

Abstract
Caspase-8, a well-characterized initiator of apoptosis, has also been found to play non-apoptotic roles in cells. In this study, we reveal that caspase-8 can induce cell death in a special way, which does not depend on activation of caspases and mitochondrial initiation. Instead, we prove that caspase-8 can cause lysosomal deacidification and thus lysosomal membrane permeabilization. V-ATPase is a multi-subunit proton pump that acidifies the lumen of lysosome. Our results demonstrate that caspase-8 can bind to the V0 domain of lysosomal Vacuolar H+-ATPase (V-ATPase), but not the V1 domain, to block the assembly of functional V-ATPase and alkalinize lysosomes. We further demonstrate that the C-terminal of caspase-8 is mainly responsible for the interaction with V-ATPase and can suffice to inhibit survival of cancer cells. Interestingly, regardless of the protein level, it is the expression rate of caspase-8 that is the major cause of cell death. Taken together, we identify a previously unrevealed caspase-8-mediated cell death pathway different form typical apoptosis, which could render caspase-8 a particular physiological function and may be potentially applied in treatments for apoptosis-resistant cancers.
Keywords: caspase-8, lysosome, V-ATPase, cell death
Graphical Abstract
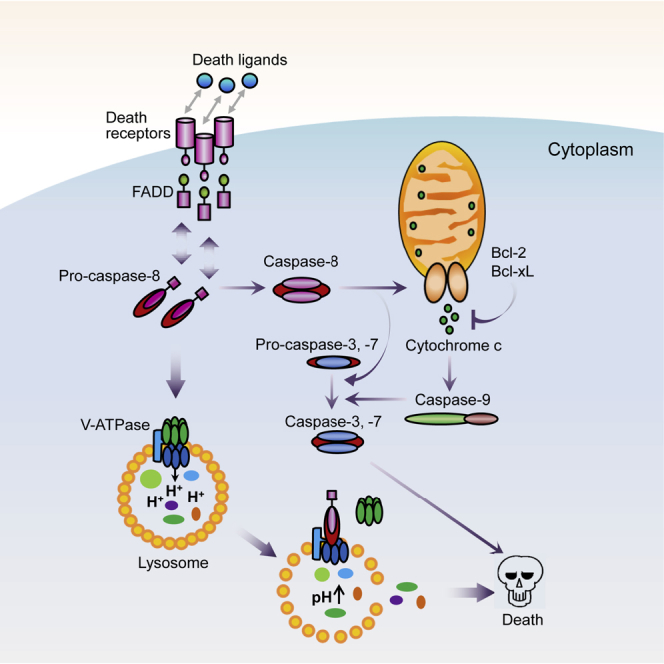
Caspase-8, a well-characterized initiator of apoptosis, has also been found to play non-apoptotic roles in cells. Zhong et al. show that overexpression of caspase-8 induces apoptosis-independent cell death in cancer cells via causing lysosomal deacidification and thus lysosomal membrane permeabilization, which could be potentially applied in treatments for apoptosis-resistant cancers.
Introduction
The equilibrium between cell division and cell death is tightly controlled, and the useless or faulty elements can effectively be eliminated by regulated cell death.1 There exist a number of patterns of cell death. Apoptosis, as the best known and characterized form of cell death, is characterized by morphologic changes such as cytoplasmic shrinkage, chromatin condensation, and DNA fragmentation, culminating with the formation of apoptotic bodies that can be removed by phagocytosis.2 In contrast to apoptosis as an inherently controlled cellular death program, necrosis is a more troubled way of dying, and it is characterized by cellular swelling and plasma membrane rupture, leading to release of the cellular contents and inflammatory response and terminating with the disposal of cell corpses in the absence of chromatin condensation and lysosomal involvement. Lysosome-dependent cell death, as another type of cell death, is initiated by perturbations of intracellular homeostasis and demarcated by lysosomal membrane permeabilization (LMP).2 It has recently become clear that there is crosstalk in the molecular mechanisms relevant to each form of cell death.
The initiation of apoptosis relies on the activation of a series of cysteine-dependent aspartyl-specific proteases known as caspases. There are two categories of apoptosis-associated caspases, the initiator caspases and the executioner caspases.3 Caspase-8, as an apical caspase, is involved in the extrinsic pathway of apoptosis in answer to ligations of the death receptors. Ligand binding induces oligomerization of receptor and recruitment of the adaptor proteins (FADD), which in turn leads to recruitment and activation of caspase-8.3 Active caspase-8 goes on to either directly initiate apoptosis by activating the executioner caspase (caspase-3 or caspase-7), or activate the intrinsic (mitochondrial) apoptotic pathway through cleaving BID.4 Then, cytochrome c is released from the mitochondria and leads to the formation of the apoptosome that consists of APAF1, cytochrome c, ATP and caspase-9, and caspase-9 activation. The active caspase-9 then cleaves and activates the executioner caspases.4 In addition, caspase-8 has also been found to play a number of non-apoptotic roles in cells. For example, caspase-8 is required for cytokine-induced proliferation of hemopoietic progenitor cells, and it has an important role in suppressing RIPK1/RIPK3-mediated necrosis to assure embryo development and erythropoiesis.5 Caspase-8 is also crucial for processing pro-interleukin (IL)-1β in response to fungi and mycobacteria, so as to protect immunity.6 In addition, caspase-8 can function within a multi-protein complex to orchestrate H2AX phosphorylation, sensing the DNA damage in hepatocytes.7 Caspase-8 has also been found to enhance cell migration and cell-matrix adhesion.8 Thus, the non-apoptosis roles of caspase-8 in cells are far beyond what we have known.
Lysosome is a membrane-bound organelle containing more than 50 soluble acid hydrolases, and its characteristic is the acidic lumen (pH 4.5–5.0), which is optimal for sets for hydrolase activity and conducive to macromolecule degradation.9 Vacuolar H+-ATPase (V-ATPase), a multi-component complex consisting of a membrane-immersed V0 domain and cytosol-exposed V1 domain, guarantees the acidic environment of lysosome and could utilize energy from ATP to transport protons from cytosol to the intralysosomal fraction against the electrochemical gradient. Therefore, V-ATPase plays a pivotal role in acidifying the lysosomes. Loss of function of V-ATPase, such as V1 domain dissociation, can lead to lysosomal alkalinization, ending up with lysosomal dysfunction and/or LMP. LMP can then allow the leakage of intralysosomal contents to trigger cell death in a caspase-dependent or caspase-independent pathway.10
It has become clear that there is a tight connection between apoptosis and lysosome-dependent cell death. Lysosomal membrane rupture can provoke apoptosis, as cathepsins can catalyze the proteolytic activation or inactivation of several substrates, such as BID, BAX, anti-apoptotic Bcl-2 family proteins, and XIAP, which are involved in mitochondrial outer membrane permeabilization (MOMP).11, 12, 13 In some circumstances, LMP appears to occur downstream of MOMP as a result of apoptotic signaling, forming an epiphenomenon of intrinsic apoptosis.14 In our study, we report that caspase-8 can directly activate lysosome-associated cell death through inhibiting V-ATPase activity and initiating LMP but not depending on caspase activation and mitochondrial initiation. These findings expand our understanding of caspases and could be applied in treatments for apoptosis-resistant cancers.
Results
Caspase-8 Overexpression Initiates Cell Death
Caspase-8, as an apical caspase, can initiate programmed cell death. In this study, we tested the effect of caspase-8 overexpression on HeLa and MCF-7 cells, two type II cell lines that relied on caspase-8-mediated cleavage and mitochondrial signal amplification.15 Gain of function of caspase-8 was performed in HeLa and MCF-7 cells and the number of cells was examined. Our results showed that caspase-8 overexpression led to significantly decreased cell number in both cancer cells (Figure 1A). Additionally, the hallmarks of classical apoptotic proteins, including cleaved caspase-8, caspase-3, caspase-7, and the poly(ADP-ribose) polymerase (PARP), were induced by caspase-8 overexpression (Figure 1B). Furthermore, a biosensor, Venus-based caspase-3-like activity indicator (VC3AI), was utilized, and it is a cyclized dead fluorescence protein but can display green fluorescence upon cleavage by activated capsases.16 As shown in Figure 1C, caspase-8 overexpression significantly activated VC3AI in MCF-7 cells based on green fluorescence, which was also confirmed by western blot analysis (Figure 1D). Annexin V/propidium iodide (PI) flow cytometric analyses also demonstrated that caspase-8 overexpression induced obvious apoptosis in HeLa and MCF-7 cells (Figure 1E; Figure S1A). Taken together, these results show that exogenously overexpressing pro-caspase-8 in cells seems to initiate typical caspase-mediated cell death, consistent with previous reports that cell death is assumed to result from the auto-activation of exogenously expressing caspase-8.17
Figure 1.

Caspase-8 Initiates Cell Death in Cancer Cells
(A) Proliferation of HeLa and MCF-7 cells expressing vector or caspase-8 in 3 days. (B) Western blot analysis of classical apoptotic proteins (cleaved caspase-8, caspase-3/caspase-7, and PARP) in HeLa and MCF-7 cells after transfection with vector or caspase-8. (C) Cell death in MCF-7/VC3AI cells transfected with vector or caspase-8. Left panel, fluorescence images of MCF-7 cells expressing VC3AI, a caspase-3/caspase-7 activation indicator. Green fluorescence indicates apoptosis. Right panel, quantification of cell death of MCF-7/VC3AI cells based on the fluorescent cells. Scale bar, 100 μm. (D) Western blot analysis of cleavage of VC3AI in MCF-7/VC3AI cells after transfection with vector or caspase-8. (E) Caspase-8 overexpression induced apoptosis, as measured by annexin V/PI flow cytometric analyses. HeLa and MCF-7 cells were transfected with vector or caspase-8, and apoptotic cells (annexin V+ PI− and annexin V+ PI+) were detected by the binding of annexin V to externalized phosphatidylserine in conjunction with PI, which is a dye excluded from viable cells. Images of MCF-7 cells are shown in Figure S1A. Each bar represents the mean ± SD for triplicate experiments.
Caspase-8 Overexpression Induces Cell Death Independently of Its Catalytic Activity and Mitochondria
We speculated that the cancer cells overexpressing a mutated caspase-8 deprived of catalytic activity would not undergo significant cell death. Therefore, we constructed a catalytically inactive point mutant of caspase-8 (casp-8CI), C360S, that was proven to have no detectable apoptotic activity,18 as well as an uncleavable double points mutant of caspase-8 (casp-8UC), D374A and D384A, that was not cleaved to form active dimers.19 Surprisingly, we found that overexpression of both mutants in HeLa and MCF-7 cells still induced cell death at levels comparable to those that resulted from the wild-type caspase-8 (Figure 2A), but caspase-3 or caspase-7 was not cleaved by both mutants (Figure 2B). This result suggests that enzyme activity is not required for the cell death caused by overexpressing caspase-8.
Figure 2.

Caspase-8 Induces Cell Death Independent of Catalytic Activity and Mitochondria
(A) Quantification of cell death of HeLa and MCF-7 cells based on the negative correspondence to ATP level. Cells were transfected with vector, caspase-8, caspase-8 catalytic inactive point mutation (casp-8CI), or caspase-8 uncleavable mutation (casp-8UC). The level of ATP was examined, and the percentage of dead cells was calculated by subtracting the percentage of living cells from 100%. Each bar represents the mean ± SD for triplicate experiments. (B) The efficiency of expression of caspase-8 or its mutations and the cleavage of caspase-3/caspase-7 in HeLa and MCF-7 cells was verified by western blot. (C) The efficiency of lentivirus-mediated Bcl-2 or Bcl-xL expression in MCF-7/VC3AI cells was verified by western blot. (D) Fluorescence images of MCF-7/VC3AI cells stably expressing vector, Bcl-2, or Bcl-xL after treatment with TNF-α (50 ng/mL) for 24 h. Scale bar, 100 μm. (E) Proliferation of MCF-7/VC3AI cells overexpressing Bcl-2 or Bcl-xL after transfection with vector or caspase-8 for 3 days. Each bar represents the mean ± SD for triplicate experiments. Scale bar, 100 μm. (F) Efficiency of caspase-8 overexpression in MCF-7/VC3AI cells overexpressing Bcl-2 or Bcl-xL was verified by western blot.
Next, we tested whether the caspase-8-induced cell death was mediated through the mitochondrial pathway that was involved in the intrinsic apoptotic pathway. The anti-apoptotic proteins, Bcl-2 and Bcl-xL, which were well-characterized to prevent mitochondria-dependent apoptosis,20 thus were overexpressed in MCF-7 cells (Figure 2C). MCF-7 cells lack caspase-3 and undergo a unique caspase-8-mitochondira-mediated cell death upon tumor necrosis factor (TNF)-α triggering.21 When MCF-7/VC3AI cells were treated with TNF-α, we observed apparent apoptosis indicated by VC3AI fluorescence that was completely blocked by overexpression of Bcl-2 or Bcl-xL (Figure 2D), demonstrating that Bcl-2 or Bcl-xL overexpression prevented MCF-7 cells from mitochondria-mediated apoptosis. However, when exogenous capsase-8 was expressed in MCF-7/VC3AI cells overexpressing Bcl-2 or Bcl-xL, it still strongly induced cell death (Figures 2E and 2F). Similar results were also obtained with HeLa cells that responded to mitochondria-mediated apoptosis by TNF-α co-treatment with cycloheximide (CHX), where CHX was used to block the activation of the nuclear factor κB (NF-κB) pathway and thus sensitized HeLa cells to TNF-α.22 HeLa cells stably expressed Bcl-2, Bcl-xL (Figures S1B and S1C), or Bcl-xL-GFP (Figures S1D–S1G) and were resistant to apoptosis induced by TNF-α plus CHX but sensitive to exogenously expressed caspase-8. Taken together, our results clearly show that exogenous expression of caspase-8 induces cell death independently of its enzymatic activity and mitochondria.
Caspase-8 Overexpression Induces Cell Death Independently of Caspase Cascades
HeLa cells implement the intrinsic apoptosis mediated through BID cleavage by activated caspase-8.23 When BID was knocked down (Figure 3A), TNF-α-induced cell death was significantly reversed in HeLa cells (Figure 3B). However, cell death induced by caspase-8 overexpression was not rescued at all by BID knockdown (Figure 3C), suggesting that blocking the downstream pathway of caspase-8 in the apoptotic pathway has little effect on cell death induced by exogenously expressed caspase-8.
Figure 3.

Bypassing the Caspase Pathway, Caspase-8 Leads to Cell Death
(A) Western blot to validate the efficiency of lentivirus-mediated knockdown of BID in HeLa cells. (B) HeLa cells with BID knockdown were treated with TNF-α (50 ng/mL) associated with CHX (10 μg/mL) for 12 h, and cell death was evaluated based on the negative correspondence to ATP level. (C) Proliferation of HeLa cells with BID knockdown after transfection with vector or caspase-8 for 3 days. (D) Proliferation of HeLa cells expressing vector or caspase-8 in the presence or absence of Z-VAD-FMK (100 mM) for 3 days. (E) Knockout of caspase-3, caspase-7, caspase-8, and caspase-9 in 4KO HeLa cell line were verified by western blot. (F) Wild-type and 4KO HeLa cells were treated with TNF-α (50 ng/mL) combined with CHX (10 μg/mL), ABT737 (10 μM), or cisplatin (50 μg/mL) for 24 h, and then stained with PI (2 μg/mL) before imaging. Red PI fluorescence indicates dead cells. The fraction of wild-type or 4KO HeLa cells with PI fluorescence is shown below. Scale bar, 100 μm. (G) Western blot analysis of introduction of caspase-8 in wild-type or 4KO HeLa cells. (H) Proliferation of wild-type and 4KO HeLa cells introduced with caspase-8 for 3 days. Each bar represents the mean ± SD for triplicate experiments. Scale bar, 100 μm.
Next, we investigated whether the pro-apoptotic caspases were involved in cell death induced by caspase-8 overexpression. We first tested whether the activities of caspases were required for exogenous caspase-8-mediated cell death. A pan-caspase inhibitor, Z-VAD-FMK,24 was used to treat HeLa cells, and we did not observed any protective effect against cell death by caspase-8 overexpression (Figure 3D). These results suggest that apoptotic caspases could not be involved in the cell death induced by exogenously expressed caspase-8.
To further exclude the involvement of caspases in exogenous caspase-8-triggerred cell death, we used the CRISPR/Cas9 system25 to knock out the critical apoptosis-required caspase-3, caspase-7, caspase-8, and caspase-9 in HeLa cells (4KO), and the 4KO HeLa cell line was confirmed by western blot (Figure 3E) and sequencing (data not shown). Knockout of these caspases increased cell growth while decreasing metastasis (Figures S2A–S2D), but it highly resisted various apoptotic stimuli, such as TNF-α/CHX, ABT737 (a Bcl-2 inhibitor),26 and cisplatin (a primary chemotherapy drug) (Figure 3F). Nonetheless, 4KO HeLa cells still remained susceptible to exogenous caspase-8 expression similar to wild-type HeLa cells (Figures 3G and 3H), indicating that caspase-8 overexpression leads to cell death, bypassing the caspase cascades. Furthermore, when these caspases were expressed back to 4KO HeLa cells, we easily obtained cells re-expressing caspase-3, caspase-7, and caspase-9 or their mutants but not caspase-8 or its mutants (Figure S3). Collectively, these results suggest that exogenous caspase-8 expression induces a unique cell death pathway, regardless of the classic caspase-dependent or mitochondria-associated apoptosis.
To further compare the typical apoptosis and apoptosis-independent cell death induced by caspase-8, we re-expressed wild-type caspase-8 or mutant casp-8CI in HeLa cells with caspase-8 knockout (casp-8 KO). As expected, both wild-type and mutant proteins induced similar levels of cell death, about 20% at 24 h post-transfection (Figure S4A), again confirming that catalytic activity is not required for exogenous capsase-8-induced cell death. In the meantime, we treated these cells at 8 h post-transfection with TNF-α plus CHX to trigger apoptosis. Our results showed that the apoptotic proteins, including cleaved caspase-8 and caspase-3, were significantly induced in the wild-type and KO cells that re-expressed wild-type caspase-8, but not in the KO cells or those that re-expressed casp-8CI (Figure S4B) after TNF-α plus CHX treatment. We also observed significant dead cells (about 80%) in the wild-type and KO cells that re-expressed wild-type caspase-8, but not in casp-8-KO cells (Figure S4A). Interestingly, regardless of stimulation by TNF-α plus CHX, casp-8-KO cells re-expressing casp-8CI underwent cell death to a similar extent (Figure S4A). These results suggest that the enzymatic activity of capsase-8 is necessary for canonical extrinsic apoptosis but not for cell death revealed in our current study.
Although caspase-8 acted as the initiator of extrinsic apoptosis, it actually repressed MLKL-mediated necroptosis.27 However, it was recently reported that the expression of enzymatically inactive capase-8 could cause embryonic lethality in mice by inducing necroptosis and pyroptosis in some specific tissues.28,29 Therefore, we next investigated whether necroptosis and pyroptosis were involved in casp-8CI-induced cell death. We treated the wild-type or 4KO HeLa cells with the RIPK1 inhibitor necrostatin-1 (Nec-1) or the MLKL inhibitor necrosulfonamide (NSA) and found that cell death in both wild-type and 4KO HeLa cells induced by casp-8CI expression was not inhibited at all by Nec-1 or NSA (Figure S5), suggesting that the cell death induced by exogenous casp-8CI is not necroptosis. In fact, the pan-caspases inhibitor Z-VAD-FMK and NSA also blocked pyroptotic death by suppressing the cleavage of gasdermin D (GSDMD), the pore-forming effector protein of pyroptosis.30,31 Taken together, these results actually exclude the involvement of pyroptosis and necroptosis.
Caspase-8 Is Functionally Linked to the Stability of Lysosomes in Cancer Cells
Interestingly, exogenous caspase-8 expression substantially triggered typical apoptotic events (Figure 1), although it elicited cell death completely independent of these pathways (Figures 2 and 3). Among cell death types reported up to date, damaged lysosomes can release hydrolases to elicit cell death and meanwhile they often activate the typical apoptotic pathways (Figure S6A).9,10,13,32. In this study, we used chloroquine (CQ), a lysosomotropic agent that diffuses through membranes to accumulate and quench the acidic pH in lysosomes and thus leads to lysosome rupture,33 to confirm that lysosomal damage indeed induced apoptotic events, including caspase activation and PARP cleavage in MCF-7 and HeLa cells (Figures S6B and S6C), precisely as for caspase-8 overexpression. Therefore, we next investigated whether lysosomes were involved in caspase-8-induced cell death. We first examined the subcellular localization of endogenous caspase-8 in HeLa cells. The immunofluorescence imaging showed that caspase-8 was partially co-localized with lysosome-associated membrane protein 1 (LAMP1), routinely a lysosome marker,34 suggesting that caspase-8 may interact with lysosomes (Figure 4A). We then tried to determine the effect of exogenously expressed caspase-8 on lysosomes. Caspase-8-GFP was transiently transfected into 4KO HeLa cells, and at 48 h post-transfection cells were stained with LysoTracker red, a red fluorescent dye that stains acidic organelles.35 We found that cells with caspase-8-GFP showed significantly decreased intensity of LysoTracker red fluorescence compared to the adjacent cells without caspase-8-GFP (Figures 4B and 4C). These results suggest that exogenously expressed caspase-8 could cause the dysfunction in lysosomes via dysregulating the acidic lumens.
Figure 4.

Caspase-8 Is Functionally Linked to the Stability of Lysosomes in Cancer Cells
(A) Subcellular localization of caspase-8 in HeLa cells. The distribution of endogenous caspase-8 or lysosomes was detected by immunofluorescence microscopy with antibodies against caspase-8 or LAMP1. DAPI staining was included to visualize the cell nucleus. Scale bar, 5 μm. (B) 4KO HeLa cells transfected with caspase-8-GFP were stained with LysoTracker red (0.5 μM) and then visualized under fluorescence microscopy. LysoTracker red staining was included to visualize the cell lysosomes, and Hoechst staining was utilized to visualize the nucleus. Scale bar, 15 μm. (C) The expression of the caspase-8-GFP in 4KO HeLa cells was verified by western blot. (D) 4KO HeLa cells re-introduced with vector or caspase-8 were stained with LysoSernser green (1 μM) for 15 min, and fluorescence was measured by a flow cytometer. The percentage of cells with decreased fluorescence intensity is indicated. Each bar represents the mean ± SD for triplicate experiments. ATPase inhibitor (1 μM of bafilomycin A) treatment for 3 h is a positive control for lysosomal alkalinization. (E) The re-introduction of caspase-8 in 4KO HeLa cells was verfied by western blot. (F) 4KO HeLa cells re-introduced with vector or caspase-8 were stained with 1.5 μg/mL of acridine orange (AO) at 37°C for 30 min, and red fluorescence was analyzed by a flow cytometer. Each bar represents the mean ± SD for triplicate experiments. (G) Fluorescence images of 4KO HeLa cells exposed to the lysosomotropic fluorophore AO, after re-introduction with vector or caspase-8. Loss of red fluorescence indicates a proton leak through the destabilized lysosomal membrane. Diminished granular red represents a decreased number of intact lysosomes. Scale bar, 5 μm. (H) Fluorescence images of 4KO HeLa cells expressing GFP-LGALS3, after re-introduction with caspase-8 or treatment with LLOMe (2 mM, 2 h). Changing of GFP-LGALS3 from diffuse to punctate after LLOMe treatment represents lysosome-specific membrane damage. Scale bar, 5 μm. The numbers of GFP dots in cells is shown below. (I) Fluorescence images of 4KO HeLa cells with antibody against cathepsin L, after re-introduction with vector or caspase-8. Scale bar, 5 μm. The maturation of cathepsin L was verified by western blot.
Furthermore, 4KO cells were transfected with caspase-8 or treated with bafilomycin A1, a V-ATPase inhibitor,36 and then stained with a LysoSensor green dye that becomes more fluorescent in an acidic environment and shows reduced fluorescence upon lysosomal alkalinization.37 Flow cytometric analyses showed that caspase-8 overexpression decreased the LysoSensor green fluorescence intensity in 4KO HeLa cells, similar to bafilomycin A1, a positive control for lysosomal alkalinization36 (Figures 4D and 4E; Figure S7A), suggesting the occurrence of lysosomal alkalinization. This speculation was further confirmed by the results with a sensitive cytochemical technique that permits visualization of the stability of lysosomes in living cells using acridine orange (AO) relocation and uptake methods. AO is a lysosomotropic base that accumulates in normal lysosomes, emitting bright red fluorescence under blue or green excitation light. When AO-loaded lysosomes are damaged, AO is released to the cytosol without fluorescence, and thus the intensity of red fluorescence in AO-exposed cells reflects the number of intact lysosomes.9 We found that caspase-8 expression, as well as bafilomycin A1, induced subdued and dispersed red fluorescence in 4KO HeLa cells (Figures 4F and 4G; Figure S7B), indicating the lysosomal damage.
As LMP resulting from lysosomal damage is a critical factor for cell death induction, we next determined the effect of exogenously expressed caspase-8 on LMP in 4KO HeLa cells using the tool of GFP-LGALS3. LGALS3/galectin-3, as a sugar-binding protein, was reported as being best suitable for LMP detection due to its widespread expression and rapid translocation to leaky lysosomes to bind lysosomal membrane sugar proteins.38 Therefore, the detection of GFP-LGALS3 puncta can indicate LMP. Our results showed that after treatment with l-leucyl-l-leucine methyl ester (LLOMe), a known LMP inducer,39 GFP-LGALS3 in 4KO HeLa cells formed puncta that were exclusively stained with LysoTracker red (Figure 4H). Importantly, caspase-8 expression induced the exactly similar GFP-LGALS3 puncta in 4KO HeLa cells (Figure 4H), strongly demonstrating the presence of lysosomal rupture. In addition, cathepsin L, the lysosomal cysteine protease, was examined by an immunofluorescence assay to determine whether the contents of lysosomes were permeated into cytoplasm. Our results showed that the granular distribution of cathepsin L was shifted into dispersion by caspase-8 expression in 4KO HeLa cells (Figure 4I). The maturation of cathepsin L was also enhanced by caspase-8 expression in 4KO HeLa cells (Figure 4I). Taken together, our results demonstrate that caspase-8 induces lysosome-associated cell death.
Caspase-8 Is Associated with the V0 Domain of V-ATPase In Vivo
V-ATPases are the ATP-dependent proton pumps that transport protons from the cytoplasm into the lumen of lysosome to maintain the acidic environment in lysosome.40 Nonfunctional V-ATPases are reported to be associated with cell death.40
To gain a mechanistic insight into the caspase-8-mediated rupture of lysosomes, total proteins from HeLa cells were extracted and co-immunoprecipitation (coIP) experiments were performed with antibodies detecting the endogenous proteins. coIP with antibodies against caspase-8 followed by immunoblotting (IB) with antibodies against subunit a or d in the V0 domain (subunit V0a or V0d), or against subunit A or C in the V1 domain (subunit V1A or V1C) of V-ATPase, revealed that caspase-8 interacts with the components of the V0 domain, but not the members in the V1 domain (Figure 5A). Reciprocally, coIP with antibodies against subunit V0a of V-ATPase followed by IB with antibodies against caspase-8 also demonstrated that caspase-8 was efficiently co-immunoprecipitated by subunit V0a of V-ATPase (Figure 5B). The interaction between caspase-8 and subunit V0a was also confirmed by coIP experiments in HeLa cells with caspase-8-GFP or FLAG-V0a expression (Figure S8). In contrast, caspase-3, caspase-7, and caspase-9 were not co-immunoprecipitated by subunit V0a of V-ATPase (Figure 5B). These results suggest that the specific structure or domain of caspase-8, which differs from that of caspase-3, caspase-7, and caspase-9, is involved in the interaction. These results support a notion that caspase-8 is associated with the V0 domain, but not the V1 domain, of V-ATPase in vivo.
Figure 5.

Caspase-8 Is Associated with the V0 Domain of V-ATPase In Vivo
(A) Whole-cell lysates from HeLa cells were immunoprecipitated with antibodies against caspase-8 or immunoglobulin G (IgG) followed by immunoblotting with the antibodies against the indicated proteins. (B) Whole-cell lysates from HeLa cells were immunoprecipitated with antibodies against subunit V0a or IgG followed by immunoblotting with the antibodies against the indicated proteins. (C) Co-localization of caspase-8 and subunit V0a of V-ATPase in HeLa and MCF-7 cells. Cells were stained with antibodies against caspase-8 or subunit V0a and analyzed by confocal microscopy. DAPI staining was utilized to visualize the nucleus. Scale bar, 5 μm. (D) Schematic diagram of caspase-8 deletion mutants. (E) 4KO cells were transfected with caspase-8 deletion mutants, and cell lysates were immunoprecipitated with antibody against GFP or IgG, followed by immunoblotting with the antibodies against the indicated proteins. (F) HeLa cells were transfected with vector or caspase-8, and cell lysates were immunoprecipitated with antibody against subunit V0a or IgG, followed by immunoblotting with the antibodies against the indicated proteins.
In addition, confocal microscopic analyses of HeLa and MCF-7 cells showed co-localization of endogenous caspase-8 and subunit V0a of V-ATPase, verifying the interaction between caspase-8 and V-ATPase in cancer cells (Figure 5C).
To further support the interaction between caspase-8 and V-ATPase, and to investigate the molecular details involved in this interaction, protein extracts from 4KO HeLa cells overexpressing GFP-fused deletion mutants of caspase-8 (Figure 5D) were immunoprecipitated with anti-GFP followed by immunoblotting with antibodies against V0a of V-ATPase. The results revealed that the region containing the P10 domain of caspase-8 was responsible for its interaction with the V0 domain of V-ATPase (Figure 5E). No interaction of subunit V0a was detected with the other mutants of caspase-8 we tested (Figure 5E). Therefore, our results identify the P10 domain of caspase-8 as responsible for interacting with the V0 domain of V-ATPase.
Assembling the V1 and V0 domains into functional V-ATPase is critical to maintain lysosomal acidification. In contrast, the dissociation of V1 from lysosomal membrane V0 can suppress V-ATPase activity.41 In order to determine whether caspase-8 could block the assembly of V-ATPase by associating with the V0 domain of V-ATPase, we performed coIP experiments on the overexpression of caspase-8. The results show that the association of V0 subunits with V1 subunits was reduced by caspase-8 expression (Figure 5F), indicating that overexpression of caspase-8 inhibits the structural integrity of V-ATPase in cancer cells.
P10 Domain of Caspase-8 Is Involved in Lysosome-Associated Cell Death
As the P10 region of caspase-8 was required for the interaction of caspase-8 with the V0 domain of ATPase, and to understand the biological significance of P10, we next investigated the effect of P10 on cell survival. To this end, 4KO HeLa cells were transfected with the GFP-fused deletion mutants of caspase-8 (Figure 5D). At 48 h after transfection, cell death was examined. Notably, overexpression of GFP-P10 of caspase-8 exerted more significant positive effects on cell death than did other deletion mutants (Figures 6A and 6B).
Figure 6.

P10 Domain of Caspase-8 Is Involved in Lysosome-Associated Cell Death
(A) 4KO HeLa cells were re-introduced with indicated deletion mutants of caspase-8 for 2 days, and cell death was evaluated based on the negative correspondence to ATP level. Each bar represents the mean ± SD for triplicate experiments. (B) The re-introduction of caspase-8 deletion mutants in 4KO HeLa cells was verified by western blot. (C) Wild-type, 4KO, or 4KO HeLa cells stably expressing GFP-P10 or P10-GFP were treated with different amounts of chloroquine to alkalized lysosomes for 24 h, and cell death was evaluated based on the negative correspondence to ATP level. Each bar represents the mean ± SD for triplicate experiments. (D) The efficiency of lentivirus-mediated GFP-P10 or P10-GFP expression in 4KO HeLa cells was verified by western blot. (E) Proposed model of caspase-8 in repressing ATPase assembling to induce cancer cell death.
To consolidate the involvement of P10 in inhibiting the V-ATPase-mediated maintenance of lysosomal homeostasis, CQ was utilized to induce lysosomal rupture in 4KO cells.33 We observed clear lysosomal swelling and LMP elicited by CQ treatment because of its capacity of leading to alkaline conditions within lysosomes, as indicated by the expanded LysoTracker red spots and the formation of GFP-LGALS3 puncta in 4KO cells (Figure S9). In response to elevated lysosomal pH by CQ, V-ATPase is apt to expedite the reversible assembly to maintain an acidic internal environment. We engineered the 4KO HeLa cells with stable overexpression of GFP-P10 or P10-GFP, and then stimulated the cells with CQ. We found that GFP-P10 or P10-GFP-overexpressed 4KO cells are more sensitive to CQ treatments (Figures 6C and 6D), indicating that P10 could functionally inhibit V-ATPase.
Therapeutic Application and Potential Physiological Role of Caspase-8-Mediated Cell Death
As shown above, we demonstrated that caspase-8 induced lysosome-associated cell death through direct expression. In this study, we utilized the Tet-On gene expression system to further determine whether the doxycycline (DOX)-induced expression of caspase-8 could lead to cell death. Our results showed that caspase-8 expression was successfully induced by DOX and also efficiently resulted in cell death in HeLa cells (Figure 7A).
Figure 7.

Therapeutic Application of Caspase-8-Mediated Cell Death in Cancer Cells
(A) Proliferation of Tet-On caspase-8 HeLa cells after treatment with DOX (5 nM) for 3 days. The cell images on the third day are shown. Scale bar, 100 μm. Induction of caspase-8 expression was verified by western blot. (B) Model of the cell suicide system (3× ERE-pE1b-TATA-rtTA-PminCMV-caspase-8). The Tet-On caspase-8 is under the control of the estrogen receptor response elements (EREs), which was followed by a transactivated minimal E1b promoter (pE1b-TATA)-driven reverse tetracycline-controlled transactivator (rtTA). Estradiol (E2) can activate ER to promote rtTA expression and further induce caspase-8 expression in the presence of DOX. (C) MCF-7 cells stably introduced the cell suicide system in (B) were treated with E2 (100 nM) and/or DOX (5 nM) for 48 h, and cell death was evaluated based on the negative correspondence to ATP level. Treatment with DMSO was regarded as control. Induction of caspase-8 expression was verified by western blot. (D) Tet-On caspase-8 HeLa cells were treated with different concentrations of DOX (1–50 nM) for 2 days, and the induction of caspase-8 was verified by western blot. Cell death was evaluated based on the negative correspondence to ATP level. Each bar represents the mean ± SD for triplicate experiments. Scale bar, 100 μm. (E) Tet-On caspase-8 HeLa cells were treated with 1 nM or 10 nM of DOX for 6, 12, or 24 h, and the expression of caspase-8 was examined by western blot. (F) Tet-On vector or Tet-On caspase-8 HeLa cells were persistently cultured with 1 nM DOX in the culture medium, and fold change in cell number in 3 days is shown. The expression of caspase-8 in Tet-On caspase-8 HeLa cells was verified by western blot.
Based on our current results, caspase-8 expression can trigger cell death independently of typical apoptotic pathways, and thus we next tried to build a cell suicide system using caspase-8 as a “suicide gene” in cancer cells (Figure 7B). Tet-On caspase-8 was constructed under the control of the estrogen receptor response elements (EREs) that was followed by a transactivated minimal E1b promoter (pE1b-TATA)-driven reverse tetracycline-controlled transactivator (rtTA) (Figure 7B). Estrogen receptor (ER) was an enhancer protein activated by steroid ligands, such as estradiol (E2), and expressed in ER+ breast cancer cells, including MCF-7. Therefore, we delivered this system to MCF-7 cells by lentivirus, and it was supposed that E2 would activate ER to promote rtTA expression, and may further induce caspase-8-mediated cell death in the presence of DOX (Figure 7B). Our results indeed showed that DOX significantly led to cell death in MCF-7 cells depending on E2, compared to the individual treatments (Figure 7C). Similarly, we could generate tumor-specific enhancer- or promoter-controlled Tet-On caspase-8 and deliver them in vivo by adeno-associated virus (AAV) or other feasible vehicles. In this way, we can convert tumor-specific enhancers or promoters to the targets of cancers by DOX.
Intriguingly, the expression of caspase-8 on E2 and/or DOX was not correlative with cell death (Figure 7C). Although E2 or DOX alone did not significantly trigger cell death, they also obviously gave rise to caspase-8 expression, which most likely resulted from the leaking expression. As such, exogenous caspase-8-induced cell death could be mediated by the expression rate but not the protein level. In order to test this speculation, we induced caspase-8 expression with different concentrations of DOX (1 nM to 50 nM) in the Tet-On caspase-8 HeLa cells. We noticed that although the expressions of caspase-8 were nearly increased to the same level after 48 h of DOX induction at all of the concentrations, 1 nM DOX induced far fewer dead cells than did others (Figure 7D). We then measured caspase-8 expression of Tet-On HeLa cells treated with 1 and 10 nM DOX at the different times and observed that 1 nM DOX-induced caspase-8 expression was indeed slower when compared to that by 10 nM DOX, although their protein levels almost reached to the flat even upon 12-h DOX induction (Figure 7E). To exclude the possibility that 1 nM DOX could take a long time to kill cells, we extended DOX treatments for days and found that cells upon induction with 10 nM DOX completely died, but those treated with 1 nM DOX survived and grew well. We then kept on culturing these cells with 1 nM DOX in the culture medium, and no significant cell death was observed. In fact, these cells proliferated normally despite that caspase-8 expression was sustained at a high level (Figure 7F). These results suggest that the fast expression rate of caspase-8 could be the main cause of induced cell death. Upon fast expression, caspase-8 could not be fully folded or partnered in time by its chaperonins, and then the immature capsase-8 probably elicits cell death.
Discussion
In this study, we reveal that caspase-8 can potentially induce cell death via interacting with the V0 domain of lysosomal V-ATPase to damage lysosomes, regardless of caspase activation and mitochondrial initiation (Figure 6E).
There was evidence that lysosomal V-ATPase activity can be controlled by different mechanisms, including regulating the assembly of V-ATPase, modulation of V-ATPase trafficking, and regulating the expression of V-ATPase subunits.41 Especially, V-ATPase activity in vivo is regulated via the unique mechanism referred to as “reversible dissociation,” a process that results in a transient pool of free cytosolic V1 and membrane integral V0 sectors that are functionally silenced.41 The reversible dissociation/reassembly can rapidly modulate V-ATPase activity in response to a variety of cues, including nutrient availability, growth factor stimulation, alkaline extracellular/vacuolar pH, activation of Ras/cyclic AMP (cAMP)/protein kinase A (PKA) pathway, and cell maturation.41 For example, increased assembly occurs in response to glucose withdrawal in a phosphatidylinositol 3-kinase (PI3K)- or mTORC1-dependent manner in mammalian cells.42 Amino acid starvation also increases V-ATPase assembly and improves V-ATPase-dependent lysosomal acidification.43 Interestingly, reassembly is reported to require the physical interaction of V-ATPase subunits with other proteins. Interaction of the glycolytic enzyme aldolase with subunits B and E of the V1 domain and subunits a of the V0 domains enhances V-ATPase assembly.44 In addition, the regulator of the ATPase of vacuolar and endosomal membranes (RAVE), a heterotrimeric complex composed of two proteins (Rav1p and Rav2p) and Skp1p, binds to subunits E, G, and C of the V1 domain and subunit a of the V0 domain of V-ATPase, and it is required for glucose-regulated assembly of V-ATPase.45 However, the proteins involved in destabilization of V-ATPase are currently unknown. In our study, we found that caspase-8 may induce dissociation of V-ATPase via the space-occupying interaction with the V0 domain to inhibit the recruitment of cytosolic V1 onto the membrane integral V0 sector. Our results identified a mechanism by which the protein-protein interaction can suppress V-ATPase activity by occupying the binding sites of V0 domains in V-ATPase and competitively inhibiting the recruitment of V1 domains.
In addition, we demonstrate that the interaction between caspase-8 and the V0 domains of V-ATPase inherently exists in the living cancer cells (Figures 5A–5C). This raises a concern why the endogenous caspase-8 does not induce cell death. It is possible that there exists a dynamic equilibrium between endogenous caspase-8 and the V0 domains of V-ATPase. Normally, endogenous caspase-8 is properly expressed, but due to fast exogenous expression, the neonatal immature caspase-8 possibly preferentially binds to V-ATPase to initiate lysosome-associated cell death.
Note that the bi-functional apoptosis regulator (BAR) can bridge caspase-8 and Bcl-2 into a protein complex to anchor caspase-8 in mitochondrial membranes.46 Therefore, it is possible that BAR is a potential detainer of caspase-8 to prevent the association of endogenous caspase-8 with V-ATPase. In addition, extrinsic apoptosis relies on the formation of a death-inducing signaling complex (DISC), which always includes FADD and caspase-8, and the presence of the FLICE-like inhibitory protein (cFLIP) in DISC determines if and how cells die. As it is clear that the procaspase-8 catalytic domain prefers heterodimerization to the cFLIP long (cFLIPL) caspase-like domain over homodimerization with catalytic domains of procaspase-8 themselves, procaspase-8 tends to be seized by cFLIPL.47 Therefore, we speculate that cFLIPL may be another captor of caspase-8. However, it is currently unclear whether BAR or cFLIPL represents the potential upstream regulator of the interaction between caspase-8 and V-ATPase in cancer cells. Beyond that, a non-apoptotic function of caspase-8 has been reported to function in the nucleus within a multi-protein complex to orchestrate H2AX phosphorylation.7 Thus, the enrichment of caspase-8 in specific subcellular organelles such as the nucleus or mitochondria may also be another potential influence factor determining the dynamic equilibrium.
Although how exactly the regulators mentioned above are coordinated in caspase-8-induced lysosome-associated cell death remains to be elucidated, our results demonstrate that the fast expression of caspase-8, not its protein level, could be the major trigger of cell death (Figures 7A–7C). This raises the possibility that the immature capsase-8 proteins, including those not fully folded or partnered with chaperonins, play a determinative role in cell death induction. Most likely, these immature proteins acquire the preferential ability to bind to some cell death-inducing proteins, such as V-ATPase. Therefore, it is possible that if some stimuli can promote endogenous caspase-8 expression, they should also give rise to cell death, complementary to other types of cell death, and may exert physiological or pathological roles.
Our results show that caspase-8 can directly induce LMP in an apoptosis-resistant cell model (Figure 4). Since LMP can trigger apoptosis by attacking mitochondria,10 lysosome-associated cell death is most often covered by apoptosis, as indicated in Figure 1. In fact, apoptotic stimuli can liberate caspase-8 in an active form, and thus during apoptosis the activated capsase-8 could also elicit lysosome-dependent cell death at the same time. For instance, in the late stage of apoptosis, apoptotic cells are often accompanied with the plasma membrane rupture,48 which could result from caspase-8-associated lysosome-mediated necrosis.
In caspase-related apoptosis, the anti-apoptotic Bcl-2 family proteins promote cancer cell survival by sequestering the pro-apoptotic proteins such as BID and BAX that act on the mitochondrial outer membrane to release cytochrome c and subsequently to activate the intrinsic apoptosis pathway through a caspase cleavage cascade.4 Indeed, several Bcl-2 antagonists are in clinical trials for cancer treatment. Nevertheless, these therapeutic agents are limited by the high expression of Mcl-1 and other molecular mechanisms that confer drug resistance.20 We report in the current study that the Bcl-2-blocked caspase pathway can be potentially circumvented by caspase-8 that interacts with V-ATPase to directly target lysosomes. Therefore, our results suggest a biologic significance that to promote caspase-8 expression is a potential therapeutic strategy for cancers with high expression of anti-apoptotic Bcl-2 family proteins, or for cancers resistant to the small-molecule inhibitors of Bcl-2. In addition, it was recently reported that mitochondria density in cells is a source of cell-to-cell variability, and cancer cells with high mitochondria density tend to resist TRAIL-induced cell death.49 Accelerating the expression of caspase-8 or its functional part could also be a plausible means to kill such subsets of cancers with high mitochondria density. We prove that the P10 fragment of caspase-8 can efficiently associate with the V0 domain of V-ATPase, induce cell death, and also sensitize cells to lysosomotropic agents. Furthermore, we build up a gene therapy system that contains Tet-On caspase-8 under the control of ER+ breast cancer-specific enhancer. Importantly, our system provides a feasible model that can expose the tumor-specific promoters or enhancers as the specific targets of cancers by DOX, which may be applied as a means of gene therapy.
Materials and Methods
Cell Culture and Transfection
HeLa, MCF-7, and 293T cells were obtained from the American Type Culture Collection (ATCC) and maintained according to ATCC recommendations. All cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) (BioInd, Israel) and 50 IU of penicillin/streptomycin (Invitrogen, USA) in a humidified atmosphere with 5% CO2 at 37°C. Transient transfections of expression plasmids in HeLa, MCF-7, and 293T cells were carried out using polyethyleneimine (PEI) (Sigma) according to the manufacturer’s recommendations.
Generation of 4KO HeLa Cell Line
pCDH-Cas9-2A-GFP-BSD was used to express Cas9. Single-guide RNAs (sgRNAs) were cloned into the pLentiGuide-Puro vector25 that had been linearized with BsmBI. Target sequence were used for each gene based on the GeCKO v2 library.25 The sgRNA sequences for human CASP were: sgCASP3, 5′-TGTTCCAAGGAATGACATCT-3′; sgCASP7, 5′-TCCTCGATACAAGATCCCAG-3′; sgCASP8, 5′-TCCTTTGCGGAATGTAGTCC-3′; and sgCASP9, 5′-GTTTCCGGTCTGAGAACCTC-3′. In order to generate the 4KO HeLa cell line, the pCDH-Cas9-2A-GFP-BSD and four single pLentiGuide-Puro-sgCASP plasmids were co-transfected into HeLa cells in six-well plates using Lipofectamine 3000. Cells were single cell sorted with a flow cytometer based on green fluorescence into the wells of a 96-well plate containing 200 μL of DMEM supplemented with 10% FBS. Cells were grown for 3 weeks, and the resultant colonies were trypsinized and expanded. Clones were validated for knockout of CASP3, CASP7, CASP8, and CASP9 by western blot and sequencing.
Gene Construction and Lentivirus Production
The cDNAs for wild-type of caspase-3, caspase-7, caspase-8, and caspase-9 were purchased from Addgene (http://www.addgene.org/protocols/plko/). Caspase-8 deletion mutants were prepared by separate fragments of full length, and point mutants of caspase-3, caspase-7, caspase-8, and caspase-9 were introduced by two-round PCR from the wild-type templates. The expression plasmids were generated by subcloning the cDNA fragment into the lentiviral expression vectors, pCDH-CMV-cDNA. The pLKO.1 lentiviral RNA interference (RNAi) expression system was used to construct lentiviral short hairpin RNA (shRNA) for genes. The sequences of shRNA used in this study included the following: shCTR, 5′-CCTAAGGTTAAGTCGCCCTCG-3′, shBID-1, 5′-GTGAGGAGCTTAGCCAGAAAT-3′; and shBID-2, 5′-CAGGGATGAGTGCATCACAAA-3′. For viral packaging, the lentiviral expression or knockdown plasmid was co-transfected together with pCMV-dR8.91 and pCMV-VSV-G into the 293T cells using PEI co-precipitation at 10:5:5 μg (for a 10-cm dish). The transfection medium containing PEI and plasmid mixture was replaced with fresh complete medium after incubation for 6 h. Media containing virus was collected 48 h later, clarified by filtration, and concentrated by ultracentrifugation. The virus was resuspended in the appropriate amount of complete growth medium and stored at −80°C. Cancer cells were infected with the viruses at the titer of 100% infection in the presence of Polybrene (10 μg/mL) for 48 h, and then cells were selected with puromycin or neomycin.
Reagents and Antibodies
Bafilomycin A1, TNF-α, PI, CHX, CQ, AO, cisplatin, DOX, and E2 were purchased from Sigma. Nec-1 was purchased from Univ. Necrosulfonamide (NSA) was purchased from Ark Pharm. LysoTracker red (L7528) and LysoSensor green (L7535) were obtained from Invitrogen. Z-VAD-FMK and ABT-737 were provided by BioVision Technologies. An annexin V-fluorescein isothiocyanate (FITC) apoptosis detection kit (556547) was purchased from BD Biosciences. Protein A/G PLUS-agarose (sc-2003) was purchased from Santa Cruz. LLOME (sc-285992) was from Santa Cruz. Puromycin and blasticidin were purchased from Invitrogen, and G418 was from Gibco. Antibodies used were as follows: anti-caspase-8 (Cell Signaling Technology, #4790, Cell Signaling Technology, #9746, Proteintech, 13423-1-AP, 1:150 for immunofluorescence [IF] and 1:1,000 for western blot); anti-caspase-3 (Cell Signaling Technology, #9662, 1:1,000 for western blot); anti-cleaved caspase-3 (Wanleit, WL01857, 1:500 for western blot); anti-caspase-7 (Cell Signaling Technology, #9494, 1:1,000 for western blot); anti-caspase-9 (Proteintech, 66169-1-Ig, 10380-1-AP, 1:1,000 for western blot); anti-V0d (Proteintech, 18274-1-AP, 1:1,000 for western blot); anti-V1C (Proteintech, 16054-1-AP, 1:1,000 for western blot); anti-V1A (Proteintech, 17115-1-AP, 1:1,000 for western blot); anti-V0a (Santa Cruz, sc-374475, 1:100 for IF and 1:600 for western blot); anti-GFP (Proteintech, 50430-2-AP, 66002-1-Ig, 1:6,000 for western blot); anti-GAPDH (Proteintech, 60004-1-Ig, 10494-1-AP, 1:8,000 for western blot); anti-β-actin (Sigma, A5316, 1:5,000 for western blot); anti-PARP (Cell Signaling Technology, 9542, 1:1,000 for western blot); anti-Bcl-2 (Abcam, #ab692, 1:1,000 for western blot); anti-Bcl-xL (Abcam, #ab77571, 1:500 for western blot); anti-LAMP1 (Abcam, #ab24170, 1:100 for IF); anti-cathepsin L (Abcam, #ab133641, 1:150 for IF); anti-mature cathepsin L (BioVision Technologies, 3741, 1:1,000 for western blot); and anti-FLAG (Proteintech, 66008-2-lg, 1:5,000 for western blot).
CoIP and Western Blot
Cellular lysates were prepared by incubating the cells in lysis buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.3% Nonidet P-40, 2 mM EDTA) containing protease inhibitor cocktail (Roche) for 40 min at 4°C, followed by centrifugation at 12,000 × g for 15 min at 4°C. The protein concentration of the lysates was determined by a bicinchoninic acid (BCA) protein assay kit (Pierce) according to the manufacturer’s protocol. For immunoprecipitation, 500 μg of protein was incubated with 2 μg of specific antibodies for 12 h at 4°C with constant rotation; 50 μL of 50% protein A or G agarose beads was added and incubated for an additional 3 h at 4°C. Beads were then washed five times using the lysis buffer. Between washes, the beads were collected by centrifugation at 1,000 × g for 3 min at 4°C. The precipitated proteins were eluted from the beads by resuspending the beads in 2× SDS-PAGE loading buffer and boiling for 10 min. The resultant materials from immunoprecipitation or cell lysates were resolved using SDS-PAGE gels and transferred onto nitrocellulose membranes. For routine western blot, cells were washed twice with cold PBS, then lysed in buffer (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM sodium vanadate, 1 mg/mL leupeptin, 1 mM phenylmethanesulfonylfluoride). Equal amounts of protein were loaded onto SDS-PAGE and transferred onto polyvinylidene fluoride (PVDF) blotting membranes. Membranes were incubated with appropriate antibodies for 1 h at room temperature or overnight at 4°C followed by incubation with a secondary antibody. Immunoreactive bands were detected by a LI-COR Biosciences Odyssey image reader (LI-COR Biosciences, USA), or they were visualized using western blotting luminol reagent (Santa Cruz) according to the manufacturer’s recommendations.
Wound-Healing Assay and Cell Invasion Assay
For the wound-healing assay, 1 × 106 cells were seeded into each well of a six-well plate and the next day the cellular monolayer was wounded by scratching with a 20-μL pipette tip. The cells were washed three times with PBS and added to serum-free medium. The distances of cell migration were calculated by subtracting the distance between the wound edges at 0, 12, 24, 36, and 48 h from the distances measured at 0 h. The transwell invasion assay was performed with transwell chambers (BD Biosciences). Cells were washed three times in PBS and resuspended in serum-free culture medium. Afterward, 8 × 104 cells in 200 μL of serum-free medium were plated onto the upper chamber of the transwell, and 500 μL of DMEM supplemented with 10% FBS was add to the lower chambers. After incubating for 15 h, cells may actively migrate from the upper to the lower side of the filter due to FBS as an attractant. Cells on the upper side were removed using cotton swabs, and the invasive cells on the lower side were stained with a three-step stain set (Thermo Fisher Scientific) and counted using a light microscope.
Colony Formation Assay
1 × 103 cells were maintained in culture media in six-well plates for 14 days, fixed with 4% paraformaldehyde, stained with 0.1% crystal violet for colony observation, and counted using a light microscope. Each experiment was performed in triplicate and repeated at last three times.
Proliferation Assay
Cells were plated in triplicate in 12-well plates at 1 × 105 cells per well in 1.5 mL of medium. After days, as indicated in experiments, wells were washed twice with PBS to remove dead cells, and then the entire contents of the well were trypsinized. Cell number was determined using a hemocytometer. For each well, the fold change in cell number relative to day 0 was presented directly or in a log2 scale.
Cell Death Assay
Dead cells were negatively corresponding to the ATP level. Therefore, the level of ATP was examined through the CellTiter-Glo luminescent cell viability assay kit (Promega, #7570), and the percentage of dead cells was calculated by subtracting the percentage of living cells from 100%. 1.5 × 105 cells per well were seeded in 12-well plates. After the desired treatment, 200 μL of CellTiter-Glo reagent was added to each well after the medium was removed. Subsequently, plates were placed on a shaker for 10 min and were then incubated at room temperature for an additional 10 min. Luminescence reading was carried out with a SpectraMax M2 reader (Molecular Devices).
Annexin V/PI Flow Cytometric Analysis
Cells were seeded in six-well plates at 1.5 × 105 cells per well. After transfection for 24 h, the suspended and scraped cells were pooled, pelleted by centrifugation, washed with ice-cold PBS, and resuspended in 200 μL of 1× binding buffer (annexin V-FITC/PI apoptosis detection kit, BD, USA). Next, the cell suspension was transferred to a 5-mL tube and incubated with 5 μL of annexin V-FITC for 15 min at 25°C followed by 5 μL of PI for 5 min at 25°C in the dark. Finally, samples were analyzed by flow cytometry within 1 h on a FACScan flow cytometer (BD Biosciences). The results are presented as the percentage of cells that were apoptotic (annexin V+ PI− or annexin V+ PI+).
Lysosomal Acidification Assessment
1.5 × 105 cells per well were grown in six-well plates, and after the desired treatment, cells were stained with AO (1.5 μg/mL) dissolved in complete growth medium for 15 min at 37°C, and then washed twice with warm PBS. The cells were rapidly imaged with a microscope (Operetta CLS, PerkinElmer). For flow cytometry analysis, a cell suspension containing 300 μL of PBS was prepared in a fluorescence-activated cell sorting (FACS) tube. Red fluorescence intensity of cells was measured by flow cytometry (FACSCanto II, BD Biosciences) in the far-red channel (phycoerythrin [PE]-Cy5). For LysoSensor green, cells were stained with 1 μM LysoSensor green for 15 min at 37°C and measured by a flow cytometer in the FITC channel. To evaluate lysosomal integrity, cells were incubated with 0.5 μM LysoTracker red at 37°C for 5 min. Hoechst was included in the final washing to stain nuclei. Images were visualized with an Olympus inverted microscope equipped with a charge-coupled camera.
Immunofluorescence
MCF-7 or HeLa cells were grown on 12-well chamber slides. After the desired treatment, cells were washed with PBS, fixed in 4% (w/v) paraformaldehyde for 20 min, permeabilized with 0.1% (v/v) Triton X-100 in PBS for 10 min, blocked with 3% BSA, and incubated with appropriate primary antibodies followed by staining with matching Alexa Fluor (AF) 488- or AF594-coupled secondary antibodies (Life Technologies, A-11008, A-11001, A-11005, A-11012, 1:150). The cells were washed four times, and a final concentration of 0.1 μg/mL DAPI (Sigma) was included in the final washing to stain nuclei. Images were visualized with a fluorescence microscope (Zeiss Imager Z2).
LMP Assay
Cells stably expressing GFP-LGALS3 were grown on 12-well plates. After the desired treatment, the cells were stained with LysoTracker red to label lysosomes. Images of LGALS3 distribution were visualized with an Olympus inverted microscope equipped with a charge-coupled camera. The filter sets for imaging were as follows: GFP, excitation (Ex) 480/30 nm, DM505, BA535/40; red fluorescent protein (RFP), Ex535/50 nm, DM575, BA590.
Statistical Analysis
Data are given as mean ± SD for triplicate experiments unless specified otherwise.
Author Contributions
B.Z., M.L., and L.L. performed experiments and analyzed data. C.B., Y.R., Y.W., Y.H., and L.Q. provided technical assistance. X.W. provided funding support and some ideas. M.L., L.L., and B.L. designed the study and wrote the manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This work was supported by Grants 81672762, 81472472, and 81902811 from the National Natural Science Foundation of China.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.01.022.
Contributor Information
Lifang Li, Email: lifangli@bjmu.edu.cn.
Binghui Li, Email: bli@ccmu.edu.cn.
Supplemental Information
References
- 1.Bröker L.E., Kruyt F.A., Giaccone G. Cell death independent of caspases: a review. Clin. Cancer Res. 2005;11:3155–3162. doi: 10.1158/1078-0432.CCR-04-2223. [DOI] [PubMed] [Google Scholar]
- 2.Galluzzi L., Vitale I., Aaronson S.A., Abrams J.M., Adam D., Agostinis P., Alnemri E.S., Altucci L., Amelio I., Andrews D.W. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25:486–541. doi: 10.1038/s41418-017-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pellegrini M., Bath S., Marsden V.S., Huang D.C., Metcalf D., Harris A.W., Strasser A. FADD and caspase-8 are required for cytokine-induced proliferation of hemopoietic progenitor cells. Blood. 2005;106:1581–1589. doi: 10.1182/blood-2005-01-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Estaquier J., Vallette F., Vayssiere J.L., Mignotte B. The mitochondrial pathways of apoptosis. Adv. Exp. Med. Biol. 2012;942:157–183. doi: 10.1007/978-94-007-2869-1_7. [DOI] [PubMed] [Google Scholar]
- 5.Rosenberg S., Zhang H., Zhang J. FADD deficiency impairs early hematopoiesis in the bone marrow. J. Immunol. 2011;186:203–213. doi: 10.4049/jimmunol.1000648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gringhuis S.I., Kaptein T.M., Wevers B.A., Theelen B., van der Vlist M., Boekhout T., Geijtenbeek T.B. Dectin-1 is an extracellular pathogen sensor for the induction and processing of IL-1β via a noncanonical caspase-8 inflammasome. Nat. Immunol. 2012;13:246–254. doi: 10.1038/ni.2222. [DOI] [PubMed] [Google Scholar]
- 7.Boege Y., Malehmir M., Healy M.E., Bettermann K., Lorentzen A., Vucur M., Ahuja A.K., Böhm F., Mertens J.C., Shimizu Y. A dual role of caspase-8 in triggering and sensing proliferation-associated DNA damage, a key determinant of liver cancer development. Cancer Cell. 2017;32:342–359.e10. doi: 10.1016/j.ccell.2017.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keller N., Ozmadenci D., Ichim G., Stupack D. Caspase-8 function, and phosphorylation, in cell migration. Semin. Cell Dev. Biol. 2018;82:105–117. doi: 10.1016/j.semcdb.2018.01.009. [DOI] [PubMed] [Google Scholar]
- 9.Brunk U.T., Neuzil J., Eaton J.W. Lysosomal involvement in apoptosis. Redox Rep. 2001;6:91–97. doi: 10.1179/135100001101536094. [DOI] [PubMed] [Google Scholar]
- 10.Wang F., Gómez-Sintes R., Boya P. Lysosomal membrane permeabilization and cell death. Traffic. 2018;19:918–931. doi: 10.1111/tra.12613. [DOI] [PubMed] [Google Scholar]
- 11.Taniguchi M., Ogiso H., Takeuchi T., Kitatani K., Umehara H., Okazaki T. Lysosomal ceramide generated by acid sphingomyelinase triggers cytosolic cathepsin B-mediated degradation of X-linked inhibitor of apoptosis protein in natural killer/T lymphoma cell apoptosis. Cell Death Dis. 2015;6:e1717. doi: 10.1038/cddis.2015.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bidère N., Lorenzo H.K., Carmona S., Laforge M., Harper F., Dumont C., Senik A. Cathepsin D triggers Bax activation, resulting in selective apoptosis-inducing factor (AIF) relocation in T lymphocytes entering the early commitment phase to apoptosis. J. Biol. Chem. 2003;278:31401–31411. doi: 10.1074/jbc.M301911200. [DOI] [PubMed] [Google Scholar]
- 13.Droga-Mazovec G., Bojic L., Petelin A., Ivanova S., Romih R., Repnik U., Salvesen G.S., Stoka V., Turk V., Turk B. Cysteine cathepsins trigger caspase-dependent cell death through cleavage of bid and antiapoptotic Bcl-2 homologues. J. Biol. Chem. 2008;283:19140–19150. doi: 10.1074/jbc.M802513200. [DOI] [PubMed] [Google Scholar]
- 14.Plotegher N., Duchen M.R. Mitochondrial dysfunction and neurodegeneration in lysosomal storage disorders. Trends Mol. Med. 2017;23:116–134. doi: 10.1016/j.molmed.2016.12.003. [DOI] [PubMed] [Google Scholar]
- 15.Uthaisang-Tanechpongtamb W., Sriyabhaya P., Wilairat P. Role of altholactone in inducing type II apoptosis signalling pathway and expression of cancer-related genes in cervical carcinoma HeLa cell line. Cell Biol. Int. 2013;37:471–477. doi: 10.1002/cbin.10059. [DOI] [PubMed] [Google Scholar]
- 16.Zhang J., Wang X., Cui W., Wang W., Zhang H., Liu L., Zhang Z., Li Z., Ying G., Zhang N., Li B. Visualization of caspase-3-like activity in cells using a genetically encoded fluorescent biosensor activated by protein cleavage. Nat. Commun. 2013;4:2157. doi: 10.1038/ncomms3157. [DOI] [PubMed] [Google Scholar]
- 17.Li M., Wu X.M., Gao J., Yang F., Zhang C.L., Ke K., Wang Y.C., Zheng Y.S., Yao J.F., Guan Y.Y. Mutations in the P10 region of procaspase-8 lead to chemotherapy resistance in acute myeloid leukemia by impairing procaspase-8 dimerization. Cell Death Dis. 2018;9:516. doi: 10.1038/s41419-018-0511-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu W.H., Johnson H., Shu H.B. Activation of NF-κB by FADD, Casper, and caspase-8. J. Biol. Chem. 2000;275:10838–10844. doi: 10.1074/jbc.275.15.10838. [DOI] [PubMed] [Google Scholar]
- 19.Keller N., Mares J., Zerbe O., Grütter M.G. Structural and biochemical studies on procaspase-8: new insights on initiator caspase activation. Structure. 2009;17:438–448. doi: 10.1016/j.str.2008.12.019. [DOI] [PubMed] [Google Scholar]
- 20.Lam L.T., Zhang H., Chyla B. Biomarkers of therapeutic response to BCL2 antagonists in cancer. Mol. Diagn. Ther. 2012;16:347–356. doi: 10.1007/s40291-012-0003-6. [DOI] [PubMed] [Google Scholar]
- 21.Scaffidi C., Fulda S., Srinivasan A., Friesen C., Li F., Tomaselli K.J., Debatin K.M., Krammer P.H., Peter M.E. Two CD95 (APO-1/Fas) signaling pathways. EMBO J. 1998;17:1675–1687. doi: 10.1093/emboj/17.6.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hellwig C.T., Kohler B.F., Lehtivarjo A.K., Dussmann H., Courtney M.J., Prehn J.H., Rehm M. Real time analysis of tumor necrosis factor-related apoptosis-inducing ligand/cycloheximide-induced caspase activities during apoptosis initiation. J. Biol. Chem. 2008;283:21676–21685. doi: 10.1074/jbc.M802889200. [DOI] [PubMed] [Google Scholar]
- 23.Li H., Zhu H., Xu C.J., Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 24.Garcia-Calvo M., Peterson E.P., Leiting B., Ruel R., Nicholson D.W., Thornberry N.A. Inhibition of human caspases by peptide-based and macromolecular inhibitors. J. Biol. Chem. 1998;273:32608–32613. doi: 10.1074/jbc.273.49.32608. [DOI] [PubMed] [Google Scholar]
- 25.Sanjana N.E., Shalem O., Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kline M.P., Rajkumar S.V., Timm M.M., Kimlinger T.K., Haug J.L., Lust J.A., Greipp P.R., Kumar S. ABT-737, an inhibitor of Bcl-2 family proteins, is a potent inducer of apoptosis in multiple myeloma cells. Leukemia. 2007;21:1549–1560. doi: 10.1038/sj.leu.2404719. [DOI] [PubMed] [Google Scholar]
- 27.Oberst A., Dillon C.P., Weinlich R., McCormick L.L., Fitzgerald P., Pop C., Hakem R., Salvesen G.S., Green D.R. Catalytic activity of the caspase-8-FLIPL complex inhibits RIPK3-dependent necrosis. Nature. 2011;471:363–367. doi: 10.1038/nature09852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fritsch M., Günther S.D., Schwarzer R., Albert M.C., Schorn F., Werthenbach J.P., Schiffmann L.M., Stair N., Stocks H., Seeger J.M. Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature. 2019;575:683–687. doi: 10.1038/s41586-019-1770-6. [DOI] [PubMed] [Google Scholar]
- 29.Newton K., Wickliffe K.E., Maltzman A., Dugger D.L., Reja R., Zhang Y., Roose-Girma M., Modrusan Z., Sagolla M.S., Webster J.D., Dixit V.M. Activity of caspase-8 determines plasticity between cell death pathways. Nature. 2019;575:679–682. doi: 10.1038/s41586-019-1752-8. [DOI] [PubMed] [Google Scholar]
- 30.Rathkey J.K., Zhao J., Liu Z., Chen Y., Yang J., Kondolf H.C., Benson B.L., Chirieleison S.M., Huang A.Y., Dubyak G.R. Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci. Immunol. 2018;3:eaat2738. doi: 10.1126/sciimmunol.aat2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang J., Liu Z., Wang C., Yang R., Rathkey J.K., Pinkard O.W., Shi W., Chen Y., Dubyak G.R., Abbott D.W., Xiao T.S. Mechanism of gasdermin D recognition by inflammatory caspases and their inhibition by a gasdermin D-derived peptide inhibitor. Proc. Natl. Acad. Sci. USA. 2018;115:6792–6797. doi: 10.1073/pnas.1800562115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boya P., Kroemer G. Lysosomal membrane permeabilization in cell death. Oncogene. 2008;27:6434–6451. doi: 10.1038/onc.2008.310. [DOI] [PubMed] [Google Scholar]
- 33.Kimura T., Takabatake Y., Takahashi A., Isaka Y. Chloroquine in cancer therapy: a double-edged sword of autophagy. Cancer Res. 2013;73:3–7. doi: 10.1158/0008-5472.CAN-12-2464. [DOI] [PubMed] [Google Scholar]
- 34.Cheng X.T., Xie Y.X., Zhou B., Huang N., Farfel-Becker T., Sheng Z.H. Revisiting LAMP1 as a marker for degradative autophagy-lysosomal organelles in the nervous system. Autophagy. 2018;14:1472–1474. doi: 10.1080/15548627.2018.1482147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pierzyńska-Mach A., Janowski P.A., Dobrucki J.W. Evaluation of acridine orange, LysoTracker Red, and quinacrine as fluorescent probes for long-term tracking of acidic vesicles. Cytometry A. 2014;85:729–737. doi: 10.1002/cyto.a.22495. [DOI] [PubMed] [Google Scholar]
- 36.Yoshimori T., Yamamoto A., Moriyama Y., Futai M., Tashiro Y. Bafilomycin A1, a specific inhibitor of vacuolar-type H+-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J. Biol. Chem. 1991;266:17707–17712. [PubMed] [Google Scholar]
- 37.Kand D., Saha T., Lahiri M., Talukdar P. Lysosome targeting fluorescence probe for imaging intracellular thiols. Org. Biomol. Chem. 2015;13:8163–8168. doi: 10.1039/c5ob00889a. [DOI] [PubMed] [Google Scholar]
- 38.Paz I., Sachse M., Dupont N., Mounier J., Cederfur C., Enninga J., Leffler H., Poirier F., Prevost M.C., Lafont F., Sansonetti P. Galectin-3, a marker for vacuole lysis by invasive pathogens. Cell. Microbiol. 2010;12:530–544. doi: 10.1111/j.1462-5822.2009.01415.x. [DOI] [PubMed] [Google Scholar]
- 39.Uchimoto T., Nohara H., Kamehara R., Iwamura M., Watanabe N., Kobayashi Y. Mechanism of apoptosis induced by a lysosomotropic agent, l-leucyl-l-leucine methyl ester. Apoptosis. 1999;4:357–362. doi: 10.1023/a:1009695221038. [DOI] [PubMed] [Google Scholar]
- 40.Colacurcio D.J., Nixon R.A. Disorders of lysosomal acidification—the emerging role of v-ATPase in aging and neurodegenerative disease. Ageing Res. Rev. 2016;32:75–88. doi: 10.1016/j.arr.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McGuire C., Stransky L., Cotter K., Forgac M. Regulation of V-ATPase activity. Front. Biosci. 2017;22:609–622. doi: 10.2741/4506. [DOI] [PubMed] [Google Scholar]
- 42.McGuire C.M., Forgac M. Glucose starvation increases V-ATPase assembly and activity in mammalian cells through AMP kinase and phosphatidylinositide 3-kinase/Akt signaling. J. Biol. Chem. 2018;293:9113–9123. doi: 10.1074/jbc.RA117.001327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stransky L.A., Forgac M. Amino acid availability modulates vacuolar H+-ATPase assembly. J. Biol. Chem. 2015;290:27360–27369. doi: 10.1074/jbc.M115.659128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lu M., Ammar D., Ives H., Albrecht F., Gluck S.L. Physical interaction between aldolase and vacuolar H+-ATPase is essential for the assembly and activity of the proton pump. J. Biol. Chem. 2007;282:24495–24503. doi: 10.1074/jbc.M702598200. [DOI] [PubMed] [Google Scholar]
- 45.Smardon A.M., Nasab N.D., Tarsio M., Diakov T.T., Kane P.M. Molecular interactions and cellular itinerary of the yeast RAVE (regulator of the H+-ATPase of vacuolar and endosomal membranes) complex. J. Biol. Chem. 2015;290:27511–27523. doi: 10.1074/jbc.M115.667634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stegh A.H., Barnhart B.C., Volkland J., Algeciras-Schimnich A., Ke N., Reed J.C., Peter M.E. Inactivation of caspase-8 on mitochondria of Bcl-xL-expressing MCF7-Fas cells: role for the bifunctional apoptosis regulator protein. J. Biol. Chem. 2002;277:4351–4360. doi: 10.1074/jbc.M108947200. [DOI] [PubMed] [Google Scholar]
- 47.Hughes M.A., Powley I.R., Jukes-Jones R., Horn S., Feoktistova M., Fairall L., Schwabe J.W., Leverkus M., Cain K., MacFarlane M. Co-operative and hierarchical binding of c-FLIP and caspase-8: a unified model defines how c-FLIP isoforms differentially control cell fate. Mol. Cell. 2016;61:834–849. doi: 10.1016/j.molcel.2016.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pietkiewicz S., Schmidt J.H., Lavrik I.N. Quantification of apoptosis and necroptosis at the single cell level by a combination of imaging flow cytometry with classical annexin V/propidium iodide staining. J. Immunol. Methods. 2015;423:99–103. doi: 10.1016/j.jim.2015.04.025. [DOI] [PubMed] [Google Scholar]
- 49.Santos L.C., Vogel R., Chipuk J.E., Birtwistle M.R., Stolovitzky G., Meyer P. Mitochondrial origins of fractional control in regulated cell death. Nat. Commun. 2019;10:1313. doi: 10.1038/s41467-019-09275-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.