

Abstract
The long noncoding RNA GUARDIN functions to protect genome stability. Inhibiting GUARDIN expression can alter cell fate decisions toward senescence or apoptosis, but the underlying molecular signals are unknown. Here, we show that GUARDIN is an essential component of a transcriptional repressor complex involving LRP130 and PGC1α. GUARDIN acts as a scaffold to stabilize LRP130/PGC1α heterodimers and their occupancy at the FOXO4 promotor. Destabilizing this complex by silencing of GUARDIN, LRP130, or PGC1α leads to increased expression of FOXO4 and upregulation of its target gene p21, thereby driving cells into senescence. We also found that GUARDIN expression was induced by rapamycin, an agent that suppresses cell senescence. FOS‐like antigen 2 (FOSL2) acts as a transcriptional repressor of GUARDIN, and lower FOSL2 levels in response to rapamycin correlate with increased levels of GUARDIN. Together, these results demonstrate that GUARDIN inhibits p21‐dependent senescence through a LRP130‐PGC1α‐FOXO4 signaling axis, and moreover, GUARDIN contributes to the anti‐aging activities of rapamycin.
Keywords: cellular senescence, GUARDIN, lncRNAs, LRP130‐PGC1α, p21
Subject Categories: Molecular Biology of Disease, RNA Biology
LncRNA GUARDIN protects genomic integrity and restrains cell entry into senescence. GUARDIN inhibits senescence via suppression of p21 independently of its role in genome maintenance.
Introduction
Senescence is an irreversible state of cellular dormancy driven by various mechanisms such as replicative exhaustion resulting from telomere shortening or uncapping, oncogene activation, and genotoxic, nutrient, and oxidative stress 1. These mechanisms commonly cumulate in the DNA damage response (DDR) leading to activation of the tumor suppressor p53 and the expression of cyclin‐dependent kinase (CDK) inhibitors such as p21 and p16 that execute the senescent response. Although cellular senescence was initially regarded as a passive cell‐autonomous anti‐proliferation program echoing normal cellular aging 2, 3, it is increasingly appreciated that senescence plays an important role in many other physiological and pathological processes including tumor suppression, neurodegeneration, and tissue remodeling 4, 5. Moreover, the senescence‐associated secretory phenotype (SASP) resulting from changes in the secretome of senescent cells contributes to regulating inflammation, tissue microenvironment, and age‐related disorders 6, 7, 8.
As an important regulatory mechanism of cellular senescence that integrates a variety of extracellular and intracellular signals 1, the mechanistic target of rapamycin (mTOR; as known as mammalian target of rapamycin) plays an important role in regulating longevity 6. Inhibition of mTOR signaling by genetic or pharmacological approaches extends lifespan of various model organisms and mice with different genetic backgrounds 7, 8, 9, 10. Indeed, treatment with the mTOR inhibitor rapamycin or its analogs (rapalogs) reduces cellular senescence 11, 12, 13, 14. Nevertheless, the molecular mechanisms involved in rapamycin‐mediated inhibition of senescence are not well understood.
An increasing number of noncoding RNAs have been found to be involved in regulation of cellular senescence 15. For example, the long noncoding RNA MIR31HG regulates the expression of p16 to modulate oncogene‐induced senescence 16, whereas HOTAIR activates cellular senescence through p53‐p21 signaling of the DDR 17. Moreover, the lncRNA OVAAL blocks cellular senescence through regulating the expression of the CDK inhibitor p27 18. We have also found that the lncRNA GUARDIN plays an essential role in maintaining genomic stability 19. Silencing of GUARDIN was shown to induce cellular senescence although the mechanism responsible remains undefined.
In this report, we demonstrate that GUARDIN serves to facilitate assembly of the complex between LRP130/PGC1α that acts as repressor complex at the FOXO4 promoter. Silencing of GUARDIN disrupts this complex leading to FOXO4‐dependent upregulation of p21, thereby driving cell entry into senescence. On the other hand, GUARDIN expression is upregulated by rapamycin treatment and this results from modulation of FOS‐like antigen 2 (FOSL2) levels. FOSL2 normally transcriptionally represses GUARDIN but downregulation of FOSL2 by rapamycin releases this inhibition to promote GUARDIN expression. Thus, GUARDIN inhibits p21‐dependent senescence induction through a signaling axis involving LRP130‐PGC1α‐FOXO4 moreover plays a functional role in the antagonistic actions of rapamycin on senescence.
Results
GUARDIN inhibits cellular senescence through suppressing transcriptional activation of p21
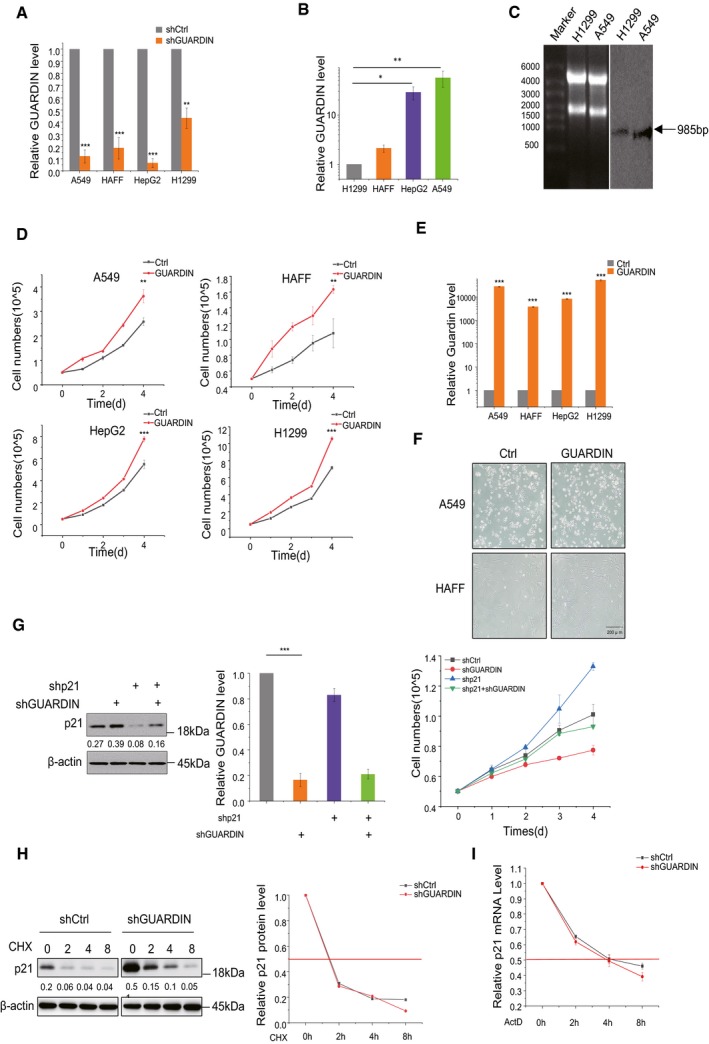
To validate the notion that GUARDIN protects cells from cellular senescence 19, we knocked down GUARDIN using shRNA in normal human adult foreskin fibroblasts (HAFF), non‐small‐cell lung carcinoma cells (A549 and H1299), and hepatocellular carcinoma HepG2 cells, respectively (Fig EV1A). We observed that shRNA silencing of GUARDIN induced senescence‐associated‐galactosidase (SA‐gal) activity (Fig 1A), the formation of senescence‐associated heterochromatin foci (SAHF) as shown by increased levels of H3K9me3 (Fig 1B), and enhanced the senescence‐associated secretory phenotype (SASP) as represented by elevated extracellular levels of IL‐6 and IL‐8 in A549 cells (Fig 1C) 20. Knockdown efficiencies varied from 50 to 90%, possibly due to the varied endogenous expression profiles of GUARDIN observed among the different cell lines (Fig EV1A and B). Alternatively, overexpression of GUARDIN accelerated cell proliferation and did not result in cellular senescence (Fig EV1D–F) consistent with prior reports 19. Northern blotting analysis of GUARDIN in H1299 and A549 cells showed a single band of 985nt (Fig EV1C), consistent with the three exon transcript reported previously 19. Thus, loss of GUARDIN induces senescence while its overexpression promotes proliferation in cell lines from different sources.
Figure EV1. Characterization and experimental manipulation of GUARDIN and the effect on p21 expression.
-
AshRNA knockdown efficiency. GUARDIN levels in A549, HAFF, HepG2, and H1299 cells were measured using qPCR.
-
BComparative levels of GUARDIN in A549, HAFF, HepG2, and H1299 cells measured using qPCR. All data normalized to H1299 levels.
-
CNorthern blotting against GUARDIN using total RNA from H1299 and A549 cells. 28S and 18S RNA loading controls (left) and GUARDIN (right) resolving as a single band.
-
DOverexpression of GUARDIN promotes cell proliferation in vitro. Cell numbers were counted in A549, HAFF, HepG2, and H1299 cells with and without GUARDIN overexpression for 4 days (d).
-
EOverexpression efficiencies. qPCR analysis of GUARDIN level in A549, HAFF, HepG2, and H1299 cells after 24‐h transduction with GUARDIN.
-
FSenescence‐associated β‐galactosidase (SA‐β‐gal) staining in A549, HAFF cells after 48‐h transduction with either negative control lentiviruses (Ctrl) or GUARDIN sense.
-
GCo‐silencing of GUARDIN and/or p21. Western blotting for p21/actin (left), qPCR assays for GUARDIN (middle), and cell proliferation assays (right) in A549 cells after 24‐h transduction with shCtrl or shGUARDIN.
-
H, ISilencing of GUARDIN does not affect p21 protein or mRNA stability. (H) A549 cells were treated with 50 μM of cycloheximide (CHX) for the indicated times before measuring p21/actin by Western blotting (left). Densitometric analysis (right) of relative p21 levels. (I) p21 mRNA levels were measured by qPCR in A549 cells treated with 5 μg/ml of actinomycin D (actD) for the indicated time points.
Figure 1. GUARDIN inhibits cellular senescence through suppression of p21.
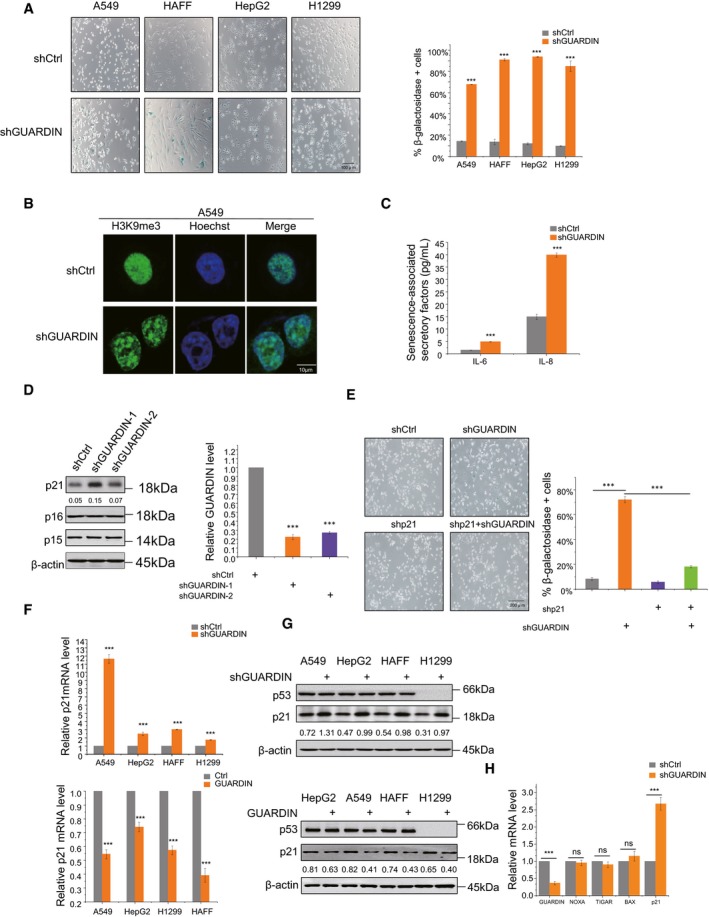
- Senescence‐associated β‐galactosidase (SA‐β‐gal) staining in A549, HAFF, HepG2, and H1299 cells after 48‐h transduction with either negative control lentiviruses (shCtrl) or those targeting GUARDIN (shGUARDIN). The graph shows percentage of SA‐β‐gal‐positive cells.
- Senescence‐associated heterochromatin foci formation (SAHF) in A549 cells with either shCtrl or shGUARDIN. Representative IF staining with H3K9me3 antibodies (green) and nuclear Hoechst counterstaining (blue).
- Secretion of SASP factors IL‐6 and IL‐8 in A549 cells with either shCtrl or shGUARDIN.
- Western blotting for p21, p16, and p15 in HepG2 cells after 48‐h transduction with shCtrl or shGUARDIN‐1, ‐2, respectively (left). Knockdown efficiencies analyzed by qPCR (right).
- SA‐β‐gal staining in A549 cells after 24‐h transduction with either shCtrl or shGUARDIN in combination with p21 shRNA (shp21). The graph shows percentage of SA‐β‐gal‐positive cells.
- qPCR measurement of p21 mRNA in A549, HepG2, HAFF, and H1299 cells with either GUARDIN shRNA knockdown (top) or GUARDIN overexpression (bottom).
- Western blotting for p53 and p21 in A549, HepG2, HAFF, and H1299 cells with either GUARDIN shRNA knockdown (top) or GUARDIN overexpression (bottom).
- qPCR assays of NOXA, TIGAR, BAX, and p21 mRNA levels in A549 cells with either shCtrl or shGUARDIN.
The induction of senescence following GUARDIN silencing was associated with upregulation of p21 protein, a factor which is well known to be involved in triggering senescence 21. In contrast, the expression of other senescence‐inducing proteins, such as p15 and p16 22, remained unaltered (Fig 1D). Indeed, p21 expression appeared critical for the induction of senescence caused by GUARDIN silencing, as co‐silencing of p21 abolished SA‐gal activity induced by GUARDIN knockdown as well as reversing the resulting inhibition of cell proliferation (Figs 1E and EV1G). Together, these results suggest that GUARDIN silencing induces senescence through upregulation of p21. Indeed, we observed increased p21 mRNA levels in cells following knockdown of GUARDIN, and consistent with this notion, overexpression of GUARDIN caused a decrease in p21 transcription levels (Fig 1F). Of note, neither GUARDIN knockdown nor overexpression affected p53 protein levels (Fig 1G), nor did these manipulations alter the mRNA levels of Noxa, TIGAR, and Bax (Fig 1H) that are known transcriptional targets of p53 23. Moreover, GUARDIN knockdown also caused upregulation of p21 and activation of senescence in p53‐null H1299 cells (Fig 1F and G). Together, these data indicated that p53 is not involved in GUARDIN‐mediated upregulation of p21.
To understand how silencing of GUARDIN could influence p21 expression, we first measured the effect of GUARDIN knockdown on the turnover of the p21 protein. Instructively, there were no changes in the half‐life time of p21 (Fig EV1H) upon GUARDIN depletion, indicating that GUARDIN did not influence the post‐translational stability of p21. Furthermore, treatment with the general transcription inhibitor actinomycin D (Act D) did not lead to noticeable changes in the turnover rate of p21 mRNA (Fig EV1I) 24, suggesting that p21 upregulation was not a consequence of an increased p21 mRNA stability; rather, it occurred through upregulated transcription. Thus, GUARDIN appears to inhibit cellular senescence through repressing transcriptional activation of p21.
GUARDIN facilitates LRP130/PGC1α interaction that mediates the transcriptional repression of p21
To investigate the mechanism responsible for GUARDIN‐mediated suppression of p21, we interrogated the protein interactome of GUARDIN using RNA pull‐down assays, and proteins that co‐precipitated with biotin‐labeled antisense probe against GUARDIN were identified by mass spectrometry (Fig 2A). Binding between GUARDIN and LRP130 was subsequently confirmed by Western blotting (Figs 2B and EV2E). Consistently, the interaction of LRP130 and GUARDIN was readily detected using RNA immunoprecipitation (RIP) assay and RNA FISH (Fig 2C and D). Moreover, subcellular fractionation assays showed that GUARDIN and LRP130 were localized to both nuclear and the cytoplasmic compartments (Fig EV2A, B and E), together indicating that LRP130 is a bona fide binding partner of GUARDIN.
Figure 2. GUARDIN facilitates LRP130‐PGC1α interaction that mediates transcriptional repression of p21.
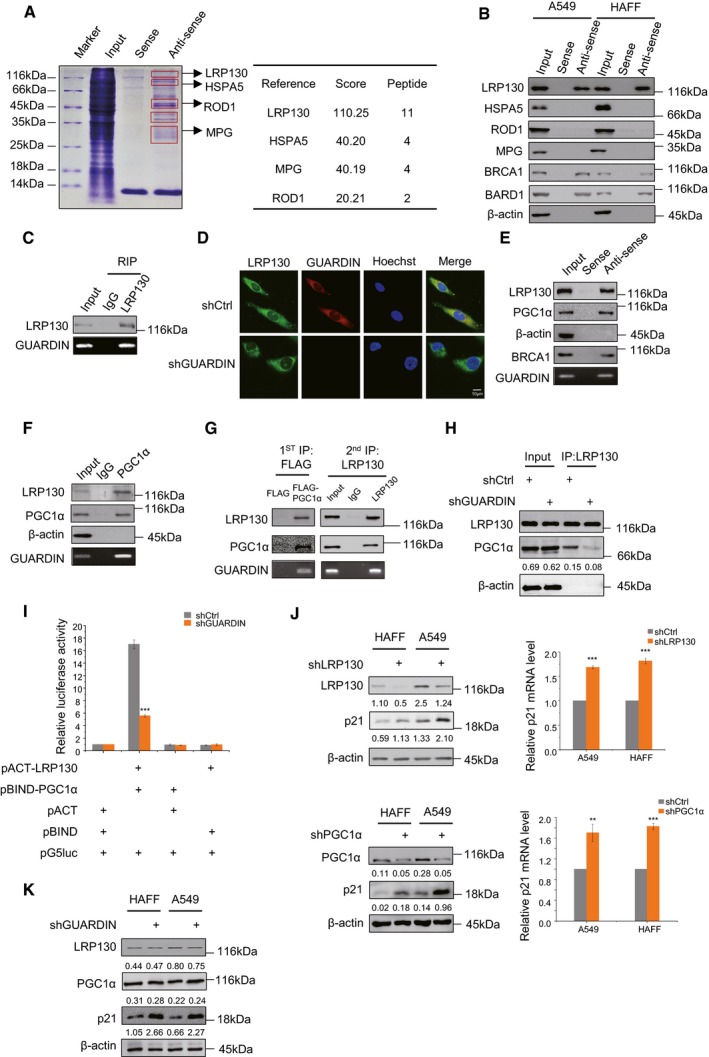
- SDS–PAGE of RNA pull‐down assays using biotin‐labeled sense/antisense probes against GUARDIN from whole‐cell lysates of A549 cells indicating putative GUARDIN‐binding proteins (left); protein identities with high probabilities were determined by mass spectrometry (right).
- RNA pull‐down assays interrogating putative GUARDIN‐associated proteins identified in (A) from whole‐cell lysates of A549 and HAFF cells. BRCA1, BARD1 served as positive controls, and β‐actin served as negative controls.
- RNA immunoprecipitation (RIP) assays against IgG/LRP130 antibodies in whole‐cell lysates of A549 cells.
- Subcellular localization of GUARDIN and its co‐localization with LRP130. RNA FISH for GUARDIN (red) and IF for LRP130 (green) in A549 cells with either shCtrl or shGUARDIN. Nucleus was counterstained with Hoechst (blue).
- RNA pull‐down assays using biotin‐labeled sense/antisense probes against GUARDIN from whole‐cell lysates of A549 cells. GUARDIN levels were measured by RT–PCR and co‐precipitated LRP130 and PGC1α detected by Western blotting. BRCA1 and β‐actin served as positive and negative controls, respectively.
- RIP assay using IgG/PGC1α antibodies from whole‐cell lysates of A549 cells. GUARDIN, LRP130, and PGC1α levels were measured as per (E).
- Two‐step IP assays in whole‐cell lysates of A549 cells transfected with FLAG‐tagged PGC1α. First‐phase IPs were conducted with FLAG antibodies (left), and following elution with FLAG peptides, eluates were further subjected to second‐phase IPs with LRP130 antibodies (right). Samples were subjected to Western blotting and qPCR analysis for LRP130, PGC1α, and GUARDIN, respectively.
- Co‐immunoprecipitation (co‐IP) between LRP130 and PGC1α in A549 cells after 48‐h transduction with shCtrl or shGUARDIN. LRP130 was precipitated, and samples were subjected to Western blotting analysis for LRP130, PGC1α and β‐actin as loading control.
- Mammalian two‐hybrid assays between pACT‐LRP130 and pBIND‐PGC1α in A549 cells after 48‐h transduction with shCtrl or shGUARDIN. Samples were subjected to the luciferase activity assays.
- LRP130/PGC1α and p21 protein expression was measured by Western blotting in A549 and HAFF cells after 48‐h transduction with shCtrl or shLRP130 (top left) or shPGC1α (bottom left) as indicated. qPCR assays for p21 mRNA levels were performed in parallel (right panels).
- Western blotting analysis of LRP130, PGC1α, and p21 protein levels in HAFF and A549 cells after 48‐h transduction with shCtrl or shGUARDIN.
Figure EV2. Characterizing the subcellular distribution of GUARDIN, LRP130, and PGC1α and the identification of their respective interacting domains.
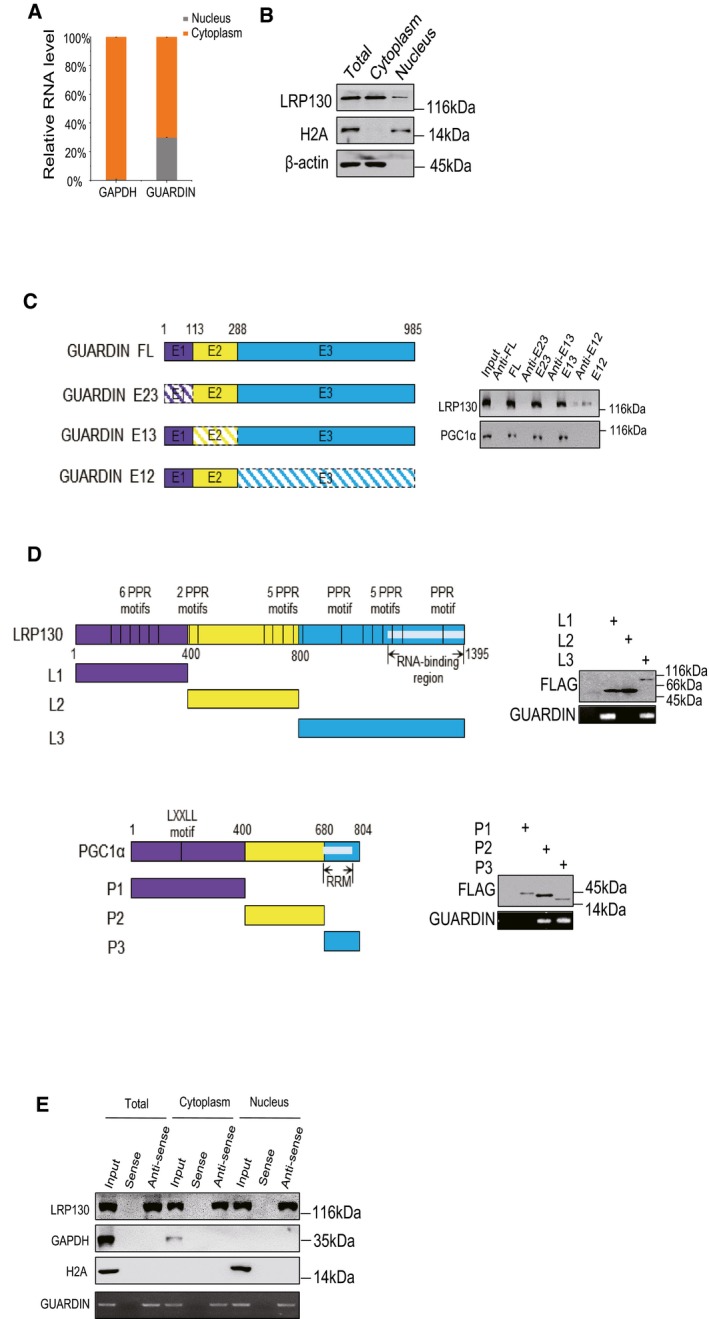
-
A, BSubcellular distribution of GUARDIN and LRP130. (A) RT–PCR assays against GUARDIN in cytoplasmic and nuclear compartments isolated from A549 cells. GAPDH served as cytoplasmic control. Values are mean ± SEM (n = 3 biological replicates). (B) Western blotting against LRP130 in distribution in subcellular fractions from (A). H2A and β‐actin served as nuclear and cytoplasmic markers, respectively.
-
CGUARDIN interacts with LRP130/PGC1α through exon3 (E3). Schematic illustration of GUARDIN and the design of exon‐deletion constructs (left). RNA pull‐down assays against whole‐cell lysates of A549 cells using in vitro‐transcribed GUARDIN or mutant RNAs (right). Western blotting assay was performed for LRP130 and PGC1α.
-
DLRP130 interacts with GUARDIN by its N‐terminal and C‐terminal domains (upper), while PGC1α interacts with GUARDIN by its C‐terminal and central region domains (lower). Schematic illustration of LRP130 and PGC1α and the design of domain truncation constructs (left). RIP assays performed against whole‐cell lysates of A549 cells transfected with indicated FLAG‐tagged LRP130/PGC1α mutant constructs (left). Western blotting assays for LRP130/PGC1α mutants using FLAG antibodies and RT–PCR assays for GUARDIN (right).
-
ERNA pull‐down assays using biotin‐labeled sense/antisense probes against GUARDIN from whole‐cell, cytoplasm, and nucleus of A549 cells. Western blotting assay was performed for LRP130, GAPDH, and H2A. H2A and GAPDH served as nuclear and cytoplasmic markers, respectively.
Source data are available online for this figure.
LRP130 is a transcription factor shown previously to functionally complex with PGC1α to exert effects on gene expression 25. RNA pull‐down assays performed with GUARDIN also showed the presence of PGC1α (Fig 2E) suggesting that GUARDIN may associate with the LRP130‐PGC1α complex. Consistently, RNA immunoprecipitation (RIP) assay showed association between GUARDIN and PGC1α (Fig 2F). To verify that GUARDIN associates with the LRP130‐PGC1α complex, we introduced FLAG‐tagged PGC1α into A549 cells and conducted two‐step IP assays from total protein extracts. In the first‐phase IP, FLAG antibodies precipitated PGC1α along with LRP130 and GUARDIN while in the second‐phase IP, LRP130 antibodies co‐precipitated PGC1α and GUARDIN (Fig 2G), indicating GUARDIN, LRP130, and PGC1α exist as a ternary complex. Instructively, GUARDIN knockdown diminished the relative amount of LRP130 associated with PGC1α as shown in co‐immunoprecipitation (Co‐IP) and mammalian two‐hybrid assays (Fig 2H and I), indicating that GUARDIN functions to facilitate interactions between LRP130 and PGC1α.
We also investigated the structural basis of the interaction of GUARDIN with LRP130 and PGC1α by using deletion‐mapping experiments. Binding assays with in vitro‐transcribed GUARDIN along with truncation mutants demonstrated that deletion of exon 3 (E3; ‐288/‐985) diminished the binding of GUARDIN to LRP130 and PGC1α (Fig EV2C). These results support the notion that E3 region of GUARDIN is the binding unit responsible for the association with LRP130 and PGC1α. Deletion‐mapping experiments with truncated mutants of LRP130 and PGC1α revealed that the N‐terminal and C‐terminal regions of LRP130 are required for its association with GUARDIN, whereas the C‐terminus and central region of PGC1α are responsible for binding to GUARDIN (Fig EV2D).
We next examined whether LRP130 and PGC1α play roles in GUARDIN‐mediated repression of p21. ShRNA knockdown of either LRP130 or PGC1α resulted in upregulation of p21 at both mRNA and protein levels (Fig 2J), recapitulating the effects of GUARDIN depletion on p21. However, neither LRP130 nor PGC1α levels were significantly impacted by GUARDIN depletion (Fig 2K). Thus, the association of LRP130 and PGC1α facilitated by GUARDIN appears necessary for GUARDIN‐mediated repression of p21 expression.
GUARDIN/LRP130/PGC1α represses transcription of FOXO4 gene
To investigate whether LRP130/PGC1α was involved directly in transcriptional regulation of p21, we interrogated the p21 promoter region for the presence of LRP130/PGC1α binding sites. Indeed, using the UCSC browser predicted LRP130/PGC1α binding sites were found embedded within DNase I hypersensitive regions (DHSs) of the p21 gene promoter (‐1870/‐1701, ‐1430/‐1221, ‐395/‐1 upstream of the transcription start site) (Fig 3A, lower part). DHSs mark diverse classes of cis‐regulatory regions, such as promoters and enhancers, and their identification represents a powerful method of identifying the location of gene regulatory elements, including promoters 28, 29, 30. Nevertheless, neither LRP130 nor PGC1α were found to directly bind to p21 promoter using ChIP assays whereas TP53, a bona fide p21 transcriptional driver, was shown to bind to the p1(‐1870/‐1701) and p4 (‐199/‐1) binding sites (Fig 3A, upper part). ABCB1 and LMNA 26, 27 served here as positive controls of LRP130 and PGC1α ChIP assays, respectively. These results implied that the upregulation of p21 by LRP13/PGC1α was unlikely to be mediated through direct transcription.
Figure 3. LRP130/PGC1α negatively regulates FOXO4 transcription.
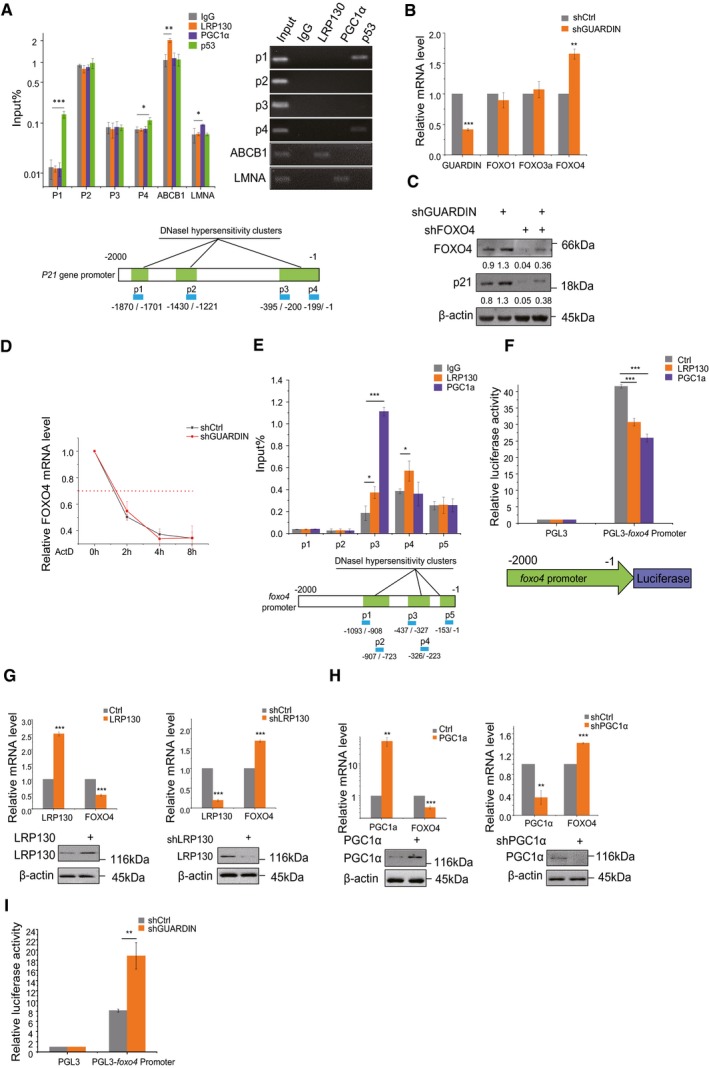
- ChIP assays detecting binding of LRP130/PGC1α to the p21 promoter using qPCR and RT–PCR (top left and right, respectively). IgG and p53 served as a negative and positive controls, respectively. Schematic illustrations of the putative LRP130/PGC1α binding sites within DNase I hypersensitive regions of the p21 promoter (bottom).
- qPCR assays for GUARDIN, FOXO1, FOXO3a, and FOXO4 in A549 cells after 24‐h transduction with shCtrl or shGUARDIN.
- Western blotting assays for FOXO4 and p21 protein expression in A549 cells after 48‐h transduction with shCtrl or shGUARDIN alone or in combination with shFOXO4. β‐actin served as loading control.
- Half‐life times of FOXO4 mRNA in A549 cells with shCtrl or shGUARDIN measured by qPCR after treating cells with 5 μg/ml of actinomycin (ActD) for the indicated times.
- ChIP assays detecting binding of LRP130/PGC1α to putative binding sites in the FOXO4 promoter using qPCR (top). Data were normalized to the IgG negative control. Schematic illustration of the LRP130/PGC1α binding sites within DNase I hypersensitive regions of the FOXO4 promoter (bottom).
- Luciferase assays conducted in A549 cells transfected with LRP130 or PGC1α using the pGL3 (negative control) or pGL3‐FOXO4 promoter reporter plasmids.
- qPCR assays for LRP130 and FOXO4 mRNA levels (upper) and Western blotting analysis for LRP130 protein level (lower) in A549 cells after 48‐h transduction with LRP130 overexpression or knockdown.
- qPCR assays for PGC1α and FOXO4 mRNA level (upper) and Western blotting analysis for PGC1α protein level (lower) in A549 cells after 48‐h transduction with PGC1α overexpression or knockdown.
- Luciferase activity assays in A549 cells transduced with shCtrl or shGUARDIN using the pGL3 (negative control) or pGL3‐FOXO4 promoter reporter plasmids.
To clarify the mechanism by which GUARDIN suppresses p21 expression and cellular senescence, we carried out comparative RNA sequencing (RNA‐seq) analysis for differentially expressed transcripts in A549 cells with and without GUARDIN shRNA knockdown (Fig EV3A). Notably, the p21 promoter region is known to contain consensus forkhead binding elements 31 with the RNA‐seq analysis revealing significant association with FoxO signaling through enrichment of the forkhead box proteins, FOXO1, FOXO3a, and FOXO4 in GUARDIN knockdown cells (Fig EV3B, Dataset EV2). Verification using qPCR showed that FOXO4 but not FOXO1 and FOXO3a mRNA levels were increased upon GUARDIN knockdown, and consistent with this notion, overexpression of GUARDIN caused a decrease in FOXO4 transcription levels (Figs 3B, EV3D and E). Importantly, knockdown of FOXO4 not only reduced p21 expression but also diminished the increase in p21 caused by GUARDIN knockdown (Fig 3C). Collectively, these results suggest that GUARDIN‐mediated repression of p21 may be due to suppression of FOXO4. Silencing of GUARDIN had no effect on the turnover rate of FOXO4 mRNA when treated with ActD (Fig 3D), suggesting that regulation of FOXO4 by GUARDIN was unlikely to affect FOXO4 mRNA stability. In support of this notion, we identified multiple consensus LRP130 and PGC1α binding sites in the DNase I hypersensitive region (‐1093/‐723, ‐437/‐223, ‐153/‐1) of the FOXO4 gene promoter (Fig 3E, bottom). ChIP assays showed that LRP130 and PGC1α bound to the P3 and to a lesser extent the P4 regions (‐437/‐327 and ‐326/‐233, respectively; Fig 3E, top).
Figure EV3. RNA‐seq‐based screening of differentially expressed genes following GUARDIN depletion identifies FOXO4.

- Differentially expressed mRNAs in A549 cells following shGUARDIN knockdown. Volcano plot comparing control versus GUARDIN knockdown in A549 cells with differentially downregulated and upregulated mRNAs shown in violet and orange, respectively.
- KEGG pathway analysis. Selected high scoring KEGG pathway enrichments using data from (A) are illustrated in the y‐axis. Q values (downregulated or upregulated) pathways are represented by color with the size of the dot indicating the number of DEGs mapped to each pathway.
- LRP130, GUARDIN, and FOXO4 expressed in 30 human tissue samples from the Genotype‐Tissue Expression (GTEx) Project.
- qPCR assays for GUARDIN and FOXO4 mRNA levels in A549 cells after 48‐h transduction with GUARDIN overexpression.
- IF assays for FOXO4 in A549 after 48‐h transduction with GUARDIN knockdown. Densitometry was performed using Image‐Pro plus 6.0 software.
To verify the interaction of LRP130 and PGC1α with the FOXO4 promoter, we constructed a luciferase reporter vector (Fig 3F, bottom). The transcriptional activity of the FOXO4 reporter significantly decreased when cells were co‐transfected with either LRP130 or PGC1α (Fig 3F, top), indicative of repression. Consistent with a repressor function, shRNA knockdown of LRP130 or PGC1α resulted in upregulation of FOXO4 mRNA expression, while the overexpression of LRP130 or PGC1α downregulated FOXO4 mRNA levels (Fig 3G and H). Silencing of GUARDIN increased the transactivation of FOXO4 promoter as shown by luciferase assay (Fig 3I). Taken together, these findings established that GUARDIN suppresses p21 expression through facilitating LRP130/PGC1α‐mediated transcriptional repression of FOXO4.
Finally, comparing LRP130, GUARDIN, and FOXO4 expression across 30 human tissue samples from the GTEx (Genotype‐Tissue Expression) Project showed the majority tissues with high expression levels of GUARDIN positively and negatively correlated with LRP130 and FOXO4, respectively (Fig EV3C), thus supporting the regulatory relationship established between GUARDIN, LRP130, and FOXO4.
GUARDIN engages the LRP130/PGC1α/FOXO4/p21 signaling axis independently of its roles in genome maintenance
GUARDIN is known to maintain genomic integrity through stabilizing BRCA1/BARD1 heterodimers and by maintaining TRF2 expression via sequestering of miR‐23a 19. To investigate whether GUARDIN‐mediated repression of p21 occurs independently of these functions, we treated cells with miR‐23a inhibitors along with knockdown of either BRCA1 or BARD1 using previously validated shRNAs 19. In contrast to shRNA‐mediated inhibition of GUARDIN, neither individual knockdown of BARD1 nor BRCA1, or the inhibition of miR‐23a, had appreciable effects on the levels of FOXO4 (Fig EV4A–C, respectively). Knockdown of GUARDIN also had no effect on miR‐23a or BARD1 levels but as anticipated from prior data 19 there were reductions in BRCA1 levels along with TRF2. With respect to effects on p21, it is known that BRCA1 is involved in the activation of p21 and that BARD1 regulates BRCA1‐mediated transactivation of the p21 promoter 32, 33. Moreover, it has been reported that TRF2 represses p21 promoter activity by promoter occupancy 34. Consistently, silencing of either BRCA1 or BARD1 along with inhibition of miR‐23a resulted in the downregulation of p21, in contrast to the increased levels of p21 resulting from shGUARDIN treatment (Fig EV4A–D). Thus, BRCA1, BARD1, and miR‐23a levels positively affect p21 expression but such effects contrast the actions of GUARDIN which acts to suppress p21. These data, together with findings that neither BRCA1, BARD1, nor miR‐23a influence FOXO4 expression, propose that GUARDIN's role in p21‐driven senescence occurs independently from its genome integrity functions.
Figure EV4. Inhibition of BARD1, BRCA1, and miR‐23a and the effects on FOXO4 and p21 expression.
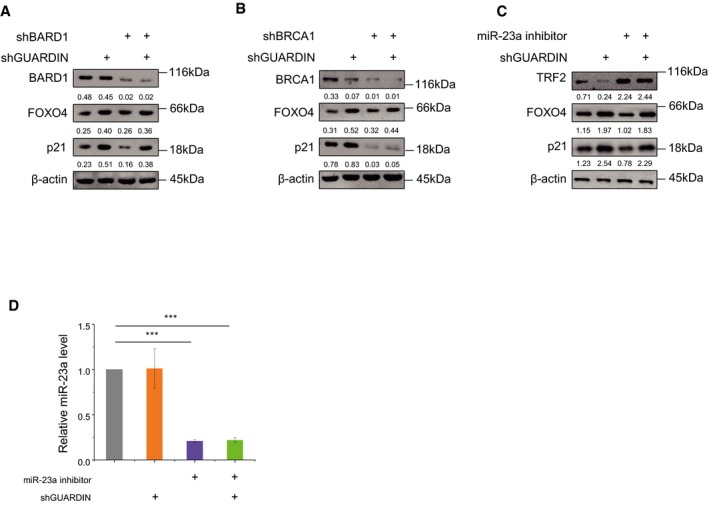
- A549 cells were treated with shCtrl or shGUARDIN alone or in combination with shBARD1 for 48 h before Western blotting to determine relative levels of BARD1, FOXO4, and p21. β‐actin served as loading control.
- A549 cells were treated with shCtrl or shGUARDIN alone or in combination with shBRCA1 for 48 h before Western blotting to determine relative levels of BRCA1, FOXO4, and p21. β‐actin served as loading control.
- A549 cells were treated with shCtrl or shGUARDIN alone or in combination with miR‐23a inhibitor before Western blotting to determine relative levels of TRF2, FOXO4, and p21. β‐actin served as loading control.
- The levels of miR‐23a were determined in cells from (C) using qPCR.
GUARDIN is transcribed by FOSL2 that is responsive to rapamycin
Rapamycin is known to inhibit senescence through a number of different mechanisms including but not only, inhibition of mTOR 11, 12. We investigated whether GUARDIN contributes to or even cooperates with rapamycin to inhibit senescence. Indeed, rapamycin treatment resulted in upregulation of GUARDIN, both in a time‐ and dose‐dependent manner (Fig 4A and B) which was associated with decreased expression of p21 and FOXO4 but not LRP130 (Fig 4C). Intriguingly, the downregulation of p21 by rapamycin was mediated through GUARDIN, since the p21‐dependent cellular senescence caused by GUARDIN depletion was reversed by rapamycin treatment (Fig 4D). Rapamycin treatment increased the relative amount of LRP130 associated with PGC1α as shown in mammalian two‐hybrid assays (Fig 4E), recapitulating the effects of GUARDIN. Therefore, GUARDIN is not only a rapamycin‐responsive lncRNA, but also it contributes to the rapamycin‐mediated inhibition of senescence. Moreover, the levels of p53 did not change upon rapamycin treatment (Fig 4C), implying that rapamycin does not regulate GUARDIN through p53.
Figure 4. Rapamycin regulates GUARDIN expression via FOSL2.
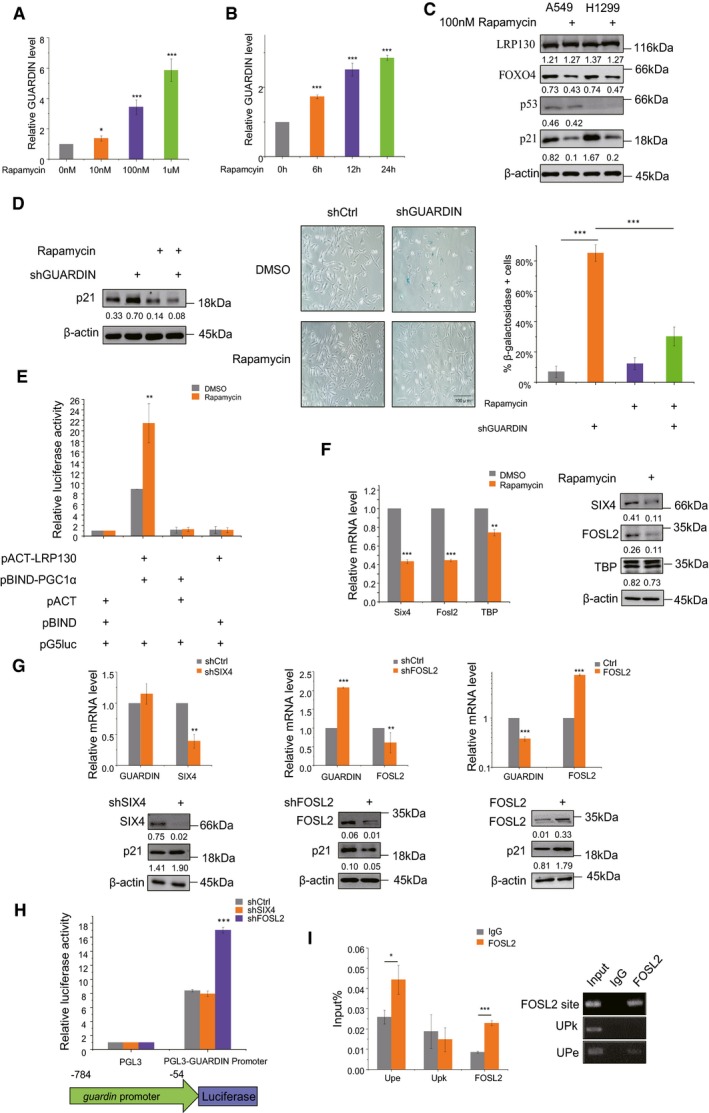
-
A, BqPCR assays for GUARDIN expression in A549 cells treated with increasing doses of rapamycin for 24 h (A) or 100 nM of rapamycin for the indicated times (B).
-
CWestern blotting assays for p53, LRP130, FOXO4, and p21 expression in A459 and H1299 cells treated with 100 nM of rapamycin or vehicle (DMSO) for 48 h.
-
DA549 cells with shCtrl or shGUARDIN were treated with 100 nM rapamycin or DMSO vehicle in the indicated combinations for 48 h. Western blotting was used to measure p21 levels (left) while conducting SA‐β‐gal staining in parallel (middle) with the percentage of senescent cells calculated from the SA‐β‐gal staining (right).
-
EMammalian two‐hybrid assays between pACT‐LRP130 and pBIND‐PGC1α in A549 cells treated with DMSO or 100 nM rapamycin. Samples were subjected to the luciferase activity assays.
-
FSIX4 and FOSL2 mRNA (left) and protein levels (right) measured by qPCR and Western blotting, respectively, in A549 cells treated with DMSO or 100 nM rapamycin. TBP served as a control.
-
GqPCR (upper) and Western blotting (lower) assays for GUARDIN, SIX4, and FOSL2 expression in A549 cells comparing shCtrl with shSIX4 (left), shFOSL2 (middle), or FOSL2 (right) after transduction for 48 h.
-
HLuciferase assays in A549 cells transduced with shCtrl, shSIX4, or shFOSL2 using pGL3 (negative control) or pGL3‐FOXO4 promoter reporter plasmids.
-
IChIP assays comparing control IgG versus FOSL2 antibodies demonstrate specific recovery of the GUARDIN promoter by RT–PCR assay. UPe and UPk served as a positive and negative controls, respectively 54.
To explore the mechanism responsible for the rapamycin‐induced upregulation of GUARDIN, we interrogated microarray gene expression data derived from tumor tissues with or without rapamycin treatment (GSE39694; Gene Expression Omnibus (GEO)). Among the 629 genes altered by treatment with rapamycin, there were 38 encoding transcription factors (Fig EV5A, Dataset EV3). Inspecting the genomic sequence in the GUARDIN promoter using the JASPAR database 35 identified putative binding sites for SIX homeobox 4 (SIX4), FOS‐like antigen 2 (FOSL2), and ATA‐binding protein (TBP) (Fig EV5B). Further assessment of the levels of these transcription factors showed that SIX4 and FOSL2 were downregulated at mRNA and protein levels, whereas TBP remained unchanged in response to rapamycin treatment (Fig 4F). Moreover, knockdown of FOSL2 but not SIX4 caused upregulation of GUARDIN and downregulation of p21 (Fig 4G), suggesting that FOSL2 expression underpinned the rapamycin‐dependent changes in GUARDIN expression. Consistently, overexpression of FOSL2 caused downregulation of GUARDIN and upregulation of p21 (Fig 4G), recapitulating the effects of rapamycin treatment on GUARDIN and p21 expression (Fig 4A–C). Additionally, FOSL2 knockdown increased the transcriptional activity of the ‐784/‐54 GUARDIN reporter construct whereas reporter levels after SIX4 shRNA were unchanged from controls (Fig 4H). In addition, ChIP assays demonstrated that FOSL2 bound to its predicted binding sites (‐503 to ‐493) in the GUARDIN promoter (Figs 4I and EV5C). Together, these results indicate that binding of FOSL2 to the GUARDIN promoter transcriptionally represses its expression. Thus, rapamycin suppresses FOSL2 and subsequently upregulates GUARDIN leading to suppression of p21 and inhibition of cellular senescence. Additionally, we compared the expression levels of GUARDIN, FOXO4, FOSL2, and LRP130 at 15 and 35 passages in HAFF cells representing early and late stages of replicative senescence (Fig EV5D). The transition to replicative senescence was associated with decreased GUARDIN, increased FOXO4 and FOSL2 but without changes in LRP130, consistent with the regulatory events identified in the preceding analysis.
Figure EV5. Utilizing RNA‐seq‐based screening to identify differentially expressed transcription factor genes following rapamycin treatment.
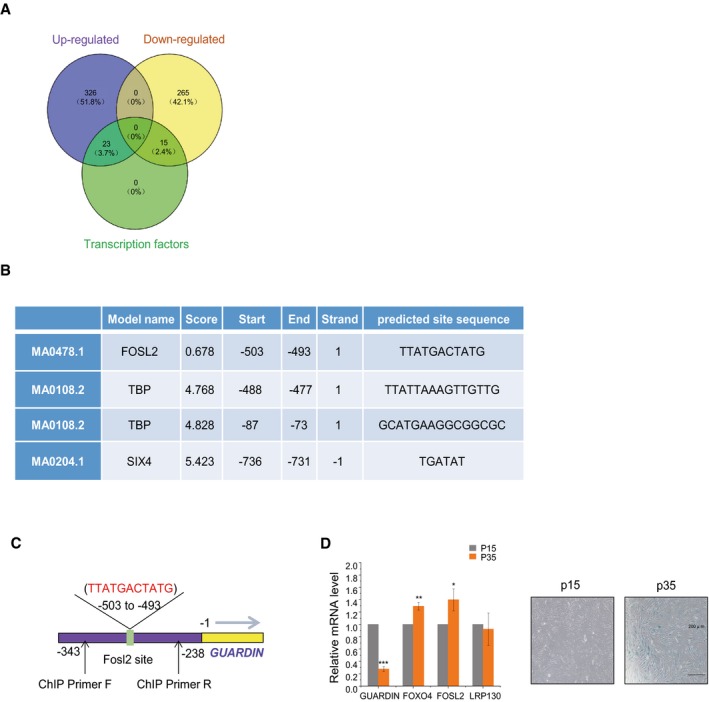
- DEGS in tumor tissues treated with rapamycin versus control groups determined by RNA‐seq. Venn diagram shows upregulated transcripts (violet) and downregulated transcripts (yellow) along with those identified as transcription factors (green).
- Transcription factor consensus binding sites present within the GUARDIN promoter.
- Schematic illustrating the putative FOSL2 binding site within the GUARDIN promoter and the location of primers for ChIP assay.
- qPCR (left) assays for GUARDIN, FOXO4, LRP130, and FOSL2 in HAFF cells while conducting SA‐β‐gal staining in parallel (right) after 15 (P15) and 35 (P35) cell passages. Values are mean ± SEM (n = 3 biological replicates); two‐tailed paired Student's t‐test (*P < 0.05, **P < 0.01, ***P < 0.001).
Discussion
We have previously shown that GUARDIN is a p53‐responsive lncRNA that is essential for cell survival and proliferation through maintaining genomic integrity in response to DNA damage 19. However, low levels of GUARDIN can still be detected in TP53‐null cells or tumors with mutations in TP53 19, although the functional significance of GUARDIN under p53 deficient conditions is still unclear. In this report, we provide evidence that GUARDIN is responsive to rapamycin and represses cellular senescence through facilitating LRP130/PGC1α‐mediated repression of FOXO4 leading to transcriptional inactivation of p21. Moreover, we show that repression of FOSL2 is responsible for upregulation of GUARDIN in response to rapamycin. These results not only reveal a p53‐independent mechanism that regulates GUARDIN expression, but also uncover a novel signaling pathway of cellular senescence induced by rapamycin.
It was previously reported that silencing of GUARDIN could induce cell senescence 19, and conceivably, this resulted from loss of one or both of GUARDIN's functions in maintaining genomic integrity, either through influencing DNA repair or by maintaining telomeric repeat binding factor 2 (TRF2) expression and the activity of the shelterin complex critical for telomeric capping 36. The latter function is relevant since telomere shortening or uncapping can also drive cellular senescence 21. However, rather than a fait accompli of genomic damage, we show here that GUARDIN plays a direct role in restraining cell entry into senescence. Indeed, senescence induced by GUARDIN knockdown was mediated by p21, as knockdown of GUARDIN upregulated p21 and co‐knockdown of p21 diminished senescence resulting from GUARDIN knockdown 19. Intriguingly, GUARDIN‐mediated regulation of p21 expression appeared to be independent of p53, although p21 is a major effector of p53 signaling and we have previously shown that GUARDIN, similar to p21, may represent an indicator of wild‐type p53 activity 19. Nevertheless, p53‐independent regulation of p21 has been widely documented 37, 38. Moreover, while the actions of GUARDIN were inextricably linked to p21, they appeared independent of other known major effectors of cellular senescence, namely p15 and p16 39, 40, 41. Our results further consolidate that notion that both GUARDIN and p21 can function independently of p53 and point to a GUARDIN‐mediated mechanism that transcriptionally represses p21 irrespective of p53 activation status.
Previously, we have found that GUARDIN acts as an RNA scaffold to facilitate the binding between BRCA1 and BARD1 in response to DNA damage 19. Intriguingly, the primary contribution of GUARDIN in suppressing senescence also involved a role in facilitating protein complexes, in this case the assembly and stability of a repressor complex formed by LRP130 and PGC1α. This was demonstrated by (i), GUARDIN, LRP130, and PGC1α formed a ternary structure; (ii), knockdown of GUARDIN reduced the association between LRP130 and PGC1α; (iii), binding of LRP130 and PGC1α to the FOXO4 promoter was associated with transcriptional repression while inhibiting LRP130, PGC1α, or GUARDIN increased its activation; and (iv), knockdown of LRP130 or PGC1α upregulated p21, recapitulating the effects of knockdown of GUARDIN. Notably, the binding interactions between BRCA1/BARD1 and LRP130/PGC1α were mediated through different regions of GUARDIN 19, indicative of distinct structural specificities embedded in the RNA sequence.
PGC1α is a transcriptional cofactor that interacts with multiple transcription factors 42 while LRP130 is a leucine‐rich repeat‐containing protein (also known as LRPPRC) functionally associated with mitophagy induction 43 but also with demonstrated transcriptional and/or translational roles in the nucleus and endoplasmic reticulum 44. The LRP130/PGC1α complex plays a critical role in glucose homeostasis and energy balance through regulation of gluconeogenic and mitochondrial genes 25, 45. Interestingly, previous studies show that PGC1α is a binding partner and coactivator of FOXO1 which drives the expression of gluconeogenic genes in liver 25, 46 while in contrast, PGC1α acts at the FOXO3 promoter to suppress its expression in skeletal muscle 47. Our results showing that the GUARDIN/LRP130/PGC1α complex acts to transcriptionally repress FOXO4 are consistent with the latter mode of regulation. Maintaining low FOXO4 levels prevents activation and upregulation of p21 expression and protects cells against senescence induction. Consistently, FOXO4 has been shown to drive oncogene‐induced senescence in the melanocytic lineage through p21 48. Moreover, the levels of FOXO4 increased during replicative senescence in fibroblasts, and among a range of normal tissues with higher GUARDIN expression, there appeared to be a negative correlation with FOXO4 expression. Indeed, FOXO4 is known to play an important role in p21 transcriptional regulation 48, 49, and notably, administration of a small interfering FOXO4 peptide reduces senescence and restores overall fitness in naturally aged mice or in fast aging mice models 50. Our results showing suppression of p21 by GUARDIN through LRP130/PGC1α‐mediated transcriptional repression of FOXO4 as an important mechanism to counteract cellular senescence implicate that promotion of GUARDIN expression may be a general approach to prevent aging through sustained repression of FOXO4.
Finally, we establish that GUARDIN expression is induced in cells in response to the mTOR inhibitor, rapamycin. This agent has long been known to inhibit cellular senescence and prolong life span in various model systems 6. However, the mechanisms involved remain less understood and are thought to be multifactorial. We found that GUARDIN contributes to rapamycin‐mediated inhibition of cellular senescence, opening a novel scenario that rapamycin protects against cellular aging through a noncoding mechanism. Instructively, treatment with rapamycin did not activate p53, consistent with senescence representing a p53‐independent aspect of GUARDIN function. The upregulation of GUARDIN by rapamycin was mediated through downregulation of FOSL2. FOSL2 was shown to exert transcriptional repression of GUARDIN through occupancy of the GUARDIN promoter although how rapamycin affects downregulation of FOSL2 is not currently known.
In summary, we have identified a signaling axis encompassing GUARDIN, LRP130/PGC1α, FOXO4, and p21 that can suppress cellular senescence (Fig 5). Manipulating GUARDIN levels by inhibition or overexpression acted to either trigger or block senescence. An important question involves how the contextual placement of GUARDIN's independent functions involving genomic maintenance versus its role in senescence. The latter involves negative control of gene expression that conceivably may act as a failsafe switch under conditions where GUARDIN expression is compromised although this thesis remains to be explored. Nevertheless, as GUARDIN is responsive to treatment with rapamycin, our findings also highlight a potential role for GUARDIN in aging‐associated diseases with perhaps practical implications in aging prevention.
Figure 5. Model for GUARDIN‐mediated regulation of cellular senescence.
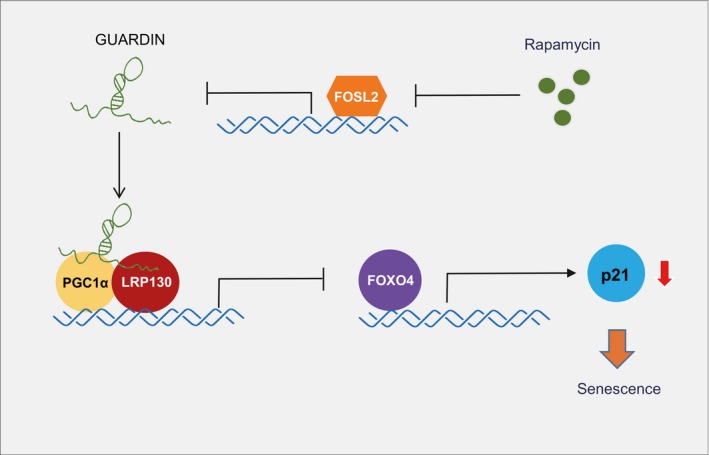
GUARDIN acts as an RNA scaffold to facilitate interaction between LRP130/PGC1α that acts to transcriptionally repress FOXO4 expression. This repression prevents FOXO4‐mediated upregulation of p21 and cell entry into senescence. GUARDIN participates in rapamycin‐induced cellular senescence, likely through downregulation by FOSL2, which is also downregulated by rapamycin.
Materials and Methods
Reagents
Reagents and antibodies were sourced as indicated: cycloheximide (Sigma); rapamycin (BBI); actinomycin D (Solarbio); streptavidin beads (Invitrogen); and A/G beads (Pierce);
Antibodies
anti‐H3K9me3 (ab176916), p15 (ab53034), p16 (ab211542), FOXO4 (ab126594), and Alexa Fluor® 488‐conjugated secondary antibodies (ab150077) from Abcam; anti‐p21 (P1484‐.2ML) and flag (F7425‐.2MG) from Sigma; anti‐IL6 (21865‐1‐AP), IL‐8 (17038‐1‐AP), LRP130 (21175‐1‐AP), HSPA5 (11587‐1‐AP), MPG (11481‐2‐AP), SIX4 (21305‐1‐AP), FOSL2 (15832‐1‐AP), and TBP (22006‐1‐AP) from Proteintech; anti‐GAPDH (AT0002) and β‐actin (AT1569) from CMC‐TAG; anti‐HUR (sc‐5261), H2A (sc‐54606), p53 (sc‐126), ROD1 (sc‐398105), and PGC1α (sc‐5816) from Santa Cruz; and HRP‐conjugated secondary antibodies against mouse and rabbit IgG (W4021,W4011) from Promega.
Cell culture
HepG2, A549, H1299, 293T, and HAFF cells were cultured in DMEM (Invitrogen) supplemented with 10% FBS/1% penicillin/streptomycin and maintained in a humidified incubator at 37°C/5% CO2. All cells were authenticated by STR profiling (GenePrint 10 System kit from Promega and AuthentiFiler PCR Amplification Kit from Thermo Fisher) and tested negative for mycoplasma contamination using the Cell Culture Contamination Detection Kit (Thermo Fisher).
RNA interference and gene overexpression
Lentiviruses for gene knockdown and overexpression experiments, respectively, were generated by transfecting HEK293T cells for 48 h with PLKO.1‐based shRNAs, pREV, pGag, and pVSVG (2:2:2:1 ratio) or psin/pCDH, pspax2, pmd2.g (2:2:1 ratio). Supernatants were 0.45 um filtered, supplemented with 8 μg/ml polybrene (Sigma) before incubating with target cells and selecting with 5 μg/ml puromycin. Targeting sequences are shown in Table EV1.
Quantitative and semi‐quantitative RT–PCR
Total RNA was isolated by TRIzol reagent (Invitrogen) and 500 ng RNA used to synthesize cDNA using PrimeScriptTM RT reagent kit (TaKaRa) according to the manufacturer's instructions. Semi‐quantitative RT–PCR was performed using 2× Taq PCR mix with 25 cycles for internal controls and 30–40 cycles for RNAs. Primer sequences are shown in Table EV1. QPCR was performed as described previously 51.
β‐Galactosidase staining and quantitation
β‐galactosidase staining was performed according to the manufacturers protocol (C0602, Beyotime). β‐galactosidase‐positive cells were scored using light microscopy, counting at least 50 cells from three random fields.
Immunofluorescence and RNA FISH
Cells grown on coverslips were fixed in 4% paraformaldehyde and permeabilized using 0.2% Triton X‐100. After blocking with 2% BSA, coverslips were incubated overnight at 4°C with primary antibodies before addition of Alexa‐488 secondary antibodies. RNA probes synthesized by T7‐mediated transcription were purified by phenol/chloroform extraction and 1 μg RNA labeled with Alexa‐546 using the ULYSIS Nucleic Acid Labeling Kit (Thermo Fisher) 52. Nuclei were counterstained with Hoechst.
ELISA
Cells were incubated in serum‐free DMEM for 24 h and IL‐6/8 levels determined in supernatants using the femtoELISA™ HRP Kit (G‐Biosciences).
Western Blotting
Whole‐cell lysates were prepared using RIPA buffer containing protease inhibitors (Beyotime) before conducting SDS–PAGE and Western blotting using enhanced chemiluminescence.
Northern Blotting
Northern blots were performed as described previously 51 against total RNA resolved on 1.5% agarose gels. Membranes were hybridized with digoxigenin‐labeled antisense GUARDIN probes synthesized using T7 RNA polymerase using the DIG Northern Starter Kit (Roche).
Biotin pull‐down assays and mass spectrometry
Biotinylated sense (negative control) and antisense biotin‐labeled DNA oligomers corresponding to GUARDIN (1 μg) were coupled to streptavidin‐coupled Dynabeads (Invitrogen). The Dynabeads were subsequently incubated with cell lysates for 4 h and eluted proteins subjected to SDS–PAGE and staining with Coomassie Brilliant Blue G‐250. Protein bands were excised and sent to Core Facility of Center for Life Sciences, USTC for mass spectrometry (MS) analysis using a Thermo‐Finnigan LTQ LC/MS‐MS. Protein IDs determined by MS are listed in Dataset EV2. All processes were performed under RNase‐free conditions.
Immunoprecipitation
Cells were lysed in IP lysis buffer (150 mM NaCl, 50 mM Tris, pH 7.4, 10% glycerol, and 1.5 mM MgCl2) supplemented with protease inhibitor cocktail before incubation for 4 h at 4°C with protein A/G beads precoated with indicated antibodies. Beads were washed three times using IP lysis buffer, eluted with heat (95°C, 10 min) and the samples analyzed by Western blotting. Where indicated, two‐step IPs were performed against cell lysates prepared using lysis buffer containing 20 mM HEPES, pH 7.8, 400 mM KCl, 5% glycerol, 5 mM EDTA, 1% NP‐40, protease inhibitors cocktail, and RNase inhibitor. The first IP used anti‐Flag antibodies before elution with Flag peptides. Ten percent of the sample was reserved for Western blotting and semi‐quantitative RT–PCR analysis, respectively, while the remaining eluate was subjected to the secondary IP. Steps were performed under RNase‐free conditions as required.
RNA immunoprecipitation
RNA immunoprecipitation (RIP) was performed as described 53. Briefly, cells were lysed in RIP buffer supplemented with RNase A inhibitor and DNase I. Soluble cell lysates were precleared with protein A/G beads alone before IP with the indicated antibodies at 4°C for 3 h. After washing, the bead‐bound immunocomplexes were eluted using elution buffer (50 mM Tris, pH 8.0, 1% SDS, and 10 mM EDTA) at 65°C for 10 min. To isolate protein‐associated RNAs from the eluted immunocomplexes, samples were treated with proteinase K, and RNAs were extracted by phenol/chloroform before RT–PCR analysis.
In vitro transcription
The T7 RNA polymerase promoter sequence was introduced into DNA templates generated by PCR to allow for in vitro transcription. PCR products were purified using the DNA Gel Extraction Kit (Axygen), and in vitro transcription performed using the T7‐Flash BiotinRNA Transcription Kit (Epicentre, biotin labeling) or TranscriptAid T7 High Yield Transcription Kit (Thermo Scientific, unlabeled) according to the manufacturer's instructions. RNA was subsequently extracted using phenol/chloroform. Primer sequences are shown in Table EV1.
ChIP assay
Cells were crosslinked with 1% formaldehyde for 10 min and ChIP assays performed using the Pierce Agarose ChIP kit (Thermo Scientific, USA) according to the manufacturer's instructions. Anti‐rabbit immunoglobulin G was used as a negative control. Bound DNA fragments were subjected to RT–PCR using the specific primers (Table EV1).
Luciferase reporter assay
Luciferase reporter assays were performed as described previously 53 using the Dual‐Luciferase Reporter Assay System (Promega, Madison, WI, USA).
Subcellular fractionation
Cells were incubated with hypotonic buffer (25 mM Tris–HCl, pH 7.4, 1 mM MgCl2, 2.5 mM KCl) on ice for 5 min. An equal volume of hypotonic buffer containing 1% NP‐40 was then added, and each sample was left on ice for another 5 min. After centrifugation at 5000 g for 5 min, the supernatant was collected as the cytosolic fraction. The pellets were resuspended in nuclear resuspension buffer (20 mM HEPES, pH 7.9, 400 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 1 mM PMSF) and incubated at 4°C for 30 min. Nuclear fractions were collected after removing insoluble membrane debris by centrifugation at 12,000 g for 10 min.
RNA‐seq
Total RNA was extracted by phenol/chloroform. RNA‐Seq was performed using an Illumina Hiseq platform (Bohao, shanghai, China). The amount of sequencing data per sample is 3G. Differential expression analysis of two groups was performed using the DESeq2 R package. DESeq2 provides statistical routines for determining differential expression in digital gene expression data using a model based on the negative binomial distribution. The resulting P‐values were adjusted using the Benjamini and Hochberg's approaches to control false discovery rate. Genes with an adjusted P < 0.05 were considered to be differentially expressed. The sequencing data were deposited in the National Center for Biotechnology Information Gene Expression Omnibus database (GSE137513).
Mammalian Two‐Hybrid assays
Complementary DNAs for PGC1α and LRP130 cloned into the pBIND and pACT vectors were transfected along with pG5luc vector (Promega) and reporter activity measured 48 h later using the Dual‐Luciferase Reporter Assay Kit (Promega). Renila measurements were used to normalize changes in firefly luciferase activity.
Quantification and statistical analysis
Statistical analysis was carried out using Microsoft Excel 2016 and GraphPad Prism 7 to assess differences between experimental groups. Densitometry was performed using Image‐Pro plus 6.0 software. Statistical significance was analyzed by two‐tailed Student's t‐test for comparisons of two samples, and two‐way ANOVA with Bonferroni's post‐test for bivariate comparisons. P‐values lower than 0.05 were considered to be statistically significant (ns, not significant, *P < 0.05, **P < 0.01, ***P < 0.001).
Author contributions
XS, SF, XL, and MW designed the research. XS and MH performed the experiments and data analysis. XDZ, JL, and RFT participated in the data analysis. XDZ, RFT, XL, and MW wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Dataset EV1
Dataset EV2
Dataset EV3
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Acknowledgements
The authors thank Dr. Qidong Li for valuable discussions and technical support. We also thank Drs. An Xu and Wanglai Hu for providing full‐length GUARDIN construct. This work was supported by grants from National Key R&D Program of China (2018YFA0107100), the National Natural Science Foundation of China (81820108021, 31871437, 31601146, 81772908 and 81970153), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB19000000), and the National Health and Medical Research Council of Australia (1147271).
EMBO Reports (2020) 21: e48796
Contributor Information
Shanshan Feng, Email: fengss2000@126.com.
Xiaoying Liu, Email: liuxiaoying@ahmu.edu.cn.
Mian Wu, Email: wumian@ustc.edu.cn.
Data availability
Raw and normalized data files for the microarray analysis have been deposited in the NCBI Gene Expression Omnibus under accession number GSE39694 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE39694) and GSE137513 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE137513).
References
- 1. Itahana K, Campisi J, Dimri GP (2004) Mechanisms of cellular senescence in human and mouse cells. Biogerontology 5: 1–10 [DOI] [PubMed] [Google Scholar]
- 2. Valenzuela CA, Quintanilla R, Moore‐Carrasco R, Brown NE (2017) The potential role of senescence as a modulator of platelets and tumorigenesis. Front Oncol 7: 188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhou X, Cao B, Lu H (2017) Negative auto‐regulators trap p53 in their web. J Mol Cell Biol 9: 62–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Munoz‐Espin D, Serrano M (2014) Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol 15: 482–496 [DOI] [PubMed] [Google Scholar]
- 5. Campisi J, d'Adda di Fagagna F (2007) Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 8: 729–740 [DOI] [PubMed] [Google Scholar]
- 6. Ehninger D, Neff F, Xie K (2014) Longevity, aging and rapamycin. Cell Mol Life Sci 71: 4325–4346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hasty P (2010) Rapamycin: the cure for all that ails. J Mol Cell Biol 2: 17–19 [DOI] [PubMed] [Google Scholar]
- 8. Richardson A, Galvan V, Lin AL, Oddo S (2015) How longevity research can lead to therapies for Alzheimer's disease: the rapamycin story. Exp Gerontol 68: 51–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kapahi P, Chen D, Rogers AN, Katewa SD, Li PW, Thomas EL, Kockel L (2010) With TOR, less is more: a key role for the conserved nutrient‐sensing TOR pathway in aging. Cell Metab 11: 453–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bjedov I, Toivonen JM, Kerr F, Slack C, Jacobson J, Foley A, Partridge L (2010) Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster . Cell Metab 11: 35–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang R, Yu Z, Sunchu B, Shoaf J, Dang I, Zhao S, Caples K, Bradley L, Beaver LM, Ho E et al (2017) Rapamycin inhibits the secretory phenotype of senescent cells by a Nrf2‐independent mechanism. Aging Cell 16: 564–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pospelova TV, Leontieva OV, Bykova TV, Zubova SG, Pospelov VA, Blagosklonny MV (2012) Suppression of replicative senescence by rapamycin in rodent embryonic cells. Cell Cycle 11: 2402–2407 [DOI] [PubMed] [Google Scholar]
- 13. Demidenko ZN, Zubova SG, Bukreeva EI, Pospelov VA, Pospelova TV, Blagosklonny MV (2009) Rapamycin decelerates cellular senescence. Cell Cycle 8: 1888–1895 [DOI] [PubMed] [Google Scholar]
- 14. Blagosklonny MV (2012) Rapalogs in cancer prevention: anti‐aging or anticancer? Cancer Biol Ther 13: 1349–1354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Abdelmohsen K, Panda A, Kang MJ, Xu J, Selimyan R, Yoon JH, Martindale JL, De S, Wood WH III, Becker KG et al (2013) Senescence‐associated lncRNAs: senescence‐associated long noncoding RNAs. Aging Cell 12: 890–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Montes M, Nielsen MM, Maglieri G, Jacobsen A, Hojfeldt J, Agrawal‐Singh S, Hansen K, Helin K, van de Werken HJG, Pedersen JS et al (2015) The lncRNA MIR31HG regulates p16(INK4A) expression to modulate senescence. Nat Commun 6: 6967 [DOI] [PubMed] [Google Scholar]
- 17. Ozes AR, Miller DF, Ozes ON, Fang F, Liu Y, Matei D, Huang T, Nephew KP (2016) NF‐kappaB‐HOTAIR axis links DNA damage response, chemoresistance and cellular senescence in ovarian cancer. Oncogene 35: 5350–5361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sang B, Zhang YY, Guo ST, Kong LF, Cheng Q, Liu GZ, Thorne RF, Zhang XD, Jin L, Wu M (2018) Dual functions for OVAAL in initiation of RAF/MEK/ERK prosurvival signals and evasion of p27‐mediated cellular senescence. Proc Natl Acad Sci USA 115: E11661–e11670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hu WL, Jin L, Xu A, Wang YF, Thorne RF, Zhang XD, Wu M (2018) GUARDIN is a p53‐responsive long non‐coding RNA that is essential for genomic stability. Nat Cell Biol 20: 492–502 [DOI] [PubMed] [Google Scholar]
- 20. Wang AS, Dreesen O (2018) Biomarkers of cellular senescence and skin aging. Front Genet 9: 247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Campisi J (2013) Aging, cellular senescence, and cancer. Annu Rev Physiol 75: 685–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kuilman T, Michaloglou C, Mooi WJ, Peeper DS (2010) The essence of senescence. Genes Dev 24: 2463–2479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rozan LM, El‐Deiry WS (2007) p53 downstream target genes and tumor suppression: a classical view in evolution. Cell Death Differ 14: 3–9 [DOI] [PubMed] [Google Scholar]
- 24. Perry RP, Kelley DE (1970) Inhibition of RNA synthesis by actinomycin D: characteristic dose‐response of different RNA species. J Cell Physiol 76: 127–139 [DOI] [PubMed] [Google Scholar]
- 25. Cooper MP, Qu L, Rohas LM, Lin J, Yang W, Erdjument‐Bromage H, Tempst P, Spiegelman BM (2006) Defects in energy homeostasis in Leigh syndrome French Canadian variant through PGC‐1alpha/LRP130 complex. Genes Dev 20: 2996–3009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Correa S, Binato R, Du Rocher B, Ferreira G, Cappelletti P, Soares‐Lima S, Pinto LF, Mencalha A, Abdelhay E (2014) ABCB1 regulation through LRPPRC is influenced by the methylation status of the GC ‐100 box in its promoter. Epigenetics 9: 1172–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jesse S, Bayer H, Alupei MC, Zugel M, Mulaw M, Tuorto F, Malmsheimer S, Singh K, Steinacker J, Schumann U et al (2017) Ribosomal transcription is regulated by PGC‐1alpha and disturbed in Huntington's disease. Sci Rep 7: 8513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mercer TR, Edwards SL, Clark MB, Neph SJ, Wang H, Stergachis AB, John S, Sandstrom R, Li G, Sandhu KS et al (2013) DNase I‐hypersensitive exons colocalize with promoters and distal regulatory elements. Nat Genet 45: 852–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Evans T, Schon E, Gora‐Maslak G, Patterson J, Efstratiadis A (1984) S1‐hypersensitive sites in eukaryotic promoter regions. Nucleic Acids Res 12: 8043–8058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang T, Marand AP, Jiang J (2016) PlantDHS: a database for DNase I hypersensitive sites in plants. Nucleic Acids Res 44: D1148–D1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang X, Tang N, Hadden TJ, Rishi AK (2011) Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta 1813: 1978–1986 [DOI] [PubMed] [Google Scholar]
- 32. MacLachlan TK, Takimoto R, El‐Deiry WS (2002) BRCA1 directs a selective p53‐dependent transcriptional response towards growth arrest and DNA repair targets. Mol Cell Biol 22: 4280–4292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fabbro M, Henderson BR (2008) BARD1 regulates BRCA1‐mediated transactivation of the p21WAF1/CIP1 and Gadd45 promoters. Cancer Lett 263: 189–196 [DOI] [PubMed] [Google Scholar]
- 34. Hussain T, Saha D, Purohit G, Kar A, Kishore Mukherjee A, Sharma S, Sengupta S, Dhapola P, Maji B, Vedagopuram S et al (2017) Transcription regulation of CDKN1A (p21/CIP1/WAF1) by TRF2 is epigenetically controlled through the REST repressor complex. Sci Rep 7: 11541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cao L, Zhang P, Li J, Wu M (2017) LAST, a c‐Myc‐inducible long noncoding RNA, cooperates with CNBP to promote CCND1 mRNA stability in human cells. Elife 6: e30433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sarek G, Vannier JB, Panier S, Petrini JHJ, Boulton SJ (2015) TRF2 recruits RTEL1 to telomeres in S phase to promote t‐loop unwinding. Mol Cell 57: 622–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Galanos P, Vougas K, Walter D, Polyzos A, Maya‐Mendoza A, Haagensen EJ, Kokkalis A, Roumelioti FM, Gagos S, Tzetis M et al (2016) Chronic p53‐independent p21 expression causes genomic instability by deregulating replication licensing. Nat Cell Biol 18: 777–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Aliouat‐Denis CM, Dendouga N, Van den Wyngaert I, Goehlmann H, Steller U, van de Weyer I, Van Slycken N, Andries L, Kass S, Luyten W et al (2005) p53‐independent regulation of p21Waf1/Cip1 expression and senescence by Chk2. Mol Cancer Res 3: 627–634 [DOI] [PubMed] [Google Scholar]
- 39. Li J, Poi MJ, Tsai M‐D (2011) Regulatory mechanisms of tumor suppressor P16INK4A and their relevance to cancer. Biochemistry 50: 5566–5582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu S, Wang X, Zhao Q, Liu S, Zhang H, Shi J, Li N, Lei X, Zhao H, Deng Z et al (2015) Senescence of human skin‐derived precursors regulated by Akt‐FOXO3‐p27(KIP(1))/p15(INK(4)b) signaling. Cell Mol Life Sci 72: 2949–2960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kim WY, Sharpless NE (2006) The regulation of INK4/ARF in cancer and aging. Cell 127: 265–275 [DOI] [PubMed] [Google Scholar]
- 42. Austin S, St‐Pierre J (2012) PGC1alpha and mitochondrial metabolism–emerging concepts and relevance in ageing and neurodegenerative disorders. J Cell Sci 125: 4963–4971 [DOI] [PubMed] [Google Scholar]
- 43. Zou J, Yue F, Jiang X, Li W, Yi J, Liu L (2013) Mitochondrion‐associated protein LRPPRC suppresses the initiation of basal levels of autophagy via enhancing Bcl‐2 stability. Biochem J 454: 447–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cui J, Wang L, Ren X, Zhang Y, Zhang H (2019) LRPPRC: a multifunctional protein involved in energy metabolism and human disease. Front Physiol 10: 595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vechetti‐Junior IJ, Bertaglia RS, Fernandez GJ, de Paula TG, de Souza RW, Moraes LN, Mareco EA, de Freitas CE, Aguiar AF, Carvalho RF et al (2016) Aerobic exercise recovers disuse‐induced atrophy through the stimulus of the LRP130/PGC‐1alpha complex in aged rats. J Gerontol A Biol Sci Med Sci 71: 601–609 [DOI] [PubMed] [Google Scholar]
- 46. Ohtake F, Takeyama K, Matsumoto T, Kitagawa H, Yamamoto Y, Nohara K, Tohyama C, Krust A, Mimura J, Chambon P et al (2003) Modulation of oestrogen receptor signalling by association with the activated dioxin receptor. Nature 423: 545–550 [DOI] [PubMed] [Google Scholar]
- 47. Sandri M, Lin J, Handschin C, Yang W, Arany ZP, Lecker SH, Goldberg AL, Spiegelman BM (2006) PGC‐1alpha protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy‐specific gene transcription. Proc Natl Acad Sci USA 103: 16260–16265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. de Keizer PL, Packer LM, Szypowska AA, Riedl‐Polderman PE, van den Broek NJ, de Bruin A, Dansen TB, Marais R, Brenkman AB, Burgering BM (2010) Activation of forkhead box O transcription factors by oncogenic BRAF promotes p21cip1‐dependent senescence. Cancer Res 70: 8526–8536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bourgeois B, Madl T (2018) Regulation of cellular senescence via the FOXO4‐p53 axis. FEBS Lett 592: 2083–2097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Baar MP, Brandt RMC, Putavet DA, Klein JDD, Derks KWJ, Bourgeois BRM, Stryeck S, Rijksen Y, van Willigenburg H, Feijtel DA et al (2017) Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell 169: 132–147.e116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhang P, Cao L, Fan P, Mei Y, Wu M (2016) LncRNA‐MIF, a c‐Myc‐activated long non‐coding RNA, suppresses glycolysis by promoting Fbxw7‐mediated c‐Myc degradation. EMBO Rep 17: 1204–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Khan MR, Xiang S, Song Z, Wu M (2017) The p53‐inducible long noncoding RNA TRINGS protects cancer cells from necrosis under glucose starvation. EMBO J 36: 3483–3500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yang F, Zhang H, Mei Y, Wu M (2014) Reciprocal regulation of HIF‐1alpha and lincRNA‐p21 modulates the Warburg effect. Mol Cell 53: 88–100 [DOI] [PubMed] [Google Scholar]
- 54. Wrann CD, Eguchi J, Bozec A, Xu Z, Mikkelsen T, Gimble J, Nave H, Wagner EF, Ong SE, Rosen ED (2012) FOSL2 promotes leptin gene expression in human and mouse adipocytes. J Clin Invest 122: 1010–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Dataset EV1
Dataset EV2
Dataset EV3
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Data Availability Statement
Raw and normalized data files for the microarray analysis have been deposited in the NCBI Gene Expression Omnibus under accession number GSE39694 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE39694) and GSE137513 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE137513).