicate similar average daily PPARγ activation (>80%, based on an in vitro PPARγ reporter gene assay), while the mid and low tesaglitazar doses resulted in ∼75% and ∼50% average daily activation, respectively (Figure S4).
Figure 3.

Tesaglitazar robustly induces browning of white fat in vivo in obese mice. (a) Study design schematic: diet-induced obese (DIO) male mice were acclimated to thermoneutrality (TN, 30 °C) for 3 weeks and treated (q.d. oral gavage) with either vehicle, rosiglitazone (Rosi, 28 μmol/kg) or tesaglitazar (Tesa low dose, Tesa mid dose and Tesa high dose: 0.2, 1.0 and 10 μmol/kg, respectively), for 2 weeks at TN (TN group) or an additional 2 weeks treatment with temperature reduced to room temperature (TN→RT group). (b) qPCR analysis of Ucp1 mRNA in iWAT, eWAT and iBAT, (c) western blot analysis and quantification of Ucp1 protein levels in iWAT and iBAT of TN→RT mice and (d) qPCR analysis of Pgc1α and Cidea mRNA in iWAT, eWAT and iBAT. Data presented as mean ± SEM (n = 4–5/group, for western blots n = 2 representative samples/group): ∗P < 0.05; ∗∗P < 0.01; ∗∗∗P < 0.001 by one-way ANOVA with Tukey's multiple comparisons test.
In DIO mice at TN, tesaglitazar dose-dependently increased Ucp1 expression in iWAT, reaching a 40-fold increase at the highest tesaglitazar dose compared to vehicle controls (Figure 3B). Ucp1 induction was even more pronounced in the TN→RT group, with a 550-fold Ucp1 induction at the highest tesaglitazar dose, suggesting that tesaglitazar and a mild cold stimulus have synergistic effects in inducing the browning of iWAT. Tesaglitazar also robustly induced Ucp1 in eWAT, in both the TN and TN→RT groups. Rosiglitazone increased Ucp1 less than 10-fold in iWAT in TN and TN→RT groups, which was much lower than the effect of tesaglitazar. However, rosiglitazone induced Ucp1 20- to 30-fold in the eWAT of both TN and TN→RT groups, on par with the tesaglitazar effect (Figure 3B). In the iBAT, mid and high dose tesaglitazar increased Ucp1 1.6-fold in TN→RT mice, and by 3-fold in TN mice compared to vehicle controls, which had lower basal Ucp1 levels than TN→RT controls. Rosiglitazone, on the other hand, did not upregulate Ucp1 in the iBAT (Figure 3B). Pgc1α and Cidea gene expression profiles followed the same pattern as Ucp1 in the different adipose tissue depots, in both TN and TN→RT groups (Figure 3D). Similar expression patterns were also observed for other brown fat-enriched genes, including Elovl3 and Cox7a1, with the most pronounced effects consistently observed in the iWAT depot (Figures. S5a–c). The lowest dose of tesaglitazar had little to no effect on the expression of the brown fat markers in any adipose depots. Importantly, the large induction of Ucp1 mRNA in iWAT following mid and high dose tesaglitazar treatment was also observed at the protein level (Figure 3C). In summary, tesaglitazar robustly induces the recruitment of thermogenic beige adipocytes in adipose tissues of both lean and obese mice.
3.4. Tesaglitazar increases energy expenditure and reduces body weight
The tesaglitazar-induced browning in DIO mice was associated with increased EE, with the high dose tesaglitazar-treated mice displaying a 12% increased EE compared to rosiglitazone and vehicle controls at TN (Figure 4A). Following brown/beige adipocyte activation by subsequently reducing the temperature to RT (TN→RT group), EE almost doubled across all treatment groups (Figure 4A–B). The temperature drop caused a further increase in EE in tesaglitazar-treated mice, with the highest dose group exhibiting a ∼10% higher EE compared to vehicle TN→RT controls (Figure 4A). Regression analysis of average EE vs. body mass shows that tesaglitazar-treated mice have significantly higher EE compared to rosiglitazone and vehicle controls, both at TN and following the temperature drop to RT (Figure 4B). Across treatments, there were no differences in locomotor activity (Figure S5g). The tesaglitazar-induced increase in EE was associated with a decrease in body weight (Figure 4C). Under TN conditions, body weight loss was evident only in the high dose tesaglitazar group. In contrast, rosiglitazone-treated mice displayed a non-significant increase in weight (Figure 4C, TN group). Following re-acclimation to RT, tesaglitazar-treated mice displayed a dose-dependent body weight loss, while vehicle-corrected weight gain in the rosiglitazone group persisted (Figure 4C, TN→RT group), due at least in part to increased food intake (Figure 4D). Food intake was temporarily reduced in the high dose tesaglitazar group but returned to similar levels as vehicle control by the end of the study. The weight loss following tesaglitazar treatment was associated with a dose-dependent decrease in adipose tissue weights and adipocyte size, while rosiglitazone treated mice had increased adipose tissue weights (Figure 4E–F). In iBAT, tesaglitazar reduced lipid content while rosiglitazone increased lipid storage (Figure 4E–F).
Figure 4.

Tesaglitazar increases energy expenditure, reduces body weight, improves metabolic control and ameliorates high fat diet-induced hepatic steatosis in obese mice. DIO male mice were used in conditions and treatments as described in Figure 3A. (a) Average energy expenditure for the full day (24 h), dark and light periods at TN (30 °C, TN group) and RT (22 °C, TN→RT group). (b) Regression plots of average energy expenditure vs. body mass. (c) Body weight over treatment time and vehicle-corrected body weight gain. (d) Food intake over treatment time. (e) Terminal iWAT, eWAT and iBAT fat pad weights. (f) Hematoxylin & Eosin staining in iBAT, iWAT and eWAT of vehicle, Rosi and Tesa high dose treated TN→RT mice. (g) Plasma glucose, (h) plasma insulin (i) QUICKI insulin sensitivity indexes, (j) plasma TG, (k) liver TG content and (l) plasma glucose levels following an insulin tolerance test (ITT, 0.75 U/kg) in vehicle, Rosi and Tesa mid dose treated TN mice. Data presented as mean ± SEM (n = 4–5/group): ∗P < 0.05; ∗∗P < 0.01; ∗∗∗P < 0.001 vs. indicated groups in a, c, e and g–k: ∗P < 0.05; ∗∗P < 0.01; ∗∗∗P < 0.001 vs. vehicle, †P < 0.05; ††P < 0.01; †††P < 0.001 vs. rosiglitazone and #P < 0.05 vs. Tesa high dose in b, d and l by one-way (a, c, e and g-k) or two-way (d and l) ANOVA or ANCOVA with body mass as covariate (b), all with Tukey's multiple comparisons test.
3.5. Tesaglitazar improves metabolic control and ameliorates high fat diet-induced hepatic steatosis
Rosiglitazone significantly decreased fasting glucose, insulin, and increased insulin sensitivity in DIO mice (Figure 4G–I). The metabolic benefits were more pronounced upon tesaglitazar treatment, with improvements observed in both the TN and TN→RT groups. In particular, the mid and high dose tesaglitazar groups demonstrated an almost complete normalization of fasting plasma glucose levels and a >10-fold reduction in fasting plasma insulin, compared to vehicle controls (Figure 4G–H), resulting in a robust increase in insulin sensitivity, as indicated by the calculated quantitative insulin sensitivity check index (QUICKI, 0.44 vs. 0.25, Figure 4I). Tesaglitazar treatment also ameliorated dyslipidemia, with a ∼2-fold reduction in both plasma triglycerides (TG) (Figure 4J) and total cholesterol levels (Figure S5f). Importantly, tesaglitazar treatment resulted in markedly reduced hepatic steatosis, with liver TG content reduced by more than 80% in the high dose tesaglitazar group at TN→RT compared to vehicle controls (Figure 4K). By contrast, rosiglitazone only modestly decreased plasma TG and cholesterol levels and did not decrease liver TG content. Plasma ketone bodies were significantly raised following tesaglitazar, but not rosiglitazone, treatment (Figure S5e), indicating an increased hepatic fatty acid oxidation. As assessed by an insulin tolerance test, both tesaglitazar (mid dose) and rosiglitazone treated mice displayed enhanced insulin sensitivity at TN. However, insulin sensitivity was significantly greater in tesaglitazar-treated mice compared to rosiglitazone-treated animals (Figure 4l).
3.6. PPARγ activation alone is sufficient to convert white into brown-like adipocytes in vitro, but PPARα activation and PPARγ activation have synergistic effects in vivo
Although tesaglitazar was superior to rosiglitazone at inducing browning in vivo, rosiglitazone and tesaglitazar were found to be equally efficacious in vitro, resulting in a 2000- to 3000-fold induction of Ucp1 and a ∼1000-fold increase in Cidea mRNA in primary mouse inguinal preadipocytes. Interestingly, the selective PPARα activator WY14643 had no effect on Ucp1 or Cidea gene expression (Figure 5A), nor did combining rosiglitazone with WY14643 in vitro further increase Ucp1 or Cidea levels compared to rosiglitazone alone (Figure 5A). A similar expression pattern was also observed in human preadipocytes: rosiglitazone and tesaglitazar treatment both resulted in similar induction of UCP1 mRNA (15- to 18-fold) and CIDEA mRNA (40- to 60-fold), WY14643 had no significant effect on the expression of those genes, and the combination of rosiglitazone and WY14643 did not result in any further increase in UCP1 or CIDEA compared to rosiglitazone or tesaglitazar alone (Figure 5C). A next to identical expression pattern was observed when comparing compound effect on differentiated mouse adipocytes and freshly isolated mature human adipocytes (Figure 5B,D), ruling out any significant or direct contribution of PPARα activation in the browning of white fat, both in preadipocytes and adipocytes.
Treating lean mice at TN with either fenofibrate (a selective PPARα activator) or rosiglitazone (a selective PPARγ activator) resulted in little to no upregulation of Ucp1 in either iWAT, eWAT, or iBAT (Figure 5E). However, dual PPARα/γ activator treatments, given either as a combination of rosiglitazone + fenofibrate (Rosi + Feno), or as tesaglitazar or saroglitazar, resulted in a marked and similar increase in Ucp1 mRNA levels across iWAT and eWAT (Figure 5E). This indicates that a combination of PPARα and PPARγ activation is necessary to promote significant browning of white fat in vivo, likely via a synergy between PPARγ action in adipose tissue and PPARα action in non-adipose tissues. Metabolic characterization of lean and DIO mice treated with PPARα, PPARγ and dual PPARα/γ activators is provided in Supplementary Figures 7 and 8.
3.7. Increased FGF21 contributes to dual PPARα/γ activation-induced browning of WAT in vivo
Fibroblast growth factor 21 (FGF21) is a predominantly liver-derived hormone that plays an important role in lipid and glucose homeostasis [38,39] and is regulated by PPARα [40]. FGF21 has been shown to induce browning of white fat [41,42]. In obese mice, tesaglitazar treatment dose-dependently increased circulating Fgf21 levels (up to 8-fold at the highest dose), as well as Fgf21 expression levels in liver and iBAT (up to 3- and 6-fold, respectively). By contrast, rosiglitazone treatment did not alter Fgf21 levels (Figure 6A). Expression of additional PPARα target genes Fabp3 and Ehhadh [29] was also strongly increased in the livers of tesaglitazar-treated mice, indicating that tesaglitazar robustly and dose-dependently activated PPARα in the liver (Figure 6A). Similar increases in Fgf21, Fabp3 and Ehhadh were also observed in lean mice following treatment with either fenofibrate, tesaglitazar, saroglitazar, or the combination of rosiglitazone + fenofibrate (Rosi + Feno) compared to vehicle controls (Figure 6B). As in obese mice, lean mice treated with rosiglitazone did not display increased Fgf21 levels or increased expression of PPARα target genes in the liver (Figure 6B). Across all dual PPARα/γ activator treatments (for example, tesaglitazar, saroglitazar and Rosi + Feno), linear regression analysis revealed a strong correlation between circulating Fgf21 levels and Ucp1 mRNA expression in iWAT, eWAT and iBAT in both lean and obese mice. This suggests that Fgf21 plays an important role in the Ucp1 induction observed with dual PPARα/γ activator treatment (Figure 6C–E). However, even though mice treated with the selective PPARα activator fenofibrate displayed similar circulating Fgf21 levels as those achieved with the dual PPARα/γ activator treatments (Figure 6B), Ucp1 transcript levels were not raised in this group (Figure 5E). This indicates that a PPARα-mediated increase in Fgf21 levels, in the absence of pharmacological PPARγ activation in adipose, is not sufficient to induce browning of white fat in vivo. To test the hypothesis that FGF21 and PPARγ synergize to induce browning of white fat, we treated lean mice with rosiglitazone, FGF21, rosiglitazone + FGF21, or tesaglitazar for two weeks in thermoneutral conditions. Rosiglitazone or FGF21 monotreatments did not significantly increase expression of Ucp1. By contrast, combined FGF21 and rosiglitazone treatment increased Ucp1 mRNA levels 30-fold in iWAT compared to vehicle controls, similar to the effect of tesaglitazar treatment. Similar gene expression pattern was observed for Cidea and Pdk4, and in eWAT (Figure 6F and Figure S9a). In vitro, combined treatment of mouse preadipocytes with Fgf21 and a low dose of rosiglitazone (inducing only a moderate Ucp1 increase) also resulted in a synergistic increase in Ucp1 mRNA levels (∼1000-fold induction), superior to the effect of either rosiglitazone (∼100-fold induction) or Fgf21 (∼10-fold induction) alone (Figure 6G). Similar effects were also observed in both human preadipocytes and mature adipocytes (Figures. S9e–f). Thus PPARγ activation and Fgf21 have synergistic effects to induce browning of white fat in mice in vivo. These effects are, at least in part, cell-autonomous via direct action on white adipocytes (Figure 6G). Together, these results indicate that PPARγ activator treatment is sufficient to promote browning of white adipocytes in vitro, with PPARα playing a negligible role. However, a combined activation of both PPARγ and PPARα is needed for robust browning in vivo, via both PPARγ action in adipose and an increase in Fgf21 levels from PPARα activation in the liver, and to a lesser extent iBAT. Increased browning of white fat leading to increased energy expenditure likely contributes to the whole-body improvements in metabolism observed with dual PPARα/γ activator treatment.
4. Discussion
Treatments that induce the browning of white fat and promote energy expenditure have recently been suggested as therapies for metabolic diseases [12,13]. Current strategies to induce the formation and activation of thermogenic adipocytes typically involve mimicking a cold stimulus via β-adrenergic receptor agonism. However, this is associated with undesirable cardiovascular effects, as β-adrenergic receptors are widely expressed across multiple tissues [7]. Thus other safer approaches are needed to clinically induce the formation and activation of thermogenic fat. Given that both PPARα and PPARγ have been implicated in browning [43], we hypothesized that dual PPARα/γ activators could possess a synergistic ability to induce browning of white fat. We found that dual PPARα/γ activators were robust Ucp1 inducers in both human and mouse preadipocytes and mature adipocytes, with tesaglitazar affording the highest Ucp1 expression of the dual PPARα/γ activators tested. In vivo, tesaglitazar treatment of both lean and obese mice robustly induced the browning of white fat, increased energy expenditure, lowered body weight, enhanced insulin sensitivity and reduced liver steatosis.
The ability of some PPARγ activators, including rosiglitazone, to induce Ucp1 has been well documented [16,[20], [21], [22], [23]]. However, to our knowledge, the effects of dual PPARα/γ activators on the browning of white fat have not previously been studied. Our results show that tesaglitazar is the most efficacious synthetic in vivo browning agent reported to date. We also show that browning of white fat via PPARα and PPARγ differs between the in vitro and in vivo settings. In vitro, PPARα activation does not induce Ucp1 expression, and PPARγ and dual PPARα/γ activators are equally efficacious inducers of Ucp1. This indicates that selective PPARγ activation is sufficient for the conversion of white into brown-like adipocytes in vitro. In vivo however, selective PPARγ activation results in only moderate browning of white fat and dual PPARα/γ activation (by either a single dual PPARα/γ molecule or a combination of selective PPARα and PPARγ activators) is necessary to induce marked browning. We propose that the superior efficacy of dual PPARα/γ activators over selective PPARγ activators is due their PPARα activation-mediated increase in circulating Fgf21 levels and subsequent synergistic effect with PPARγ activation in adipose tissues. Indeed, tesaglitazar treatment resulted in a marked and dose-dependent increase in Fgf21 expression in both liver and iBAT and increased plasma Fgf21 levels. Plasma Fgf21 levels significantly and positively correlated with Ucp1 levels in adipose tissues in all in vivo studies performed with dual PPARα/γ activation. Furthermore, combined FGF21 and rosiglitazone treatment leads to a synergistic browning of white fat in vivo, similar to the effect of tesaglitazar treatment. The liver is the main site of FGF21 production and it is transcriptionally induced in response to fasting and PPARα activation [38] and pharmacological FGF21 treatment has been shown to induce browning of white fat [41,42]. In the present study, elevated circulating levels of Fgf21 are observed following both selective PPARα activator treatment (fenofibrate) and dual PPARα/γ activator treatments (tesaglitazar, saroglitazar or the combination of rosiglitazone + fenofibrate) but not selective PPARγ activator treatment (rosiglitazone). However, PPARα activator treatment does not significantly induce browning of white fat, despite an 8-fold elevation of plasma Fgf21 levels. Similarly, in vitro treatment of preadipocytes or adipocytes with FGF21 results in little to no increase in UCP1 mRNA levels. However, a synergistic increase in UCP1 is observed when cells are treated with the combination of FGF21 and rosiglitazone, indicating that FGF21 has a direct effect on the browning of white adipocytes, however, only when PPARγ is activated. An additional contribution from FGF21 is also likely to occur via the central nervous system. FGF21 can cross the blood–brain barrier and act on the paraventricular hypothalamus to increase β-adrenergic signaling in sympathetic ganglia, resulting in increased adrenergic activity in tissues such as WAT and BAT [44,45].
In obese mice housed at TN, tesaglitazar treatment significantly decreased body weight, improved insulin sensitivity and dyslipidemia, and reduced hepatic steatosis. This is due in part to the browning of white fat, as tesaglitazar increased energy expenditure in both lean and obese mice even under thermoneutral conditions, with no difference in locomotor activity. RT is considered a mild cold challenge for mice [37], resulting in a β-adrenergic receptor-mediated increase and activation of beige and brown adipocytes [46]. Switching the mice to RT for an additional 2 weeks after an initial 2 weeks of tesaglitazar treatment at TN (TN→RT group), resulted in an additional induction of Ucp1 in the iWAT depot, compared to tesaglitazar treated TN mice (TN group). In lean mice, maximal tesaglitazar-induced browning was observed after 2 weeks, with no additional increase in Ucp1 following 3 more weeks of treatment. Together, this suggests that tesaglitazar and a mild cold stimulus (RT) act in synergy to induce maximal browning of white fat. Induction of thermogenic genes after treatment with the β3-adrenergic receptor agonist CL316,243 has been reported to be lessened in PPARα-null mice, indicating that PPARα plays a role in cold-induced browning [47]. Moreover, in obese mice treated with rosiglitazone (present study) at TN, reducing the temperature below TN did not result in further induction of Ucp1 expression in WAT compared to TN conditions. Notably, the relatively modest rosiglitazone-induced browning did not result in increased energy expenditure, and body weight even increased, at least in part due to increased food intake. This is consistent with previous findings showing that, in spite of an increased capacity for Ucp1-mediated uncoupled respiration [18,19], in vivo browning of WAT induced by treatment with selective PPARγ activators is not associated with increased energy expenditure [21,27]. Other studies have shown that selective PPARγ activators attenuate the endogenous β-adrenergic stimulated activation of adipocytes in vivo [[48], [49], [50]]. While the tesaglitazar-mediated increase in energy expenditure is likely due to the superior browning of white fat compared to selective PPARγ treatment, it could also be in part PPARα-mediated, via FGF21-dependent increased lipid and glucose metabolism and/or increased sympathetic nerve activity [45,51]. Ucp1 was also slightly increased in iBAT following tesaglitazar treatment. While the fold increase of Ucp1 expression in iBAT is much smaller compared to that observed in iWAT (550-fold vs. 5.5-fold), it represents a large change in overall Ucp1 expression levels, given the considerably higher basal expression of Ucp1 in BAT compared to WAT [2]. Therefore the tesaglitazar-induced increase in Ucp1 expression in iBAT may also contribute to the effects of tesaglitazar on energy expenditure and metabolism.
Despite identical in vitro substance concentrations across all dual PPARα/γ compounds tested in the present study, the level of Ucp1 induction differed substantially between compounds with tesaglitazar being the most robust Ucp1 inducer. Comparing absolute in vitro potency values between studies is difficult since assays are designed differently. Nonetheless, one may compare the ratio between PPARγ/PPARα potency (summarized in Table S2). Tesaglitazar, ragaglitazar, imiglitazar, aleglitazar and muraglitazar are reported to have similar PPARα/PPARγ potency ratios [[52], [53], [54], [55], [56]]. Saroglitazar on the other hand, has considerably higher selectivity towards PPARα compared to PPARγ [57], and chiglitazar and reglitazar have higher selectivity towards PPARγ compared to PPARα [58,59]. Thus neither potency nor PPARα/γ selectivity appears to explain the difference in UCP1-inducing properties of the tested dual PPARα/γ compounds in vitro. Selective modulation of the receptors, as opposed to activation/agonism, as measured in a reporter gene assay, may be more important to induce a brown fat gene transcription program in adipocytes [60]. PPARγ-mediated browning of WAT occurs via SIRT1-, PRDM16-, C/EBPα-, and PGC-1α-dependent mechanisms [16,[24], [25], [26]]. Post-translational modifications of PPARγ also play a role in browning of white fat [27]. Thus, it is possible that tesaglitazar induces “optimal” conformational or post-translational changes at the receptor level, facilitating the interaction between key transcriptional coregulators and factors, ultimately promoting a brown fat phenotype.
Tesaglitazar (Galida™) was developed by AstraZeneca with the intention to treat glucose and lipid abnormalities in patients with type 2 diabetes. Although very promising anti-diabetic effects have been reported in both rodents [61] and humans [62], the development of tesaglitazar was discontinued during phase III, because of a poor benefit–risk profile [63]. Despite the challenges of developing activators of the PPAR family of transcription factors, the present study provides clear evidence that it may be possible to develop compounds inducing the robust browning of white fat and metabolic improvements, via dual modulation of PPARα and PPARγ. A combination of a safe PPARγ activator together with FGF21 could also be considered as therapeutic approach for the treatment of diabetes.
5. Conclusions
Our study clarifies the respective roles of PPARα and PPARγ in the browning of white fat in vitro and in vivo. In vitro, selective PPARγ activation is sufficient for the conversion of white into brown-like adipocytes, with PPARα playing a negligible role. However, a combined activation of both PPARγ and PPARα is needed for robust browning in vivo; via both PPARγ action in adipose and an increase in FGF21 levels from PPARα activation in the liver, and to a lesser extent iBAT. To our knowledge, tesaglitazar is the most efficacious synthetic in vivo browning agent reported to date and is therefore a valuable new tool to study the function of beige adipocytes and the process of browning of white fat in vivo. Our study also identifies a novel opportunity to develop compounds able to robustly convert white into brown adipocytes, through the modulation of multiple PPARs.
Funding and acknowledgements
This work was supported by AstraZeneca Gothenburg Sweden; Knut and Alice Wallenberg Foundation and the Wallenberg Centre for molecular and translational medicine, University of Gothenburg, Sweden. T.K. M.H. and S.M. are/were fellows of the AstraZeneca postdoc programme. The authors acknowledge the Animal Sciences & Technologies (AST), AstraZeneca Gothenburg for their excellent support in the in vivo studies.
Author contributions
T.K, M.H., S.M., D.N., I.A., A.A, A.L. and J.B. performed and analyzed the in vivo experiments. M.H., S.M and L.B performed and analyzed the in vitro experiments. P.G. performed the modeling of pharmacokinetic data. T.K., M.H., V.O., C.M., G.O. and J.B. conceived the study. T.K., M.H. and J.B. interpreted the data. T.K. and J.B. wrote the manuscript, with contributions from M.H., P.G. and G.O. All authors critically reviewed and edited the manuscript. J.B. supervised the study.
Data availability
Data supporting this study are available from the corresponding author upon reasonable request.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.molmet.2020.02.007.
Conflict of interest
None declared.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Frayn K. Adipose tissue as a buffer for daily lipid flux. Diabetologia. 2002;45:1201–1210. doi: 10.1007/s00125-002-0873-y. [DOI] [PubMed] [Google Scholar]
- 2.Cannon B., Nedergaard J. Brown adipose tissue: Function and Physiological Significance Physiological Reviews. 2004;84:277–359. doi: 10.1152/physrev.00015.2003. [DOI] [PubMed] [Google Scholar]
- 3.Cypess A.M., Lehman S., Williams G., Tal I., Rodman D., Goldfine A.B. Identification and importance of brown adipose tissue in adult humans New England. Journal of Medicine. 2009;360:1509–1517. doi: 10.1056/NEJMoa0810780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saito M., Okamatsu-Ogura Y., Matsushita M., Watanabe K., Yoneshiro T., Nio-Kobayashi J. High incidence of metabolically active brown adipose tissue in healthy adult humans: effects of cold exposure and adiposity. Diabetes. 2009;58:1526–1531. doi: 10.2337/db09-0530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Marken Lichtenbelt W.D., Vanhommerig J.W., Smulders N.M., Drossaerts J.M., Kemerink G.J., Bouvy N.D. Cold-activated brown adipose tissue in healthy men New England. Journal of Medicine. 2009;360:1500–1508. doi: 10.1056/NEJMoa0808718. [DOI] [PubMed] [Google Scholar]
- 6.Virtanen K.A., Lidell M.E., Orava J., Heglind M., Westergren R., Niemi T. Functional brown adipose tissue in healthy adults. New England Journal of Medicine. 2009;360:1518–1525. doi: 10.1056/NEJMoa0808949. [DOI] [PubMed] [Google Scholar]
- 7.Cypess A.M., Weiner L.S., Roberts-Toler C., Elía E.F., Kessler S.H., Kahn P.A. Activation of human brown adipose tissue by a β3-adrenergic receptor agonist. Cell Metabolism. 2015;21:33–38. doi: 10.1016/j.cmet.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cousin B., Cinti S., Morroni M., Raimbault S., Ricquier D., Penicaud L. Occurrence of brown adipocytes in rat white adipose tissue: molecular and morphological characterization. Journal of Cell Science. 1992;103:931–942. doi: 10.1242/jcs.103.4.931. [DOI] [PubMed] [Google Scholar]
- 9.Wu J., Boström P., Sparks L.M., Ye L., Choi J.H., Giang A.-H. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell. 2012;150:366–376. doi: 10.1016/j.cell.2012.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shabalina I.G., Petrovic N., de Jong J.M., Kalinovich A.V., Cannon B., Nedergaard J. UCP1 in brite/beige adipose tissue mitochondria is functionally. Thermogenic Cell Reports. 2013;vol. 5:1196–1203. doi: 10.1016/j.celrep.2013.10.044. [DOI] [PubMed] [Google Scholar]
- 11.Seale P., Conroe H.M., Estall J., Kajimura S., Frontini A., Ishibashi J. Prdm16 determines the thermogenic program of subcutaneous white adipose tissue in mice. Journal of Clinical Investigation. 2011;121:96–105. doi: 10.1172/JCI44271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peng X.-R., Gennemark P., O'Mahony G., Bartesaghi S. Unlock the thermogenic potential of adipose tissue: pharmacological modulation and implications for treatment of diabetes and obesity. Frontiers in Endocrinology. 2015;6:174. doi: 10.3389/fendo.2015.00174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Emont M.P., Kim D.-i., Wu J. 2018. Development, activation, and therapeutic potential of thermogenic adipocytes Biochimica et Biophysica Acta (BBA)-Molecular and Cell Biology of Lipids. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Desvergne B., Wahli W. Peroxisome proliferator-activated receptors. Nuclear Control of Metabolism Endocrine Reviews. 1999;20:649–688. doi: 10.1210/edrv.20.5.0380. [DOI] [PubMed] [Google Scholar]
- 15.Petrovic N., Shabalina I.G., Timmons J.A., Cannon B., Nedergaard J. Thermogenically competent nonadrenergic recruitment in brown preadipocytes by a PPARγ agonist. American Journal of Physiology. Endocrinology and Metabolism. 2008;295:E287–E296. doi: 10.1152/ajpendo.00035.2008. [DOI] [PubMed] [Google Scholar]
- 16.Vernochet C., Peres S.B., Davis K.E., McDonald M.E., Qiang L., Wang H. C/EBPα and the corepressors CtBP1 and CtBP2 regulate repression of select visceral white adipose genes during induction of the brown phenotype in white adipocytes by peroxisome proliferator-activated receptor. γ agonists Molecular and cellular biology. 2009;29:4714–4728. doi: 10.1128/MCB.01899-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elabd C., Chiellini C., Carmona M., Galitzky J., Cochet O., Petersen R. Human Multipotent Adipose-Derived Stem Cells Differentiate into Functional Brown Adipocytes Stem cells. 2009;27:2753–2760. doi: 10.1002/stem.200. [DOI] [PubMed] [Google Scholar]
- 18.Petrovic N., Walden T.B., Shabalina I.G., Timmons J.A., Cannon B., Nedergaard J. Chronic peroxisome proliferator-activated receptor γ (PPARγ) activation of epididymally derived white adipocyte cultures reveals a population of thermogenically competent, UCP1-containing adipocytes molecularly distinct from classic brown adipocytes. Journal of Biological Chemistry. 2010;285:7153–7164. doi: 10.1074/jbc.M109.053942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bartesaghi S., Hallen S., Huang L., Svensson P.-A., Momo R.A., Wallin S. Thermogenic activity of UCP1 in human white fat-derived beige adipocytes. Molecular Endocrinology. 2015;29:130–139. doi: 10.1210/me.2014-1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fukui Y., Masui S.-i., Osada S., Umesono K., Motojima K. A new thiazolidinedione, NC-2100, which is a weak PPAR-gamma activator, exhibits potent antidiabetic effects and induces uncoupling protein 1 in white adipose tissue of KKAy obese mice. Diabetes. 2000;49:759–767. doi: 10.2337/diabetes.49.5.759. [DOI] [PubMed] [Google Scholar]
- 21.Sell H., Berger J.P., Samson P., Castriota G., Lalonde J.e., Deshaies Y. Peroxisome proliferator-activated receptor γ agonism increases the capacity for sympathetically mediated thermogenesis in lean and ob/ob mice. Endocrinology. 2004;145:3925–3934. doi: 10.1210/en.2004-0321. [DOI] [PubMed] [Google Scholar]
- 22.Wilson-Fritch L., Nicoloro S., Lazar M.A., Chui P.C., Leszyk J., Straubhaar J. Mitochondrial remodeling in adipose tissue associated with obesity and treatment with rosiglitazone. Journal of Clinical Investigation. 2004;114:1281–1289. doi: 10.1172/JCI21752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rong J.X., Qiu Y., Hansen M.K., Zhu L., Zhang V., Xie M. Adipose mitochondrial biogenesis is suppressed in db/db and high-fat diet–fed mice and improved by rosiglitazone. Diabetes. 2007;56:1751–1760. doi: 10.2337/db06-1135. [DOI] [PubMed] [Google Scholar]
- 24.Pardo R., Enguix N., Lasheras J., Feliu J.E., Kralli A., Villena J.A. Rosiglitazone-induced mitochondrial biogenesis in white adipose tissue is independent of peroxisome proliferator-activated receptor γ coactivator-1α. PloS One. 2011;6 doi: 10.1371/journal.pone.0026989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qiang L., Wang L., Kon N., Zhao W., Lee S., Zhang Y. Brown remodeling of white adipose tissue by. SirT1-dependent deacetylation of Pparγ Cell. 2012;150:620–632. doi: 10.1016/j.cell.2012.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ohno H., Shinoda K., Spiegelman B.M., Kajimura S. PPARγ agonists induce a white-to-brown fat conversion through stabilization of PRDM16 protein. Cell Metabolism. 2012;15:395–404. doi: 10.1016/j.cmet.2012.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang H., Liu L., Lin J.Z., Aprahamian T.R., Farmer S.R. Browning of white adipose tissue with roscovitine induces a distinct population of UCP1+ adipocytes. Cell Metabolism. 2016;24:835–847. doi: 10.1016/j.cmet.2016.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferré P. The biology of peroxisome proliferator-activated receptors: relationship with lipid metabolism and insulin sensitivity. Diabetes. 2004;53:S43–S50. doi: 10.2337/diabetes.53.2007.s43. [DOI] [PubMed] [Google Scholar]
- 29.Rakhshandehroo M., Knoch B., Müller M., Kersten S. 2010. Peroxisome proliferator-activated receptor alpha target genes PPAR research 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hondares E., Rosell M., Dıaz-Delfın J., Olmos Y. Peroxisome proliferator-activated receptor (PPAR) induces PPAR coactivator 1 (PGC-1) gene expression and contributes to thermogenic activation of brown fat: involvement of PRDM16. Journal of Biological Chemistry. 2011;286:43112–43122. doi: 10.1074/jbc.M111.252775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barberá M.J., Schlüter A., Pedraza N., Iglesias R., Villarroya F., Giralt M. Peroxisome proliferator-activated receptor α activates transcription of the brown fat uncoupling protein-1 gene a link between regulation of the thermogenic and lipid oxidation pathways in the brown fat cell. Journal of Biological Chemistry. 2001;276:1486–1493. doi: 10.1074/jbc.M006246200. [DOI] [PubMed] [Google Scholar]
- 32.Rachid T.L., Silva-Veiga F.M., Graus-Nunes F., Bringhenti I., Mandarim-de-Lacerda C.A., Souza-Mello V. Differential actions of PPAR-α and PPAR-β/δ on beige adipocyte formation: a study in the subcutaneous white adipose tissue of obese male mice. PloS One. 2018;13 doi: 10.1371/journal.pone.0191365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rachid T.L., Penna-de-Carvalho A., Bringhenti I., Aguila M.B., Mandarim-de-Lacerda C.A., Souza-Mello V. Fenofibrate (PPARalpha agonist) induces beige cell formation in subcutaneous white adipose tissue from diet-induced male obese mice. Molecular and Cellular Endocrinology. 2015;402:86–94. doi: 10.1016/j.mce.2014.12.027. [DOI] [PubMed] [Google Scholar]
- 34.Harms M.J., Ishibashi J., Wang W., Lim H.-W., Goyama S., Sato T. Prdm16 is required for the maintenance of brown adipocyte identity and function in adult mice. Cell Metabolism. 2014;19:593–604. doi: 10.1016/j.cmet.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harms M.J., Li Q., Lee S., Zhang C., Kull B., Hallen S. Mature human white adipocytes cultured under membranes maintain identity, function. And Can Transdifferentiate Into Brown-like Adipocytes Cell Reports. 2019;27:213–225. doi: 10.1016/j.celrep.2019.03.026. e215. [DOI] [PubMed] [Google Scholar]
- 36.Warner A., Kjellstedt A., Carreras A., Böttcher G., Peng X.-R., Seale P. Activation of β3-adrenoceptors increases in vivo free fatty acid uptake and utilization in brown but not white fat depots in high-fat-fed rats. American Journal of Physiology. Endocrinology and Metabolism. 2016;311:E901–E910. doi: 10.1152/ajpendo.00204.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fischer A.W., Cannon B., Nedergaard J. Optimal housing temperatures for mice to mimic the thermal environment of humans: an experimental study. Molecular metabolism. 2018;7:161–170. doi: 10.1016/j.molmet.2017.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Inagaki T., Dutchak P., Zhao G., Ding X., Gautron L., Parameswara V. Endocrine regulation of the fasting response by PPARα-mediated induction of fibroblast growth factor 21. Cell Metabolism. 2007;5:415–425. doi: 10.1016/j.cmet.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 39.Potthoff M.J., Kliewer S.A., Mangelsdorf D.J. Endocrine fibroblast growth factors 15/19 and 21: from feast to famine. Genes & Development. 2012;26:312–324. doi: 10.1101/gad.184788.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Badman M.K., Pissios P., Kennedy A.R., Koukos G., Flier J.S., Maratos-Flier E. Hepatic fibroblast growth factor 21 is regulated by PPARα and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metabolism. 2007;5:426–437. doi: 10.1016/j.cmet.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 41.Kleiner S., Douris N., Fox E.C., Mepani R.J., Verdeguer F., Wu J. FGF21 regulates PGC-1α and browning of white adipose tissues in adaptive thermogenesis. Genes & Development. 2012;26:271–281. doi: 10.1101/gad.177857.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coskun T., Bina H.A., Schneider M.A., Dunbar J.D., Hu C.C., Chen Y. Fibroblast growth factor 21 corrects obesity in mice. Endocrinology. 2008;149:6018–6027. doi: 10.1210/en.2008-0816. [DOI] [PubMed] [Google Scholar]
- 43.Seale P. Transcriptional regulatory circuits controlling brown fat development and activation Diabetes. 2015;64:2369–2375. doi: 10.2337/db15-0203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Douris N., Stevanovic D.M., Fisher F.M., Cisu T.I., Chee M.J., Nguyen N.L. Central fibroblast growth factor 21 browns white fat via sympathetic action in male mice. Endocrinology. 2015;156:2470–2481. doi: 10.1210/en.2014-2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Owen B.M., Ding X., Morgan D.A., Coate K.C., Bookout A.L., Rahmouni K. FGF21 acts centrally to induce sympathetic nerve activity, energy expenditure. And Weight Loss Cell Metabolism. 2014;20:670–677. doi: 10.1016/j.cmet.2014.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kalinovich A.V., de Jong J.M., Cannon B., Nedergaard J. UCP1 in adipose tissues: two steps to full browning. Biochimie. 2017;134:127–137. doi: 10.1016/j.biochi.2017.01.007. [DOI] [PubMed] [Google Scholar]
- 47.Barquissau V., Beuzelin D., Pisani D.F., Beranger G.E., Mairal A., Montagner A. White-to-brite conversion in human adipocytes promotes metabolic reprogramming towards fatty acid anabolic and catabolic pathways. Molecular Metabolism. 2016;5:352–365. doi: 10.1016/j.molmet.2016.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bakopanos E., Silva J.E. Thiazolidinediones inhibit the expression of beta3-adrenergic receptors at a transcriptional level. Diabetes. 2000;49:2108–2115. doi: 10.2337/diabetes.49.12.2108. [DOI] [PubMed] [Google Scholar]
- 49.Festuccia W.T., Oztezcan S., Laplante M., Berthiaume M., Michel C., Dohgu S. Peroxisome proliferator-activated receptor-γ-mediated positive energy balance in the rat is associated with reduced sympathetic drive to adipose tissues and thyroid status. Endocrinology. 2008;149:2121–2130. doi: 10.1210/en.2007-1553. [DOI] [PubMed] [Google Scholar]
- 50.Festuccia W.T., Blanchard P.-G., Turcotte V., Laplante M., Sariahmetoglu M., Brindley D.N. The PPARγ agonist rosiglitazone enhances rat brown adipose tissue lipogenesis from glucose without altering glucose uptake. American Journal of Physiology - Regulatory, Integrative and Comparative Physiology. 2009;296:R1327–R1335. doi: 10.1152/ajpregu.91012.2008. [DOI] [PubMed] [Google Scholar]
- 51.Goto T., Hirata M., Aoki Y., Iwase M., Takahashi H., Kim M. The hepatokine FGF21 is crucial for peroxisome proliferator-activated receptor-α agonist-induced amelioration of metabolic disorders in obese mice. Journal of Biological Chemistry. 2017;292:9175–9190. doi: 10.1074/jbc.M116.767590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cronet P., Petersen J.F., Folmer R., Blomberg N., Sjöblom K., Karlsson U. Structure of the PPARα and-γ ligand binding domain in complex with AZ 242; ligand selectivity and agonist activation in the. PPAR family Structure. 2001;9:699–706. doi: 10.1016/s0969-2126(01)00634-7. [DOI] [PubMed] [Google Scholar]
- 53.Chakrabarti R., Vikramadithyan R.K., Misra P., Hiriyan J., Raichur S., Damarla R.K. Ragaglitazar: a novel PPARα & PPARγ agonist with potent lipid-lowering and insulin-sensitizing efficacy in animal models. British Journal of Pharmacology. 2003;140:527–537. doi: 10.1038/sj.bjp.0705463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sakamoto J., Kimura H., Moriyama S., Imoto H., Momose Y., Odaka H. A novel oxyiminoalkanoic acid derivative, TAK-559, activates human peroxisome proliferator-activated receptor subtypes. European Journal of Pharmacology. 2004;495:17–26. doi: 10.1016/j.ejphar.2004.05.020. [DOI] [PubMed] [Google Scholar]
- 55.Bénardeau A., Benz J., Binggeli A., Blum D., Boehringer M., Grether U. Aleglitazar, a new, potent, and balanced dual PPARα/γ agonist for the treatment of type II diabetes. Bioorganic & Medicinal Chemistry Letters. 2009;19:2468–2473. doi: 10.1016/j.bmcl.2009.03.036. [DOI] [PubMed] [Google Scholar]
- 56.Devasthale P.V., Chen S., Jeon Y., Qu F., Shao C., Wang W. Design and synthesis of N-[(4-Methoxyphenoxy) carbonyl]-N-[[4-[2-(5-methyl-2-phenyl-4-oxazolyl) ethoxy] phenyl] methyl] glycine [Muraglitazar/BMS-298585], a novel peroxisome proliferator-activated receptor α/γ dual agonist with efficacious glucose and lipid-lowering activities. Journal of Medicinal Chemistry. 2005;48:2248–2250. doi: 10.1021/jm0496436. [DOI] [PubMed] [Google Scholar]
- 57.Jain M.R., Giri S.R., Trivedi C., Bhoi B., Rath A., Vanage G. vol. 3. 2015. (Saroglitazar, a novel PPARα/γ agonist with predominant PPARα activity, shows lipid-lowering and insulin-sensitizing effects in preclinical models Pharmacology research & perspectives). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li P.P., Shan S., Chen Y.T., Ning Z.Q., Sun S.J., Liu Q. The PPARα/γ dual agonist chiglitazar improves insulin resistance and dyslipidemia in MSG obese rats. British Journal of Pharmacology. 2006;148:610–618. doi: 10.1038/sj.bjp.0706745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shibata T., Matsui K., Nagao K., Shinkai H., Yonemori F., Wakitani K. Pharmacological profiles of a novel oral antidiabetic agent, JTT-501. An Isoxazolidinedione Derivative European Journal of Pharmacology. 1999;364:211–219. doi: 10.1016/s0014-2999(98)00832-2. [DOI] [PubMed] [Google Scholar]
- 60.Cock T.A., Houten S.M., Auwerx J. Peroxisome proliferator-activated receptor-γ: too much of a good thing causes harm. EMBO Reports. 2004;5:142–147. doi: 10.1038/sj.embor.7400082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wallenius K., Kjellstedt A., Thalén P., Löfgren L., Oakes N.D. 2013. The PPARα/γ agonist, tesaglitazar, improves insulin mediated switching of tissue glucose and free fatty acid utilization in vivo in the obese Zucker rat PPAR research 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fagerberg B., Edwards S., Halmos T., Lopatynski J., Schuster H., Stender S. Tesaglitazar, a novel dual peroxisome proliferator-activated receptor α/γ agonist, dose-dependently improves the metabolic abnormalities associated with insulin resistance in a non-diabetic population. Diabetologia. 2005;48:1716–1725. doi: 10.1007/s00125-005-1846-8. [DOI] [PubMed] [Google Scholar]
- 63.Wilding J.P., Gause-Nilsson I., Persson A. Tesaglitazar, as add-on therapy to sulphonylurea, dose-dependently improves glucose and lipid abnormalities in patients with type 2 diabetes diabetes and vascular disease. Research: Ideas for Today's Investors. 2007;4:194–203. doi: 10.3132/dvdr.2007.040. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data supporting this study are available from the corresponding author upon reasonable request.