

Abstract
Checkpoint-inhibiting antibodies elicit impressive clinical responses, but still face several issues. The current study evaluated whether DNA-based delivery can broaden the application of checkpoint inhibitors, specifically by pursuing cost-efficient in vivo production, facilitating combination therapies, and exploring administration routes that lower immune-related toxicity risks. We therefore optimized plasmid-encoded anti-CTLA-4 and anti-PD-1 antibodies, and studied their pharmacokinetics and pharmacodynamics when delivered alone and in combination via intramuscular or intratumoral electroporation in mice. Intramuscular electrotransfer of these DNA-based antibodies induced complete regressions in a subcutaneous MC38 tumor model, with plasma concentrations up to 4 and 14 μg/mL for anti-CTLA-4 and anti-PD-1 antibodies, respectively, and antibody detection for at least 6 months. Intratumoral antibody gene electrotransfer gave similar anti-tumor responses as the intramuscular approach. Antibody plasma levels, however, were up to 70-fold lower and substantially more transient, potentially improving biosafety of the expressed checkpoint inhibitors. Intratumoral delivery also generated a systemic anti-tumor response, illustrated by moderate abscopal effects and prolonged protection of cured mice against a tumor rechallenge. In conclusion, intramuscular and intratumoral DNA-based delivery of checkpoint inhibitors both enabled long-term anti-tumor responses despite distinct systemic antibody exposure, highlighting the potential of the tumor as delivery site for DNA-based therapeutics.
Keywords: antibody gene transfer, CTLA-4, PD-1, plasmid DNA, intramuscular electroporation, intratumoral electroporation
Graphical Abstract
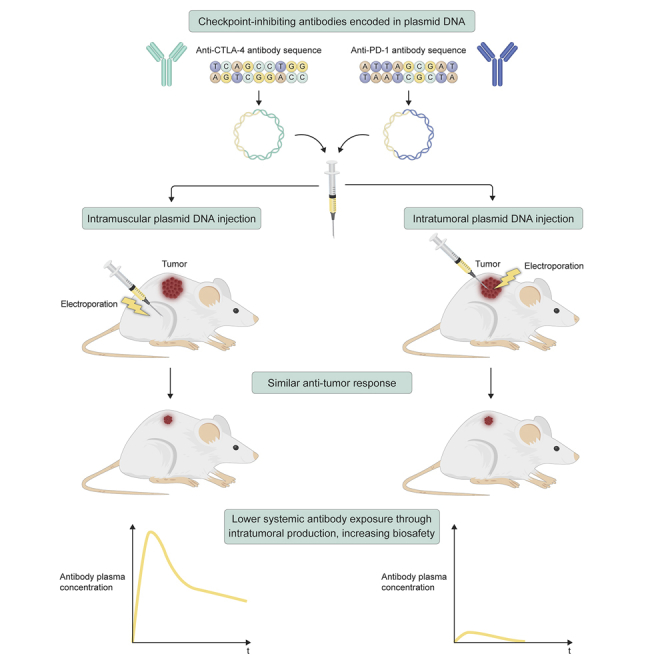
Jacobs and colleagues evaluated DNA-based delivery of checkpoint inhibitors as an alternative to conventional antibody protein therapy. Intramuscular and intratumoral gene transfer both mediated local and systemic anti-tumor responses. Delivery to the tumor resulted in significantly lower systemic antibody exposure, improving the biosafety of DNA-based combination therapies.
Introduction
Checkpoint-inhibiting monoclonal antibodies (mAbs) have shown impressive results in several cancer indications, but the complex production, significant portion of refractory patients, and severe toxicity risks hamper a broader application.1 To improve response rates, more than a thousand immunotherapy combinations are currently under clinical evaluation. These combination therapies, however, increase costs and the risk for immune-related adverse events.2,3 Overall, these issues highlight the need for innovations in mAb production and delivery.
Antibody gene transfer aims to administer the mAb-encoding nucleotides instead of the recombinant protein, which enables the patient’s body to produce and secrete the mAb for a prolonged period of time.4 Viral vectors, plasmid DNA (pDNA), and mRNA have successfully been applied to express therapeutic mAb (combinations) in vivo. Among these available options, we focus on pDNA as expression platform because of the ease of production, limited immunogenicity, and favorable biosafety profile.4 We recently demonstrated proof of concept for DNA-based intramuscular gene transfer of tumor-targeting mAbs in mice and sheep.5,6 As in our experiments, pDNA is typically administered in vivo in combination with electroporation, a technique that employs electrical pulses to temporally increase the permeability of cell membranes at the application site.7 Compared with viral vectors and mRNA, this allows a more targeted and controlled transfection of the tissue of interest. Electroporation has already been extensively used in the clinic, e.g., in the context of DNA vaccines and electrochemotherapy,7 and can target both superficial and deep-seated sites, e.g., by catheter-based electroporation devices.8 DNA-based antibody gene electrotransfer has shown preclinical efficacy in oncology and infectious, auto-immune, and cardiovascular diseases,4 and is under clinical evaluation (ClinicalTrials.gov: NCT011384109 and NCT03831503).
Thus far, DNA-based antibody gene transfer studies have exclusively focused on the muscle or liver as site of transfection, resulting in systemic mAb exposure.4 pDNA delivery directly into the tumor, in contrast, leads to in situ transgene expression and more restricted exposure. Intratumoral electrotransfer of DNA-based interleukin 12 (IL-12), for example, resulted in high IL-12 concentrations in the tumor and limited detection in the circulation, enabling local and systemic therapeutic responses in both preclinical and clinical trials.10,11 For checkpoint inhibitors, similar findings were reported for the intratumoral delivery of mAb proteins12, 13, 14, 15 and mAb-armed viruses,16,17 which are both under clinical evaluation.12,13,18 Gene transfer of immunomodulatory mAbs has indeed mainly been performed with viral vectors. To our knowledge, only two recent studies reported the DNA-based delivery of checkpoint inhibitors,19,20 but these focused exclusively on the intramuscular route and single-agent treatments.
The current study aimed to establish preclinical proof of concept for intramuscular and intratumoral DNA-based electrotransfer of single and combined checkpoint-inhibiting mAbs, and compare the resulting pharmacokinetics and pharmacodynamics in a well-characterized mouse tumor model. Anti-cytotoxic T-lymphocyte associated protein 4 (CTLA-4) and anti-programmed cell death protein 1 (PD-1) mAbs served as a model, given their regulatory approved combination,21,22 high frequency of immune-related adverse events after systemic infusion,1,3 and safe and effective intratumoral delivery as mAb proteins12, 13, 14, 15 or viral-vectored gene therapy.16,17
Results
Engineering of the Expression Cassette and Antibody Sequences Increased Antibody Expression
A DNA-based anti-mouse CTLA-4 mAb was engineered based on the validated murine 9D9 clone, with some framework modifications and a murine IgG2ab (C57BL/6 haplotype)23 constant region (Table 1). Selby et al.24 previously showed that this isotype variant elicited enhanced anti-tumor responses compared with the original IgG2b isotype. Similar to our previous studies,5 heavy chain (HC) and light chain (LC) sequences were cloned into a single pDNA construct p(aCTLA-4-HC+LC) by means of two CAG promoters in tandem. Intramuscular electrotransfer of p(aCTLA-4-HC+LC) in C57BL/6J mice resulted in long-term mAb expression, with mAb plasma concentrations up to 2 μg/mL and detectable levels for at least 5 months (mean concentration of 130 ng/mL; Figure 1A). When two separate CAG-driven pDNA constructs p(aCTLA-4-HC) and p(aCTLA-4-LC) were used to express HC and LC, respectively, in vitro mAb expression increased 4-fold compared with the single construct (p < 0.0001; Figure 1B). Accordingly, an equimolar mixture of p(aCTLA-4-HC) and p(aCTLA-4-LC) was used in all subsequent experiments and referred to as p(aCTLA-4) (Table 1).
Table 1.
Characteristics of Anti-CTLA-4 and Anti-PD-1 mAb-Encoding pDNA Constructs
| Promoter | Variable Regions | Isotype | Plasmid Design | Plasmid Backbone | |
|---|---|---|---|---|---|
| p(aCTLA-4-HC+LC) | CAG (two in tandem) | murine 9D9 | IgG2ab | single pDNA | pNull |
| p(aCTLA-4) = p(aCTLA-4-HC) + p(aCTLA-4-LC) | CAG | murine 9D9 | IgG2ab | equimolar mixture of HC-pDNA and LC-pDNA | pNull |
| p(CAG-caPD-1-HC2a+LC) | CAG (two in tandem) | rat RMP1-14 | IgG2aa | single pDNA | pNull |
| p(CMV-caPD-1-HC1) + p(CMV-caPD-1-LC) | CMV | rat RMP1-14 | IgG1 | equimolar mixture of HC-pDNA and LC-pDNA | pControl |
| p(CMV-maPD-1) = p(CMV-maPD-1-HC1) + p(CMV-maPD-1-LC) | CMV | murinized RMP1-14 | IgG1 | equimolar mixture of HC-pDNA and LC-pDNA | pControl |
| p(CAG-maPD-1) = p(CAG-maPD-1-HC1) + p(CAG-maPD-1-LC) | CAG | murinized RMP1-14 | IgG1 | equimolar mixture of HC-pDNA and LC-pDNA | pNull |
HC, heavy chain; LC, light chain; pDNA, plasmid DNA.
Figure 1.

Engineering of a DNA-Based Anti-CTLA-4 mAb and DNA-Based Anti-PD-1 mAb
mAb concentrations were determined in plasma after a single intramuscular (i.m.) electrotransfer of 60 μg pDNA in C57BL/6J mice (A, C, and D), and in cell supernatant (SN) after transfection of five million 293F Freestyle suspension cells with 5 μg pDNA (B). (A) Validation of anti-CTLA-4 mAb expression by p(aCTLA-4-HC+LC) (n = 5 mice). (B) Comparison of p(aCTLA-4-HC) + p(aCTLA-4-LC) and an equimolar amount of p(aCTLA-4-HC+LC). Data were analyzed with an unpaired t test (n = 8 or 9 biological replicates). (C) Comparison of p(CAG-caPD-1-HC2a+LC), p(CMV-caPD-1-HC1) + p(CMV-caPD-1-LC), and p(CMV-maPD-1-HC1) + p(CMV-maPD-1-LC). Asterisks indicate the statistical difference between the p(CMV-caPD-1) and p(CMV-maPD-1) group, analyzed with an unpaired t test (n = 5 or 10 mice per group). (D) Comparison of the CMV-based and CAG-based pDNA constructs to express the murinized IgG1 anti-PD-1 mAb. mAb plasma concentrations were compared with an unpaired t test (n = 10 mice per group). All data are represented as mean + SEM (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Because we had no access to the sequences of a murine anti-mouse PD-1 mAb, a DNA-based chimeric anti-PD-1 mAb p(CAG-caPD-1-HC2a+LC) was generated, containing the variable regions of the rat RMP1-14 clone and a murine IgG2aa (BALB/c haplotype)23 backbone (Table 1). Despite efficient mAb expression in vitro, mAb plasma levels in C57BL/6J mice showed a rapid decline after intramuscular p(CAG-caPD-1-HC2a+LC) electrotransfer and were undetectable after 2 weeks (Figure 1C) due to anti-drug antibody formation (data not shown). We therefore shifted to another chimeric anti-PD-1 mAb with the same variable regions but an IgG1 isotype, which was initially available in another cassette configuration: p(CMV-caPD-1-HC1) and p(CMV-caPD-1-LC) (Table 1). Replacing the IgG2aa by the murine IgG1 HC backbone avoided the induction of anti-drug antibodies and allowed for long-term mAb detection after intramuscular electrotransfer (Figure 1C). Murinization of the rat variable regions in p(CMV-maPD-1-HC1) and p(CMV-maPD-1-LC) [referred to as p(CMV-maPD-1) when combined; Table 1] significantly increased anti-PD-1 mAb expression in vivo (1.4- to 4.6-fold; p < 0.05; Figure 1C). One mouse reached mAb plasma levels up to 60 μg/mL, which explains the high variance in Figure 1C. In an independent experiment, the murinized anti-PD-1 mAb was still detectable in plasma at concentrations ranging from 400 ng/mL to 2 μg/mL 7 months after a single intramuscular p(CMV-maPD-1) electrotransfer. Cloning the murinized HC and LC sequences into the CAG-driven pDNA backbone p(CAG-maPD-1-HC1) and p(CAG-maPD-1-LC) [hereafter referred to as p(CAG-maPD-1) when combined; Table 1] further increased the anti-PD-1 mAb plasma levels (1.3- to 6.0-fold; p < 0.05 to p < 0.0001; Figure 1D). In summary, peak anti-PD-1 mAb plasma concentrations could be increased up to 8-fold through combined optimization of the mAb sequences and mAb expression cassette.
Intramuscular DNA-Based Delivery of Immunomodulatory Antibody Combinations Enabled Complete Tumor Regressions and Long-Term Anti-tumor Protection
The efficacy of the DNA-based anti-CTLA-4 and anti-PD-1 mAbs, alone or in combination, was verified by intramuscular delivery. In a first experiment, prophylactic intramuscular pDNA electrotransfer was performed in C57BL/6J mice, 7 days before subcutaneous (s.c.) MC38 tumor cell injection. Mice received p(aCTLA-4) or p(CMV-maPD-1) in the left tibialis anterior muscle or received both DNA-based mAbs in the left and right leg, respectively. The higher-expressing p(CAG-maPD-1) construct was not yet available at that time. Control mice received an equimolar amount of an empty plasmid pNull, devoid of an expression cassette, in the right and left tibialis anterior. One day later, electrotransfer of the corresponding pDNA constructs was repeated in the gastrocnemius muscle. Prophylactic intramuscular antibody gene transfer significantly delayed tumor growth compared with untreated mice (p < 0.0001), resulting in 42% complete responders with p(aCTLA-4) and 17% with p(CMV-maPD-1) (Figure 2A; Figure S1A). Combined DNA-based mAb delivery even enabled 67% complete responders, thereby significantly improving survival compared with p(CMV-maPD-1) alone (p < 0.05; Figure S1A). pNull had no effect on tumor growth (Figure 2A; Figure S1A). Resulting mAb plasma concentrations peaked up to 4 and 14 μg/mL for anti-CTLA-4 and anti-PD-1 mAbs, respectively, 2 weeks after intramuscular pDNA electrotransfer and were still detectable 6 months later (mean concentrations of 340 and 690 ng/mL for anti-CTLA-4 mAbs, and 1.8 and 3.0 μg/mL for anti-PD-1 mAbs; Figure 2B). In a second experiment, intramuscular antibody gene transfer was performed 3 days after MC38 cell injection (Figure 2C; Figure S1B). In this therapeutic setting, p(CMV-maPD-1) alone did not affect survival compared with untreated mice. p(aCTLA-4), in contrast, elicited significant tumor growth delay (p < 0.0001) with 10% complete responders. Combined with p(CMV-maPD-1), 30% of the mice showed complete tumor regressions, an improvement that did not reach statistical significance compared with p(aCTLA-4) alone. mAb response rates were thus less pronounced than after prophylactic intramuscular antibody gene transfer.
Figure 2.

Prophylactic and Therapeutic Intramuscular Electrotransfer of DNA-Based Checkpoint Inhibitors
C57BL/6J mice received an intramuscular (i.m.) pDNA electrotransfer in the tibialis anterior 7 days before (A, B, and D) or 3 days after (C, indicated by arrow) s.c. MC38 tumor cell injection. One day later, pDNA electrotransfer was repeated in the gastrocnemius muscle. Each electrotransfer, the single-treatment groups received 60 μg p(aCTLA-4) or p(CMV-maPD-1) in the left leg, and the combination-treatment group received both DNA-based mAbs in different legs. Control mice got an equimolar amount of pNull in both legs or were left untreated. (A and C) Tumor growth and number of complete responders (CR). Tumor volumes were compared with one-way ANOVA on day 19 after tumor cell injection, when the first untreated mice had to be sacrificed (n = 10 or 12 mice per group). (B) Anti-CTLA-4 (black lines) and anti-PD-1 (gray lines) mAb plasma concentrations in the single-treatment groups (solid lines) and combination-treatment group (dashed lines; n = 12 mice per group). (D) MC38 tumor rechallenge in complete responders of prophylactic intramuscular DNA-based mAb therapy, 23 weeks after the first tumor cell injection. The number of mice that became tumor-free (TF) after rechallenge is indicated. Tumor volumes were compared with age-matched naive mice with an unpaired t test (n = 14 or 15 mice per group). All data are represented as mean + SEM (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). For Kaplan-Meier survival curves, see Figure S1.
To evaluate whether intramuscular DNA-based delivery of checkpoint inhibitors confers long-term anti-cancer protection, complete responders of the prophylactic intramuscular approach were rechallenged with MC38 cells 23 weeks after the first tumor cell injection. Nine out of 15 rechallenged mice again achieved complete tumor regressions, whereas as expected no complete responders were observed in the naive control mice (follow-up period of 8 weeks; Figure 2D). At the time of rechallenge, mAb plasma concentrations varied between 150 ng/mL and 1.3 μg/mL for anti-CTLA-4 mAbs and 230 ng/mL and 7.1 μg/mL for anti-PD-1 mAbs (Figure 2B). Although these systemic mAb levels did not correlate with the anti-tumor responses after rechallenge, one cannot distinguish whether the delayed tumor growth was caused by an anti-tumor immune memory or by the circulating mAbs. Nevertheless, these experiments show that both single and combined intramuscular electrotransfer of DNA-based checkpoint inhibitors resulted in long-term mAb expression and effective, long-term anti-tumor responses.
Intratumoral DNA-Based Anti-CTLA-4 Antibody Delivery Caused Significant Regressions in Treated Tumors
To evaluate the efficacy of intratumoral DNA-based antibody gene transfer, DNA-based anti-CTLA-4 mAbs were delivered at either three or six doses in the s.c. MC38 model using a validated intratumoral electroporation protocol (Figure S2).25,26 These dose regimens were based on pilot data showing only limited effects with a single intratumoral pDNA administration (data not shown). Three and six p(aCTLA-4) doses gave similar, significant anti-tumor responses compared with untreated tumors (p < 0.0001; Figure 3A). Doubling the number of pDNA doses increased mAb plasma titers (p < 0.05 and p < 0.01 for week 3 plasma levels and peak plasma levels, respectively; Figure S3A), which overall correlated with the observed anti-tumor response (p < 0.01, R2 = 0.327; Figure 3B). Both p(aCTLA-4) regimens led to complete responders, albeit limited in number (9% in each group), with six doses leading to a significant improved survival compared with the empty plasmid pNull (p < 0.05; Figure S3B). In line with previous reports,27 intratumoral electrotransfer of the plasmid backbone had some effect on tumor growth, mainly caused by the injection of the pDNA, rather than by the electrical pulses (Figure S4). This anti-tumor effect of pNull was more pronounced at six doses than at three doses (p < 0.05 and p < 0.01 for tumor growth and survival, respectively), but did not result in complete regressions (Figure 3A; Figure S3B). Because three pDNA doses gave the biggest differentiation between p(aCTLA-4) and pNull, this regimen was applied in all subsequent intratumoral gene transfer experiments.
Figure 3.

Dose Comparison for Intratumoral p(aCTLA-4) or pNull Electrotransfer
MC38-bearing C57BL/6J mice received three or six intratumoral electroporations (indicated by black and gray arrows, respectively) of 60 μg p(aCTLA-4) or an equimolar amount of pNull. Control mice were left untreated. (A) Tumor growth and number of complete responders (CR). Tumor volumes, represented as mean + SEM, were compared with one-way ANOVA on day 19 after tumor cell injection, when the first mice had to be sacrificed (*p < 0.05, **p < 0.01, ****p < 0.0001). (B) Correlation between tumor volumes and anti-CTLA-4 mAb plasma concentrations on day 19 after tumor cell injection. R2 indicates the square of the Pearson correlation coefficient (n = 10 or 11 mice per group). For mAb plasma concentrations over time and Kaplan-Meier survival curves, see Figure S3.
To assess how intratumoral delivery of mAb proteins compares with DNA-based mAbs, we set up an exploratory study. MC38-bearing mice (n = 4 per group) received three intratumoral injections of either 3, 1, 0.3, or 0.1 μg anti-CTLA-4 mAb protein. One microgram protein gave comparable peak mAb plasma levels as three intratumoral doses of 60 μg p(aCTLA-4) (Figure S5A). Initial tumor responses were also similar, but concomitantly with a rapid decline of mAb plasma concentrations (below 50 ng/mL), tumors started regrowing (Figure S5B). This suggests that prolonged in situ mAb expression by intratumoral gene transfer might provide improved outcomes over intratumoral mAb protein administration.
Intratumoral Electrotransfer of DNA-Based Checkpoint Inhibitors Resulted in Similar Efficacy as Intramuscular Electrotransfer, Despite Lower Systemic Antibody Exposure
To test whether intratumoral DNA-based gene transfer can be of benefit for combination therapies, combined delivery of DNA-based anti-CTLA-4 and anti-PD-1 mAbs was examined in the s.c. MC38 model. Mice received three intratumoral electroporations with 60 μg of either p(aCTLA-4) or p(CAG-maPD-1) in 25 μL D-PBS, or both together in 50 μL D-PBS. Control mice were treated with pNull, injected in 50 μL D-PBS at a pDNA dose equimolar to the combined DNA-based mAb treatment. p(CAG-maPD-1) had a limited impact on tumor growth (Figure 4A; Figure S6), as was previously observed with the intramuscular evaluation of p(CMV-maPD-1) in established tumors (Figure 2C; Figure S1B). p(aCTLA-4), in contrast, led to 10% complete responders, and the combination treatment to 20% (Figure 4A; Figure S6). Overall, the response rates were similar compared with the therapeutic intramuscular approach, although mAb plasma concentrations were 10- to 70-fold lower (p < 0.0001) and more transient (Figure 4B). pNull initially elicited an anti-tumor response similar to that of the DNA-based mAbs, but again did not result in complete tumor regressions (Figure 4A; Figure S6). Taken together, these results indicate that intratumoral DNA-based electrotransfer can be applied to combine different mAbs. Further fine-tuning of the individual doses and choice of the most effective combinations remains required to improve anti-tumor responses.
Figure 4.

Combined Intratumoral Electrotransfer of DNA-Based Checkpoint Inhibitors
MC38-bearing C57BL/6J mice received three intratumoral (i.t.) electroporations (indicated by arrows) of 60 μg p(aCTLA-4) or p(CAG-maPD-1) in 25 μL D-PBS, or the combination of p(aCTLA-4) and p(CAG-maPD-1) or an equimolar amount of pNull in 50 μL D-PBS. One group received no treatment. (A) Tumor growth and number of complete responders (CR). Tumor volumes were compared with one-way ANOVA on day 18 after tumor cell injection, when the first mice had to be sacrificed (**p < 0.01, ****p < 0.0001). (B) Anti-CTLA-4 (black lines) and anti-PD-1 (gray lines) mAb plasma concentrations in the single-treatment groups (solid lines) and combination-treatment group (dashed lines). All data are represented as mean + SEM (n = 10 mice per group). For Kaplan-Meier survival curves, see Figure S6.
Intratumoral DNA-Based Delivery of Immunomodulatory Antibodies Induced a Systemic, Long-Term Anti-tumor Response
Because systemic efficacy is highly relevant for in situ therapies, intratumoral p(aCTLA-4) electrotransfer was evaluated in mice bearing two contralateral s.c. MC38 tumors. The primary tumor received three pDNA doses, whereas the smaller, contralateral tumor was left untreated. p(aCTLA-4) significantly delayed tumor growth of primary tumors compared with an equimolar amount of pNull (p < 0.01) and untreated mice (p < 0.0001), with 33% complete regressions (Figure 5A). In contralateral tumors, a moderate but significant abscopal effect was observed (p < 0.05; Figure 5B), even though anti-CTLA-4 mAb plasma levels did not exceed 100 ng/mL. In 1 mouse out of 12, both tumors completely regressed. pNull, in contrast, had no effect on the distant, non-treated tumor. These results indicate that intratumoral DNA-based anti-CTLA-4 mAb delivery can elicit systemic anti-tumor responses, despite limited systemic mAb exposure.
Figure 5.

Systemic Anti-tumor Effects of Intratumoral DNA-Based Delivery of Checkpoint Inhibitors
(A and B) C57BL/6J were s.c. injected with 1 × 106 MC38 cells in the right flank (primary tumor), and 0.25 × 106 MC38 cells in the left flank 3 days later (contralateral tumor). The primary tumor (A) received three intratumoral electrotransfers of 60 μg p(aCTLA-4) or an equimolar amount of pNull (indicated by arrows), or was left untreated. The contralateral tumor (B) received no treatment. Complete tumor regressions (CR) are separately indicated for the primary and contralateral tumor. Tumor volumes of the p(aCTLA-4) group and pNull group were compared with the untreated mice on day 19 after primary tumor cell injection and compared with each other on day 21 with one-way ANOVA. On days 19 and 21, the first mice of the untreated group and the first mice of the p(aCTLA-4) and pNull group had to be sacrificed, respectively (n = 12 mice per group). (C) MC38 tumor rechallenge in complete responders of intratumoral DNA-based mAb therapy, 11–14 weeks after the first tumor cell injection. Data of multiple tumor rechallenge experiments are pooled, including mice that were cured by DNA-based mAb monotherapy and combination therapy. The number of mice that became tumor-free (TF) after rechallenge is indicated. Tumor volumes were compared with age-matched naive mice with an unpaired t test (n = 10 or 21 mice per group in total). All data are represented as mean + SEM (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Complete responders from the different intratumoral gene transfer studies, including mice cured by DNA-based mAb monotherapy and combination therapy, were subsequently rechallenged with MC38 tumor cells, 11 to 14 weeks after the first tumor cell injection. In contrast with the intramuscular gene transfer approach, mAb plasma levels were all below 50 ng/mL at the time of rechallenge. In all rechallenge experiments, tumor growth was significantly delayed compared with tumor growth in age-matched controls (p < 0.001 to p < 0.0001), with 2 out of 10 mice achieving again complete tumor regressions (follow-up period of 11 and 17 weeks; Figure 5C). Taken together, this suggests that intratumoral delivery of DNA-based checkpoint inhibitors generates a long-term systemic anti-tumor immune memory, thereby protecting mice against a future challenge with the same tumor.
Discussion
To overcome the hurdles associated with checkpoint-inhibiting mAbs, we present DNA-based antibody gene electrotransfer as an alternative to the conventional production and parenteral delivery of mAb proteins. The current study aimed to establish preclinical proof of concept for the DNA-based delivery of immunomodulatory mAb combinations in the muscle and, for the first time, directly into the tumor.
First, we engineered and optimized DNA-encoded anti-CTLA-4 and anti-PD-1 mAbs. Despite contradictory reports about the optimal plasmid design,28,29 mAb expression significantly increased when HC and LC were expressed by separate pDNA constructs instead of a single pDNA. In mice, intramuscular delivery of our DNA-based murine anti-CTLA-4 mAb enabled long-term in vivo mAb expression. The DNA-based chimeric IgG2aa anti-PD-1 mAb led to only short-term mAb detection in plasma after intramuscular electrotransfer, due to anti-drug antibodies toward the expressed mAb. Not the rat variable sequences, but the BALB/c IgG2aa backbone23 appeared to be the most immunogenic region, as demonstrated by the absence of anti-drug antibodies against, and long-term expression of, the chimeric IgG1 anti-PD-1 mAb. Previous work by Belmar et al.30 supports this hypothesis. Murinization of the rat variable regions further improved anti-PD-1 mAb expression, which highlights the importance of similarity between the expressed mAb and mouse germline sequences.
Using the optimized pDNA constructs, we evaluated the efficacy and pharmacokinetics of intramuscular DNA-based delivery of checkpoint-inhibiting mAb combinations in the well-characterized MC38 tumor model. In both a prophylactic and therapeutic setting, combined DNA-based anti-CTLA-4 and anti-PD-1 mAb delivery resulted in more complete responders compared with each DNA-based mAb alone. As expected, prophylactic antibody gene transfer gave better responses than the therapeutic approach, caused by the shifted plasma concentration-time profiles of the expressed mAbs. Importantly, 5 months after treatment, most cured mice were still resistant to a rechallenge with MC38 cells. The DNA-based murinized anti-PD-1 mAb was effective only when given prophylactically, which can be explained by the lower intrinsic anti-tumor effect of the original rat anti-PD-1 mAb compared with our anti-CTLA-4 mAb, as observed in a pilot study with the mAb proteins (Figure S7). To improve the efficacy of the DNA-based murinized anti-PD-1 mAb in future experiments, a D265A mutation could be introduced in the IgG1 backbone, which cancels all Fcγ receptor binding,31 or higher mAb plasma levels could be targeted, e.g., by increasing the pDNA dose or further optimizing the expression cassette. In the current study, mean plasma levels up to 4 and 14 μg/mL were reached for anti-CTLA-4 and anti-PD-1 mAbs, respectively, similar to previous DNA-based delivery reports,19,20 and maintained at a high ng/mL to low μg/mL range after 6 months. Peak plasma levels were also of the same order of magnitude as mean trough concentrations reached in clinical trials (20 and 30–90 μg/mL for anti-CTLA-4 and anti-PD-1 mAbs, respectively).21,22,32 In these studies, however, severe adverse events occurred in up to 15% of the patients, with even higher toxicity risks at the highest anti-CTLA-4 mAb doses.33,34
Due to the potential toxicity associated with systemic exposure to checkpoint-inhibiting mAbs,33 we explored alternatives to intramuscular gene transfer, e.g., DNA-based delivery directly into the tumor. In case of IL-12, for example, intratumoral gene electrotransfer led to regressions of both treated and untreated lesions, while avoiding toxicity associated with systemic delivery of this cytokine.10,11 To the best of our knowledge, the current study is the first to evaluate the tumor as a site of delivery for DNA-based mAbs. In addition to local regressions, intratumoral gene electrotransfer of checkpoint inhibitors induced mild abscopal effects in distant lesions and delayed tumor growth in cured mice after tumor rechallenge, suggesting a long-term systemic anti-tumor immune response. Importantly, systemic mAb exposure was considerably lower compared with intramuscular pDNA delivery (up to 70-fold) and with the exposure reported in some intratumoral protein-based studies.14,15 The impact of this lower exposure on immune-related adverse events was not documented here, because such toxicity assessment requires more specific mouse strains.35
We demonstrated that intratumoral DNA-based gene transfer can enable mAb combination therapies, yielding a higher complete response rate than each DNA-based single treatment. Differences in tumor growth or survival were not significant between the different groups, except for the untreated mice, which might be explained by the dose-dependent anti-tumor effect of the pDNA backbone pNull. Indeed, it has been shown before that intratumoral electrotransfer of control plasmids causes cell death, immune activation, and thereby tumor regressions,27 making differentiation to intratumoral DNA-based mAb therapy harder in the current study. Still, the long-term benefits of the latter were obvious, because we did not observe complete tumor regressions, nor systemic anti-tumor responses with pNull. Reportedly, the anti-tumor response to such control plasmids strongly depends on the applied electroporation protocol and tumor model. Immunologically hot tumors,27 like the s.c. MC38 model used in the current study, are more sensitive than cold tumors.36 The clinical relevance of the empty plasmid effect is still unclear and requires further research. Overall, the current study highlights both the potential and the hurdles for intratumoral antibody gene electrotransfer. Moving forward, we will further characterize this approach, also in additional tumor models, and explore more effective or appropriate combinations of DNA-based biologicals. Combinations that are too toxic to be administered systemically or that therapeutically benefit from prolonged local exposure can thereby be of specific interest. Immunological characterization, e.g., of intratumoral T cell infiltration, can support candidate selection and provide valuable insights in the underlying mechanisms of action of intratumoral gene transfer. In addition, clinical translation requires further optimization of the currently used pDNA constructs (e.g., via antibiotic resistance-free backbones)37 and implementation of clinical-grade electroporation setups.10,38
In conclusion, we optimized the in vivo expression of two DNA-based checkpoint inhibitors through engineering of the plasmid design, expression cassette, and mAb sequences. Both intramuscular and intratumoral delivery gave significant anti-tumor responses, despite the distinct mAb exposure. The tumor thereby emerged as an appealing delivery site for cost-efficient DNA-based mAb (combination) therapies, enabling local and systemic efficacy with favorable exposure. Pending further characterization and validation, this approach could provide an alternative when conventional administration is not adequate.
Materials and Methods
pDNA Constructs
For the construction of a DNA-based murine anti-mouse CTLA-4 mAb, cDNA sequences of the 9D9 clone were derived from the literature.39 Based on alignment to the mouse IGHV1-19*01 and IGKV1-117*01 germline sequences, three amino acids were modified in the variable regions of the 9D9 HC and LC, respectively. These sequences were then grafted onto a murine IgG2ab HC and kappa LC backbone, and codon-optimized for expression in mice. HC and LC were cloned by Icosagen (Tartu, Estonia) into separate pDNA constructs [p(aCTLA-4-HC) and p(aCTLA-4-LC), respectively], each containing an ampicillin resistance gene, a pUC origin of replication, a CAG promoter, and a TK poly(A) sequence. Additionally, a single construct was generated expressing both HC and LC by means of a dual CAG cassette in tandem [p(aCTLA-4-HC+LC)].
A DNA-based anti-mouse PD-1 mAb was similarly constructed as p(aCTLA-4-HC+LC). Here, the rat variable regions of the RMP1-14 clone (kindly provided by Prof. Hideo Yagita, Juntendo University School of Medicine, Tokyo, Japan) were grafted onto a murine IgG2aa HC and kappa LC backbone. These were then cloned into a single construct containing two CAG promoters [p(CAG-caPD-1-HC2a+LC)] by Icosagen. To reduce the immunogenicity of the expressed anti-PD-1 mAb, pDNA encoding a chimeric mAb with the same variable regions but a murine IgG1 backbone [p(CMV-caPD-1-HC1), p(CMV-caPD-1-LC)] was purchased from Absolute Antibody (Redcar, UK). Additionally, Absolute Antibody murinized the chimeric IgG1 anti-PD-1 mAb sequences, i.e., mutations were introduced in the variable regions to improve similarity to mouse germline sequences [p(CMV-maPD-1-HC1), p(CMV-maPD-1-LC)]. The latter pDNA constructs consisted of an ampicillin resistance gene, a pUC origin of replication, and a CMV-driven expression cassette, expressing either the HC or LC and ending with an SV40 poly(A) sequence. Finally, the murinized anti-PD-1 sequences were cloned into the CAG-driven pDNA backbone, with separate plasmids expressing HC and LC [p(CAG-maPD-1-HC1) and p(CAG-maPD-1-LC), respectively]. Characteristics of the anti-CTLA-4 and anti-PD-1 mAb-encoding pDNA constructs are summarized in Table 1.
Two empty plasmids served as controls. pNull contains only the ampicillin resistance gene and pUC origin of replication, and was provided by Icosagen. pControl was cloned in-house by deletion of the CMV-driven expression cassette from the Absolute Antibody pDNA backbone. pDNA production and purification were performed as previously described.5
Mice and Tumor Experiments
C57BL/6J mice were bred at the KU Leuven Animal Research Center or purchased from Charles River Laboratories (Saint Germain Nuelles, France). All experiments were approved by the KU Leuven Animal Ethics Committee (P130/2017).
The MC38 cell line (ENH204-FP), derived from C57BL/6 murine colon adenocarcinoma cells, was purchased from Kerafast (Boston, MA, USA) in March 2017 and tested for Mycoplasma contamination. Cells were grown in Dulbecco’s modified Eagle medium, supplemented with 10% heat-inactivated fetal bovine serum, 0.1 mM non-essential amino acids, 1 mM sodium pyruvate, 10 mM HEPES, and 50 U/mL penicillin/streptomycin (Thermo Fischer Scientific, Waltham, MA, USA), at 37°C in a humidified incubator at 5% CO2, starting from a master stock frozen after three passages in March 2017. Following three to six additional passages, 1 × 106 MC38 cells in 100 μL D-PBS (14190-094, Thermo Fischer Scientific) were injected s.c. in the right flank of 6- to 8-week-old female C57BL/6J mice. To study abscopal effects, 0.25 × 106 MC38 cells in 100 μL D-PBS were injected s.c. in the left flank 3 days after the first tumor cell injection. For rechallenge experiments, mice with complete tumor regressions received a s.c. injection of 1 × 106 MC38 cells in 100 μL D-PBS in the left flank 11–23 weeks after the first tumor cell injection. Tumors were measured three times per week using an electronic caliper (500-712-20; Mitutoyo, Kawasaki, Japan). Tumor volume was calculated using the formula a × b2 × 0.5, in which a represents the tumor length and b the width. Mice were sacrificed when total tumor volume exceeded 2,000 mm3.
Intramuscular and Intratumoral Electroporation
Intramuscular pDNA electrotransfer was performed in the tibialis anterior muscle of 7- to 8-week-old female mice as previously described,5 unless otherwise stated. In brief, muscles were pretreated with 40 μL of 0.4 U/μL hyaluronidase from bovine testes (reconstituted in sterile saline or D-PBS; H4272; Sigma, St. Louis, MO, USA). Approximately 1 h later, 60 μg pDNA formulated in 30 μL D-PBS was injected intramuscularly immediately followed by electroporation. The electroporation protocol comprised three series of four 20-ms square-wave pulses of 120 V/cm with a 50-ms interval between the pulses and polarity switching after two pulses. Intratumoral pDNA electrotransfer was performed 4–7 days after tumor cell injection, when s.c. tumors were palpable, using a previously described preclinical protocol.25,26 A total of 60 μg pDNA formulated in 30 μL D-PBS was injected in the tumor, unless otherwise stated, immediately followed by two series of four 5-ms square-wave pulses of 600 V/cm in perpendicular directions at a frequency of 1 Hz. When separate pDNA constructs were administered encoding either the mAb HC or LC, these were first mixed at a 1:1 molar ratio. Electrical pulses were delivered by the preclinical NEPA21 Electroporator (Sonidel, Dublin, Ireland) with CUY650P5 tweezer electrodes (Sonidel) at a fixed width of 5 mm. To improve contact of the electrodes with the skin and to control tissue impedance, the skin and electrodes were covered with Signa Electrode Gel (15-25; Parker Laboratories, Fairfield, NJ, USA) or Eco Ultrasound Transmission Gel (G0066; Fiab, Vicchio, Italy) before intramuscular and intratumoral electroporation, respectively. Pulse current and total energy were verified with the NEPA21 readout.
In Vitro Antibody Production
Anti-CTLA-4 and anti-PD-1 mAbs were produced in vitro in 293F Freestyle suspension cells (R79007; Thermo Fischer Scientific), as previously described.5 Cell media were collected 3–5 days after transfection, followed by centrifugation, 0.2-μm filtration of the supernatant, and storage at −20°C. Expressed mAbs were purified as described before5 and used as calibrator in the respective ELISAs or used for intratumoral and intraperitoneal injection in mice.
ELISA
Blood was collected by retro-orbital bleeding of mice, processed to plasma, and stored at −20°C. Anti-CTLA-4 and anti-PD-1 mAbs were quantified in plasma and cell supernatant using two separate in-house developed ELISAs. Ninety-six-well plates were coated overnight at 4°C with 100 μL of 250 ng/mL mouse CTLA-4/Fc protein (50503-M02H; Sino Biological, Beijing, China) or 2 μg/mL mouse PD-1/Fc chimera protein (1021-PD-100; R&D Systems, Minneapolis, MN, USA) in PBS, respectively. Then, plates were blocked with 200 μL of 1% bovine serum albumin (BSA; A3294-50G; Sigma) in PBS at room temperature for 2 h. Cell supernatant samples were diluted in PTA (0.1% BSA and 0.002% Tween 80 in PBS), plasma samples and calibrator in PTAE (5 mM EDTA disodium salt dihydrate in PTA), and 100 μL of each was incubated in the wells at room temperature for 1 h. The calibration curve consisted of serial 2-fold dilutions of in-house-produced and purified mAb proteins (anti-CTLA-4 mAb: 125 to 0.98 ng/mL; anti-PD-1 mAb: 120 to 0.94 ng/mL). Captured anti-CTLA-4 or anti-PD-1 mAbs were detected with 100 μL rabbit anti-mouse IgG-horseradish peroxidase (HRP) (RAM/IgG(H+L)/PO; Nordic-MUbio, Susteren, Netherlands) or goat anti-mouse IgG-HRP (170-6516; Bio-Rad, Hercules, CA, USA), respectively, diluted 1:10,000 in PTA and incubated at room temperature for 1 h. Finally, plates were developed with 160 μL of 400 μg/mL O-phenylenediamine and 0.04% H2O2 in citrate buffer (pH 5) at room temperature. After 30–45 min, the reaction was stopped with 50 μL of 4 M H2SO4, and absorption was measured at 490 nm using an ELx808 Absorbance Microplate Reader (BioTek Instruments, Winooski, VT, USA). The presence of antibodies toward the expressed anti-PD-1 mAbs was assessed by drug-tolerant affinity capture elution (ACE) assays set up as previously described.40
Statistics
At the start of experiments, mice were randomized based on tumor volume and/or weight. Statistical analyses were performed with GraphPad Prism 7.02 (GraphPad Software, San Diego, CA, USA). Data were presented as mean + standard error of the mean (SEM) and compared using the unpaired t test or one-way ANOVA with a Tukey’s test for multiple comparison, depending on the number of groups. Kaplan-Meier survival curves were analyzed with the log-rank (Mantel-Cox) test with a Holm’s test for multiple comparisons. Two-sided p values below 0.05 were considered significant.
Author Contributions
L.J., N.G., P.D., and K.H. contributed to the design of the study. L.J. and E.D.S. performed the experiments. L.J., E.D.S., N.G., P.D., and K.H. interpreted the results. L.J. wrote the manuscript, which was reviewed and edited by P.D. and K.H. All authors read and approved the manuscript for publication.
Conflicts of Interest
K.H. received consulting fees from OncoSec Medical (San Diego, CA, USA). All other authors declare no competing interests.
Acknowledgments
This research is supported by Research Foundation - Flanders (FWO: PhD mandate 1133220N to L.J.; research project G0E2117N to P.D. and K.H.), KU Leuven (C2 grant: C22/15/024 to P.D. and K.H.), and Flanders Innovation & Entrepreneurship (VLAIO: IWT.150743 to K.H.). We would also like to thank Dr. Andres Männik (Icosagen, Tartu, Estonia) for his guidance in plasmid design, and Prof. Gregor Serša, Prof. Maja Čemažar, and Dr. Špela Kos (Institute of Oncology, Ljubljana, Slovenia) for sharing their expertise in intratumoral electroporation.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.02.007.
Contributor Information
Paul Declerck, Email: paul.declerck@kuleuven.be.
Kevin Hollevoet, Email: kevin.hollevoet@kuleuven.be.
Supplemental Information
References
- 1.Ribas A., Wolchok J.D. Cancer immunotherapy using checkpoint blockade. Science. 2018;359:1350–1355. doi: 10.1126/science.aar4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andrews A. Treating with Checkpoint Inhibitors-Figure $1 Million per Patient. Am. Health Drug Benefits. 2015;8:9. [PMC free article] [PubMed] [Google Scholar]
- 3.Schmidt C. The benefits of immunotherapy combinations. Nature. 2017;552:S67–S69. doi: 10.1038/d41586-017-08702-7. [DOI] [PubMed] [Google Scholar]
- 4.Hollevoet K., Declerck P.J. State of play and clinical prospects of antibody gene transfer. J. Transl. Med. 2017;15:131. doi: 10.1186/s12967-017-1234-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hollevoet K., De Smidt E., Geukens N., Declerck P. Prolonged in vivo expression and anti-tumor response of DNA-based anti-HER2 antibodies. Oncotarget. 2018;9:13623–13636. doi: 10.18632/oncotarget.24426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hollevoet K., De Vleeschauwer S., De Smidt E., Vermeire G., Geukens N., Declerck P. Bridging the Clinical Gap for DNA-Based Antibody Therapy Through Translational Studies in Sheep. Hum. Gene Ther. 2019;30:1431–1443. doi: 10.1089/hum.2019.128. [DOI] [PubMed] [Google Scholar]
- 7.Heller R., Heller L.C. Gene electrotransfer clinical trials. Adv. Genet. 2015;89:235–262. doi: 10.1016/bs.adgen.2014.10.006. [DOI] [PubMed] [Google Scholar]
- 8.Egeland C., Baeksgaard L., Johannesen H.H., Löfgren J., Plaschke C.C., Svendsen L.B., Gehl J., Achiam M.P. Endoscopic electrochemotherapy for esophageal cancer: a phase I clinical study. Endosc. Int. Open. 2018;6:E727–E734. doi: 10.1055/a-0590-4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patel P.M., Ottensmeier C.H., Mulatero C., Lorigan P., Plummer R., Pandha H., Elsheikh S., Hadjimichael E., Villasanti N., Adams S.E. Targeting gp100 and TRP-2 with a DNA vaccine: Incorporating T cell epitopes with a human IgG1 antibody induces potent T cell responses that are associated with favourable clinical outcome in a phase I/II trial. OncoImmunology. 2018;7:e1433516. doi: 10.1080/2162402X.2018.1433516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Daud A.I., DeConti R.C., Andrews S., Urbas P., Riker A.I., Sondak V.K., Munster P.N., Sullivan D.M., Ugen K.E., Messina J.L., Heller R. Phase I trial of interleukin-12 plasmid electroporation in patients with metastatic melanoma. J. Clin. Oncol. 2008;26:5896–5903. doi: 10.1200/JCO.2007.15.6794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Canton D.A., Shirley S., Wright J., Connolly R., Burkart C., Mukhopadhyay A., Twitty C., Qattan K.E., Campbell J.S., Le M.H. Melanoma treatment with intratumoral electroporation of tavokinogene telseplasmid (pIL-12, tavokinogene telseplasmid) Immunotherapy. 2017;9:1309–1321. doi: 10.2217/imt-2017-0096. [DOI] [PubMed] [Google Scholar]
- 12.Ray A., Williams M.A., Meek S.M., Bowen R.C., Grossmann K.F., Andtbacka R.H., Bowles T.L., Hyngstrom J.R., Leachman S.A., Grossman D. A phase I study of intratumoral ipilimumab and interleukin-2 in patients with advanced melanoma. Oncotarget. 2016;7:64390–64399. doi: 10.18632/oncotarget.10453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ellmark P., Mangsbo S.M., Furebring C., Norlén P., Tötterman T.H. Tumor-directed immunotherapy can generate tumor-specific T cell responses through localized co-stimulation. Cancer Immunol. Immunother. 2017;66:1–7. doi: 10.1007/s00262-016-1909-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Hooren L., Sandin L.C., Moskalev I., Ellmark P., Dimberg A., Black P., Tötterman T.H., Mangsbo S.M. Local checkpoint inhibition of CTLA-4 as a monotherapy or in combination with anti-PD1 prevents the growth of murine bladder cancer. Eur. J. Immunol. 2017;47:385–393. doi: 10.1002/eji.201646583. [DOI] [PubMed] [Google Scholar]
- 15.Hebb J.P.O., Mosley A.R., Vences-Catalán F., Rajasekaran N., Rosén A., Ellmark P., Felsher D.W. Administration of low-dose combination anti-CTLA4, anti-CD137, and anti-OX40 into murine tumor or proximal to the tumor draining lymph node induces systemic tumor regression. Cancer Immunol. Immunother. 2018;67:47–60. doi: 10.1007/s00262-017-2059-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hamilton J.R., Vijayakumar G., Palese P. A Recombinant Antibody-Expressing Influenza Virus Delays Tumor Growth in a Mouse Model. Cell Rep. 2018;22:1–7. doi: 10.1016/j.celrep.2017.12.025. [DOI] [PubMed] [Google Scholar]
- 17.Ballesteros-Briones M.C., Martisova E., Casales E., Silva-Pilipich N., Buñuales M., Galindo J., Mancheño U., Gorraiz M., Lasarte J.J., Kochan G. Short-Term Local Expression of a PD-L1 Blocking Antibody from a Self-Replicating RNA Vector Induces Potent Antitumor Responses. Mol. Ther. 2019;27:1892–1905. doi: 10.1016/j.ymthe.2019.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Replimune . 2020. Replimune Provides 2019 Year End Corporate Update and Reviews Expected 2020 Milestones.https://ir.replimune.com/news-releases/news-release-details/replimune-provides-2019-year-end-corporate-update-and-reviews [Google Scholar]
- 19.Duperret E.K., Trautz A., Stoltz R., Patel A., Wise M.C., Perales-Puchalt A., Smith T., Broderick K.E., Masteller E., Kim J.J. Synthetic DNA-Encoded Monoclonal Antibody Delivery of Anti-CTLA-4 Antibodies Induces Tumor Shrinkage In Vivo. Cancer Res. 2018;78:6363–6370. doi: 10.1158/0008-5472.CAN-18-1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perales-Puchalt A., Duperret E.K., Muthumani K., Weiner D.B. Simplifying checkpoint inhibitor delivery through in vivo generation of synthetic DNA-encoded monoclonal antibodies (DMAbs) Oncotarget. 2019;10:13–16. doi: 10.18632/oncotarget.26535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.European Medicines Agency . 2020. Ipilimumab (Yervoy)—Summary of Product Characteristics.https://www.ema.europa.eu/en/documents/product-information/yervoy-epar-product-information_en.pdf [Google Scholar]
- 22.European Medicines Agency . 2020. Nivolumab (Opdivo)—Summary of Product Characteristics.https://www.ema.europa.eu/en/documents/product-information/opdivo-epar-product-information_en.pdf [Google Scholar]
- 23.Schreier P.H., Bothwell A.L., Mueller-Hill B., Baltimore D. Multiple differences between the nucleic acid sequences of the IgG2aa and IgG2ab alleles of the mouse. Proc. Natl. Acad. Sci. USA. 1981;78:4495–4499. doi: 10.1073/pnas.78.7.4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Selby M.J., Engelhardt J.J., Quigley M., Henning K.A., Chen T., Srinivasan M., Korman A.J. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol. Res. 2013;1:32–42. doi: 10.1158/2326-6066.CIR-13-0013. [DOI] [PubMed] [Google Scholar]
- 25.Cemazar M., Golzio M., Sersa G., Hojman P., Kranjc S., Mesojednik S., Rols M.P., Teissie J. Control by pulse parameters of DNA electrotransfer into solid tumors in mice. Gene Ther. 2009;16:635–644. doi: 10.1038/gt.2009.10. [DOI] [PubMed] [Google Scholar]
- 26.Todorovic V., Kamensek U., Sersa G., Cemazar M. Changing electrode orientation, but not pulse polarity, increases the efficacy of gene electrotransfer to tumors in vivo. Bioelectrochemistry. 2014;100:119–127. doi: 10.1016/j.bioelechem.2013.12.002. [DOI] [PubMed] [Google Scholar]
- 27.Bosnjak M., Jesenko T., Kamensek U., Sersa G., Lavrencak J., Heller L., Cemazar M. Electrotransfer of Different Control Plasmids Elicits Different Antitumor Effectiveness in B16.F10 Melanoma. Cancers (Basel) 2018;10:37. doi: 10.3390/cancers10020037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kitaguchi K., Toda M., Takekoshi M., Maeda F., Muramatsu T., Murai A. Immune deficiency enhances expression of recombinant human antibody in mice after nonviral in vivo gene transfer. Int. J. Mol. Med. 2005;16:683–688. [PubMed] [Google Scholar]
- 29.Andrews C.D., Luo Y., Sun M., Yu J., Goff A.J., Glass P.J., Padte N.N., Huang Y., Ho D.D. In Vivo Production of Monoclonal Antibodies by Gene Transfer via Electroporation Protects against Lethal Influenza and Ebola Infections. Mol. Ther. Methods Clin. Dev. 2017;7:74–82. doi: 10.1016/j.omtm.2017.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Belmar N.A., Chan S.W., Fox M.I., Samayoa J.A., Stickler M.M., Tran N.N., Akamatsu Y., Hollenbaugh D., Harding F.A., Alvarez H.M. Murinization and H Chain Isotype Matching of the Anti-GITR Antibody DTA-1 Reduces Immunogenicity-Mediated Anaphylaxis in C57BL/6 Mice. J. Immunol. 2017;198:4502–4512. doi: 10.4049/jimmunol.1601512. [DOI] [PubMed] [Google Scholar]
- 31.Dahan R., Sega E., Engelhardt J., Selby M., Korman A.J., Ravetch J.V. FcγRs Modulate the Anti-tumor Activity of Antibodies Targeting the PD-1/PD-L1 Axis. Cancer Cell. 2015;28:285–295. doi: 10.1016/j.ccell.2015.08.004. [DOI] [PubMed] [Google Scholar]
- 32.European Medicines Agency . 2019. Pembrolizumab (Keytruda)—Summary of Product Characteristics.https://www.ema.europa.eu/en/documents/product-information/keytruda-epar-product-information_en.pdf [Google Scholar]
- 33.Feng Y., Roy A., Masson E., Chen T.T., Humphrey R., Weber J.S. Exposure-response relationships of the efficacy and safety of ipilimumab in patients with advanced melanoma. Clin. Cancer Res. 2013;19:3977–3986. doi: 10.1158/1078-0432.CCR-12-3243. [DOI] [PubMed] [Google Scholar]
- 34.Wang X., Feng Y., Bajaj G., Gupta M., Agrawal S., Yang A., Park J.S., Lestini B., Roy A. Quantitative Characterization of the Exposure-Response Relationship for Cancer Immunotherapy: A Case Study of Nivolumab in Patients With Advanced Melanoma. CPT Pharmacometrics Syst. Pharmacol. 2017;6:40–48. doi: 10.1002/psp4.12133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu J., Blake S.J., Smyth M.J., Teng M.W. Improved mouse models to assess tumour immunity and irAEs after combination cancer immunotherapies. Clin. Transl. Immunology. 2014;3:e22. doi: 10.1038/cti.2014.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dolinsek T., Markelc B., Bosnjak M., Blagus T., Prosen L., Kranjc S., Stimac M., Lampreht U., Sersa G., Cemazar M. Endoglin silencing has significant antitumor effect on murine mammary adenocarcinoma mediated by vascular targeted effect. Curr. Gene Ther. 2015;15:228–244. doi: 10.2174/1566523215666150126115501. [DOI] [PubMed] [Google Scholar]
- 37.Kamensek U., Tesic N., Sersa G., Cemazar M. Clinically Usable Interleukin 12 Plasmid without an Antibiotic Resistance Gene: Functionality and Toxicity Study in Murine Melanoma Model. Cancers (Basel) 2018;10:60. doi: 10.3390/cancers10030060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spanggaard I., Snoj M., Cavalcanti A., Bouquet C., Sersa G., Robert C., Cemazar M., Dam E., Vasseur B., Attali P. Gene electrotransfer of plasmid antiangiogenic metargidin peptide (AMEP) in disseminated melanoma: safety and efficacy results of a phase I first-in-man study. Hum. Gene Ther. Clin. Dev. 2013;24:99–107. doi: 10.1089/humc.2012.240. [DOI] [PubMed] [Google Scholar]
- 39.Allison J., Curran M. 2017. Methods and Compositions for Localized Secretion of Anti-CTLA-4 Antibodies. US Patent US20170114364A9. filed March 30, 2007, and published April 27, 2017. [Google Scholar]
- 40.Van Stappen T., Vande Casteele N., Van Assche G., Ferrante M., Vermeire S., Gils A. Clinical relevance of detecting anti-infliximab antibodies with a drug-tolerant assay: post hoc analysis of the TAXIT trial. Gut. 2018;67:818–826. doi: 10.1136/gutjnl-2016-313071. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.