

Summary
Tumor acquired radioresistance remains as the major limit in cancer radiotherapy (RT). Rab25, a receptor recycling protein, has been reported to be enhanced in tumors with aggressive phenotype and chemotherapy resistance. In this study, elevated Rab25 expression was identified in an array of radioresistant human cancer cell lines, in vivo radioresistant xenograft tumors. Clinical investigation confirmed that Rab25 expression was also associated with a worse prognosis in patients with lung adenocarcinoma (LUAD) and nasopharyngeal carcinoma (NPC). Enhanced activities of EGFR were observed in both NPC and LUAD radioresistant cells. Rab25 interacts with EGFR to enhance EGFR recycling to cell surface and to decrease degradation in cytoplasm. Inhibition of Rab25 showed synergized radiosensitivity with reduced aggressive phenotype. This study provides the clinical and experimental evidence that Rab25 is a potential therapeutic target to alleviate the hyperactive EGFR signaling and to prevent RT-acquired tumor resistance in patients with LUAD and NPC.
Subject Areas: Radiation Biology, Molecular Biology, Cell Biology, Cancer
Graphical Abstract
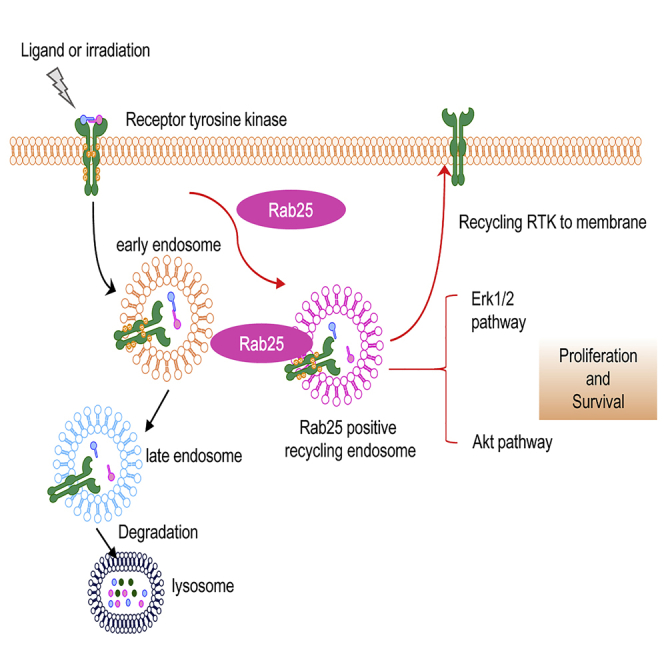
Highlights
-
•
High expression of Rab25 is linked to radioresistance and EMT in lung cancer and NPC
-
•
Rab25 promotes cell therapeutic resistance via RTK-mediated signaling pathways
-
•
Rab25 elevates EGFR recycling rate onto the membrane in therapeutic-resistant cells
-
•
Targeting Rab25 combined with IR might be a potent treatment for the patients with cancer
Radiation Biology; Molecular Biology; Cell Biology; Cancer
Introduction
More than 50% of solid tumors including lung adenocarcinoma (LUAD) and nasopharyngeal carcinoma (NPC) are treated by radiotherapy (RT). However, to enhance the RT efficacy it remains urgent to invent effective targets to eliminate the acquired resistance in treated tumor cells (Huang et al., 2013, Aravindan et al., 2014, Begg et al., 2011). Among the major signaling pathways dominated in cancer cells, the activation of receptor tyrosine kinases (RTKs) has been identified to be the most frequent oncogenic event that drives proliferation and malignant phenotype in tumor cells (Casaletto and Mcclatchey, 2012) and the activated EGFR is tightly associated with tumor response and prognosis after RT (Jakobsen et al., 2016, Higgins et al., 2016). Targeting RTK suppresses tumor growth and synergizes the efficacy of RT (Camidge et al., 2014). Although recycling of EGFR via endosomal pathway is able to enhance oncogenic potency and tumor aggressive phenotype (Shapira et al., 2013, Ye et al., 2016), the mechanism of EGFR recycling underlying tumor adaptive radioresistance remains unsolved.
Both the endocytosis and recycling of EGFR could be enhanced when it is combined with its ligand EGF or under stress condition (Ceresa, 2012, Tan et al., 2016). However, it is unclear how the two processes are coordinated in EGFR-targeted therapy using small molecule tyrosine kinase inhibitors (TKIs) or EGFR-specific antibodies (Chung et al., 2011, Ricordel et al., 2018). To treat tumors with the acquired resistance to TKIs, RT among the others is a choice especially for treatment of local lesions. It has been identified that after endocytosis, the ligand-bound EGFR is arrested in the endosomes via p38/MAPK, which has been linked with the cell death induced by UV, TNFα, or cisplatin (Zwang and Yarden, 2006, Tomas et al., 2015). However, the mechanism regulating the traffic and recycling of EGFR in RT-induced tumor adaptive resistance remains elusive.
Rab25, a member of the Rab11 family, predominantly expresses in epithelial cells and plays a key role in recycling of endosome compartment (Hehnly and Doxsey, 2014). Enhanced GTPase activity is detected in Rab25 compared with other recycling-endosome-related Rab proteins (Casanova et al., 1999). Accumulating evidence suggests that Rab25 is actively involved in carcinogenesis and aggressive phenotype of tumors (Cheng et al., 2012, Dozynkiewicz et al., 2012) and inhibition of Rab25 reduces the aggressive growth in cancer cells (Mitra et al., 2017). Integrin α5β1 is reported to transport by Rab25-positive endosome (Caswell et al., 2007). Rab25 collaborated with CLIC3 to recycle α5β1 in cells of pancreatic ductal adenocarcinoma to promote tumor aggression (Dozynkiewicz et al., 2012). Although both Rab25 and EGFR are suggested to be involved in tumor response to therapies with gefitinib and migration (Jeong et al., 2018, Jo et al., 2014), the interplay between these two factors in signaling tumor acquired radioresistance is to be investigated.
Using radioresistant tumor models from two human cancers mostly treated by RT, this study revealed a previously unknown mechanism by which Rab25 enhances the aberrant transportation of EGFR in radiation-adapted cancer cells. Rab25 is overexpressed in the radioresistant cancer cells and in radioresistant xenograft tumors treated by in vivo RT and correlated with poorer RT response and disease-free survival rate. High expression of Rab25 in the radioresistant cells enhanced the transportation of EGFR to cell surface upon ligand stimulation, and blockage of Rab25 reduced the clonogenicity and aggressive phenotype of radioresistant cancer cells. These provide the evidence indicating that Rab25 plays a critical role in radiation-induced aberrant transportation of EGFR, and thus the Rab25-EGFR pathway is a potential therapeutic target to re-sensitize radioresistant cancer cells.
Results
Rab25 Is Correlated with Tumor Response to RT
To identify key factors associated with NPC radioresistance, a profile of 84 cell death-related genes (Qiagen) was analyzed in CNE2R versus its wild-type counterpart CNE2 (Guo et al., 2003, Li et al., 2013, Fu et al., 2019). This pair of cells showed different morphology, EMT potential, and radiation-induced apoptotic cell death (Figures S1A–S1C). Among the short list of genes upregulated in radioresistant NPC cell CNE2R, Rab25, the only protein involved in cargo recycling, showed a 7-fold increase in comparison with CNE2 cells (Figure S1D). The enhanced Rab25 protein levels were then further identified in CNE2R and in three radioresistant LUAD (A549R, H358R, and H157R) cells You et al., 2014 (Figure 1A) and two chemo-resistant cancer cell lines, ovarian cancer SKOV3R and NPC CNE1-TR cells Hou et al., 2017, Zhang et al., 2012, Zhou et al., 2015 (Figure S3A). We also observed a significant increased expression of Rab25 in lung xenografts that received one dose of radiation (Figure 1B), suggesting that radiation might induce Rab25 expression.
Figure 1.

Induction of Rab25 Is Involved in Acquired Tumor Radioresistance
(A) Increased Rab25 expression in radioresistant lung cancer cells (H157R, H358R, and A549R) and NPC cells (CNE2R) derived from the surviving residues of corresponding wild-type counterparts treated by radiation with fractionated doses.
(B) Schematic (top) and IHC staining of Rab25 in A549 xenografts treated with or without local irradiation, 2 Gy per day for 2 days (bottom). The average Rab25-positive cells in tumors were quantified and are shown in the right bar graph. Scale bar, 50 μm. n = 3, mean ± SD, ∗p < 0.05.
(C) Schematic diagram for establishment of radioresistant xenograft model. CNE2 cells were injected subcutaneously into the right flanks of nude mice, and when tumors reached a volume of approximately 200 mm3, radiotherapy was delivered to the local tumor (5 Gy per day for 2 days; total dose = 10 Gy). On day 5 after last irradiation, the tumors were removed and one part of tumor tissues was fixed for IHC analysis and another part of tumor tissues was inoculated subcutaneously into the right flank of another mouse (P1). When the volume of the re-planted tumors in the P1 mice reached approximately 200 mm3, radiotherapy was delivered again with the same dose, and such treatment was repeated for 11 cycles (P11). The radiosensitivity of tumors from P1 and P11 mice was measured by transplanting the tumors from P1 and P11 mice and irradiating with 5 Gy × 2 when tumor reached 200 mm3.
(D) Representative IHC staining of Rab25 in P1 and P11tumors (left). The average Rab25-positive cells in tumors were quantified (six fields were randomly selected for each xenograft) and shown in the right. Scale bar, 50 μm. n = 18, mean ± SD, ∗∗p < 0.01.
To elucidate why expression of Rab25 could be induced by a single dose of radiation, we assume that such a quick induction of Rab25 expression may well be mediated at a transcriptional level. Thus, we sought candidate transcriptional factors in the 500-bp promoter region of Rab25 gene by a prediction database JASPAR (http://jaspar.genereg.net/) and found that there were two binding sites of Stat5 in the promoter of Rab25 (Figure S1E). As reported by us (Fu et al., 2019) and others (Hasselbach et al., 2005, Maranto et al., 2018), STAT5 is one of the important transcriptional factors responsive to irradiation. In CNE2R cells, expression of Stat5a or Stat5b was much higher than in CNE2 cell and could be induced by a single dose of radiation (Figures S1F and S1G). We also observed an induction for protein levels of both Rab25 and Stat5 in CNE2 cell after a single-dose radiation (Figure S1H). Knockdown of Stat5a and Stat5b by small interfering RNA (siRNA) in CNE2R cell dramatically decreased the expression of Rab25 (Figure S1I). The above results suggested that Stat5 might be an important transcriptional factor for Rab25 and might regulate the radiation-induced expression of Rab25.
We constructed a radioresistant xenografts model to study whether Rab25 expression is linked to radioresistance in vivo. Briefly, we injected CNE2 cells subcutaneously into the flank of nude mice (P0), and when xenografts volume reached 200 mm3, we passaged the xenograft into another mice (P1). From P1 passage, xenografts were irradiated at 5 Gy two times when their volume reached 200 mm3. Xenografts had received a total dose of 110 Gy when we transplanted tumor for 11 passages (Figure 1C). Compared with P1 (only transplant for one passage), P11 tumor growth became non-responsive to radiotherapy after 5 Gy∗2 treatments (Figure S2A). The enhanced Rab25 immunohistochemistry (IHC) staining was observed in three of four P11 xenografts, whereas no enhanced IHC staining was detected in four P0 xenografts, demonstrating that Rab25 can be up-regulated in tumors that received radiotherapy in vivo, or in other words, Rab25 was enhanced in radioresistant tumors (Figures 1D). In addition, the Rab25 IHC staining in sham P4 tumor (just transplanted for four passages without irradiation) was not increased, suggesting that transplantation alone might not induce Rab25 expression (Figure S2B).
To further examine Rab25-mediated tumor aggressive phenotype, IHC staining of Rab25 was conducted on 101 tumor tissues of patients with NPC who received upfront standard radiotherapy in Xiangya Hospital in recent 5 years. According to the treatment outcomes evaluated using magnetic resonance imaging (MRI) and the Response Evaluation Criteria in Solid Tumors (RECIST) used in the clinic, patients were categorized into three groups: complete response (CR), partial response (PR), and stable disease (SD). Rab25 expression was then evaluated in these three groups. All 17 SD samples (100%) and 33 of 47 (70.2%) PR samples showed strong Rab25 staining, whereas only 8 of 37 (21.6%) of the CR samples were Rab25 positive (score higher than 2, Figures 2A and S2C). However, Rab25 expression was not significantly correlated with histological stage, lymph node metastasis, age, or tumor size (Table S1), further suggesting that Rab25 is a potential biomarker for predicting the radiotherapy response. An analysis based on the Cancer Genome Atlas (TCGA) revealed that Rab25 is highly expressed in various tumors (Figure 2B and Table S2), including LUAD, which exhibits much higher expression of Rab25 than adjacent normal tissues (Figure 2B). The data from TCGA showed that both overall survival (OS) and disease-free survival (DFS) of the patients with LUAD with high Rab25 expression had worse outcomes than those with low Rab25 expression (Figures 2C and 2D), which suggested a role of Rab25 in tumor progression and therapeutic response.
Figure 2.

Rab25 Is Linked with Poor Prognosis and Less Responsiveness of Patients with NPC to Radiotherapy
(A) RT responsiveness of 101 patients with NPC was correlated with Rab25 expression levels; clinic response of all patients was scored as complete response (CR), partial response (PR), and stable disease (SD), and expression levels of Rab25 were scored with four levels (0–4) illustrated in left. Scale bar, 50 μm. (B) Elevated Rab25 expression in a large scale of human cancers (red) compared with normal tissues (gray). The graph was generated from the GEPIA website (http://gepia.cancer-pku.cn/) and was based on the transcriptional level of Rab25 in an array of human cancer from multiple cohorts of TCGA study. The median expression of Rab 25 presented as transcripts per million (TPM) was chosen in each cancer type, and the case number of each cancer type was illustrated in Table S2. Right, Rab25 expression summarized from 483 patients with LUAD and 347 normal lung tissues (mean ± SD, ∗p < 0.05). (C) Overall survival or (D) disease-free survival of patients with LUAD generated using the log rank test based on Rab25 expression in LUAD tissues from the TCGA cohort.
Down-Expression of Rab25 Suppresses RTKs-Mediated Signaling Pathways
RTK-mediated signaling is among the most frequently activated pathways that regulate cancer progression and therapeutic response. We observed that the content of EGFR was increased in radio- or chemo-resistant cells, whereas the downstream pathways, including Akt and Erk signaling, were hyperactive (Figures 3A and S3A). In NPC clinical samples, the EGFR expression level and the treatment response were closely correlated in CR and SD groups (Figures 3B and S3B), whereas the levels of EGFR and Rab25 expression was positively associated, indicating these two molecules contributed to NPC radioresponse (Pearson r = 0.4, p = 0.00005; Figure 3C).
Figure 3.

Rab25 Is Involved in the Activation of EGFR Pathway
(A) Immunoblotting of EGFR and phosphorylation status of Akt at Ser473 and Erk at Thr202 and Tyr204 in three pairs of lung radioresistant cells.
(B) Scores of EGFR IHC staining were shown as percentage for EGFR IHC analysis with 4 as the highest and 0 the lowest expression. Representative IHC imaging were shown in Figure S3B.
(C) Co-expression of Rab25 and EGFR in 101 NPC tissues corresponding to the patients grouped as CR, PR, and SD. The intensity of IHC staining is scored from 0 to 1 (for “no signal”) to 4 (for “strongest signal”). Rab25 and EGFR staining intensity from each patient was compared as two datasets in a column table. R value was calculated using GraphPad Prism software and the p value was calculated by two-tailed analysis and confidence interval is 95%.
(D) The relative phosphorylation of RTKs in H358R cells and H358R transfected with siRab25. Total proteins were extracted and screened simultaneously using R&D Systems Human Phospho-Receptor Tyrosine Kinase (RTK) Array Kit. The spot intensities were quantified using ImageJ software. The values for control cells (blue box) were set as 1, and the values of siRab25 (red box) were shown relative to the control, n = 3, mean ± SD, ∗∗p < 0.01.
(E) Expression of phosphorylated EGFR, HER2, c-Met, and the associated downstream signal molecules was analyzed by PathScan RTK signaling antibody array kit in CNE2R cell (control) and CNE2R transfected with Rab25 siRNA. The spot intensities were quantified using ImageJ software. The values for control cells (blue) were set as 1, and the values of siRNA (red) were shown relative to the control. Results represent mean ± SD for three analysis. ∗p < 0.05, ∗∗p < 0.01.
(F) Down-expression of Rab25 reduced the expression of EGFR and the activation of associated signaling pathways in radioresistant cells.
Previous studies (Caswell et al., 2007) and our current data demonstrated that Rab25 could regulate the cellular anti-anoikis ability controlled by RTK-mediated signaling. Thus, we suspected that Rab25 might regulate activities of RTKs and their downstream signaling pathways. In radioresistant cells with silenced Rab25, protein array analyses showed that the levels of some RTK molecules, including RYK, EphB6, EphB2, and Met, had significantly decreased (Figures 3D, 3E, S3C) and the downstream signaling activation was inhibited after Rab25 suppression in CNE2R cells (Figure 3E). Furthermore, a decrease in not only EGFR content but also components of the downstream signaling pathways was verified in NPC CNE2R cell and LUAD A549R cell (Figure 3F).
Rab25 Is Involved in Endosomes Transport of EGFR
We wondered that Rab25 may play a key role in signaling radioresistance via EGFR transportation. Using structured illumination microscopy (SIM) super resolution technology, we observed several red circles (represents Rab25 positive endosome) contained green dots (represents EGFR molecule) and red dots were co-localized with green dots (Figure 4A). We then carried out an immunoprecipitation assay using anti-Rab25 antibody in CNE2R cells and observed the association between Rab25 and EGFR (Figure 4B). To further validate their association, we performed Duolink in situ proximity ligation assay (PLA) and detected numerous EGFR/Rab25 interaction signals representing Rab25 in close proximity to EGFR. The signals for co-localization (red dots) were enhanced at 1 h post irradiation, suggesting radiation promoted the interactions between Rab25 and EGFR (Figure 4C).
Figure 4.

Rab25 Binds and Prevents EGFR Degradation
(A) Images of confocal microscopy of A549R cells stained with antibodies of Rab25 and EGFR and DAPI. White arrows indicate the merged signals. Scale bar of right picture is 2.5 μm.
(B) Rab25 was immunoprecipitated from CNE2R cells, followed by immunoblotting of EGFR and Rab25.
(C) CNE2R cells treated with sham or 5 Gy radiation for 24 h were stained with EGFR and Rab25 antibodies and subjected to Duolink in situ PLA. Scale bar, 25 μm. Bar diagrams, the number of interactions per cell. Mean ± SD, ∗∗p < 0.01.
(D) Images of confocal microscopy of cells stained with antibodies of EEA1 and EGFR.
(E) Co-localization of EGFR and early endosome marker EEA1. Mean ± SD, ∗∗p<0.01.
(F) Immunoblotting of EGFR in CNE2R cells transfected with control or shRab25 and treated with either vehicle (0.01% dimethyl sulfoxide) or 10uM cycloheximide (Chx) for the indicated time.
To characterize the effect of Rab25-positive endosomes on EGFR intracellular endosome distribution, we analyzed the co-localization rate of EGFR with early endosome marker EEA1 in radioresistant cells after downregulating Rab25. Rab25 knockdown led to a significant reduction of EGFR in EEA1-positive early endosomes (Figures 4D and 4E), implying that Rab25 might prevent EGFR degradation by routing EGFR to EEA1-positive early endosomes. Consistently, when treating cells with cycloheximide (CHX) to inhibit EGFR synthesis, we observed that the half-life of EGFR was shortened in Rab25-depleted cells (Figure 4F). These results suggest that Rab25-positive endosomes transport EGFR away from late endosomes to prevent its degradation and to maintain active signaling.
Rab25 Transports EGFR onto the Cell Surface upon EGF Stimulation
Next, we examined whether high expression of Rab25 could enhance the membrane content of EGFR. The radioresistant cancer cells exhibited higher level of EGFR on cell surface than that on parental cells (Figure 5A), and EGFR immunostaining of Rab25-knockdown CNE2R cells showed that Rab25 knockdown caused significant reduction of membrane distribution of EGFR (Figure S4A). Then we tracked the membrane content of EGFR at different times after stimulating cell with EGF. The membrane content of EGFR in CNE2R or A549R cells was consistently much higher than that in parental cells after EGF stimulation (Figure 5B). In addition, overexpression or knockdown of Rab25 markedly impacted the tyrosine phosphorylation of EGFR (Figures 5C and 5D), further indicating that Rab25 influenced EGFR activity.
Figure 5.

Rab25 Enhances EGFR Recycle and Increases Cell Surface Content of EGFR
(A) Immunoblotting of EGFR on cell surface in A549 and A549R cells. PMCA1 was used as plasma membrane loading control, and Hsp90 was used as cytosol protein control.
(B–D) (B) The kinetics of ligand-induced internalization of EGFR in radioresistant CNE2R cell or A549R cell. Phosphorylated tyrosine (p-Tyr) was immunoprecipitated from CNE2R cells with knockdown (C) or overexpression (D) of Rab25, followed with immunoblotting of EGFR.
(E) Endocytotic rate of EGF in CNE2R cell or CNE2R cells with down-expression of Rab25. The amount of alex-488 conjugated EGF was quantified by flow cytometry.
(F) Quantification of the cell surface content of EGFR in cells or in cells with down-expression of Rab25. EGFR cell surface content was performed every 5 min after stimulation with 1 ng/mL EGF. Right, flow cytometry analysis at each time point was plotted as histogram.
(G) Left, cell surface EGFR recycling rate in H358 or H358R cells pretreated with 1 ng/mL EGF for 10 min before which cells had been starved for 6 h. Right: flow cytometry analysis at each time point was plotted as histogram.
(H and I) (H) Cell surface EGFR recycling rate in CNE2 and CNE2 cells stably transfected with Rab25 or in ovarian carcinoma taxol-resistant SKOV3R cells with knockdown of Rab25 (I). Mean ± SD, ∗∗p < 0.01.
(J) EGF-induced activation of RTK-mediated signaling pathways in A549R cells and A549R with siRNA-mediated knockdown of Rab25. Cells were first incubated for 6 h in serum-free medium and then treated with 1 ng/mL EGF for indicated time.
(K) Immunoblotting of radiation-induced activation of RTK pathways in A549R cells or in A549R with siRNA-mediated knockdown of Rab25. Cells were irradiated at 4 Gy.
To clarify whether membrane content alteration of EGFR mediated by Rab25 was on account of the variation in ligand-induced endocytosis, we stimulated cells with two doses of Alexa Fluor 488-EGF (1 or 100 ng/mL) to monitor EGF uptake in CNE2R cells with or without Rab25 knockdown. Flow cytometry analysis showed that, regardless of the EGF concentration, no difference of the rate of EGF uptake was observed between the two types of cells, indicating that Rab25-mediated regulation of the membrane EGFR content was not due to a variation in the coated vesicle formation rate (Figure 5E). The membrane content of EGFR was decreased when we knockdown Rab25 in CNE2R or A549R cell (Figure 5F), which was supported in other therapeutic-resistant cell lines, including radioresistant H358R and H157R LUAD cells and Taxol-resistant ovarian carcinoma SKOV3R cells (Figures S4B–S4E).
We then examined whether the transportation route of internalized EGFR was influenced by Rab25. A “pulse-chase” experiment Kondapalli et al., 2015 was performed to determine the rate of recycling of internalized EGFR. Cells were incubated with a fixed concentration of EGF (1 ng/mL) for 10 or 30 min (stimulation), washed with acid to remove surface EGF, and subsequently transferred the cells back to 37°C for 10–40 min (rest). At the end of the chase, the amount of cell surface EGFR was quantified by FACS analysis. The results showed that during the “chasing phase” the surface content of EGFR in radioresistant H358R cells was quickly recovered to the original level after 10 min, whereas the content of EGFR in parental cells was only less than 60% of the original level (Figure 5G). In Rab25-overexpressing and control NPC cells, both cells had decreased by 25% of the initial value after EGF stimulation for 30 min. During the chasing phase, a small fraction of EGFR molecules was recycled to the cell surface in control cells, and the EGFR membrane content remained at a low level. In contrast, after 40 min of chasing, the membrane content of EGFR nearly reverted to the initial level in Rab25-overexpressing cells (Figure 5H). In addition, suppression of Rab25 in SKOV3R cells severely impaired the trafficking of EGFR onto the cell surface (Figure 5I). We next examined whether hyperactive EGFR signaling in radioresistant cells was dependent on Rab25 expression. After knockdown Rab25 by lenti-viral transduction of Rab25-specific shRNA in radioresistant cells, we found that the phosphorylation of EGFR and activation of Akt and Erk declined when the cells were treated with ligand or radiation (Figures 5J, 5K, and S4F–S4H) and re-expression of Rab25 in Rab25 knockdown cells compensated the declination, suggesting that Rab25 actively participates in regulating RTK protein levels and activity in response to irradiation or ligands. Taken together, these results indicate that Rab25-endosomes are responsible for the enhanced level and activity of EGFR in therapeutic-resistant cancer cells.
Blocking Rab25 Sensitizes Radioresistant Cells with Reversed EMT
The high expression of Rab25 in radioresistant cells indicated its close association with radioresistance. FACS analysis showed an increase in the extent of apoptosis in Rab25-knockdown cells at 24 and 36 h (Figure 6A) after cells received a 4-Gy dose radiation. Colony formation assay further demonstrated that Rab25 depletion markedly suppressed the clonogenicity of CNE2R cells and A549R cells after radiation treatment, especially when the cells received a 6- or 8-Gy dose (Figures 6B and S5A). We also examined the effect of Rab25 knockdown on radiosensitivity of radioresistant H358R cells and chemo-sensitivity of taxol-resistant CNE1-TR cells and observed that both cells became sensitive to therapeutic treatment upon Rab25 knockdown (Figures S5B and S5C). Thus, these results revealed that Rab25 expression contributed to therapeutic resistance of cancer cells. In addition, we also employed a TKI erlotinib to treat PC9 cell to study whether EGFR inhibition could sensitize cancer cells to irradiation. After erlotinib treatment, FACS analysis showed that cells became sensitive to radiation, suggesting that TKIs treatment might increase cell radiosensitivity (Figure S5D).
Figure 6.

Blocking Rab25 Enhances Cell Death and Radio-Sensitization In Vitro and In Vivo
(A) Apoptosis rate in CNE2R or CNE2R cells transfected with shRab25 24 or 36 h after 4 Gy radiation.
(B) Clonogenic survival of radiation in radioresistant cells with shRab25-mediated knockdown. The clonogenicity was calculated by normalizing to the control cells that received 0 Gy IR, and the normalized clonogenic survival rates are shown on the bottom (n = 3, mean ± SD, ∗p < 0. 05, ∗∗p < 0. 01).
(C) Apoptosis rate after cells cultured in ultra-low attached wells for indicated time.
(D) Images and capacity of trans-well invasion (au, arbitrary units; n = 3; mean ± SD, ∗∗p < 0.01).
(E) Mammosphere formation assay (n = 3 in each group, mean ± SD, ∗∗p < 0.01), scale bar, 50 μm.
(F) Radiosensitivity of xenograft tumors generated with control and CNE2R-shRab25 cells and treated with 2 Gy at day 12 and day 13 (n = 5; mean ± SD, ∗∗p < 0.01).
(G) Representative images of IHC staining for Rab25 in NPC xenografts with or without irradiation. The average Rab25-positive cells in tumors were quantified and shown in the right bar graph. Scale bar, 50 μm. Mean ± SD, ∗p < 0.05, ∗∗p < 0.01.
(H) Representative images of IHC staining for EGFR in NPC xenografts with or without irradiation. The average EGFR-positive cells in tumors were quantified and shown in the right bar graph. Scale bar, 50 μm. mean ± SD, ∗∗p < 0.01.
Most cancer cells exhibit an EMT-like transition when they acquire resistance to therapies. The loss of epithelial features and acquisition of mesenchymal characteristics enable resistant cells to spread more quickly and be more invasive. We observed that, after knocking down of Rab25, the expression of EMT markers in radioresistant CNE2R cells could be reversed (Figure S5E) and cells were sensitive to induction of apoptosis and anoikis (Figures 6C, S5F, and S5G) with inhibited invasiveness (Figure 6D). These observations were further confirmed in taxol-resistant CNE1-TR cells and radioresistant H358R cells transfected with Rab25-specific siRNAs, which showed increased cell apoptosis rates induced by ultra-low attachment treatment. In addition, the sphere formation ability of CNE2R cells was dramatically decreased after Rab25 knockdown (Figure 6E).
Blocking Rab25 Resensitizes Radioresistant Tumors
We wonder whether Rab25 is a therapeutic target to breakdown tumor radioresistance. Using in vivo xenograft tumors of CNE2R-shcontrol or CNE2R-shRab25 cells, we found that silencing of Rab25 gene expression significantly suppress tumor growth (Figure 6F, blue line versus green line). From day 16, the radiation-treated group of shRab25 xenografts showed an obvious reduction in tumor growth rate compared with other three groups (Figure 6F). The knockdown efficiency of Rab25 in these xenografts was confirmed using IHC staining (Figure 6G), and down-expression of EGFR (Figure 6H) was observed in xenografts knockdown Rab25. These results demonstrate that Rab25-mediated EGFR enhancement is critically required for tumor cells to survive radiation.
Discussion
This study reveals a potential therapeutic target of Rab25-mediated EGFR enhancement in radioresistant LUAD and NPC cells. Rad25 has been linked with a poor prognosis in several cancer types (Cheng et al., 2004, Zhang et al., 2013a), and our current results further demonstrate a function of Rab25 in recycling EGFR, which is responsible for the acquired radioresistance in LUAD and NPC cells. Rab25 interacts with EGFR to reduce the degradation of EGFR leading to the enhancement of EGFR richness on cell surface causing an increased RTK-mediated signaling and aggressive phenotype of the radioresistant cancer cells. These findings suggest a model of Rab25-mediated EGFR recycling in acquired radioresistance, in which the formation of Rab25-EGFR conjugates in the cytoplasm is able to reduce the degradation of eternized EGFR and to recycle them to the cell surface required for the surviving tumor cells to keep the essential proliferation capacity and regrow under genotoxic stress conditions such as radiotherapy.
Although long being discovered, few studies has revealed the exact function of Rab25. The association between Rab25 and wild-type EGFR was first observed in lung cancer, where Rab25 inhibition promoted the endocytosis of EGFR and thus contributed to gefitinib sensitization (Jo et al., 2014). Jeong et al. recently reported that Rab25-induced tumor EMT was regulated by integrin/EGFR/VEGF-A/Snail axis, suggesting Rab25-EGFR interaction plays an important role in tumor migration (Jeong et al., 2018). In this paper, we first and directly proved that Rab25 regulated EGFR content and activation by fast recycling EGFR to cell surface upon ligand or stress stimulation, adding a significant information on tumor adaptive response to therapeutic radiation. Not only EGFR content but also phosphorylation status of EGFR was influenced by Rab25 expression. EGFR recruitment into endosomes was dependent on Grb2 (Johannessen et al., 2006, Jiang et al., 2003), which in this study was observed to co-localize with Rab25. In addition, GAB1, a scaffold protein in amplification of EGFR-mediated PI3K activation (Mattoon et al., 2004), was found in Rab25-EGFR co-localized complex, suggesting that EGFR located in Rab25-endosome was active in signaling transduction.
Rab25-mediated EGFR recycling will also provide mechanistic insights in revealing tumor acquired resistance to TKIs. EGFR TKIs alone or in combination with other treatment strategies are now the recommended treatment for patients with lung cancer harboring mutant EGFR. However, patients with mutant EGFR eventually develop acquired TKI resistance and patients harboring wild-type EGFR show poor response to first-generation TKIs and serious side effect for second- or third-generation TKIs (Liao et al., 2015). Moreover, EGFR mutation in non-small-cell lung cancer of adenocarcinoma histology ranges from roughly 12%–47% (Midha et al., 2015), implying that there are a lot of patients with lung cancer with wild-type EGFR who cannot benefit from TKIs treatment. Our finding that wild-type EGFR was tightly controlled by Rab25 provided a possibility to develop generation of TKIs, especially after identification of Rab25-EGFR interaction motif. In addition, we observed that other RTK-like receptors including Met, RYK, EphB2, and EphB6 were also regulated by Rab25, suggesting that Rab25-endosome might be one of the main RTK trafficking route in radioresistant cells. Considering the cross talk between different RTK members, we believed Rab25 might be a promising target for inhibiting RTK-mediated signaling pathway.
In this study, we illustrated that Rab25 was acquired for radioresistance in two major solid tumors treated by radiotherapy, which was linked with EMT enhancement and survival advantage under anchorage-independent environment. The EMT plasticity of a cancer cell is a major feature of therapy-resistant tumors. The adaptive tumor resistance and increased metastasis constitute the major failure of anti-cancer therapy causing 90% of cancer mortalities (Ahmed et al., 2018) in which acquired EMT-like phenotype and enhanced survival ability are linked with RTK-mediated pathway (Buchheit et al., 2014). RTK-mediated signaling pathways are often hyperactive in EMT program, which were activated by growth factors and then rendered the tight junction of cells (Forster, 2008, Ray and Jablons, 2009). The EGFR content on the cell surface is functionally important for activating EGFR downstream signaling. Here, we observed a rapid recovery of the EGFR surface level in Rab25 over-expressing cells. High expression of Rab25 greatly promotes its binding to EGFR and accelerated the recovery of EGFR content on cell membrane. Downregulation of Rab25 led to a reversal of EMT-like markers and reduced anti-anoikis ability, indicating that EMT occurrence in therapeutic-resistant cells is partially Rab25 dependent. Our current results provide new insights into the mechanisms of how therapeutic-resistant cells acquire EMT plasticity, suggesting that Rab25 is a potential effective target to block EGFR signaling and tumor proliferation.
By analyzing samples from 101 patients with NPC, we demonstrate that the expression of Rab25 in NPC was highly related to the radiotherapy response. However, the level of Rab25 expression did not seem to be associated with the histological grade, tumor size, or regional nodal metastasis. In patients with lung cancer, the same is true that high expression of Rab25 renders a poorer survival rate. The majority of patients with lung cancer are clinically diagnosed at advanced stages with a poor survival rate (Zappa and MOUSA, 2016). Particularly, these patients who lack surgical indications are recommended to receive radiotherapy (Postmus et al., 2017). For NPC, owing to its anatomical features and relative sensitivity to radiation, radiotherapy has been a first-line treatment (Zhang et al., 2013b). Although radiotherapy can control local tumor, radioresistance always occurs and becomes a challenging obstacle for curative lung cancer and NPC treatment. This highlights the Rab25-dependent effect on radiotherapy efficacy in patients with cancer.
Given the importance of Rab25 and the EGFR signaling pathway in human cancers in general, Rab25 may also represent a predictive marker for the cancer radiotherapy response and a potential candidate target for enhancing therapeutic efficacy. Further elucidating the molecular mechanisms underlying Rab25 controlled activation and duration of EGFR signaling may invent additional targets for desensitization of resistant tumor cells.
Limitation of the Study
In this study, we found that hyperactivation of RTK-mediated signaling pathways in therapeutic-resistant cells was due to rapid recycling of EGFR mediated by Rab25. Although a positive correlation between Rab25 expression and other RTKs such as MET and IGF1R was also observed, the detailed clarification was not performed, which warrants further investigation.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We acknowledge Dr. David Gandara and Dr. Megan Daily at University of California Davis for discussion on clinic lung cancer treatment and the TCGA database for providing the data and analyses in this article. This study was partially supported by the grants from National Natural Science Foundation of China, 81602679 (to L.Z.), 81530084 (to L.-Q.S.) and 81672509 (to B.X.), and 81602406 (to Z.L.), 81874200, 81572750, and US National Cancer Institute Grants (CA152313 to J.J.L.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U. S. C. Section 1734 solely to indicate this fact.
Author Contributions
Conception and design: L.-Q.S., L.Z., J.J.L. Development of methodology: L.Z., M.F. Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): L.Z., B.X., Y.Q., J.Z., Z.L. Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): L.Z., L.-Q.S., J.J.L., B.X. Writing, review, and/or revision of the manuscript: L.Z., L.-Q.S., J.J.L., B.X. Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): M.F., Y.D., D.J., Y.Q., R.T., J.H.
Declaration of Interests
No potential conflicts of interest were disclosed.
Published: April 24, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.100997.
Contributor Information
Jian Jian Li, Email: jijli@ucdavis.edu.
Lun-Quan Sun, Email: lunquansun@csu.edu.cn.
Supplemental Information
References
- Ahmed K.A., Scott J.G., Arrington J.A., Naghavi A.O., Grass G.D., Perez B.A., Caudell J.J., Berglund A.E., Welsh E.A., Eschrich S.A. Radiosensitivity of lung metastases by primary histology and implications for stereotactic body radiation therapy using the genomically adjusted radiation dose. J. Thorac. Oncol. 2018;13:1121–1127. doi: 10.1016/j.jtho.2018.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravindan N., Aravindan S., Pandian V., Khan F.H., Ramraj S.K., Natt P., Natarajan M. Acquired tumor cell radiation resistance at the treatment site is mediated through radiation-orchestrated intercellular communication. Int. J. Radiat. Oncol. Biol. Phys. 2014;88:677–685. doi: 10.1016/j.ijrobp.2013.11.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begg A.C., Stewart F.A., Vens C. Strategies to improve radiotherapy with targeted drugs. Nat. Rev. Cancer. 2011;11:239–253. doi: 10.1038/nrc3007. [DOI] [PubMed] [Google Scholar]
- Buchheit C.L., Weigel K.J., Schafer Z.T. Cancer cell survival during detachment from the ECM: multiple barriers to tumour progression. Nat. Rev. Cancer. 2014;14:632–641. doi: 10.1038/nrc3789. [DOI] [PubMed] [Google Scholar]
- Camidge D.R., Pao W., Sequist L.V. Acquired resistance to TKIs in solid tumours: learning from lung cancer. Nat. Rev. Clin. Oncol. 2014;11:473–481. doi: 10.1038/nrclinonc.2014.104. [DOI] [PubMed] [Google Scholar]
- Casaletto J.B., Mcclatchey A.I. Spatial regulation of receptor tyrosine kinases in development and cancer. Nat. Rev. Cancer. 2012;12:387–400. doi: 10.1038/nrc3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova J.E., Wang X., Kumar R., Bhartur S.G., Navarre J., Woodrum J.E., Altschuler Y., Ray G.S., Goldenring J.R. Association of Rab25 and Rab11a with the apical recycling system of polarized Madin-Darby canine kidney cells. Mol. Biol. Cell. 1999;10:47–61. doi: 10.1091/mbc.10.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caswell P.T., Spence H.J., Parsons M., White D.P., Clark K., Cheng K.W., Mills G.B., Humphries M.J., Messent A.J., Anderson K.I. Rab25 associates with alpha5beta1 integrin to promote invasive migration in 3D microenvironments. Dev. Cell. 2007;13:496–510. doi: 10.1016/j.devcel.2007.08.012. [DOI] [PubMed] [Google Scholar]
- Ceresa B.P. Spatial regulation of epidermal growth factor receptor signaling by endocytosis. Int. J. Mol. Sci. 2012;14:72–87. doi: 10.3390/ijms14010072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng K.W., Agarwal R., Mitra S., Lee J.S., Carey M., Gray J.W., Mills G.B. Rab25 increases cellular ATP and glycogen stores protecting cancer cells from bioenergetic stress. EMBO Mol. Med. 2012;4:125–141. doi: 10.1002/emmm.201100193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng K.W., Lahad J.P., Kuo W.L., Lapuk A., Yamada K., Auersperg N., Liu J., Smith-Mccune K., Lu K.H., Fishman D. The RAB25 small GTPase determines aggressiveness of ovarian and breast cancers. Nat. Med. 2004;10:1251–1256. doi: 10.1038/nm1125. [DOI] [PubMed] [Google Scholar]
- Chung J.H., Rho J.K., Xu X., Lee J.S., Yoon H.I., Lee C.T., Choi Y.J., Kim H.R., Kim C.H., Lee J.C. Clinical and molecular evidences of epithelial to mesenchymal transition in acquired resistance to EGFR-TKIs. Lung Cancer. 2011;73:176–182. doi: 10.1016/j.lungcan.2010.11.011. [DOI] [PubMed] [Google Scholar]
- Dozynkiewicz M.A., Jamieson N.B., Macpherson I., Grindlay J., van den Berghe P.V., von Thun A., Morton J.P., Gourley C., Timpson P., Nixon C. Rab25 and CLIC3 collaborate to promote integrin recycling from late endosomes/lysosomes and drive cancer progression. Dev. Cell. 2012;22:131–145. doi: 10.1016/j.devcel.2011.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster C. Tight junctions and the modulation of barrier function in disease. Histochem. Cell Biol. 2008;130:55–70. doi: 10.1007/s00418-008-0424-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu S., Li Z., Xiao L., Hu W., Zhang L., Xie B., Zhou Q., He J., Qiu Y., Wen M. Glutamine synthetase promotes radiation resistance via facilitating nucleotide metabolism and subsequent DNA damage repair. Cell Rep. 2019;28:1136–1143 e4. doi: 10.1016/j.celrep.2019.07.002. [DOI] [PubMed] [Google Scholar]
- Guo G., Yan-Sanders Y., Lyn-Cook B.D., Wang T., Tamae D., Ogi J., Khaletskiy A., Li Z., Weydert C., Longmate J.A. Manganese superoxide dismutase-mediated gene expression in radiation-induced adaptive responses. Mol. Cell Biol. 2003;23:2362–2378. doi: 10.1128/MCB.23.7.2362-2378.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselbach L., Haase S., Fischer D., Kolberg H.C., Sturzbecher H.W. Characterisation of the promoter region of the human DNA-repair gene Rad51. Eur. J. Gynaecol Oncol. 2005;26:589–598. [PubMed] [Google Scholar]
- Hehnly H., Doxsey S. Rab11 endosomes contribute to mitotic spindle organization and orientation. Dev. Cell. 2014;28:497–507. doi: 10.1016/j.devcel.2014.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins G.S., Krause M., Mckenna W.G., Baumann M. Personalized radiation oncology: epidermal growth factor receptor and other receptor tyrosine kinase inhibitors. Recent Results Cancer Res. 2016;198:107–122. doi: 10.1007/978-3-662-49651-0_5. [DOI] [PubMed] [Google Scholar]
- Hou Y., Zhu Q., Li Z., Peng Y., Yu X., Yuan B., Liu Y., Yin L., Jiang Z., Li J. The FOXM1-ABCC5 axis contributes to paclitaxel resistance in nasopharyngeal carcinoma cells. Cell Death Dis. 2017;8:e2659. doi: 10.1038/cddis.2017.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S., LI C., Armstrong E.A., Peet C.R., Saker J., Amler L.C., Sliwkowski M.X., Harari P.M. Dual targeting of EGFR and HER3 with MEHD7945A overcomes acquired resistance to EGFR inhibitors and radiation. Cancer Res. 2013;73:824–833. doi: 10.1158/0008-5472.CAN-12-1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsen K.R., Demuth C., Sorensen B.S., Nielsen A.L. The role of epithelial to mesenchymal transition in resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Transl Lung Cancer Res. 2016;5:172–182. doi: 10.21037/tlcr.2016.04.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong B.Y., Cho K.H., Jeong K.J., Park Y.Y., Kim J.M., Rha S.Y., Park C.G., Mills G.B., Cheong J.H., Lee H.Y. Rab25 augments cancer cell invasiveness through a beta1 integrin/EGFR/VEGF-A/Snail signaling axis and expression of fascin. Exp. Mol. Med. 2018;50:e435. doi: 10.1038/emm.2017.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X., Huang F., Marusyk A., Sorkin A. Grb2 regulates internalization of EGF receptors through clathrin-coated pits. Mol. Biol. Cell. 2003;14:858–870. doi: 10.1091/mbc.E02-08-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo U., Park K.H., Whang Y.M., Sung J.S., Won N.H., Park J.K., Kim Y.H. EGFR endocytosis is a novel therapeutic target in lung cancer with wild-type EGFR. Oncotarget. 2014;5:1265–1278. doi: 10.18632/oncotarget.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannessen L.E., Pedersen N.M., Pedersen K.W., Madshus I.H., Stang E. Activation of the epidermal growth factor (EGF) receptor induces formation of EGF receptor- and Grb2-containing clathrin-coated pits. Mol. Cell Biol. 2006;26:389–401. doi: 10.1128/MCB.26.2.389-401.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondapalli K.C., Llongueras J.P., Capilla-Gonzalez V., Prasad H., Hack A., Smith C., Guerrero-Cazares H., Quinones-Hinojosa A., Rao R. A leak pathway for luminal protons in endosomes drives oncogenic signalling in glioblastoma. Nat. Commun. 2015;6:6289. doi: 10.1038/ncomms7289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G., Liu Y., Su Z., Ren S., Zhu G., Tian Y., Qiu Y. MicroRNA-324-3p regulates nasopharyngeal carcinoma radioresistance by directly targeting WNT2B. Eur. J. Cancer. 2013;49:2596–2607. doi: 10.1016/j.ejca.2013.03.001. [DOI] [PubMed] [Google Scholar]
- Liao B.C., Lin C.C., Yang J.C. Second and third-generation epidermal growth factor receptor tyrosine kinase inhibitors in advanced nonsmall cell lung cancer. Curr. Opin. Oncol. 2015;27:94–101. doi: 10.1097/CCO.0000000000000164. [DOI] [PubMed] [Google Scholar]
- Maranto C., Udhane V., Hoang D.T., Gu L., Alexeev V., Malas K., Cardenas K., Brody J.R., Rodeck U., Bergom C. STAT5A/B Blockade Sensitizes Prostate Cancer to Radiation through Inhibition of RAD51 and DNA Repair. Clin. Cancer Res. 2018;24:1917–1931. doi: 10.1158/1078-0432.CCR-17-2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattoon D.R., Lamothe B., Lax I., Schlessinger J. The docking protein Gab1 is the primary mediator of EGF-stimulated activation of the PI-3K/Akt cell survival pathway. BMC Biol. 2004;2:24. doi: 10.1186/1741-7007-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midha A., Dearden S., Mccormack R. EGFR mutation incidence in non-small-cell lung cancer of adenocarcinoma histology: a systematic review and global map by ethnicity (mutMapII) Am. J. Cancer Res. 2015;5:2892–2911. [PMC free article] [PubMed] [Google Scholar]
- Mitra S., Montgomery J.E., Kolar M.J., Li G., Jeong K.J., Peng B., Verdine G.L., Mills G.B., Moellering R.E. Stapled peptide inhibitors of RAB25 target context-specific phenotypes in cancer. Nat. Commun. 2017;8:660. doi: 10.1038/s41467-017-00888-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postmus P.E., Kerr K.M., Oudkerk M., Senan S., Waller D.A., Vansteenkiste J., Escriu C., Peters S. Early and locally advanced non-small-cell lung cancer (NSCLC): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2017;28:iv1–iv21. doi: 10.1093/annonc/mdx222. [DOI] [PubMed] [Google Scholar]
- Ray M., Jablons D. Lung cancer metastasis novel biological mechanisms and impact on clinical practice. Hallmarks of Metastasis. 2009:29–46. [Google Scholar]
- Ricordel C., Friboulet L., Facchinetti F., Soria J.C. Molecular mechanisms of acquired resistance to third-generation EGFR-TKIs in EGFR T790M-mutant lung cancer. Ann. Oncol. 2018;29:i28–i37. doi: 10.1093/annonc/mdx705. [DOI] [PubMed] [Google Scholar]
- Shapira I., Lee A., Vora R., Budman D.R. P53 mutations in triple negative breast cancer upregulate endosomal recycling of epidermal growth factor receptor (EGFR) increasing its oncogenic potency. Crit. Rev. Oncol. Hematol. 2013;88:284–292. doi: 10.1016/j.critrevonc.2013.05.003. [DOI] [PubMed] [Google Scholar]
- Tan X., Lambert P.F., Rapraeger A.C., Anderson R.A. Stress-induced EGFR trafficking: mechanisms, functions, and therapeutic implications. Trends Cell Biol. 2016;26:352–366. doi: 10.1016/j.tcb.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomas A., Vaughan S.O., burgoyne T., Sorkin A., Hartley J.A., Hochhauser D., Futter C.E. Wash and Tsg101/ALIX-dependent diversion of stress-internalized EGFR from the canonical endocytic pathway. Nat. Commun. 2015;6:7324. doi: 10.1038/ncomms8324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Q.H., Zhu W.W., Zhang J.B., Qin Y., Lu M., Lin G.L., Guo L., Zhang B., Lin Z.H., Roessler S. GOLM1 modulates EGFR/RTK cell-surface recycling to drive hepatocellular carcinoma metastasis. Cancer Cell. 2016;30:444–458. doi: 10.1016/j.ccell.2016.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You S., Li R., Park D., Xie M., Sica G.L., Cao Y., Xiao Z.Q., Deng X. Disruption of STAT3 by niclosamide reverses radioresistance of human lung cancer. Mol. Cancer Ther. 2014;13:606–616. doi: 10.1158/1535-7163.MCT-13-0608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zappa C., Mousa S.A. Non-small cell lung cancer: current treatment and future advances. Transl Lung Cancer Res. 2016;5:288–300. doi: 10.21037/tlcr.2016.06.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Wei J., Lu J., Tong Z., Liao B., Yu B., Zheng F., Huang X., Chen Z., Fang Y. Overexpression of Rab25 contributes to metastasis of bladder cancer through induction of epithelial-mesenchymal transition and activation of Akt/GSK-3beta/Snail signaling. Carcinogenesis. 2013;34:2401–2408. doi: 10.1093/carcin/bgt187. [DOI] [PubMed] [Google Scholar]
- Zhang L., Chen Q.Y., Liu H., Tang L.Q., Mai H.Q. Emerging treatment options for nasopharyngeal carcinoma. Drug Des. Devel Ther. 2013;7:37–52. doi: 10.2147/DDDT.S30753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Li W., Li H., Ma Y., He G., Tan G. Genomic methylation profiling combined with gene expression microarray reveals the aberrant methylation mechanism involved in nasopharyngeal carcinoma taxol resistance. Anticancer Drugs. 2012;23:856–864. doi: 10.1097/CAD.0b013e3283548d73. [DOI] [PubMed] [Google Scholar]
- Zhou Z., Zhang L., Xie B., Wang X., Yang X., Ding N., Zhang J., Liu Q., Tan G., Feng D., Sun L.Q. FOXC2 promotes chemoresistance in nasopharyngeal carcinomas via induction of epithelial mesenchymal transition. Cancer Lett. 2015;363:137–145. doi: 10.1016/j.canlet.2015.04.008. [DOI] [PubMed] [Google Scholar]
- Zwang Y., Yarden Y. p38 MAP kinase mediates stress-induced internalization of EGFR: implications for cancer chemotherapy. EMBO J. 2006;25:4195–4206. doi: 10.1038/sj.emboj.7601297. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.