

Abstract
Mitochondria are cellular organelles that orchestrate a vast range of biological processes, from energy production and metabolism to cell death and inflammation. Despite this seemingly symbiotic relationship, mitochondria harbour within them a potent agonist of innate immunity: their own genome. Release of mitochondrial DNA into the cytoplasm and out into the extracellular milieu activates a plethora of different pattern recognition receptors and innate immune responses, including cGAS‐STING, TLR9 and inflammasome formation leading to, among others, robust type I interferon responses. In this Review, we discuss how mtDNA can be released from the mitochondria, the various inflammatory pathways triggered by mtDNA release and its myriad biological consequences for health and disease.
Keywords: cell death, inflammation, immunity, mitochondria, mtDNA
Subject Categories: Autophagy & Cell Death, Immunology,
Mitochondrial DNA is increasingly recognized as a potent agonist of innate immunity. This review discusses how mitochondrial DNA can be released from mitochondria and trigger the activation of different pro‐inflammatory signaling pathways.
Glossary
- 5hmC
5‐Hydroxymethylcytosine
- 5mC
5‐Methylcytosine
- AGS
Aicardi–Goutieres syndrome
- AIM2
Absent in melanoma 2
- APC
Antigen‐presenting cell
- ASC
Apoptosis‐associated speck‐like protein containing a CARD
- ATP
Adenosine triphosphate
- BAK
Bcl‐2 homologous antagonist/killer
- BAX
Bcl‐2‐associated X protein
- BID
BH3 interacting‐domain death agonist
- CARD
Caspase activation and recruitment domain
- CD47
Cluster of differentiation 47
- CDN
Cyclic dinucleotide
- cGAMP
Cyclic guanosine monophosphate–adenosine monophosphate
- cGAS
Cyclic GMP‐AMP synthase
- CLR
C‐type lectin receptor
- CMPK2
Cytidine/Uridine monophosphate kinase 2
- DAMP
Damage‐associated molecular pattern
- DC
Dendritic cell
- DNase
Deoxyribonuclease
- dsDNA
Double‐stranded DNA
- ER
Endoplasmic reticulum
- EV
Extracellular vesicle
- GTP
Guanosine‐5′‐triphosphate
- HMGB1
High‐mobility group protein 1
- HSV‐1
Herpes simplex virus‐1
- IAP
Inhibitor of apoptosis protein
- IFNAR
Interferon‐α/β receptor
- IFN‐β
Interferon‐β
- IFN‐γ
Interferon‐γ
- IL‐18
Interleukin‐18
- IL‐1R
Interleukin‐1 receptor
- IL‐1β
Interleukin‐1β
- IL‐6
Interleukin‐6
- IRF3
Interferon regulatory factor 3
- ISG
Interferon‐stimulated gene
- K+
Potassium
- LPS
Lipopolysaccharide
- LRR
Leucine‐rich repeat
- MAPK
Mitogen‐activated protein kinase
- MAVS
Mitochondrial anti‐viral signalling protein
- MDA5
Melanoma differentiation‐associated protein 5
- MEF
Mouse embryonic fibroblast
- MiDAS
Mitochondrial dysfunction‐associated senescence
- MI
Myocardial infarction
- MOMP
Mitochondrial outer membrane permeabilisation
- mPTP
Mitochondrial permeability transition pore
- mtDNA
Mitochondrial DNA
- NASH
Non‐alcoholic fatty liver disease
- NET
Neutrophil extracellular trap
- NF‐κB
Nuclear factor kappa‐light‐chain‐enhancer of activated B cells
- NLRC4
NLR Family CARD Domain Containing 4
- NLR
Nucleotide oligomerisation domain‐like receptor
- NLRP1
NLR Family Pyrin Domain Containing 1
- NLRP3
NACHT, LRR and PYD domain‐containing protein 3
- NOD
Nucleotide oligomerisation domain
- ODN
Oligodeoxynucleotide
- OPA1
Optic Atrophy 1 Mitochondrial Dynamin Like GTPase
- PAMP
Pathogen‐associated molecular pattern
- pDC
Plasmacytoid dendritic cell
- PD‐L1
Programmed death‐ligand 1
- PINK1
Phosphatase and tensin homolog‐induced kinase 1
- PMA
Phorbol 12‐myristate 13‐acetate
- PNPase
Polynucleotide phosphorylase
- PRR
Pattern recognition receptor
- PYD
Pyrin domain
- RAGE
Receptor for advanced glycation endproducts
- RIG‐I
Retinoic acid‐inducible gene I
- RIP1
Receptor‐interacting serine/threonine‐protein kinase 1
- RLR
Retinoic acid‐inducible gene‐I‐like receptors
- RNP IC
Ribonucleotide immune complex
- ROS
Reactive oxygen species
- SAMDH1
Sterile alpha motif domain and HD domain‐containing protein 1
- SIRS
Systemic inflammatory response syndrome
- SLE
Systemic lupus erythematosus
- ssDNA
Single‐stranded DNA
- STING
Stimulator of interferon genes
- SUV3
Suppressor of Var1
- TBK1
TANK‐binding kinase 1
- TFAM
Transcription factor A, mitochondrial
- TLR9
Toll‐like receptor 9
- TLR
Toll‐like receptor
- TNF
Tumour necrosis factor
- TREX1
Three Prime Repair Exonuclease 1
- tRNA
Transfer RNA
- VDAC
Voltage‐dependent anion channel
Introduction
Serving as a first line of defence, the innate immune system guards us against a plethora of insults and invading microorganisms. Infection by pathogenic agents is detected in cells by pattern recognition receptors (PRRs) which recognise specific pathogen‐associated molecular patterns (PAMPs). PRRs can be broadly classified into four distinct groups: NOD‐like receptors (NLRs), Toll‐like receptors (TLRs), retinoic acid‐inducible gene‐I (RIG‐I)‐like receptors (RLRs) and C‐type lectin receptors (CLRs) 1. Upon detection of a PAMP, PRRs initiate a multitude of different signalling pathways, which culminate in the up‐regulation of various type I interferons, pro‐inflammatory chemokines and cytokines. These prime the adaptive immune system and create a hostile environment for the microorganism in which to survive. Additionally, damage‐associated molecular patterns (DAMPs) are immune triggers that arise from the cell itself, such as proteins or DNA, and can activate innate immune pathways 2.
Mitochondria first appeared in eukaryotic cells about two billion years ago as α‐proteobacterium, in what is thought to be an endosymbiotic relationship 3, 4. Over time, these bacteria evolved to become the much‐studied organelle that we know today, playing crucial roles in metabolism, calcium homeostasis and cell death. Nevertheless, they have maintained an independent genome, which encodes 37 genes, comprised of 13 mRNAs forming key components of the oxidative phosphorylation system, in addition to 2 ribosomal RNA components and 22 tRNAs 3, 4. An estimated 1,000 proteins are located in the mitochondria, all of which, except those encoded by mtDNA, are translated in the cytosol and imported into the mitochondria 5.
Mitochondrial DNA itself is a circular molecule of double‐stranded (ds)DNA. Transcription of both the heavy and light strand results in long, full‐length transcripts which are processed by RNase enzymes to produce mature mRNA, tRNA and ribosomal RNA. In mammals, the polymerase responsible for mtDNA replication is DNA polymerase γ, but as POLγ cannot replicate dsDNA, the DNA helicase Twinkle is required to act directly before to unwind the DNA structure. Newly synthesised single‐stranded (ss)DNA is bound by mitochondrial single‐stranded DNA‐binding protein to prevent secondary structure formation and attack by nucleases. Mitochondrial DNA replication has recently been reviewed extensively elsewhere 6; here, we focus on the unique aspects of mtDNA which make it immunostimulatory. We will then discuss how mtDNA which is ejected from the mitochondria under specific circumstances can activate different innate immune pathways, including cGAS‐STING signalling, inflammasomes and Toll‐like receptors. We will also focus on the role of mtDNA in the formation of neutrophil extracellular traps (NETs) and the transfer of mtDNA between cells.
Mitochondrial DNA as a stimulator of the immune system
Potentially stemming from its bacterial origin, mitochondrial DNA is sensed as “foreign”, suggesting that it is seen differently to “self” DNA in cells. One example of this can be seen in its methylation status, where many studies have reported mtDNA to be hypomethylated compared to nuclear DNA 7, 8, despite the presence of DNA methyltransferases in the mitochondria 9, 10. Some groups have reported aberrant methylation patterns of mtDNA, including 5‐methylcytosine (5mC) and 5‐hydroxymethylcytosine (5hmC) at CpG motifs 9, 10, 11, 12, 13, 14, although others have proposed technical limitations to this work and using more sensitive techniques report that mtDNA is devoid of CpG methylation 15. Clearly, more effort is required in determining the precise degree of methylation in mtDNA, but if studies showing an absence of CpG methylation are correct, then mtDNA would harbour unmethylated CpG motifs similar to bacterial DNA, which could potentially activate pattern recognition receptors such as TLR9, absent in melanoma 2 (AIM2) and cGAS 15, 16, 17, 18. Mitochondrial DNA replication and transcription itself may represent a rich source of potential activators of DNA pattern recognition receptors; for example, RNA:DNA hybrids form during transcription, in addition to long stretches of ssDNA and R‐loops composed of RNA:DNA hybrids with a non‐template ssDNA which can be recognised by cGAS 16.
Mitochondrial DNA exists in the mitochondrial matrix in close proximity to the electron transport chain, a major source of reactive oxygen species. Due to this, it is particularly vulnerable to oxidation, resulting in mtDNA mutations which can contribute to the pathogenesis of cancer 17, diabetes 18 and ageing 19. It was thought the cell had limited capacity to repair mtDNA; however, multiple repair pathways are now well characterised 20. Mitochondrial DNA is often schematically represented as a plasmid structure; however, this is an over‐simplification. Rather, super‐resolution imaging has revealed that it is densely compacted into nucleoids consisting of one copy of mtDNA and a number of different proteins 21, the most notable of which is mitochondrial transcription factor A (mtTFA, commonly referred to as TFAM). It might be assumed that the compaction of mtDNA into protein structures shields DNA from recognition, but this is not the case as we shall discuss further in this Review, and in fact, a number of studies have shown that TFAM itself might be immunostimulatory 13, 14.
In a landmark study in 2004, Collins et al 22 found that injecting mtDNA into the joints of mice resulted in localised inflammation and arthritis. Further investigation revealed that the inflammation was dependent on the presence of oxidatively damaged bases in the mtDNA, as injection of an oligodeoxynucleotide (ODN) with the same sequence but without the oxidised residue had no effect. The observation that mtDNA can elicit potent immune responses opened a whole new field of research, and it is now appreciated that mtDNA can stimulate many PRRs, including cGAS, TLR9 and inflammasomes (Fig 1). Release of mtDNA from mitochondria and subsequent recognition by PRRs occurs during many cellular processes, including infection, cell death and neurodegeneration, and this will be the focus of the rest of this Review.
Figure 1. Overview of pro‐inflammatory signalling pathways engaged by mitochondrial DNA .
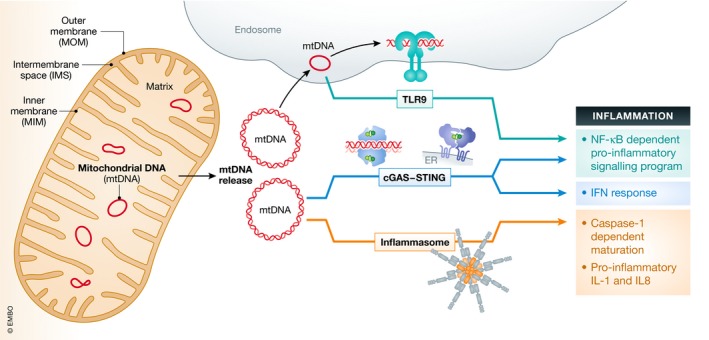
Mitochondrial DNA (mtDNA) can trigger various pro‐inflammatory signalling pathways by endosomal localised TLR9 or via cytosolic cGAS‐STING or via cytosolic inflammasome (AIM2 or NLRP3). Top: TLR9 binds mtDNA in the endosome eliciting an NF‐κB‐dependent pro‐inflammatory signalling program. Middle: cGAS recognises mtDNA in the cytosol and activates endoplasmic reticulum (ER)‐localised STING triggering an interferon response. Bottom: mtDNA‐dependent inflammasome activity leads to caspase‐1‐dependent maturation or pro‐inflammatory IL‐1 and IL‐8.
mtDNA‐dependent activation of cGAS‐STING signalling
mtDNA release in infection
Through necessity, cells have evolved elegant systems to detect the presence of invading pathogenic DNA. Cyclic GMP‐AMP synthase (cGAS) is one such direct detector, which binds dsDNA to form a dimer 23, 24. cGAS then undergoes a conformational change which facilitates the conversion of ATP and GTP into 2′3′‐cyclic GMP‐AMP (cGAMP) 25, 26, 27, 28, 29, 30, 31. cGAMP is a second messenger, which binds the endoplasmic reticulum (ER)‐resident protein stimulator of interferon genes (STING) inducing a conformational change in its C‐terminal tail. TANK‐binding kinase 1 (TBK1) is recruited to STING which phosphorylates it and the transcription factor interferon regulatory factor 3 (IRF3), eliciting the transcription of hundreds of interferon stimulatory genes (ISGs) that are potently anti‐viral 32 (Fig 2). cGAS was assumed to be primarily cytosolic to avoid persistent activation by self‐DNA in the nucleus, but recent work has shown it to be present in the nucleus 33, 34 and at the plasma membrane 35. A recent attempt to resolve these discrepancies by Volkmann et al 36 reveals a more complex model than the cytosolic DNA sensing paradigm. The authors show that the majority of cGAS protein is nuclear, and they propose a model where cGAS must be “desequestered” prior to its full activation. However, it remains unclear how cytosolic DNA can be detected by cGAS, if cGAS is tethered in the nuclear compartment. Three independent studies were the first to show that mtDNA released from mitochondria is able to activate cGAS‐STING signalling 37, 38, 39. White et al and Rongvaux et al explored mtDNA release in the context of cell death (discussed later in this Review), whereas West et al provided evidence that TFAM deficiency promotes mitochondrial stress and mis‐packaged mtDNA, resulting in their ejection into the cytoplasm where they bind and activate cGAS initiating a type I interferon response 39 (Fig 2). Of pathophysiological relevance, infection with Herpes simplex virus‐1 (HSV‐1) or vesicular stomatitis virus (VSV) results in mtDNA stress, TFAM depletion and mtDNA entrance into the cytoplasm. The cytoplasmic mtDNA is then sensed by cGAS, triggering cGAS‐STING signalling leading to the up‐regulation of a plethora of interferon genes, conferring an anti‐viral state on the cell. Importantly, Tfam +/− cells, which exhibit mtDNA stress, are more resistant to infection with HSV‐1 or VSV than wild‐type cells, as they have heightened ISG expression owing to mtDNA release. Mechanistically, the HSV‐1 virus encodes a nuclease, UL12.5, which localises to the mitochondria and degrades mtDNA, resulting in complete loss of mtDNA in infected cells 40, 41. Removal of mtDNA in infected cells does not appear to impact HSV replication 42. Furthermore, exonuclease activity is required for effective viral DNA production to maintain cell‐to‐cell infectivity, though whether this is related to UL12.5's mtDNA‐targeted nuclease activity is unknown 43.
Figure 2. mtDNA‐dependent activation of cGAS‐STING signalling.
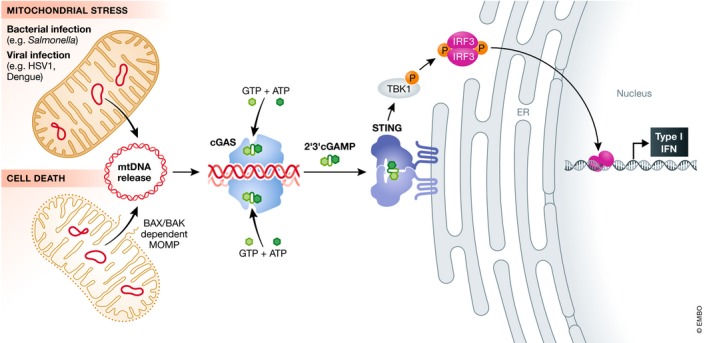
Various mitochondrial stresses including bacterial or viral infection can lead to mtDNA release. Alternatively, activation of BAX and BAK leads to outer mitochondrial membrane permeabilisation (MOMP) and mtDNA release. Once cytoplasmic, mtDNA can bind the DNA sensing protein cGAS that catalyses the production of the secondary messenger 2′3′ cyclic GMP–AMP (2′3′cGAMP) from ATP and GTP. cGAMP binds the adaptor molecule STING on the ER leading to activation of TBK1 kinase. Active TBK1 phosphorylates the transcription factor IRF3 initiating a type I interferon response.
Curiously, infection with RNA viruses, such as dengue virus, also elicits a cGAS‐STING response, despite cGAS being a DNA‐specific PRR 44. Several studies have now shown that dengue virus causes the release of predominantly oxidised mtDNA into the cytosol, where it can activate both cGAS 45, 46 and TLR9 47. Dengue virus has evolved strategies to circumvent cytosolic mtDNA‐induced cGAS signalling during infection by encoding proteases which target cGAS and STING for degradation, thus ensuring persistence of the virus 46, 48, 49.
Infection with the bacterial pathogen Mycobacterium tuberculosis triggers cGAS activation and subsequent IRF3‐dependent type I interferon response 50, 51, 52. This was assumed to be solely due to detection of mycobacterium DNA, but other studies have identified a role for mitochondrial stress and ensuing release of mtDNA into the cytoplasm 53. This observation is strain‐dependent but does propose a role for mitochondrial stress and dynamics on the M. tuberculosis‐induced release of mtDNA. Previous work has observed cytochrome c release from mitochondria in cells infected with M. tuberculosis, indicating that there may be a possible role for BAX/BAK‐dependent mitochondrial permeabilisation (discussed in detail later) in infection‐related mtDNA release 54 (Fig 2).
Pathogen‐infected cells often secrete IL‐1β due to inflammasome activation. A recent report by Aarreberg et al discovers a link between IL‐1β secretion in infected cells, which can then activate a cGAS‐STING‐dependent type I interferon response in surrounding bystander cells. Interestingly, IL‐1β stimulation of bystander cells increases mitochondrial mass, decreases mitochondrial membrane potential and induces mtDNA release 55. However, mtDNA release is observed in the absence of detectable cytochrome c release and cell death, suggesting that this is not the mechanism of mtDNA release, although it does not rule out limited mitochondrial permeabilisation seen by us and others in the context of infection (see below). This is not the first time IL‐1R signalling has been implicated in cell‐intrinsic defence 56, 57, 58, but it is the first to suggest that mtDNA release plays a key role in the initiation of cGAS‐STING signalling in the bystander cells.
mtDNA activation of cGAS‐STING during cell death
During programmed cell death, the pro‐apoptotic proteins BAX and BAK permeabilise the mitochondrial outer membrane to allow the passage of pro‐apoptotic molecules to move from the inner membrane space into the cytosol, where they can initiate a caspase cascade, resulting in a rapid cell death 59. White et al and Rongvaux et al showed that in the absence of apoptotic caspase activation, mtDNA activates cGAS in a promiscuous manner, which in vivo leads to mildly elevated IFN‐β protein levels in blood, though a level sufficient to induce the expression of interferon‐stimulated genes 37, 38 (Fig 3). This suggests that apoptotic caspases play a crucial role in dampening type I interferon responses in dying cells, maintaining the “immune‐silent” nature of apoptosis (Fig 3). Further work has shown that apoptotic caspases directly cleave cGAS, IRF3 and mitochondrial anti‐viral signalling protein (MAVS), key proteins required for the production of type I interferon 60, supporting the notion that caspases dampen the immune response during cell death. High‐resolution imaging studies have further expanded our understanding of how mtDNA is released from the mitochondria during cell death. We and others recently showed that BAX and BAK can permeabilise the mitochondrial outer membrane, but in the context of caspase inhibition these pores grow dramatically, sufficient to allow inner membrane herniation and extrusion of mtDNA 61, 62, 63 (Fig 3). We found that under caspase‐inhibited conditions, mitochondrial permeabilisation leads to down‐regulation of inhibitor of apoptosis proteins (IAPs), NF‐κB‐inducing kinase (NIK) activation and an NF‐κB transcriptional program, in addition to mtDNA release‐induced cGAS‐STING activation 64. The cytokines and chemokines up‐regulated via NF‐κB after mitochondrial permeabilisation can serve to promote macrophage activation 64, 65. This leads to robust anti‐tumour effects, highlighting a potential therapeutic role for caspase inhibition in cancer treatment 64. Collectively, these results help to reconcile how predominantly cytosolic cGAS can be activated by mtDNA during cell death. Nevertheless, a number of unresolved questions remain. Firstly, is inner membrane permeabilisation a regulated process, and if so, how? A rapid inner membrane permeabilisation of sufficient size to allow the passage of small ions is observed minutes after outer membrane permeabilisation 61, but is insufficient to allow mtDNA nucleoid extrusion and is probably only transient, as inner membrane potential can be maintained after outer membrane permeabilisation 66, 67, 68, 69. Secondly, there are cell type differences in the degree of inner membrane permeabilisation, as different studies report varying degrees of mtDNA release during cell death 61, 62, implying that specific cell‐intrinsic factors play a role in inner membrane permeabilisation. Finally, the physiological relevance of cell death‐related mtDNA release is unknown. Most cell types undergo rapid and complete caspase‐dependent apoptosis in vivo, presumably limiting any potential for mtDNA‐driven inflammation during cell death. However, some cell types, for instance cardiomyocytes, display deficient caspase activity downstream of mitochondrial permeabilisation 70. Such cells might generate a greater type I anti‐viral interferon response after mitochondrial permeabilisation. Alternatively, cGAMP might transfer from apoptotic to healthy cells, serving as an “early warning” defence system, instructing healthy cells to transcribe genes important for their survival (Fig 4) 71, 72.
Figure 3. BAX/BAK‐dependent initiation of inflammation.
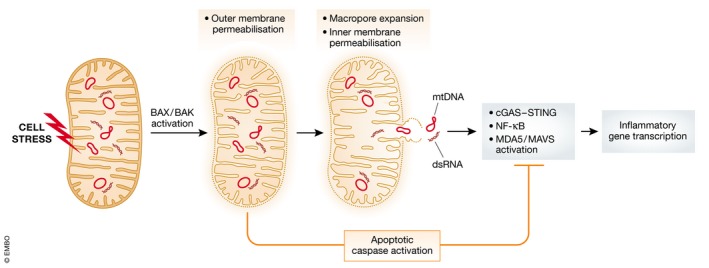
Following a pro‐apoptotic stress, BAX and BAK are activated leading to mitochondrial outer membrane permeabilisation. This enables the release of caspase‐activating proteins from the mitochondrial intermembrane space. Following this, macropores form on the mitochondrial outer membrane causing extrusion and permeabilisation of the inner membrane. This enables release of mtDNA. Mitochondrial double‐stranded RNA (dsRNA) can also be released. Collective release of these molecules triggers inflammation via MAVS, cGAS‐STING and NF‐κB. Caspase activity is anti‐inflammatory, in part, through direct cleavage and inactivation of inflammatory signalling molecules.
Figure 4. Non‐cell autonomous effects of mtDNA .
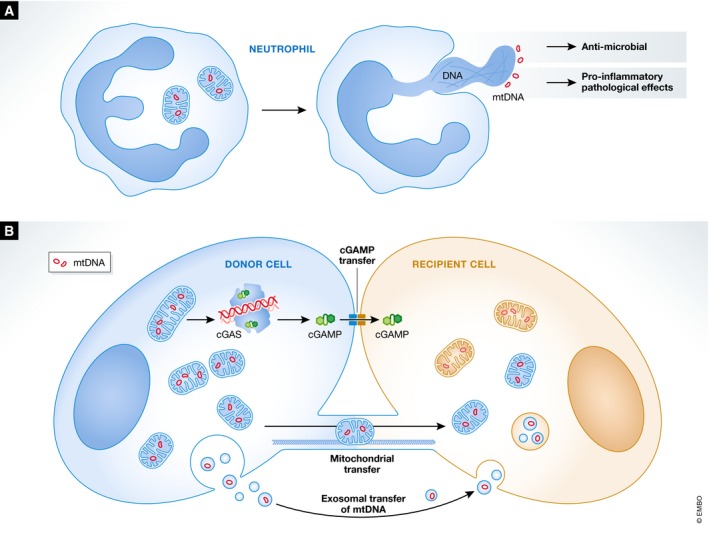
(A) Upon pathogen encounter, neutrophils can extrude DNA (both nuclear and mitochondrial) that forms an extracellular trap for extracellular microbes. Due to pro‐inflammatory properties, these DNA neutrophil extracellular traps (NETs) can also have pathological effects in diseases such as lupus. (B) mtDNA can transfer via exosomes or in intact mitochondria to neighbouring cells, impacting on the metabolism and survival of the recipient cell. Inflammatory responses to mtDNA can also have non‐cell autonomous effects. The cGAS‐induced secondary messenger cGAMP has been shown to transfer via gap junctions eliciting anti‐viral interferon responses in neighbouring cells.
In addition to DNA, mitochondria also possess dsRNA which is known to be potently immunogenic 73. Mitochondrial dsRNA arises from transcription of both the heavy and lights strands of mtDNA; however, although the light strand is rapidly degraded the heavy strand is not, and nearly all the dsRNA detected in the cytoplasm are of mitochondrial origin. The mitochondrial helicase SUV3 and polynucleotide phosphorylase PNPase dampen the accumulation of dsRNA, but when these are depleted, dsRNA accumulates in the cytoplasm where it activates a type I interferon response driven by the dsRNA receptor MDA5 74. Silencing of BAX and BAK suppresses the type I interferon response, strongly suggesting that BAX/BAK‐dependent mitochondrial outer membrane permeabilisation is responsible for mitochondrial dsRNA escape into the cytoplasm 74 Furthermore, patients with mutations leading to a decrease in PNPT1, the gene that encodes PNPase protein, exhibit greater accumulation of dsRNA and elevated interferon levels in their serum 74.
Mitochondrial outer membrane permeabilisation is a rapid and complete event, spreading to all mitochondria in a cell. Following formation of BAX/BAK pores, pro‐apoptotic proteins such as cytochrome c are released from the intermembrane space where they initiate the caspase cascade, culminating in cell death. However, we have found that under conditions of sub‐lethal stress, a limited number of mitochondria in a cell can undergo permeabilisation, called minority MOMP, leading to genomic instability and transformation 75. A recent report by Brokatsky et al reveals a link between pathogen invasion and activation of mitochondrial cell death machinery 76. In this study, it was found that various pathogens can induce limited mitochondrial permeabilisation. It remains unclear how pathogens can trigger minority MOMP, but nevertheless they can, resulting in mtDNA release (presumably through BAX/BAK pores), stimulating cGAS‐STING activation and cytokine secretion 76.
How else might mtDNA be released from mitochondria? Another potential mechanism for mtDNA release from mitochondria is through the mitochondrial permeability transition pore (mPTP) 77, 78. The exact composition of the pore is unclear, although there seems to be consensus that cyclophilin D is present 79. The mPTP spans the mitochondrial inner membrane and forms in response to high mitochondrial calcium concentration and various other cellular stresses. However, the mPTP is predicted to only allow the efflux of molecules smaller than 1.5 kDa, much smaller than a mtDNA nucleoid 80, 81. In line with this, studies have shown that only fragments of mtDNA can pass through the mPTP 77, 82, 83. It remains possible that sustained opening of the pore can lead to swelling of the mitochondria and subsequent rupture of the inner membrane, which would permit the efflux of mtDNA into the cytoplasm. The involvement of mPTP in mtDNA release during cell death has been ruled out 61, but chitosan, a vaccine adjuvant, appears to induce a cGAS‐STING‐ and mPTP‐dependent type I interferon response. This is possibly due to mtDNA release, though a direct role for mtDNA has not been rigorously assessed 84. An intriguing recent report suggests that cells experiencing mitochondrial stress caused by the lack of mitochondrial endonuclease G release mtDNA through pores formed by oligomers of the voltage‐dependent anion channel (VDAC) 85. As mitochondrial DNA release is thought to play a role in the pathogenesis of lupus 86, 87, a role for VDAC pore formation was tested in an in vivo model of lupus‐like disease. Using the VDAC1 oligomerisation inhibitor VBIT‐4, the authors were able to reduce lupus‐like symptoms in lupus‐prone mice, providing a rationale to target VDAC‐mediated mtDNA release in this disease 85.
Therapeutic targeting of mtDNA‐dependent cGAS‐STING activity
There is currently intense interest in the development of inhibitors and activators of the cGAS‐STING pathway, depending on the disease. In humans, the systemic inflammatory disease Aicardi–Goutières syndrome (AGS) is characterised by mutations in a number of different genes involved in DNA sensing 88. For example, TREX1, a DNA exonuclease, is frequently mutated in human patients with AGS and systemic lupus erythematosus (SLE) 89, 90, 91, and co‐deletion of cGAS, STING, Interferon‐α/β receptor (IFNAR) or IRF3 rescues this phenotype 92, 93, 94, 95, 96, 97, 98. Accumulation of cytosolic DNA appears to be a defining characteristic of AGS and SLE, as deletions in DNA‐ and RNA‐related genes including SAMDH1, a DNA exonuclease and RnaseH2 are frequent 99, 100, 101, 102. Gain‐of‐function mutations in STING itself lead to an up‐regulation of type I interferon responses and lupus‐like symptoms in patients 103, 104. DNase II deficiency in humans leads to autoinflammation with increased type I IFN 105 and in mice causes arthritis 106. This is thought to be due to the lack of self‐DNA degradation in dead cells engulfed by macrophages resulting in sustained cGAS‐STING stimulation 98, 106, 107, and AIM2 inflammasome formation 108, 109 with a possible contribution of endosomal TLRs 108. Myocardial infarction (MI) is another condition known to involve a strong inflammatory component. King et al 110 showed that ischaemic cell death and engulfment by macrophages drives an IRF3‐dependent type I IFN response. Genetic or pharmacological disruption of cGAS‐STING signalling in mice improved their outcomes post‐MI, proposing this signalling axis as suitable for therapeutic intervention in patients 110, 111. While it is not clear if this is due to mtDNA release per se, increased mtDNA in plasma from patients with heart disease has been frequently observed 112, 113, 114. Clearly, inhibiting the cGAS‐STING pathway in these disease settings might be beneficial to patients. Small molecules targeting both cGAS 115, 116 and STING 117 have been developed, with STING antagonists emerging as the most promising. Blocking the IFNAR receptor to block interferon signalling in SLE patients had seemed like a viable therapeutic route; however, late‐stage clinical trials in this area have failed, prompting more investigation of how important interferon signalling is in the pathogenesis of SLE.
The ability to turn immunologically “cold” tumours “hot” and make them more responsive to immunotherapy is a desirable outcome in cancer treatment. Efficient T‐cell responses to tumour cells is a critical step to durable cancer treatment control 118. STING is required for spontaneous CD8+ T‐cell priming in vivo 119. Mechanistically, dying tumour cells transfer their DNA to antigen‐presenting dendritic cells when phagocytosed, eliciting cGAS‐STING‐IRF3 signalling leading to an anti‐tumour T‐cell response 119, 120, 121. Activation of STING by addition of exogenous cGAMP can also enhance anti‐tumour immunity after irradiation 120, the first evidence that therapeutic activation of STING may improve cancer therapy. This effect was later shown to be exclusive to dendritic cells over macrophages; blockade of the “don't eat‐me” signal CD47 results in increased tumour‐originated mtDNA in the cytosol of DCs and is required for the cross‐priming and type I IFN response mediated through cGAS 122. Dying tumour cells transfected with exogenous cytosolic DNA, viral DNA or cyclic dinucleotides (CDNs) have a greater capacity to activate STING signalling in antigen‐presenting cells, enhancing T‐cell priming and expansion of anti‐tumour T cells 123. Therefore, it is also possible that mtDNA may act as a STING activator in antigen‐presenting cells (APCs) under certain circumstances, for example when apoptotic caspases are inhibited. Another example of immune cell communication is in the interaction of T cells with antigen‐presenting dendritic cells. Upon formation of an immunological synapse between these two cell types, T cells shed extracellular vesicles (EVs) containing genomic and mtDNA. These EVs are taken up by the dendritic cell, triggering a cGAS‐STING‐dependent anti‐viral response, conferring resistance to subsequent viral infection 124. In the context of cancer treatment, it is plausible that apoptotic cell‐containing dendritic cells could stimulate a similar effect in T cells, generating longer lived dendritic cells for more durable treatment responses 125. Together, these data and many others provide a rationale for enhancing STING signalling in cancer treatment, and this is currently under active investigation 126, 127.
mtDNA release, cGAS‐STING and neurodegeneration
Under normal, homeostatic conditions, damaged or stressed mitochondria are eliminated from the cell by a type of mitochondrial‐selective autophagy called mitophagy 128. Mutations in proteins involved in mitophagy pathways can contribute to neurodegeneration. This is perhaps best evidenced for PINK1/Parkin‐dependent mitophagy. For instance, loss‐of‐function mutations in the PINK1/Parkin pathway of mitophagy associate with early onset Parkinson's disease 129, 130, 131, 132, 133. In a simplified view, the kinase PINK1 is activated on dysfunctional mitochondria where it phosphorylates ubiquitin. Phospho‐ubiquitin allosterically activates the E3 ubiquitin ligase Parkin leading to enhanced mitochondrial ubiquitination that serves as an autophagic signal to remove the damaged mitochondrion 134, 135, 136. Parkinson's disease is associated with neuroinflammation 137, and the serum from Parkinson's patients is often enriched for pro‐inflammatory cytokines, including TNF, IL‐1β, IFNɣ and IL‐6 138, 139. However, many of the studies elucidating the mechanistic basis of Parkinson's have been performed in cultured cell lines, and despite much effort, the in vivo relevance of PINK1/Parkin‐mediated mitophagy was not well understood, particularly since mice that lack either PINK1 or Parkin exhibit no Parkinson's‐like disease phenotypes 140, 141, 142. Knowing that defective mitochondria can release innate immune‐activating DAMPs, Sliter et al 143 investigated the effect of exhaustive exercise or mtDNA mutation on inflammation. When challenged with exhaustive exercise, Parkin −/− or Pink1 −/− mice displayed higher serum levels of pro‐inflammatory IL‐6 and IFN‐β when compared to wild‐type mice, in addition to increased levels of uncleared mitochondria. Remarkably, this could be completely rescued by deletion of STING or administering IFNAR‐blocking antibody to mice, strongly suggesting that mtDNA released from damaged mitochondria that are not cleared is responsible for the inflammation observed in Parkinson's patients. Interestingly, the authors also observed increased circulating mtDNA in Parkin −/− mice following exhaustive exercise, meaning that the mtDNA is not only extruded from mitochondria but also exits the cell. Mutator mice expressing a proofreading‐defective mtDNA polymerase (PolG) accumulate mutations in mtDNA, which instead of causing neurodegeneration results in dopaminergic neuron loss and defective movement. While no difference in inflammatory cytokine levels was noted between wild‐type, Parkin −/− or mutator mice, Parkin −/−;mutator mice do have higher serum cytokine levels. Again, cytokine levels and the movement disorder could be completely rescued by co‐deletion of STING, reinforcing the cGAS‐STING axis as the major player in Parkinson's‐associated inflammation. However, further work is needed to elucidate the absolute requirement for mtDNA over nuclear DNA and the precise mechanism of how mtDNA is released from the mitochondria.
mtDNA as an inflammasome activator
Inflammasomes are multi‐subunit complexes which form in response to exogenous PAMPs and DAMPs 144. One of four receptors—absent in melanoma 2 (AIM2), NOD, LRR and Pyrin domain‐containing protein 1 (NLRP1), NLRP3 or NLR family CARD domain‐containing protein 4 (NLRC4), bind to the adaptor molecule ASC forming a platform for the dimerisation, autoprocessing and activation of caspase‐1. Active caspase‐1 can then process pro‐IL‐1β and pro‐IL‐18 into their mature form so they can be secreted (see Fig 1). The first report of mtDNA acting as an activator of inflammasomes came in 2011 when Nakahira et al 145 reported that depletion of proteins involved in autophagy leads to an accumulation of dysfunctional, persistent mitochondria exhibiting excessive ROS. These mitochondria were more prone to extrude mtDNA into the cytoplasm upon stimulation with lipopolysaccharide (LPS) or ATP, dependent on the ability to from NLRP3 inflammasomes. Interestingly, Nakahira et al suggested that as well as acting downstream of mtDNA release, NLRP3 also acts upstream, to facilitate mPTP formation on the mitochondria allowing mtDNA release. However, as already discussed, whether mPTP is sufficient to allow mtDNA translocation from the mitochondrial matrix into the cytoplasm is debatable. Extending this work, the following year Shimada et al 146 reported that during macrophage apoptosis mtDNA is released and binds NLRP3. Notably, NLRP3 appears to have a preference for oxidised mtDNA, clarifying the observations that ROS plays a crucial role in inflammasome activation 147. Linking these observations, deletion of the autophagy receptor p62 prevents mitophagic clearance of mitochondria damaged by NLRP3 agonists, exacerbating inflammasome formation and IL‐1β secretion 148. More recent work has pointed to newly synthesised, oxidised mtDNA as the species which binds NLRP3 149. Zhong et al discovered that levels of the mitochondrial deoxyribonucleotide kinase CMPK2 increase upon LPS stimulation. CMPK2 catalyses a step in the synthesis of the nucleotide cytidine triphosphate, which is rate‐limiting for mtDNA synthesis. Elevated dCTP levels in turn increase mtDNA replication, which is oxidised by ROS and released into the cytoplasm where it can activate NLRP3 and stimulate IL‐1β secretion. However, the role of NLRP3 as a direct sensor of DNA is contentious, as many disparate signals have been reported as the common signal for NLRP3 activation 144. Indeed, recent work from the Chen laboratory has shown that dispersal of the trans‐Golgi network following K+ efflux is the likely common trigger 150.
Supporting the notion that inflammasomes and caspase activity can act upstream of mtDNA release, there are reports that caspases cause mitochondrial damage. For example, inflammasome‐activated caspase‐1 has been reported to damage mitochondria and promote the release of cytochrome c, indicative of mitochondrial outer membrane permeabilisation 151. The authors suggest that this is due to mPTP formation, although a role for BAX and BAK was not rigorously assessed in this work. Impairment of mitophagy was also implicated, as Parkin was found to be a substrate of caspase‐1 in macrophages, leading to an accumulation of damaged, ROS‐producing macrophages 151. Furthermore, during infection‐related ER stress, NLRP3 (but not the adaptor protein ASC or caspase‐1) is involved in caspase‐2 activation and cleavage of the pro‐apoptotic protein BID, promoting mitochondrial permeabilisation 152.
Neutrophil extracellular traps
So far, we have mainly discussed the cell autonomous role of mtDNA release; however, it is becoming clear that mtDNA can also be extruded from the mitochondria, into the cytoplasm and outward further into the extracellular space. One interesting example of this is in the generation of neutrophil extracellular traps, and in particular the role of mtDNA in their formation (Fig 4).
Neutrophils are the first line of attack in infection, capable of engulfing pathogens and degranulating, the process of releasing soluble anti‐microbials. In 2004, Brinkmann and colleagues discovered that upon stimulation with IL‐8, phorbol myristate acetate (PMA) or LPS, neutrophils extruded vast fibrous networks, which they termed neutrophil extracellular traps (NETs) 153. Analysis of these NETs showed that they contained a variety of microbial‐killing proteins, including elastase, cathepsin G and myeloperoxidase. However, they also contain DNA, as noted by reactivity with antibodies against histones and DNA intercalating dyes. Successive work showed that NETs were also enriched for mtDNA 154, 155, 156. NET formation has been well studied in patients with systemic lupus erythematosus (SLE), an autoimmune condition hallmarked by the appearance of autoantibodies against dsDNA and RNA‐protein complexes, resulting in elevated type I interferon responses. A number of studies show that mtDNA is part of NETs formed in SLE. Caielli et al 87 found that in healthy neutrophils, mitochondria with oxidative damage are removed not via mitophagy, but by extruding their mitochondrial matrix contents, including TFAM‐mtDNA nucleoids, into the extracellular space. These TFAM‐mtDNA nucleoids are devoid of oxidised DNA, and so do not activate plasmacytoid dendritic cells (pDCs) and thus are not immunogenic. Healthy neutrophils remove oxidised mtDNA by signalling PKA phosphorylation of TFAM which initiates its degradation and by shuttling oxidised mtDNA into lysosomes. In contrast, neutrophils in SLE have reduced PKA activation and so do not degrade TFAM as efficiently, leading to the extrusion of immunogenic oxidised mtDNA 87. Another report reveals ROS to be an important mediator for neutrophils to produce oxidised mtDNA‐containing NETs in response to stimulation by ribonucleotide immune complexes (RNP ICs) 86. The authors also found that injecting this DNA was pro‐inflammatory and dependent on the STING pathway revealing a dual role for mitochondria in providing the source of DNA for NETs and oxidising it for maximal interferogenic response in SLE 86 (Fig 4). Sustained IFNα signalling in SLE is also known to deregulate mitochondrial metabolism in monocytes, leading to reduced autophagy and an accumulation of mtDNA in the cytoplasm. This leads to cGAS‐STING activation which promotes secretion of TNF and IL‐6 and the expansion of self‐DNA autoreactive lymphocytes 157. It is now also appreciated that other cell types, including lymphocytes and eosinophils, can secrete mtDNA‐containing webs which act to prime type I interferon responses in peripheral blood mononuclear cells 158, 159.
Nuclear DNA is prepared for expulsion as NETs through a highly regulated process involving decondensation of chromatin and citrullination of histones. Furthermore, plasma membrane permeabilisation is also regulated, inevitably leading to cell death. Within minutes of stimulation, neutrophils rapidly produce NETs, whereas the death of neutrophils (dubbed “NETosis”) occurs ~2 h after 160. While these two phenomena are often conflated in the literature, the timing argues against a general lytic mechanism of mtDNA release. In fact, release of mtDNA as NETs seems to be energy‐dependent 161. The precise mechanism of mtDNA escape during NET formation remains to be elucidated; one possibility is that it may be due to BAX/BAK pore formation on the mitochondrial outer membrane 61, 62, although this seems unlikely as this would induce a rapid cell death.
mtDNA and TLR9
The Toll‐like family of receptors (TLR) recognise a plethora of different bacterial features to instigate innate immunity. TLR9 recognises hypomethylated CpG motifs found in bacteria. TLR9 is expressed primarily in monocytes, macrophages, plasmocytoid dendritic cells and B lymphocytes. In resting cells, TLR9 resides on the endoplasmic reticulum, but recognition of DNA occurs in the endolysosomes (see Fig 1) 162, 163, 164, 165. DNA‐bound TLR9 recruits MyD88 which activates MAPK and NF‐κB, inducing an inflammatory response. In common with bacterial DNA, mtDNA is hypomethylated at CpG motifs, making it a potent activator of TLR9 166, 167. mtDNA detection by TLR9 was first noted in 2010 by Zhang et al, who observed that during systemic inflammatory response syndrome (SIRS) mtDNA was released into the blood where it can activate TLR9 on neutrophils 168, 169. In the heart, autophagy is required to remove damaged mitochondria and maintain heart function during hemodynamic stress 170. However, when DNase II, a lysosomal DNase, is deleted from cardiac cells, the mice succumb faster following heart pressure overload 171. Delving deeper into the mechanism, the authors found that this was due to an increase in mtDNA which has escaped degradation, thus activating a TLR9‐dependent inflammatory response 171. Mitochondrial DNA released from dying cells or as part of NETs can form a complex with the anti‐microbial peptide LL‐37. This mtDNA:LL‐37 complex evades degradation by DNase II and can activate TLR9 on pDCs, neutrophils and endothelial cells to exacerbate atherosclerosis 172. High‐mobility group box 1 (HMGB1) is a DNA‐binding protein released from necrotic 173 and cytokine‐stimulated cells 174. HMBG1 binds a receptor, called RAGE, leading to inflammatory signalling. In particular, HMGB1 has been shown to be released from pDCs following stimulation with CpG oligodeoxynucleotides (ODNs). CpG‐ODNs can bind and activate TLR9, but when complexed with HMGB1 the inflammatory response is augmented through HMGB1 activation of RAGE 175. In an analogous manner, TFAM co‐operates with mtDNA released from necrotic cells to increase pro‐inflammatory signalling in pDCs through RAGE and TLR9 176, 177.
TLR9 has been particularly well studied in liver pathologies. In liver cancer, hypoxia triggers the translocation of mtDNA and HMGB1 into the cytoplasm of cancer cells to activate TLR9, resulting in tumour cell proliferation 178. TLR9 is crucial for the development of acetaminophen‐induced hepatotoxicity 179 and fibrosis 180. Development of non‐alcoholic steatohepatitis (NASH) involves innate immunity, with hepatic stellate cells and macrophage‐like Kupffer cells being particularly relevant. Mice fed a choline‐deficient amino acid‐defined diet develop NASH, whereas TLR9−/− mice do not, implicating TLR9 as a requirement for NASH development 181. The precise ligand for TLR9‐derived liver disease was poorly understood, although the observation that NASH patients had higher mitochondrial mass, but reduced respiration, suggested that mitochondria may play a role 182. In line with these observations, Garcia‐Martinez et al 183 found that mice and human patients with NASH exhibited higher levels of oxidised mtDNA in hepatocytes and plasma. As the oxidisation of mtDNA increases its ability to activate TLR9, the authors confirmed that was the case. Importantly, mice dosed with a TLR9 antagonist displayed reduced symptoms of NASH, validating the importance of mtDNA release and TLR9 signalling in the pathogenesis of NASH 183. NASH is characterised by different forms of cell death, most prominently apoptosis 184 and necrosis 185, 186. In hepatocytes, mitochondrial permeabilisation results in an increase of DNase II activity, and knockdown of DNase II switches the mode of cell death to a RIP1‐dependent non‐apoptotic form 187. Importantly, this is due to the release of mtDNA after mitochondrial permeabilisation, which triggers TLR9 signalling and subsequent IFNβ secretion. In mice fed a high‐fat diet, a model of NASH, DNase II activity is diminished, providing a mechanistic link as to how necrosis of hepatocytes can augment NASH symptoms in patients 187. It is unclear why the release of mtDNA triggers either cGAS‐STING or TLR9 signalling in different studies; however, it may be due to different cell types, length or oxidation status of mtDNA, activity of DNA nucleases or different cellular compartments.
Mutations in OPA1, a protein required for mitochondrial inner membrane fusion and cristae formation, have been reported to cause mtDNA instability 188, 189, 190, 191. Deletion of OPA1 in skeletal muscle, a tissue with high metabolic demands, predictably results in mitochondrial dysfunction, mtDNA stress and inflammation leading to reduced growth and early death in mice 192, 193. Interestingly however, OPA1 deletion leads to disruption of mitophagy due to impaired autophagic flux resulting in higher levels of dysfunctional mitochondria in these tissues 194. When mtDNA localisation was examined, following OPA1 deletion there is high co‐localisation of mtDNA and TLR9, implicating TLR9 as the driver of OPA1‐deletion inflammation 194.
Transfer of mtDNA between cells
So far, this Review has mainly focussed on the cell‐intrinsic biological effects of mtDNA release. However, it is possible that released mtDNA nucleoids could move from one cell to another, thus “spreading” the inflammatory signal across a population of cells. It is now well established that mitochondria, including mtDNA, can be transferred between cells (Fig 4). A seminal study in 1989 was the first to describe such a phenomena, where cells devoid of mtDNA (ρ0 cells) and thus lacking respiratory competence could be repopulated with mitochondria from other cell lines 195. More recent work has shown that following stroke, whole mitochondria can be transferred from astrocytes to neurons, a process proven to be beneficial to recovery 196. In cancer models, ρ0 cells have delayed tumour growth, likely due to defects in energy production. Horizontal transfer of mitochondria from cells in the tumour microenvironment restored respiration in ρ0 cells and instigated tumour growth 197. Horizontal transfer of mitochondria could occur through a number of different mechanisms. Firstly, cancer cells can form tunnelling nanotubes with cells in the tumour microenvironment, through which cytoplasmic contents, including mitochondria, can be transferred 198. Tunnelling nanotubes form between endothelial cells and cancer cells to transfer mitochondria, conferring chemoresistance to the cancer cell 199 but also between early apoptotic cells and healthy cells, where mitochondrial transfer can reverse apoptosis 200. Secondly, mtDNA has been proposed to be packaged into extracellular vesicles (EVs). Specifically, cancer‐associated fibroblasts can package entire mitochondrial genomes into EVs which then fuse with cancer cells to transfer mtDNA. Importantly, the size of these EVs, ~100 nm, is far below the size of a mitochondria, so making it unlikely that an entire mitochondria is transferred in this manner 201. However, mtDNA nucleoids are within these size constraints 21. It is important to note that other studies see transfer of entire mitochondria between cells, and so whether these or just mtDNA genomes are transferred is controversial 202. Thirdly, mitochondria can be directly transferred between cells through connexin 43 gap junctions, as had been seen between bone marrow‐derived stromal cells and pulmonary alveoli during lung injury 203. Interestingly, transplanting tumour or embryonic stem cells into hosts with the same nuclear DNA background but different mtDNA from allogenic mouse strains resulted in rejection 204. Mechanistically, this is dependent on MyD88, the adaptor molecule required for TLR9 signalling, suggesting that TLR9 may be the PRR in this situation 204. However, whether or how mtDNA is released from these cells is unknown, but it is clear that allogenic mtDNA can trigger innate immune pathways. This hints at the intriguing notion that inflammation could spread between cells via detection of mtDNA, perhaps through connexin 43 gap junctions, in a similar manner to the observation that cGAMP can transfer to activate STING in neighbouring cells 71 (Fig 4). Contrary to this is data showing that cell‐free mtDNA (for example, as seen in sepsis) can actually suppress inflammation 205. Increased serum concentration of mtDNA is associated with a poorer outcome in sepsis patients, and injection of mtDNA in mice suppresses the adaptive immune response in a TLR9‐dependent manner 205. Immunosuppressive markers, such as an increase in PD‐L1 expression in the spleen, are seen in mice injected with mtDNA, which is reflective of what is seen in sepsis patients 205. Clearly, there is conflicting data on the immunostimulatory or immunosuppressive role of cell‐free mtDNA, which may depend on pathophysiological context; nevertheless, release of mtDNA appears to potently affect the immune system.
Conclusions and future perspectives
Mitochondria are multi‐faceted organelles orchestrating key events in both life and death. They represent a rich source of DAMPs which can potently trigger the innate immune system, such as ATP, formyl peptides and mtDNA. Possibly stemming from its bacterial origin, mtDNA is particularly effective at initiating inflammatory and anti‐viral signalling.
The last number of years has seen an explosion in interest in how mitochondria initiate innate immunity in the context of pathogen invasion, cell death and pathology. However, many of these studies leave us with unresolved questions as to precisely how mtDNA is extruded from the mitochondria. In the context of cell death, it is now clear that BAX and BAK form the pores on the mitochondrial outer membrane through which the inner membrane herniates, leading to mtDNA release, although how the inner membrane permeabilises is as yet not fully resolved 61, 62. Many other studies have suggested that the mPTP is involved in various contexts, but again this is controversial. Clearly, further investigation is required, whether to determine a more universal role for BAX/BAK‐dependent mtDNA release, utilising our current knowledge of the nature of the mPTP, or whether an altogether unknown mechanism is involved.
It is also apparent that cellular context will determine how mtDNA causes inflammation. cGAS‐STING signalling seems to be widely available across most cell types, a notable exception being some transformed cells. However, TLR9 protein expression appears to be restricted to immune cells, as does expression of inflammasome components. Perhaps most interesting will be determining what the outcomes of triggering innate immunity with cytosolic or cell‐free mtDNA are. For example, in the context of cell death, does production of cGAMP in apoptotic cells transfer to healthy apoptotic cells via gap junctions to promote a death‐resistant state, in a manner similar to what has been observed in astrocytes 71? Pathogen invasion stimulates a limited degree of mtDNA release by hijacking the apoptotic machinery, so it is plausible to see how this might act as a cell‐intrinsic warning system, but it will be fascinating to understand how this functions in the context of a whole tissue. Furthermore, can we leverage what we have learnt about anti‐viral signalling during cell death to enhance anti‐cancer therapy by inhibiting caspases? Likewise, will our understanding of how mtDNA and STING function in neurodegeneration lead to novel therapeutic strategies to enhance healthy ageing 143? Along these lines, mitochondrial dysfunction has been shown to induce a specific form of senescence termed MiDAS (mitochondrial dysfunction‐associated senescence) 206—given the links between ageing, senescence and inflammation, it is tempting to hypothesise that mtDNA plays a role in the initiation of this phenotype.
A broad spectrum of pathologies, from cancer, to autoimmunity and ageing all have aberrant mtDNA release as a driver or contributor of disease. Future work aimed at understanding how mtDNA is involved will no doubt afford us new therapeutic avenues with which to treat patients.
In need of answers.
Why do different tissue and cell types respond to cytosolic mtDNA through different pathways?
Can mtDNA release be harnessed therapeutically for treatment of inflammatory diseases or cancer?
Where is cGAS located in the cell?
What are the physiological, non‐lethal effects of mtDNA release into the cytoplasm?
Conflict of interest
The authors declare that they have no conflict of interest.
Acknowledgements
Research in our laboratory is supported by funding from Cancer Research UK and Prostate Cancer UK.
EMBO Reports (2020) 21: e49799
See the Glossary for abbreviations used in this article.
Contributor Information
Joel S Riley, Email: joel.riley@glasgow.ac.uk.
Stephen WG Tait, Email: stephen.tait@glasgow.ac.uk.
References
- 1. Brubaker SW, Bonham KS, Zanoni I, Kagan JC (2015) Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol 33: 257–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gong T, Liu L, Jiang W, Zhou R (2019) DAMP‐sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol 20: 95–112 [DOI] [PubMed] [Google Scholar]
- 3. Roger AJ, Muñoz‐Gómez SA, Kamikawa R (2017) The origin and diversification of mitochondria. Curr Biol 27: R1177–R1192 [DOI] [PubMed] [Google Scholar]
- 4. Hampl V, Čepička I, Eliáš M (2019) Was the mitochondrion necessary to start eukaryogenesis? Trends Microbiol 27: 96–104 [DOI] [PubMed] [Google Scholar]
- 5. Fox TD (2012) Mitochondrial protein synthesis, import, and assembly. Genetics 192: 1203–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Holt IJ, Reyes A (2012) Human mitochondrial DNA replication. Cold Spring Harb Perspect Biol 4: a012971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mechta M, Ingerslev LR, Fabre O, Picard M, Barrès R (2017) Evidence suggesting absence of mitochondrial DNA methylation. Front Genet 8: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nass MM (1973) Differential methylation of mitochondrial and nuclear DNA in cultured mouse, hamster and virus‐transformed hamster cells. In vivo and in vitro methylation. J Mol Biol 80: 155–175 [DOI] [PubMed] [Google Scholar]
- 9. Bellizzi D, D'Aquila P, Scafone T, Giordano M, Riso V, Riccio A, Passarino G (2013) The control region of mitochondrial DNA shows an unusual CpG and non‐CpG methylation pattern. DNA Res 20: 537–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shock LS, Thakkar PV, Peterson EJ, Moran RG, Taylor SM (2011) DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc Natl Acad Sci USA 108: 3630–3635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Infantino V, Castegna A, Iacobazzi F, Spera I, Scala I, Andria G, Iacobazzi V (2011) Impairment of methyl cycle affects mitochondrial methyl availability and glutathione level in Down's syndrome. Mol Genet Metab 102: 378–382 [DOI] [PubMed] [Google Scholar]
- 12. Dzitoyeva S, Chen H, Manev H (2012) Effect of aging on 5‐hydroxymethylcytosine in brain mitochondria. Neurobiol Aging 33: 2881–2891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ghosh S, Sengupta S, Scaria V (2016) Hydroxymethyl cytosine marks in the human mitochondrial genome are dynamic in nature. Mitochondrion 27: 25–31 [DOI] [PubMed] [Google Scholar]
- 14. Sun Z, Terragni J, Jolyon T, Borgaro JG, Liu Y, Yu L, Guan S, Wang H, Sun D, Cheng X et al (2013) High‐resolution enzymatic mapping of genomic 5‐hydroxymethylcytosine in mouse embryonic stem cells. Cell Rep 3: 567–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hong EE, Okitsu CY, Smith AD, Hsieh C‐L (2013) Regionally specific and genome‐wide analyses conclusively demonstrate the absence of CpG methylation in human mitochondrial DNA. Mol Cell Biol 33: 2683–2690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mankan AK, Schmidt T, Chauhan D, Goldeck M, Höning K, Gaidt M, Kubarenko AV, Andreeva L, Hopfner K‐P, Hornung V (2014) Cytosolic RNA:DNA hybrids activate the cGAS‐STING axis. EMBO J 33: 2937–2946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chatterjee A, Mambo E, Sidransky D (2006) Mitochondrial DNA mutations in human cancer. Oncogene 25: 4663–4674 [DOI] [PubMed] [Google Scholar]
- 18. Jiang W, Li R, Zhang Y, Wang P, Wu T, Lin J, Yu J, Gu M (2017) Mitochondrial DNA mutations associated with type 2 diabetes mellitus in Chinese Uyghur population. Sci Rep 7: 16989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Larsson N‐G (2010) Somatic mitochondrial DNA mutations in mammalian aging. Annu Rev Biochem 79: 683–706 [DOI] [PubMed] [Google Scholar]
- 20. Kazak L, Reyes A, Holt IJ (2012) Minimizing the damage: repair pathways keep mitochondrial DNA intact. Nat Rev Mol Cell Biol 13: 659–671 [DOI] [PubMed] [Google Scholar]
- 21. Kukat C, Wurm CA, Spåhr H, Falkenberg M, Larsson N‐G, Jakobs S (2011) Super‐resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proc Natl Acad Sci USA 108: 13534–13539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Collins LV, Hajizadeh S, Holme E, Jonsson I‐M, Tarkowski A (2004) Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J Leukoc Biol 75: 995–1000 [DOI] [PubMed] [Google Scholar]
- 23. Sun L, Wu J, Du F, Chen X, Chen ZJ (2013) Cyclic GMP‐AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339: 786–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wu J, Sun L, Chen X, Du F, Shi H, Chen C, Chen ZJ (2013) Cyclic GMP‐AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339: 826–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang X, Wu J, Du F, Xu H, Sun L, Chen Z, Brautigam CA, Zhang X, Chen ZJ (2014) The cytosolic DNA sensor cGAS forms an oligomeric complex with DNA and undergoes switch‐like conformational changes in the activation loop. Cell Rep 6: 421–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Diner EJ, Burdette DL, Wilson SC, Monroe KM, Kellenberger CA, Hyodo M, Hayakawa Y, Hammond MC, Vance RE (2013) The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell Rep 3: 1355–1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gao P, Ascano M, Wu Y, Barchet W, Gaffney BL, Zillinger T, Serganov AA, Liu Y, Jones RA, Hartmann G et al (2013) Cyclic [G(2′,5′)pA(3′,5′)p] is the metazoan second messenger produced by DNA‐activated cyclic GMP‐AMP synthase. Cell 153: 1094–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Civril F, Deimling T, de Oliveira Mann CC, Ablasser A, Moldt M, Witte G, Hornung V, Hopfner K‐P (2013) Structural mechanism of cytosolic DNA sensing by cGAS. Nature 498: 332–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gao P, Ascano M, Zillinger T, Wang W, Dai P, Serganov AA, Gaffney BL, Shuman S, Jones RA, Deng L et al (2013) Structure‐function analysis of STING activation by c[G(2′,5′)pA(3′,5′)p] and targeting by antiviral DMXAA. Cell 154: 748–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kranzusch PJ, Lee AS‐Y, Berger JM, Doudna JA (2013) Structure of human cGAS reveals a conserved family of second‐messenger enzymes in innate immunity. Cell Rep 3: 1362–1368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang X, Shi H, Wu J, Zhang X, Sun L, Chen C, Chen ZJ (2013) Cyclic GMP‐AMP containing mixed phosphodiester linkages is an endogenous high‐affinity ligand for STING. Mol Cell 51: 226–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang C, Shang G, Gui X, Zhang X, Bai X‐C, Chen ZJ (2019) Structural basis of STING binding with and phosphorylation by TBK1. Nature 567: 394–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gentili M, Lahaye X, Nadalin F, Nader GPF, Lombardi EP, Herve S, De Silva NS, Rookhuizen DC, Zueva E, Goudot C et al (2019) The N‐terminal domain of cGAS determines preferential association with centromeric DNA and innate immune activation in the nucleus. Cell Rep 26: 2377–2393e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu H, Zhang H, Wu X, Ma D, Wu J, Wang L, Jiang Y, Fei Y, Zhu C, Tan R et al (2018) Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature 563: 131–136 [DOI] [PubMed] [Google Scholar]
- 35. Barnett KC, Coronas‐Serna JM, Zhou W, Ernandes MJ, Cao A, Kranzusch PJ, Kagan JC (2019) Phosphoinositide interactions position cGAS at the plasma membrane to ensure efficient distinction between self‐ and viral DNA. Cell 176: 1432–1446.e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Volkman HE, Cambier S, Gray EE, Stetson DB (2019) Tight nuclear tethering of cGAS is essential for preventing autoreactivity. Elife 8: 394–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. White MJ, McArthur K, Metcalf D, Lane RM, Cambier JC, Herold MJ, van Delft MF, Bedoui S, Lessene G, Ritchie ME et al (2014) Apoptotic caspases suppress mtDNA‐induced STING‐mediated type I IFN production. Cell 159: 1549–1562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rongvaux A, Jackson R, Harman CCD, Li T, West AP, de Zoete MR, Wu Y, Yordy B, Lakhani SA, Kuan C‐Y et al (2014) Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 159: 1563–1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. West AP, Khoury‐Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff DA et al (2015) Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520: 553–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Saffran HA, Pare JM, Corcoran JA, Weller SK, Smiley JR (2007) Herpes simplex virus eliminates host mitochondrial DNA. EMBO Rep 8: 188–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Corcoran JA, Saffran HA, Duguay BA, Smiley JR (2009) Herpes simplex virus UL12.5 targets mitochondria through a mitochondrial localization sequence proximal to the N terminus. J Virol 83: 2601–2610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Duguay BA, Saffran HA, Ponomarev A, Duley SA, Eaton HE, Smiley JR, Sandri‐Goldin RM (2014) Elimination of mitochondrial DNA is not required for herpes simplex virus 1 replication. J Virol 88: 2967–2976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Grady LM, Szczepaniak R, Murelli RP, Masaoka T, Le Grice SFJ, Wright DL, Weller SK (2017) The exonuclease activity of herpes simplex virus 1 UL12 is required for production of viral DNA that can be packaged to produce infectious virus. J Virol 91: 194–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schoggins JW, MacDuff DA, Imanaka N, Gainey MD, Shrestha B, Eitson JL, Mar KB, Richardson RB, Ratushny AV, Litvak V et al (2014) Pan‐viral specificity of IFN‐induced genes reveals new roles for cGAS in innate immunity. Nature 505: 691–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sun B, Sundström KB, Chew JJ, Bist P, Gan ES, Tan HC, Goh KC, Chawla T, Tang CK, Ooi EE (2017) Dengue virus activates cGAS through the release of mitochondrial DNA. Sci Rep 7: 1148–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Aguirre S, Luthra P, Sanchez‐Aparicio MT, Maestre AM, Patel J, Lamothe F, Fredericks AC, Tripathi S, Zhu T, Pintado‐Silva J et al (2017) Dengue virus NS2B protein targets cGAS for degradation and prevents mitochondrial DNA sensing during infection. Nat Microbiol 2: 17037–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lai JH, Wang MY, Huang CY, Wu CH, Hung LF, Yang C‐Y, Ke P‐Y, Luo SF, Liu SJ, Ho LJ (2018) Infection with the dengue RNA virus activates TLR9 signaling in human dendritic cells. EMBO Rep 19: e46182–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Aguirre S, Maestre AM, Pagni S, Patel JR, Savage T, Gutman D, Maringer K, Bernal‐Rubio D, Shabman RS, Simon V et al (2012) DENV inhibits type I IFN production in infected cells by cleaving human STING. PLoS Pathog 8: e1002934–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yu C‐Y, Chang T‐H, Liang J‐J, Chiang R‐L, Lee Y‐L, Liao C‐L, Lin Y‐L (2012) Dengue virus targets the adaptor protein MITA to subvert host innate immunity. PLoS Pathog 8: e1002780–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wassermann R, Gulen MF, Sala C, Perin SG, Lou Y, Rybniker J, Schmid‐Burgk JL, Schmidt T, Hornung V, Cole ST et al (2015) Mycobacterium tuberculosis differentially activates cGAS‐ and inflammasome‐dependent intracellular immune responses through ESX‐1. Cell Host Microbe 17: 799–810 [DOI] [PubMed] [Google Scholar]
- 51. Collins AC, Cai H, Li T, Franco LH, Li X‐D, Nair VR, Scharn CR, Stamm CE, Levine B, Chen ZJ et al (2015) Cyclic GMP‐AMP synthase is an innate immune DNA sensor for Mycobacterium tuberculosis . Cell Host Microbe 17: 820–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Watson RO, Bell SL, MacDuff DA, Kimmey JM, Diner EJ, Olivas J, Vance RE, Stallings CL, Virgin HW, Cox JS (2015) The cytosolic sensor cGAS detects Mycobacterium tuberculosis DNA to induce type I interferons and activate autophagy. Cell Host Microbe 17: 811–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wiens KE, Ernst JD (2016) The mechanism for type I interferon induction by Mycobacterium tuberculosis is bacterial strain‐dependent. PLoS Pathog 12: e1005809–e1005820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Abarca‐Rojano E, Rosas‐Medina P, Zamudio‐Cortéz P, Mondragón‐Flores R, Sánchez‐García FJ (2003) Mycobacterium tuberculosis virulence correlates with mitochondrial cytochrome c release in infected macrophages. Scand J Immunol 58: 419–427 [DOI] [PubMed] [Google Scholar]
- 55. Aarreberg LD, Esser‐Nobis K, Driscoll C, Shuvarikov A, Roby JA, Gale M (2019) Interleukin‐1β induces mtDNA release to activate innate immune signaling via cGAS‐STING. Mol Cell 74: 801–815.e806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Orzalli MH, Smith A, Jurado KA, Iwasaki A, Garlick JA, Kagan JC (2018) An antiviral branch of the IL‐1 signaling pathway restricts immune‐evasive virus replication. Mol Cell 71: 825–840.e826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mayer‐Barber KD, Andrade BB, Oland SD, Amaral EP, Barber DL, Gonzales J, Derrick SC, Shi R, Kumar NP, Wei W et al (2014) Host‐directed therapy of tuberculosis based on interleukin‐1 and type I interferon crosstalk. Nature 511: 99–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Copenhaver AM, Casson CN, Nguyen HT, Duda MM, Shin S (2015) IL‐1R signaling enables bystander cells to overcome bacterial blockade of host protein synthesis. Proc Natl Acad Sci USA 112: 7557–7562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bock FJ, Tait SWG (2019) Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol 17: 608 [DOI] [PubMed] [Google Scholar]
- 60. Ning X, Wang Y, Jing M, Sha M, Lv M, Gao P, Zhang R, Huang X, Feng J‐M, Jiang Z (2019) Apoptotic caspases suppress type I interferon production via the cleavage of cGAS, MAVS, and IRF3. Mol Cell 74: 19–31.e7 [DOI] [PubMed] [Google Scholar]
- 61. Riley JS, Quarato G, Cloix C, Lopez J, O'Prey J, Pearson M, Chapman J, Sesaki H, Carlin LM, Passos JF et al (2018) Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. EMBO J 37: e99238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. McArthur K, Whitehead LW, Heddleston JM, Li L, Padman BS, Oorschot V, Geoghegan ND, Chappaz S, Davidson S, San Chin H et al (2018) BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 359: eaao6047 [DOI] [PubMed] [Google Scholar]
- 63. Ader NR, Hoffmann PC, Ganeva I, Borgeaud AC, Wang C, Youle RJ, Kukulski W (2019) Molecular and topological reorganizations in mitochondrial architecture interplay during Bax‐mediated steps of apoptosis. Elife 8: 303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Giampazolias E, Zunino B, Dhayade S, Bock F, Cloix C, Cao K, Roca A, Lopez J, Ichim G, Proïcs E et al (2017) Mitochondrial permeabilization engages NF‐κB‐dependent anti‐tumour activity under caspase deficiency. Nat Cell Biol 19: 1116–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Abe T, Barber GN (2014) Cytosolic‐DNA‐mediated, STING‐dependent proinflammatory gene induction necessitates canonical NF‐κB activation through TBK1. J Virol 88: 5328–5341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. von Ahsen O, Renken C, Perkins G, Kluck RM, Bossy‐Wetzel E, Newmeyer DD (2000) Preservation of mitochondrial structure and function after Bid‐ or Bax‐mediated cytochrome c release. J Cell Biol 150: 1027–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Waterhouse NJ, Goldstein JC, von Ahsen O, Schuler M, Newmeyer DD, Green DR (2001) Cytochrome c maintains mitochondrial transmembrane potential and ATP generation after outer mitochondrial membrane permeabilization during the apoptotic process. J Cell Biol 153: 319–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bossy‐Wetzel E, Newmeyer DD, Green DR (1998) Mitochondrial cytochrome c release in apoptosis occurs upstream of DEVD‐specific caspase activation and independently of mitochondrial transmembrane depolarization. EMBO J 17: 37–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Huber HJ, Dussmann H, Kilbride SM, Rehm M, Prehn JHM (2011) Glucose metabolism determines resistance of cancer cells to bioenergetic crisis after cytochrome‐c release. Mol Syst Biol 7: 470–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Potts MB, Vaughn AE, McDonough H, Patterson C, Deshmukh M (2005) Reduced Apaf‐1 levels in cardiomyocytes engage strict regulation of apoptosis by endogenous XIAP. J Cell Biol 171: 925–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Chen Q, Boire A, Jin X, Valiente M, Er EE, López‐Soto A, Jacob LS, Patwa R, Shah H, Xu K et al (2016) Carcinoma–astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 533: 493–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Marcus A, Mao AJ, Lensink‐Vasan M, Wang L, Vance RE, Raulet DH (2018) Tumor‐derived cGAMP triggers a STING‐mediated interferon response in non‐tumor cells to activate the NK cell response. Immunity 49: 754–763.e754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Karikó K, Buckstein M, Ni H, Weissman D (2005) Suppression of RNA recognition by Toll‐like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity 23: 165–175 [DOI] [PubMed] [Google Scholar]
- 74. Dhir A, Dhir S, Borowski LS, Jimenez L, Teitell M, Rötig A, Crow YJ, Rice GI, Duffy D, Tamby C et al (2018) Mitochondrial double‐stranded RNA triggers antiviral signalling in humans. Nature 560: 238–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ichim G, Lopez J, Ahmed SU, Muthalagu N, Giampazolias E, Delgado ME, Haller M, Riley JS, Mason SM, Athineos D et al (2015) Limited mitochondrial permeabilization causes DNA damage and genomic instability in the absence of cell death. Mol Cell 57: 860–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Brokatzky D, Dörflinger B, Haimovici A, Weber A, Kirschnek S, Vier J, Metz A, Henschel J, Steinfeldt T, Gentle IE et al (2019) A non‐death function of the mitochondrial apoptosis apparatus in immunity. EMBO J 38: e100907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Patrushev M, Kasymov V, Patrusheva V, Ushakova T, Gogvadze V, Gaziev AI (2006) Release of mitochondrial DNA fragments from brain mitochondria of irradiated mice. Mitochondrion 6: 43–47 [DOI] [PubMed] [Google Scholar]
- 78. Patrushev M, Kasymov V, Patrusheva V, Ushakova T, Gogvadze V, Gaziev A (2004) Mitochondrial permeability transition triggers the release of mtDNA fragments. Cell Mol Life Sci 61: 3100–3103 [DOI] [PubMed] [Google Scholar]
- 79. Izzo V, Bravo‐San Pedro JM, Sica V, Kroemer G, Galluzzi L (2016) Mitochondrial permeability transition: new findings and persisting uncertainties. Trends Cell Biol 26: 655–667 [DOI] [PubMed] [Google Scholar]
- 80. Bernardi P (1999) Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol Rev 79: 1127–1155 [DOI] [PubMed] [Google Scholar]
- 81. Halestrap AP, McStay GP, Clarke SJ (2002) The permeability transition pore complex: another view. Biochimie 84: 153–166 [DOI] [PubMed] [Google Scholar]
- 82. García N, García JJ, Correa F, Chávez E (2005) The permeability transition pore as a pathway for the release of mitochondrial DNA. Life Sci 76: 2873–2880 [DOI] [PubMed] [Google Scholar]
- 83. García N, Chávez E (2007) Mitochondrial DNA fragments released through the permeability transition pore correspond to specific gene size. Life Sci 81: 1160–1166 [DOI] [PubMed] [Google Scholar]
- 84. Carroll EC, Jin L, Mori A, Muñoz‐Wolf N, Oleszycka E, Moran HBT, Mansouri S, McEntee CP, Lambe E, Agger EM et al (2016) The vaccine adjuvant chitosan promotes cellular immunity via DNA sensor cGAS‐STING‐dependent induction of type I interferons. Immunity 44: 597–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kim J, Gupta R, Blanco LP, Yang S, Shteinfer‐Kuzmine A, Wang K, Zhu J, Yoon HE, Wang X, Kerkhofs M et al (2019) VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus‐like disease. Science 366: 1531–1536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Lood C, Blanco LP, Purmalek MM, Carmona‐Rivera C, De Ravin SS, Smith CK, Malech HL, Ledbetter JA, Elkon KB, Kaplan MJ (2016) Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus‐like disease. Nat Med 22: 146–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Caielli S, Athale S, Domic B, Murat E, Chandra M, Banchereau R, Baisch J, Phelps K, Clayton S, Gong M et al (2016) Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J Exp Med 213: 697–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Crow YJ, Manel N (2015) Aicardi‐Goutières syndrome and the type I interferonopathies. Nat Rev Immunol 15: 429–440 [DOI] [PubMed] [Google Scholar]
- 89. Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, Black DN, van Bokhoven H, Brunner HG, Hamel BC et al (2006) Mutations in the gene encoding the 3′‐5′ DNA exonuclease TREX1 cause Aicardi‐Goutières syndrome at the AGS1 locus. Nature Genet 38: 917–920 [DOI] [PubMed] [Google Scholar]
- 90. Namjou B, Kothari PH, Kelly JA, Glenn SB, Ojwang JO, Adler A, Alarcón‐Riquelme ME, Gallant CJ, Boackle SA, Criswell LA et al (2011) Evaluation of the TREX1 gene in a large multi‐ancestral lupus cohort. Genes Immun 12: 270–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Lee‐Kirsch MA, Gong M, Chowdhury D, Senenko L, Engel K, Lee Y‐A, de Silva U, Bailey SL, Witte T, Vyse TJ et al (2007) Mutations in the gene encoding the 3′‐5′ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat Genet 39: 1065–1067 [DOI] [PubMed] [Google Scholar]
- 92. Stetson DB, Ko JS, Heidmann T, Medzhitov R (2008) Trex1 prevents cell‐intrinsic initiation of autoimmunity. Cell 134: 587–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Yan N, Regalado‐Magdos AD, Stiggelbout B, Lee‐Kirsch MA, Lieberman J (2010) The cytosolic exonuclease TREX1 inhibits the innate immune response to human immunodeficiency virus type 1. Nat Immunol 11: 1005–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Gall A, Treuting P, Elkon KB, Loo Y‐M, Gale M Jr, Barber GN, Stetson DB (2012) Autoimmunity initiates in nonhematopoietic cells and progresses via lymphocytes in an interferon‐dependent autoimmune disease. Immunity 36: 120–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Ablasser A, Hemmerling I, Schmid‐Burgk JL, Behrendt R, Roers A, Hornung V (2014) TREX1 deficiency triggers cell‐autonomous immunity in a cGAS‐dependent manner. J Immunol 192: 5993–5997 [DOI] [PubMed] [Google Scholar]
- 96. Ahn J, Ruiz P, Barber GN (2014) Intrinsic Self‐DNA triggers inflammatory disease dependent on STING. J Immunol 193: 4634–4642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Gray EE, Treuting PM, Woodward JJ, Stetson DB (2015) Cutting edge: cGAS is required for lethal autoimmune disease in the Trex1‐deficient mouse model of Aicardi‐Goutières syndrome. J Immunol 195: 1939–1943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Gao D, Li T, Li X‐D, Chen X, Li Q‐Z, Wight‐Carter M, Chen ZJ (2015) Activation of cyclic GMP‐AMP synthase by self‐DNA causes autoimmune diseases. Proc Natl Acad Sci USA 112: E5699–E5705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Crow YJ, Leitch A, Hayward BE, Garner A, Parmar R, Griffith E, Ali M, Semple C, Aicardi J, Babul‐Hirji R et al (2006) Mutations in genes encoding ribonuclease H2 subunits cause Aicardi‐Goutières syndrome and mimic congenital viral brain infection. Nat Genet 38: 910–916 [DOI] [PubMed] [Google Scholar]
- 100. Rice GI, Bond J, Asipu A, Brunette RL, Manfield IW, Carr IM, Fuller JC, Jackson RM, Lamb T, Briggs TA et al (2009) Mutations involved in Aicardi‐Goutières syndrome implicate SAMHD1 as regulator of the innate immune response. Nat Neurosci 41: 829–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Crow YJ, Chase DS, Lowenstein Schmidt J, Szynkiewicz M, Forte GMA, Gornall HL, Oojageer A, Anderson B, Pizzino A, Helman G et al (2015) Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am J Med Genet A 167A: 296–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Günther C, Kind B, Reijns MAM, Berndt N, Martinez‐Bueno M, Wolf C, Tüngler V, Chara O, Lee Y‐A, Hübner N et al (2015) Defective removal of ribonucleotides from DNA promotes systemic autoimmunity. J Clin Invest 125: 413–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Montealegre Sanchez GA, Tenbrock K, Wittkowski H, Jones OY, Kuehn HS et al (2014) Activated STING in a vascular and pulmonary syndrome. N Engl J Med 371: 507–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Jeremiah N, Neven B, Gentili M, Callebaut I, Maschalidi S, Stolzenberg M‐C, Goudin N, Frémond M‐L, Nitschke P, Molina TJ et al (2014) Inherited STING‐activating mutation underlies a familial inflammatory syndrome with lupus‐like manifestations. J Clin Invest 124: 5516–5520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Rodero MP, Tesser A, Bartok E, Rice GI, Mina Della E, Depp M, Beitz B, Bondet V, Cagnard N, Duffy D et al (2017) Type I interferon‐mediated autoinflammation due to DNase II deficiency. Nat Commun 8: 2176–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Kawane K, Ohtani M, Miwa K, Kizawa T, Kanbara Y, Yoshioka Y, Yoshikawa H, Nagata S (2006) Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature 443: 998–1002 [DOI] [PubMed] [Google Scholar]
- 107. Ahn J, Gutman D, Saijo S, Barber GN (2012) STING manifests self DNA‐dependent inflammatory disease. Proc Natl Acad Sci USA 109: 19386–19391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Baum R, Sharma S, Carpenter S, Li Q‐Z, Busto P, Fitzgerald KA, Marshak‐Rothstein A, Gravallese EM (2015) Cutting edge: AIM2 and endosomal TLRs differentially regulate arthritis and autoantibody production in DNase II‐deficient mice. J Immunol 194: 873–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Jakobs C, Perner S, Hornung V (2015) AIM2 drives joint inflammation in a Self‐DNA triggered model of chronic polyarthritis. PLoS ONE 10: e0131702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. King KR, Aguirre AD, Ye Y‐X, Sun Y, Roh JD, Ng RP, Kohler RH, Arlauckas SP, Iwamoto Y, Savol A et al (2017) IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat Med 23: 1481–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Cao DJ, Schiattarella GG, Villalobos E, Jiang N, May HI, Li T, Chen ZJ, Gillette TG, Hill JA (2018) Cytosolic DNA sensing promotes macrophage transformation and governs myocardial ischemic injury. Circulation 137: 2613–2634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Wang L, Xie L, Zhang Q, Cai X, Tang Y, Wang L, Hang T, Liu J, Gong J (2015) Plasma nuclear and mitochondrial DNA levels in acute myocardial infarction patients. Coron Artery Dis 26: 296–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Bliksøen M, Mariero LH, Ohm IK, Haugen F, Yndestad A, Solheim S, Seljeflot I, Ranheim T, Andersen GØ, Aukrust P et al (2012) Increased circulating mitochondrial DNA after myocardial infarction. Int J Cardiol 158: 132–134 [DOI] [PubMed] [Google Scholar]
- 114. Liu X‐M, Du L‐L (2015) A selective autophagy pathway takes an unconventional route. Autophagy 11: 2381–2382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Vincent J, Adura C, Gao P, Luz A, Lama L, Asano Y, Okamoto R, Imaeda T, Aida J, Rothamel K et al (2017) Small molecule inhibition of cGAS reduces interferon expression in primary macrophages from autoimmune mice. Nat Commun 8: 750–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Lama L, Adura C, Xie W, Tomita D, Kamei T, Kuryavyi V, Gogakos T, Steinberg JI, Miller M, Ramos‐Espiritu L et al (2019) Development of human cGAS‐specific small‐molecule inhibitors for repression of dsDNA‐triggered interferon expression. Nat Commun 10: 2261–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Haag SM, Gulen MF, Reymond L, Gibelin A, Abrami L, Decout A, Heymann M, van der Goot FG, Turcatti G, Behrendt R et al (2018) Targeting STING with covalent small‐molecule inhibitors. Nature 559: 269–273 [DOI] [PubMed] [Google Scholar]
- 118. Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, Schreiber RD (2001) IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 410: 1107–1111 [DOI] [PubMed] [Google Scholar]
- 119. Woo S‐R, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MYK, Duggan R, Wang Y, Barber GN, Fitzgerald KA et al (2014) STING‐dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41: 830–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, Li X‐D, Mauceri H, Beckett M, Darga T et al (2014) STING‐dependent cytosolic DNA sensing promotes radiation‐induced type I interferon‐dependent antitumor immunity in immunogenic tumors. Immunity 41: 843–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Klarquist J, Hennies CM, Lehn MA, Reboulet RA, Feau S, Janssen EM (2014) STING‐mediated DNA sensing promotes antitumor and autoimmune responses to dying cells. J Immunol 193: 6124–6134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Xu MM, Pu Y, Han D, Shi Y, Cao X, Liang H, Chen X, Li X‐D, Deng L, Chen ZJ et al (2017) Dendritic cells but not macrophages sense tumor mitochondrial DNA for cross‐priming through signal regulatory protein α signaling. Immunity 47: 363–373.e365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Ahn J, Xia T, Rabasa Capote A, Betancourt D, Barber GN (2018) Extrinsic phagocyte‐dependent STING signaling dictates the immunogenicity of dying cells. Cancer Cell 33: 862–873.e865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Torralba D, Baixauli F, Villarroya‐Beltrí C, Fernández‐Delgado I, Latorre‐Pellicer A, Acín‐Pérez R, Martín‐Cófreces NB, Jaso‐Tamame ÁL, Iborra S, Jorge I et al (2018) Priming of dendritic cells by DNA‐containing extracellular vesicles from activated T cells through antigen‐driven contacts. Nat Commun 9: 2658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Kitai Y, Kawasaki T, Sueyoshi T, Kobiyama K, Ishii KJ, Zou J, Akira S, Matsuda T, Kawai T (2017) DNA‐containing exosomes derived from cancer cells treated with topotecan activate a STING‐dependent pathway and reinforce antitumor immunity. J Immunol 198: 1649–1659 [DOI] [PubMed] [Google Scholar]
- 126. Berger G, Marloye M, Lawler SE (2019) Pharmacological modulation of the STING pathway for cancer immunotherapy. Trends Mol Med 25: 412–427 [DOI] [PubMed] [Google Scholar]
- 127. Ramanjulu JM, Pesiridis GS, Yang J, Concha N, Singhaus R, Zhang S‐Y, Tran J‐L, Moore P, Lehmann S, Eberl HC et al (2018) Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature 564: 439–443 [DOI] [PubMed] [Google Scholar]
- 128. Riley JS, Tait SWG (2016) Mechanisms of mitophagy: putting the powerhouse into the doghouse. Biol Chem 397: 617–635 [DOI] [PubMed] [Google Scholar]
- 129. Valente EM, Salvi S, Ialongo T, Marongiu R, Elia AE, Caputo V, Romito L, Albanese A, Dallapiccola B, Bentivoglio AR (2004) PINK1 mutations are associated with sporadic early‐onset parkinsonism. Ann Neurol 56: 336–341 [DOI] [PubMed] [Google Scholar]
- 130. Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392: 605–608 [DOI] [PubMed] [Google Scholar]
- 131. Lücking CB, Dürr A, Bonifati V, Vaughan J, De Michele G, Gasser T, Harhangi BS, Meco G, Denèfle P, Wood NW et al (2000) Association between early‐onset Parkinson's disease and mutations in the parkin gene. N Engl J Med 342: 1560–1567 [DOI] [PubMed] [Google Scholar]
- 132. Abbas N, Lücking CB, Ricard S, Dürr A, Bonifati V, De Michele G, Bouley S, Vaughan JR, Gasser T, Marconi R et al (1999) A wide variety of mutations in the parkin gene are responsible for autosomal recessive parkinsonism in Europe. French Parkinson's Disease Genetics Study Group and the European Consortium on Genetic Susceptibility in Parkinson's Disease. Hum Mol Genet 8: 567–574 [DOI] [PubMed] [Google Scholar]
- 133. West A, Periquet M, Lincoln S, Lücking CB, Nicholl D, Bonifati V, Rawal N, Gasser T, Lohmann E, Deleuze J‐F et al (2002) Complex relationship between Parkin mutations and Parkinson disease. Am J Med Genet 114: 584–591 [DOI] [PubMed] [Google Scholar]
- 134. Gladkova C, Maslen SL, Skehel JM, Komander D (2018) Mechanism of parkin activation by PINK1. Nature 559: 410–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ (2015) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524: 309–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Wauer T, Simicek M, Schubert A, Komander D (2015) Mechanism of phospho‐ubiquitin‐induced PARKIN activation. Nature 524: 370–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Vivekanantham S, Shah S, Dewji R, Dewji A, Khatri C, Ologunde R (2015) Neuroinflammation in Parkinson's disease: role in neurodegeneration and tissue repair. Int J Neurosci 125: 717–725 [DOI] [PubMed] [Google Scholar]
- 138. Dobbs RJ, Charlett A, Purkiss AG, Dobbs SM, Weller C, Peterson DW (1999) Association of circulating TNF‐alpha and IL‐6 with ageing and parkinsonism. Acta Neurol Scand 100: 34–41 [DOI] [PubMed] [Google Scholar]
- 139. Mogi M, Harada M, Riederer P, Narabayashi H, Fujita K, Nagatsu T (1994) Tumor necrosis factor‐alpha (TNF‐alpha) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci Lett 165: 208–210 [DOI] [PubMed] [Google Scholar]
- 140. Goldberg MS, Fleming SM, Palacino JJ, Cepeda C, Lam HA, Bhatnagar A, Meloni EG, Wu N, Ackerson LC, Klapstein GJ et al (2003) Parkin‐deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem 278: 43628–43635 [DOI] [PubMed] [Google Scholar]
- 141. Perez FA, Palmiter RD (2005) Parkin‐deficient mice are not a robust model of parkinsonism. Proc Natl Acad Sci USA 102: 2174–2179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Kitada T, Pisani A, Porter DR, Yamaguchi H, Tscherter A, Martella G, Bonsi P, Zhang C, Pothos EN, Shen J (2007) Impaired dopamine release and synaptic plasticity in the striatum of PINK1‐deficient mice. Proc Natl Acad Sci USA 104: 11441–11446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Sliter DA, Martinez J, Hao L, Chen X, Sun N, Fischer TD, Burman JL, Li Y, Zhang Z, Narendra DP et al (2018) Parkin and PINK1 mitigate STING‐induced inflammation. Nature 561: 258–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Broz P, Dixit VM (2016) Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol 16: 407–420 [DOI] [PubMed] [Google Scholar]
- 145. Nakahira K, Haspel JA, Rathinam VAK, Lee S‐J, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP et al (2011) Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12: 222–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM et al (2012) Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36: 401–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Zhou R, Yazdi AS, Menu P, Tschopp J (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469: 221–225 [DOI] [PubMed] [Google Scholar]
- 148. Zhong Z, Umemura A, Sanchez‐Lopez E, Liang S, Shalapour S, Wong J, He F, Boassa D, Perkins G, Ali SR et al (2016) NF‐κB restricts inflammasome activation via elimination of damaged mitochondria. Cell 164: 896–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Zhong Z, Liang S, Sanchez‐Lopez E, He F, Shalapour S, Lin X‐J, Wong J, Ding S, Seki E, Schnabl B et al (2018) New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 560: 198–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Chen J, Chen ZJ (2018) PtdIns4P on dispersed trans‐Golgi network mediates NLRP3 inflammasome activation. Nature 564: 71–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Yu J, Nagasu H, Murakami T, Hoang H, Broderick L, Hoffman HM, Horng T (2014) Inflammasome activation leads to Caspase‐1‐dependent mitochondrial damage and block of mitophagy. Proc Natl Acad Sci USA 111: 15514–15519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Bronner DN, Abuaita BH, Chen X, Fitzgerald KA, Nuñez G, He Y, Yin X‐M, O'Riordan MXD (2015) Endoplasmic reticulum stress activates the inflammasome via NLRP3‐ and caspase‐2‐driven mitochondrial damage. Immunity 43: 451–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A (2004) Neutrophil extracellular traps kill bacteria. Science 303: 1532–1535 [DOI] [PubMed] [Google Scholar]
- 154. Wang H, Li T, Chen S, Gu Y, Ye S (2015) Neutrophil extracellular trap mitochondrial DNA and its autoantibody in systemic lupus erythematosus and a proof‐of‐concept trial of metformin. Arthritis Rheumatol 67: 3190–3200 [DOI] [PubMed] [Google Scholar]
- 155. McIlroy DJ, Jarnicki AG, Au GG, Lott N, Smith DW, Hansbro PM, Balogh ZJ (2014) Mitochondrial DNA neutrophil extracellular traps are formed after trauma and subsequent surgery. J Crit Care 29: 1133.e1–.e5 [DOI] [PubMed] [Google Scholar]
- 156. Becker Y, Loignon R‐C, Julien A‐S, Marcoux G, Allaeys I, Lévesque T, Rollet‐Labelle E, Benk‐Fortin H, Cloutier N, Melki I et al (2019) Anti‐mitochondrial autoantibodies in systemic lupus erythematosus and their association with disease manifestations. Sci Rep 9: 4530–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Gkirtzimanaki K, Kabrani E, Nikoleri D, Polyzos A, Blanas A, Sidiropoulos P, Makrigiannakis A, Bertsias G, Boumpas DT, Verginis P (2018) IFNα impairs autophagic degradation of mtDNA promoting autoreactivity of SLE monocytes in a STING‐dependent fashion. Cell Rep 25: 921–933.e925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Yousefi S, Gold JA, Andina N, Lee JJ, Kelly AM, Kozlowski E, Schmid I, Straumann A, Reichenbach J, Gleich GJ et al (2008) Catapult‐like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat Med 14: 949–953 [DOI] [PubMed] [Google Scholar]
- 159. Ingelsson B, Söderberg D, Strid T, Söderberg A, Bergh A‐C, Loitto V, Lotfi K, Segelmark M, Spyrou G, Rosén A (2018) Lymphocytes eject interferogenic mitochondrial DNA webs in response to CpG and non‐CpG oligodeoxynucleotides of class C. Proc Natl Acad Sci USA 115: E478–E487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon H‐U (2009) Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ 16: 1438–1444 [DOI] [PubMed] [Google Scholar]
- 161. Amini P, Stojkov D, Felser A, Jackson CB, Courage C, Schaller A, Gelman L, Soriano ME, Nuoffer J‐M, Scorrano L et al (2018) Neutrophil extracellular trap formation requires OPA1‐dependent glycolytic ATP production. Nat Commun 9: 2958–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Takeshita F, Leifer CA, Gursel I, Ishii KJ, Takeshita S, Gursel M, Klinman DM (2001) Cutting edge: role of Toll‐like receptor 9 in CpG DNA‐induced activation of human cells. J Immunol 167: 3555–3558 [DOI] [PubMed] [Google Scholar]
- 163. Ahmad‐Nejad P, Häcker H, Rutz M, Bauer S, Vabulas RM, Wagner H (2002) Bacterial CpG‐DNA and lipopolysaccharides activate Toll‐like receptors at distinct cellular compartments. Eur J Immunol 32: 1958–1968 [DOI] [PubMed] [Google Scholar]
- 164. Leifer CA, Kennedy MN, Mazzoni A, Lee C, Kruhlak MJ, Segal DM (2004) TLR9 is localized in the endoplasmic reticulum prior to stimulation. J Immunol 173: 1179–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Latz E, Schoenemeyer A, Visintin A, Fitzgerald KA, Monks BG, Knetter CF, Lien E, Nilsen NJ, Espevik T, Golenbock DT (2004) TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat Immunol 5: 190–198 [DOI] [PubMed] [Google Scholar]
- 166. Cardon LR, Burge C, Clayton DA, Karlin S (1994) Pervasive CpG suppression in animal mitochondrial genomes. Proc Natl Acad Sci USA 91: 3799–3803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Pollack Y, Kasir J, Shemer R, Metzger S, Szyf M (1984) Methylation pattern of mouse mitochondrial DNA. Nucleic Acids Res 12: 4811–4824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ (2010) Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464: 104–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Zhang Q, Itagaki K, Hauser CJ (2010) Mitochondrial DNA is released by shock and activates neutrophils via p38 map kinase. Shock 34: 55–59 [DOI] [PubMed] [Google Scholar]
- 170. Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M et al (2007) The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med 13: 619–624 [DOI] [PubMed] [Google Scholar]
- 171. Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, Oyabu J, Murakawa T, Nakayama H, Nishida K et al (2012) Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 485: 251–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Zhang Z, Meng P, Han Y, Shen C, Li B, Hakim MA, Zhang X, Lu Q, Rong M, Lai R (2015) Mitochondrial DNA‐LL‐37 complex promotes atherosclerosis by escaping from autophagic recognition. Immunity 43: 1137–1147 [DOI] [PubMed] [Google Scholar]
- 173. Scaffidi P, Misteli T, Bianchi ME (2002) Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418: 191–195 [DOI] [PubMed] [Google Scholar]
- 174. Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L et al (1999) HMG‐1 as a late mediator of endotoxin lethality in mice. Science 285: 248–251 [DOI] [PubMed] [Google Scholar]
- 175. Tian J, Avalos AM, Mao S‐Y, Chen B, Senthil K, Wu H, Parroche P, Drabic S, Golenbock D, Sirois C et al (2007) Toll‐like receptor 9‐dependent activation by DNA‐containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol 8: 487–496 [DOI] [PubMed] [Google Scholar]
- 176. Julian MW, Shao G, Bao S, Knoell DL, Papenfuss TL, VanGundy ZC, Crouser ED (2012) Mitochondrial transcription factor A serves as a danger signal by augmenting plasmacytoid dendritic cell responses to DNA. J Immunol 189: 433–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Julian MW, Shao G, VanGundy ZC, Papenfuss TL, Crouser ED (2013) Mitochondrial transcription factor A, an endogenous danger signal, promotes TNFα release via RAGE‐ and TLR9‐responsive plasmacytoid dendritic cells. PLoS ONE 8: e72354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Liu X, He Y, Li F, Huang Q, Kato TA, Hall RP, Li C‐Y (2015) Caspase‐3 promotes genetic instability and carcinogenesis. Mol Cell 58: 284–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Imaeda AB, Watanabe A, Sohail MA, Mahmood S, Mohamadnejad M, Sutterwala FS, Flavell RA, Mehal WZ (2009) Acetaminophen‐induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J Clin Invest 119: 305–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Watanabe A, Hashmi A, Gomes DA, Town T, Badou A, Flavell RA, Mehal WZ (2007) Apoptotic hepatocyte DNA inhibits hepatic stellate cell chemotaxis via toll‐like receptor 9. Hepatology 46: 1509–1518 [DOI] [PubMed] [Google Scholar]
- 181. Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoyama T, Ohnishi H, Olefsky JM, Brenner DA, Seki E (2010) Toll‐like receptor 9 promotes steatohepatitis by induction of interleukin‐1beta in mice. Gastroenterology 139: 323–34.e327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Koliaki C, Szendroedi J, Kaul K, Jelenik T, Nowotny P, Jankowiak F, Herder C, Carstensen M, Krausch M, Knoefel WT et al (2015) Adaptation of hepatic mitochondrial function in humans with non‐alcoholic fatty liver is lost in steatohepatitis. Cell Metab 21: 739–746 [DOI] [PubMed] [Google Scholar]
- 183. Garcia‐Martinez I, Santoro N, Chen Y, Hoque R, Ouyang X, Caprio S, Shlomchik MJ, Coffman RL, Candia A, Mehal WZ (2016) Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J Clin Invest 126: 859–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Feldstein AE, Canbay A, Angulo P, Taniai M, Burgart LJ, Lindor KD, Gores GJ (2003) Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology 125: 437–443 [DOI] [PubMed] [Google Scholar]
- 185. Afonso MB, Rodrigues PM, Carvalho T, Caridade M, Borralho P, Cortez‐Pinto H, Castro RE, Rodrigues CMP (2015) Necroptosis is a key pathogenic event in human and experimental murine models of non‐alcoholic steatohepatitis. Clin Sci 129: 721–739 [DOI] [PubMed] [Google Scholar]
- 186. Gautheron J, Vucur M, Reisinger F, Cardenas DV, Roderburg C, Koppe C, Kreggenwinkel K, Schneider AT, Bartneck M, Neumann UP et al (2014) A positive feedback loop between RIP3 and JNK controls non‐alcoholic steatohepatitis. EMBO Mol Med 6: 1062–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Saito Y, Hikita H, Nozaki Y, Kai Y, Makino Y, Nakabori T, Tanaka S, Yamada R, Shigekawa M, Kodama T et al (2019) DNase II activated by the mitochondrial apoptotic pathway regulates RIP1‐dependent non‐apoptotic hepatocyte death via the TLR9/IFN‐β signaling pathway. Cell Death Differ 26: 470–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Amati‐Bonneau P, Valentino ML, Reynier P, Gallardo ME, Bornstein B, Boissière A, Campos Y, Rivera H, la de Aleja JG, Carroccia R et al (2008) OPA1 mutations induce mitochondrial DNA instability and optic atrophy “plus” phenotypes. Brain 131: 338–351 [DOI] [PubMed] [Google Scholar]
- 189. Hudson G, Amati‐Bonneau P, Blakely EL, Stewart JD, He L, Schaefer AM, Griffiths PG, Ahlqvist K, Suomalainen A, Reynier P et al (2008) Mutation of OPA1 causes dominant optic atrophy with external ophthalmoplegia, ataxia, deafness and multiple mitochondrial DNA deletions: a novel disorder of mtDNA maintenance. Brain 131: 329–337 [DOI] [PubMed] [Google Scholar]
- 190. Kim JY, Hwang J‐M, Ko HS, Seong M‐W, Park BJ, Park SS (2005) Mitochondrial DNA content is decreased in autosomal dominant optic atrophy. Neurology 64: 966–972 [DOI] [PubMed] [Google Scholar]
- 191. Yu‐Wai‐Man P, Sitarz KS, Samuels DC, Griffiths PG, Reeve AK, Bindoff LA, Horvath R, Chinnery PF (2010) OPA1 mutations cause cytochrome c oxidase deficiency due to loss of wild‐type mtDNA molecules. Hum Mol Genet 19: 3043–3052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Tezze C, Romanello V, Desbats MA, Fadini GP, Albiero M, Favaro G, Ciciliot S, Soriano ME, Morbidoni V, Cerqua C et al (2017) Age‐associated loss of OPA1 in muscle impacts muscle mass, metabolic homeostasis, systemic inflammation, and epithelial senescence. Cell Metab 25: 1374–1389.e1376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Pereira RO, Tadinada SM, Zasadny FM, Oliveira KJ, Pires KMP, Olvera A, Jeffers J, Souvenir R, Mcglauflin R, Seei A et al (2017) OPA1 deficiency promotes secretion of FGF21 from muscle that prevents obesity and insulin resistance. EMBO J 36: 2126–2145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Rodríguez Nuevo A, Díaz Ramos A, Noguera E, Díaz Sáez F, Duran X, Muñoz JP, Romero M, Plana N, Sebastián D, Tezze C et al (2018) Mitochondrial DNA and TLR9 drive muscle inflammation upon Opa1 deficiency. EMBO J 37: e96553–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. King MP, Attardi G (1989) Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science 246: 500–503 [DOI] [PubMed] [Google Scholar]
- 196. Hayakawa K, Esposito E, Wang X, Terasaki Y, Liu Y, Xing C, Ji X, Lo EH (2016) Transfer of mitochondria from astrocytes to neurons after stroke. Nature 535: 551–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Tan AS, Baty JW, Dong L‐F, Bezawork‐Geleta A, Endaya B, Goodwin J, Bajzikova M, Kovarova J, Peterka M, Yan B et al (2015) Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab 21: 81–94 [DOI] [PubMed] [Google Scholar]
- 198. Rustom A, Saffrich R, Markovic I, Walther P, Gerdes H‐H (2004) Nanotubular highways for intercellular organelle transport. Science 303: 1007–1010 [DOI] [PubMed] [Google Scholar]
- 199. Pasquier J, Guerrouahen BS, Thawadi Al H, Ghiabi P, Maleki M, Abu‐Kaoud N, Jacob A, Mirshahi M, Galas L, Rafii S et al (2013) Preferential transfer of mitochondria from endothelial to cancer cells through tunneling nanotubes modulates chemoresistance. J Transl Med 11: 94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Wang X, Gerdes H‐H (2015) Transfer of mitochondria via tunneling nanotubes rescues apoptotic PC12 cells. Cell Death Differ 22: 1181–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Sansone P, Savini C, Kurelac I, Chang Q, Amato LB, Strillacci A, Stepanova A, Iommarini L, Mastroleo C, Daly L et al (2017) Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy‐resistant breast cancer. Proc Natl Acad Sci USA 114: E9066–E9075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Dong L‐F, Kovarova J, Bajzikova M, Bezawork‐Geleta A, Svec D, Endaya B, Sachaphibulkij K, Coelho AR, Sebkova N, Ruzickova A et al (2017) Horizontal transfer of whole mitochondria restores tumorigenic potential in mitochondrial DNA‐deficient cancer cells. Elife 6: 529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Islam MN, Das SR, Emin MT, Wei M, Sun L, Westphalen K, Rowlands DJ, Quadri SK, Bhattacharya S, Bhattacharya J (2012) Mitochondrial transfer from bone‐marrow‐derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat Med 18: 759–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Ishikawa K, Toyama‐Sorimachi N, Nakada K, Morimoto M, Imanishi H, Yoshizaki M, Sasawatari S, Niikura M, Takenaga K, Yonekawa H et al (2010) The innate immune system in host mice targets cells with allogenic mitochondrial DNA. J Exp Med 207: 2297–2305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Schäfer ST, Franken L, Adamzik M, Schumak B, Scherag A, Engler A, Schönborn N, Walden J, Koch S, Baba HA et al (2016) Mitochondrial DNA: an endogenous trigger for immune paralysis. Anesthesiology 124: 923–933 [DOI] [PubMed] [Google Scholar]
- 206. Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A, Shirakawa K, Lim HW, Davis SS, Ramanathan A et al (2016) Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab 23: 303–314 [DOI] [PMC free article] [PubMed] [Google Scholar]