

N 6‐methyladenosine (m6A) has emerged as a crucial molecular code to influence pluripotency and differentiation of diverse stem cells. A study in this issue now shows that, in intestinal stem cells, the m6A reader protein YTHDF1 directs translational control of stemness in both m6A‐ and Wnt‐dependent manner [1].
Subject Categories: Cancer, Signal Transduction, Protein Biosynthesis & Quality Control
N 6‐methyladenosine has emerged as a crucial mark that influences pluripotency and differentiation. A study in this issue shows that the m6A reader YTHDF1 directs translational control of intestinal stemness in a Wnt‐dependent manner.
The intestinal epithelium is one of the few active cellular compartments harboring efficient self‐renewing capacity in mammals, making it quickly adaptive to microenvironmental perturbations. Intestinal stem cells (ISCs), located at the base of crypts, are responsible for intestinal homeostasis and regeneration through a tightly regulated balance between proliferation and differentiation, thereby maintaining the integrity and viability of epithelial tissues. As an evolutionarily conserved pathway, Wnt/β‐catenin signaling is generally considered as a prominent driving force in ISC maintenance and epithelial renewal, and deregulation of this pathway is closely linked to colorectal disease and cancer 2. In unstimulated cells (Wnt off), the key transcriptional coactivator β‐catenin is targeted for proteasomal degradation by a cytoplasmic “destruction complex” comprised of AXIN, adenomatous polyposis coli (APC), and glycogen synthase kinase 3 (GSK3) (Fig 1). Upon activation of Wnt signaling through ligand binding to the transmembrane receptor FZD and its coreceptor LRP5/6, the deactivation of the destruction complex facilitates the stabilization and nuclear translocation of β‐catenin, leading to T‐cell factor/lymphoid enhancer factor (TCF/LEF)‐mediated transcriptional activation of Wnt target genes 3. Despite the well‐established role of β‐catenin in Wnt signaling, additional layers of regulatory signals remain poorly understood. In this issue, Han et al 1 describe a previously unappreciated mechanism required for the amplification of Wnt signaling during ISC homeostasis, which involves YTHDF1‐ and m6A‐dependent translational control of the key transcriptional activator TCF4.
Figure 1. YTHDF1‐mediated translational control of TCF4 reinforces Wnt‐driven intestinal regeneration and tumorigenesis.
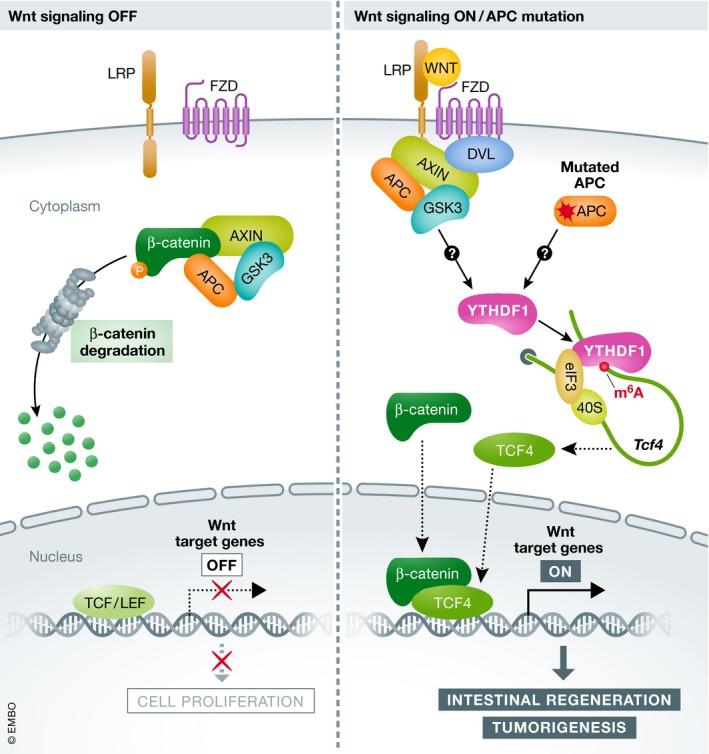
Without Wnt activation (Wnt off—left), cytoplasmic β‐catenin is recruited to the “destruction complex” consisting of AXIN, APC and GSK3 for proteasomal degradation, resulting in TCF/LEF‐mediated repression of Wnt target genes. In the state of activated Wnt (Wnt on—right), by ligand binding or APC mutation, the key coactivator β‐catenin is stabilized and translocated into the nucleus. In parallel, Wnt promotes translation of YTHDF1, which subsequently favors the translation of the transcriptional activator TCF4. YTHDF1‐mediated translational control of TCF4 and β‐catenin stabilization then synergistically contribute to the activated transcription of Wnt target genes, leading to intestinal homeostasis and tumorigenesis.
In mammalian cells, m6A is introduced into RNA molecules in a co‐transcriptional manner by a heterodimeric METTL3‐METTL14 methytransferase complex. The m6A mark can be removed by two major demethylases, FTO, and ALKBH5, creating a dynamic epitranscriptomic landscape. Through binding to different YTHDF “reader” proteins, m6A serves as a critical modulator of gene expression in diverse biological processes 4. To investigate the potential role of m6A in Wnt‐driven intestinal homeostasis, Han et al initially examined expression levels of the m6A machinery in mouse intestinal crypts. Besides a slight increase of METTL3 expression, the authors observed an unexpected induction of the m6A “reader” YTHDF1 upon Wnt activation 1. Consistently, YTHDF1 exhibited higher levels in intestinal tumor tissues harboring Apc mutations than in non‐tumor tissues. A recent study suggested that Ythdf1 gene copy number is positively correlated with its expression in colorectal cancer in which Apc mutations are frequently observed 5. However, Han et al 1 found that Wnt‐induced YTHDF1 induction mainly occurs at the translational level, as evidenced by the elevated levels of Ythdf1 mRNA in the polysome fractions following Wnt activation. As an independent validation, inhibiting Wnt signaling by APC overexpression led to reduced YTHDF1 translation. The exact mechanism underlying Wnt‐mediated translational control of YTHDF1 remains to be hammered out.
To understand the physiological impact of increased YTHDF1 expression in response to Wnt signaling, Han et al created an intestinal‐specific YTHDF1 knock‐out mouse model, but found negligible phenotypes regarding physiological traits of the intestine. Only after irradiation damage, YTHDF1 did become important for cell proliferation during intestinal regeneration 1. This requirement of YTHDF1 after stress is reminiscent of recent reports about the potential role of YTHDF1 in hippocampus‐dependent learning and memory 6, as well as hypoxia adaptation 7. The authors verified the Wnt‐dependent YTHDF1 functionality by phenotypic analysis of intestinal tumorigenesis and pluripotency. Indeed, depletion of YTHDF1 not only impaired Wnt‐driven intestinal tumor formation, but also reduced stemness of both mouse intestinal stem cells and human colorectal cancer stem cells 1.
A growing body of evidence has demonstrated pivotal but complicated roles of m6A in stem cell pluripotency. For instance, a study by Wang et al 8 revealed that METTL3 depletion promotes embryonic stem cell (ESC) differentiation through stabilizing developmental regulators by decreasing m6A levels. Another study proposed that METTL3 targets m6A‐containing mRNAs such as Nanog for degradation, resulting in reduced pluripotency 9. These seemingly discrepant effects of m6A are possibly due to distinct cellular contexts, but there is little doubt about the crucial role of m6A in stem cell homeostasis. In the case of ISCs, YTHDF1 clearly exerts its functionality via m6A, as the mutant protein without m6A‐binding domain failed to restore the pluripotent defects of Ythdf1 knock‐out ISCs. Further supporting the crucial role of m6A in ISC maintenance, impairment of METTL3 restrained the growth of organoids under active Wnt signaling 1.
YTHDF1 has been shown to promote translation of certain m6A‐containing transcripts via recruiting translation initiation complexes 4. Consistent with this notion, YTHDF1 knockdown decreased the translational efficiency of a subgroup of m6A‐marked target mRNAs including the key components in Wnt/β‐catenin signaling pathway 1. Several lines of evidence indicate Tcf4 as the most prominent mRNA target of YTHDF1. First, Tcf4 mRNA bears multiple m6A sites across the whole transcript as revealed by m6A‐seq. Second, YTHDF1 binds to Tcf4 as evidenced by RIP‐seq. Third, translation efficiency of Tcf4 undergoes tight regulation by YTHDF1 as confirmed by Ribo‐seq 1. TCF4 has been identified as a major transcriptional mediator of classic Wnt/β‐catenin signaling by cooperating with β‐catenin 3. Thus, YTHDF1‐mediated translational control of TCF4 and nuclear translocation of β‐catenin independently but coordinately contribute to the signaling outputs of the Wnt pathway. Further supporting TCF4 as the critical downstream target of YTHDF1 under physiological conditions, overexpression of TCF4 greatly rescued stemness defects of Ythdf1‐deleted ISCs 1. Collectively, m6A‐dependent translational control of TCF4 reinforces Wnt/β‐catenin‐driven intestinal homeostasis.
By focusing on Wnt signaling, Han et al extended the role of m6A in stem cell biology. It is intriguing that YTHDF1 is dispensable for physiological intestinal development, but becomes essential during intestinal regeneration. It has been well documented that m6A levels fluctuate depending on different cellular stages or growth conditions. As a generic m6A reader, YTHDF1 can hardly achieve mRNA specificity. It is conceivable that the functional balance between m6A levels and the amount of readers could help to achieve targeting specificity above the threshold. Indeed, activated Wnt signaling triggers YTHDF1 induction at the translational level 1. Elevated YTHDF1 then promotes the translation of TCF4, the master transcription factor in response to Wnt signaling (Fig 1). This positive feedback loop accelerates Wnt‐mediated regeneration and tumorigenesis. Since YTHDF1 is required for Wnt‐driven tumorigenesis rather than normal intestinal proliferation, it is an ideal therapeutic target for colorectal cancer. Given the recently identified role of YTHDF1 in antigen presentation 10, inactivating YTHDF1 would open new perspectives for colorectal cancer therapy by simultaneously inhibiting intestinal tumorigenesis and priming anti‐tumor immune response.
EMBO Reports (2020) 21: e50097
See also: https://doi.org/10.15252/embr.201949229 (April 2020)
References
- 1. Han B, Yan S, Wei S et al (2020) EMBO Rep 21: e49229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Santos AJM, Lo YH, Mah AT et al (2018) Trends Cell Biol 28: 1062–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Merenda A, Fenderico N, Maurice MM (2019) Trends Cell Biol 30: 60–73 [DOI] [PubMed] [Google Scholar]
- 4. Frye M, Harada BT, Behm M et al (2018) Science 361: 1346–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bai Y, Yang C, Wu R et al (2019) Front Oncol 9: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shi H, Zhang X, Weng YL et al (2018) Nature 563: 249–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shi Y, Fan S, Wu M et al (2019) Nat Commun 10: 4892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang Y, Li Y, Toth JI et al (2014) Nat Cell Biol 16: 191–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Geula S, Moshitch‐Moshkovitz S, Dominissini D et al (2015) Science 347: 1002–1006 [DOI] [PubMed] [Google Scholar]
- 10. Han D, Liu J, Chen C et al (2019) Nature 566: 270–274 [DOI] [PMC free article] [PubMed] [Google Scholar]