

Abstract
Mesenchymal stem cells (MSCs) have shown great promise in inflammatory bowel disease (IBD) treatment, owing to their immunosuppressive capabilities, but their therapeutic effectiveness is sometimes thwarted by their low efficiency in entering the inflamed colon and variable immunomodulatory ability in vivo. Here, we demonstrated a new methodology to manipulate MSCs to express CX3C chemokine receptor 1 (CX3CR1) and interleukin-25 (IL-25) to promote their delivery to the inflamed colon and enhance their immunosuppressive capability. Compared to MSCs without treatment, MSCs infected with a lentivirus (LV) encoding CX3CR1 and IL-25 (CX3CR1&IL-25-LV-MSCs) exhibited enhanced targeting to the inflamed colon and could further move into extravascular space of the colon tissues via trans-endothelial migration in dextran sodium sulfate (DSS)-challenged mice after MSC intravenous injection. The administration of the CX3CR1&IL-25-LV-MSCs achieved a better therapeutic effect than that of the untreated MSCs, as indicated by pathological indices and inflammatory markers. Antibody-blocking studies indicated that the enhanced therapeutic effects of dual-functionalized MSCs were dependent on CX3CR1 and IL-25 function. Overall, this strategy, which is based on enhancing the homing and immunosuppressive abilities of MSCs, represents a promising therapeutic approach that may be valuable in IBD therapy.
Keywords: inflammatory bowel disease, dual-functionalized mesenchymal stem cells, CX3CR1, IL-25
Graphical Abstract
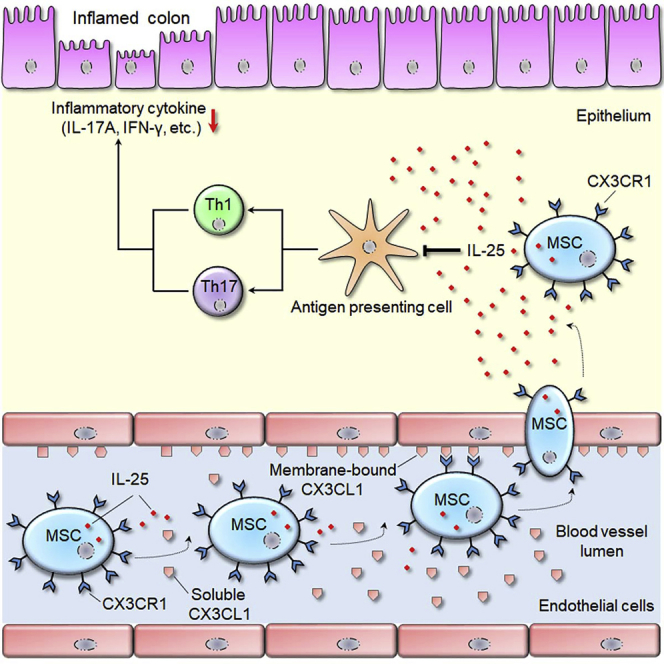
Fu et al. report a new methodology to manipulate MSCs to express CX3CR1 and IL-25, and the engineered MSCs can effectively protect against experimental colitis. Based on enhancing the homing and immunosuppressive abilities of MSCs, this strategy represents a promising approach to improve the therapeutic outcome in inflammatory bowel diseases.
Introduction
Inflammatory bowel disease (IBD) is a chronic relapsing inflammatory disorder within the gastrointestinal tract.1 Although the precise etiology of IBD remains unclear, it is generally accepted that interactions between genetic susceptibility and environmental triggers lead to chronic intestinal inflammation.2,3 Notably, the expansion of mucosal leukocytes and the expression of related inflammatory cytokines lead to damage the gut mucosal barrier, which seems to play a crucial role in the pathogenesis of IBD.4,5
Current strategies for biological therapy such as application of monoclonal antibodies (mAbs) against immune cells, inflammatory cytokines, or immunomodulatory factors have shown great promise in clinical IBD studies.6,7 Owing to their immunosuppressive capabilities and homing property, mesenchymal stem cells (MSCs) have potential in IBD treatment.8 However, the immunomodulatory properties of MSCs are highly variable in a complex pathophysiological environment.9 Moreover, the efficiency of MSC delivery to inflamed sites is poor when these cells are systemically administered.10 Therefore, modifying MSCs to express therapeutic and targeting molecules can potentially be a useful approach to improve their therapeutic efficacy in inflammation-related diseases.
In the present study, interleukin 25 (IL-25) and CX3C chemokine receptor 1 (CX3CR1) were chosen as the therapeutic and targeting molecules, respectively. IL-25 is an important regulatory cytokine that plays a key role on colonic immune tolerance via suppressing mucosal Th1/Th17 responses.11 CX3CL1, which is significantly upregulated in the inflamed colon tissues, can recruit circulating cells expressing CX3CR1 into colon tissues and may further promote trans-endothelial migration of MSCs.12 Particularly, we infected bone marrow-derived MSCs (BM-MSCs) from rats with a lentivirus (LV) encoding both CX3CR1 and IL-25 and evaluated phenotypic changes and immunosuppressive and migratory capabilities of the engineered MSCs in vitro. Then, biodistribution and therapeutic effect of these dual-functionalized MSCs were investigated in a dextran sodium sulfate (DSS)-induced murine colitis model (Figure 1). Moreover, the influence of the modified MSCs on colonic mucosal T cell responses was studied via qRT-PCR, cytokine analysis, flow cytometry analysis, and immunofluorescence staining.
Figure 1.

A Schematic Diagram of the Experimental Approach for Engineered MSCs in Colitis
Results
Determination of CX3CR1 and IL-25 Expression Levels in Engineered MSCs
The construction process used to produce engineered MSCs is shown in Figure 1. As shown in Figure 2A, MSCs infected with LV encoding CX3CR1 and IL-25 (CX3CR1&IL-25-LV-MSCs) exhibited the strongest CX3CR1 expression at 96 h after infection. MSCs infected with CX3CR1-LV or CX3CR1&IL-25-LV showed higher CX3CR1 expression than blank MSCs or empty-LV-infected MSCs (Figure 2B). Consistent with the western blotting results, flow cytometry analysis results indicated that infection with CX3CR1-LV (96.13% CX3CR1 positive) or CX3CR1&IL-25-LV (94.48% CX3CR1 positive) resulted in enhanced surface expression of CX3CR1, whereas empty-LV or IL-25-LV infection showed no effect on CX3CR1 expression (Figures 2C and 2D). Time-course experiments demonstrated that IL-25 expression in CX3CR1&IL-25-LV-MSCs reached a peak at day 4 post-LV infection (Figure 2E). Either IL-25-LV or CX3CR1&IL-25-LV infection could enhance IL-25 production (Figure 2F). Immunofluorescence staining also indicated strong expression of CX3CR1 and IL-25 in CX3CR1&IL-25-LV-infected cells (Figure 2G).
Figure 2.

MSC Expression of CX3CR1 and IL-25 following LV Infection
(A) The expression level of CX3CR1 was examined in MSCs after infection with a LV encoding both CX3CR1 and IL-25 at different time points. (B) The expression level of CX3CR1 was examined in MSCs after treatment with different types of LVs at 96 h post-infection. (C) Representative flow cytometry images of CX3CR1 expression levels in MSCs administered the indicated treatments. (D) The quantification results of CX3CR1 positive cells in MSCs. (E) The protein levels of IL-25 in MSCs treated with CX3CR1&IL-25-LV at different time points were assayed by ELISA. (F) The protein levels of IL-25 in MSCs 96 h after different types of LV infection were assayed by ELISA. (G) Immunofluorescence staining for CX3CR1 and IL-25 was performed with empty-LV-infected MSCs and CX3CR1&IL-25-LV-MSCs (96 h post-infection; red indicates CX3CR1, green indicates IL-25, and blue indicates DAPI nuclear staining). Scale bars, 10 μm. For cell experiments, five samples were analyzed per condition, and the experiments were performed in triplicate. The western blotting data shown are representative of three individual analyses. Values are expressed as the mean ± SEM. ***p < 0.001; n.d., not detected.
Phenotypic Stability and Immunogenicity of MSCs following LV Infection
Results using microscopy indicated that LV infection showed no effects on the morphology of MSCs (Figure 3A). In addition, the results of flow cytometry analysis indicated that the expression levels of leukocyte markers (CD11b and CD45) and MSC markers (CD29 and CD90) were not significantly changed among MSCs that received different treatments (Figure 3B; Figure S1A). The results of the Cell Counting Kit-8 (CCK-8; Dojindo Laboratory) assay showed no significant change in the number of MSCs after LV infection (Figure 3C). To assess the immunogenicity of different LV-infected MSCs, flow cytometry analysis was further applied, and the results revealed that the expression levels of major histocompatibility complex class I and class II (MHC-I and MHC-II, respectively) and costimulatory molecules (CD40, CD80, or CD86) were not significantly varied after MSCs received different treatments (Figure 3D; Figure S1B). After co-culture with LV-infected MSCs, the stimulation index (SI) of lymphocytes from mice with DSS-induced colitis was not different from that of the negative control group (lymphocytes without any treatment) (Figure 3E). These results demonstrated that the engineered MSCs were hypoimmunogenic and might be beneficial to avoid immune rejection.
Figure 3.

Phenotypic Characterization and Immunogenicity Analysis of following LV Infection
(A) The morphologies of MSCs treated with different types of LVs at passage 2. Scale bars, 20 μm. For cell experiments, five samples were analyzed per condition, and the experiments were performed in triplicate. (B) Flow cytometry analysis of immune cell markers (CD11b and CD45) and stem cell markers (CD29 and CD90) in MSCs given different treatments. (C) Cell proliferation rates of MSCs given the indicated treatments, as determined using a CCK-8 assay. NS, no significant change, compared between MSCs and CX3CR1&IL-25-LV-MSCs. (D) Flow cytometry analysis of markers related to immunogenicity (CD80, CD86, CD40, MHC-I, and MHC-II) in MSCs with LV infection. (E) Lymphocytes from mice with DSS-induced colitis were co-cultured with LV-infected MSCs for 72 h, and then the proliferation rates of lymphocytes were determined by a CCK-8 assay. Stimulation index (SI): for co-culture groups, SI = (sample OD − basal OD)/negative control OD; for lymphocytes and lymphocytes + Con A groups, SI = sample OD/negative control OD. Values are expressed as the mean ± SEM. ***p < 0.001. NS, no significant change.
Migratory and Immunosuppressive Capabilities of Engineered MSCs In Vitro
An MSC chemotaxis assay was performed to investigate the migratory ability of engineered MSCs in vitro. Migration analysis demonstrated that the chemotactic responses of MSCs infected with different LVs exhibited no differences when the lower chamber contained only culture medium without the addition of CX3CL1. However, the addition of a recombinant CX3CL1 protein into the lower chamber could greatly enhance the migration of CX3CR1-LV- or CX3CR1&IL-25-LV-MSCs (Figures 4A and 4B). We also noticed that the addition of CX3CL1 could mildly promote the migration of blank, empty-LV-, or IL-25-LV-treated MSCs. It is possible that recombinant CX3CL1 protein has cross-reactivity and can recruit MSCs that have a basal level of CX3CR1. To examine the potential immunosuppressive capability of engineered MSCs, supernatants from MSCs were added to lipopolysaccharide (LPS)-activated antigen-presenting cells (APCs). ELISAs indicated that the supernatants of both IL-25-LV- and CX3CR1&IL-25-LV-MSCs could suppress IL-12p70 and IL-23 secretion by APCs relatively effectively, whereas the addition of an anti-IL-25 mAb to the supernatants of IL-25-LV- and CX3CR1&IL-25-LV-MSCs recovered the production of IL-12/IL-23 in APCs (Figure 4C). To examine the influences of the different populations of treated MSCs on Th cell differentiation, naive CD4+ T cells were isolated and cultured with or without the medium from the aforementioned treated APCs for 4 days, and the protocol is shown in Figure 4D. Flow cytometry analysis indicated that the supernatants from LPS-activated APCs could promote the polarization of Th1 and Th17 cells, whereas the addition of the supernatants from the APCs treated with IL-25-LV-infected- or CX3CR1&IL-25-LV-MSC medium could effectively reduce the percentages of interferon (IFN)-γ+ Th1 cells and IL-17A+ Th17 cells. Furthermore, the supernatant from CX3CR1&IL-25-LV-MSCs with an added anti-IL-25 mAb recovered the percentages of IFN-γ+ Th1 cells and IL-17A+ Th17 cells (Figure 4E; Figure S2).
Figure 4.

Migratory Ability and Immunomodulatory Capability Assays with Engineered MSCs
(A) The cell migration of MSCs given different treatments (infected with empty-LV, IL-25-LV, CX3CR1-LV, or CX3CR1&IL-25-LV) was examined by Transwell migration assays after the cells were plated in the upper chamber, CX3CL1 was added in the bottom chamber, and the system was incubated for 24 h. Scale bars, 20 μm. (B) Cells were counted in 5 randomly selected fields, and cell counts were averaged. (C) The production of IL-12/IL-23 in LPS-activated APCs was detected after a 12-h exposure to the supernatants of MSCs infected with different types of LVs or the aforementioned supernatant supplemented with an anti-IL-25 mAb or rat IgG (2 μg/mL). (D) Naive CD4+ T cells magnetically purified from spleens harvested from C57BL/6 mice were cultured with the aforementioned treated medium from APCs for 96 h. (E) Representative flow cytometry plots are shown after gating on live CD4+ T cells, followed by gating on CD4+ IFN-γ+ cells or CD4+ IL-17A+ cells. ST, supernatant. For cell experiments, five samples were analyzed per condition, and the experiments were performed in triplicate. Values are expressed as the mean ± SEM *p < 0.05; **p < 0.01; ***p < 0.001.
Tissue Distribution of Engineered MSCs in Mice with DSS-Induced Colitis
We further examined the CX3CL1 levels in tissues from healthy mice and mice with DSS-induced colitis. Results of an ELISA indicated that the concentration of CX3CL1 in the inflamed colon was higher than that in other organs or that in healthy colon (Figure 5A). To examine the biodistribution of engineered MSCs, fluorescence images of organs from mice were performed after intravenous injection with MSCs at 2 h, 24 h, and 8 days. As shown in Figures 5B–5D, compared with empty-LV-MSCs, CX3CR1&IL-25-LV-MSCs remarkably enhanced the accumulation of MSCs in the inflamed colon at 2 h, 24 h, and 8 days after cell injection. High levels of CX3CR1&IL-25-LV-MSCs could be observed in liver and lung at all time points after cell injection, implying that engineered MSCs might be passively entrapped in these organs. However, the amounts of CX3CR1&IL-25-LV-MSCs in the colon were even higher than or comparable with that in the liver and lung at 24 h and 8 days after cell injection (Figures S3C–S3F), suggesting that CX3CR1&IL-25-LV-MSCs can persist in colon long enough to exert their anti-colitis effect. Consistent with the fluorescence images of colon, the intensities of fluorescence of frozen sections of mouse colon also indicated that a large amount of CX3CR1&IL-25-LV-MSCs were localized in the colon (Figure 5C). Furthermore, CD31 immunofluorescence staining was performed on the colon sections from mice with DSS-induced colitis treated with 1,1'-dioctadecyl-3,3,3',3'-tetramethylindocarbocyanine perchlorate (DiI)-labeled CX3CR1&IL-25-LV-MSCs 2 h post-injection. As shown in Figure 5E, CX3CR1&IL-25-LV-MSCs could be observed in extravascular space around the vessel in the inflamed colon tissues. These results imply that CX3CL1 expressed on the endothelial cell surface may promote the trans-endothelial migration of CX3CR1&IL-25-LV-MSCs.
Figure 5.

The Biodistribution of Engineered MSCs
(A) The levels of CX3CL1 in different organs from control mice and mice with DSS-induced colitis. (B and C) Fluorescence quantitative analysis (B) and frozen sections of colon from mice with colitis 2 h, 24 h, and 8 days after fluorescently labeled LV-treated MSCs injection (C) (red indicates MSCs, and blue indicates DAPI nuclear staining; scale bars, 100 μm). (D) Fluorescence images of the colon from mice with colitis at 2 h, 24 h, and 8 days after the administration of different modified MSCs (MSCs 96 h post-LV infection, empty-LV-treated MSCs, or CX3CR1&IL-25-LV-MSCs). (E) Frozen sections of the colons from mice with DSS-induced colitis treated with DiI-labeled CX3CR1&IL-25-LV-MSCs 2 h post-injection were stained with endothelial cell markers (red indicates DiI-labeled CX3CR1&IL-25-LV-MSCs, green indicates CD31, and blue indicates DAPI nuclear staining). Scale bars: 200×, 100 μm; 1,000×, 20 μm. The white arrow represented extravascular DiI-labeled CX3CR1&IL-25-LV-MSCs. BV, blood vessel. Values are expressed as the mean ± SEM (n = 7 mice per group). **p < 0.01; ***p < 0.001; NS, no significant change.
Engineered MSCs Protected against DSS-Induced Colitis
Mice that consumed a 5% DSS solution developed rapid-onset colitis marked by weight loss, diarrhea, and bloody stool, resulting in a mortality rate of 80% (Figure 6A). Moreover, DSS-treated mice given CX3CR1&IL-25-LV-MSCs rapidly recovered lost body weight and exhibited low disease activity index (DAI) scores (Figures 6B and 6D). Macroscopic examination of colons obtained 16 days after model establishment showed striking hyperemia, inflammation, and a shortened colon length. Colons from mice treated with CX3CR1&IL-25-LV-MSCs showed no significant sign of macroscopic inflammation and a longer colon length (Figures 6C and 6E). Myeloperoxidase (MPO) activity, which is positively associated with colonic neutrophil infiltration, was also reduced significantly (Figure 6F). Histological examination showed that DSS-induced colitis affected all layers of the colon, and the effects included submucosal edema, muscle thickening, and strong granulocyte infiltration (Figure 6G). When compared with other groups, the group treated with CX3CR1&IL-25-LV-MSCs showed notable improvements in histological characteristics (Figures 6G and 6H). In addition, the effects of CX3CR1&IL-25-LV-MSCs on mucosal Th1 and Th17 responses were investigated. The mRNA levels of Tbet and-Rorγ in colonic CD4+ T cells were downregulated in the CX3CR1&IL-25-LV-MSC-treated group compared to the control group (Figure S4A). The results of Figure S4B indicated that the engineered MSCs significantly decreased the levels of colonic cytokines (IL-12p70, IL-23, IFN-γ, and IL-17A) and increased IL-25 production. Additionally, the expression of the pro-inflammatory cytokines (IL-1β, IL-6, and tumor necrosis factor alpha [TNF-α]) in inflamed colon was suppressed, which indicated that the dual-functionalized MSCs also impaired innate responses in vivo (Figure S4B). Through immunofluorescence staining for CD4 and IFN-γ/IL-17A, massive Th1/Th17 cell infiltration was observed in the colon sections from mice with DSS-induced colitis. When CX3CR1&IL-25-LV-MSCs were administered, reductions in Th1 (IFN-γ+ CD4+) and Th17 (IL-17A+ CD4+) cells in the lamina propria of colon were observed (Figure S4C).
Figure 6.

Engineered MSCs Protected Mice against DSS-Induced Colitis
Mice were intravenously injected with different types of MSCs (96 h post-infection) on days 4, 6, and 8 (three times in total). (A and B) Survival analysis was performed (A), and body weight was measured daily to monitor colitis severity (B). (C) Colons excised from mice with DSS-induced colitis were photographed. (D and E) The DAI was determined (D), and the colon length was measured (E). (F) Colonic MPO activity was examined. (G and H) Colon sections from mice that had undergone different treatments were examined by H&E staining (G), and histopathological scoring was analyzed (H). Scale bars, 100 μm. Values are expressed as the mean ± SEM (n = 7 mice per group). *p < 0.05; **p < 0.01; ***p < 0.001.
The immune rejection responses induced by xenogeneic MSCs were further studied in mice with DSS-induced colitis. Results in Figures S5A–S5F indicated that intravenous administration of rat MSCs did not change the proportion of CD4+ or CD8+ T cells or B cells in peripheral blood. The CD4/CD8 ratio, a marker of immune activation and immune senescence,13 was not changed in mice with colitis after MSC treatment (Figure S5G). Moreover, cytokine analysis in organs indicated that MSCs did not affect the levels of cytokines that may be involved in immune rejection in different organs, including heart, liver, spleen, lung, and kidney at 2 h, 24 h (data not shown), and 8 days after MSC injection (Figure S5H). These results indicated that the engineered rat MSCs did not induce detectable immune rejection responses in mice with colitis in this study.
Anti-colitis Effect of Engineered MSCs Depends on the Function of CX3CR1 and IL-25
Previous studies indicated that MSCs could express IL-10, prostaglandin E2 (PGE2), and insulin-like growth factor 1 (IGF-1), which might contribute to tissue repair and anti-inflammation.14, 15, 16 Therefore, the paracrine factors of IL-10, PGE2, and IGF-1 were detected in LV-infected MSCs. As shown in Figure S6, LV infection showed no influence on the levels of IGF-1, PGE2, and IL-10, indicating that the enhanced anti-colitis effect of CX3CR1&IL-25-LV-MSCs might not be contributed by the change of paracrine factors.
To examine whether the anti-colitis effect of the dual-functionalized MSCs was dependent on the function of CX3CR1 and IL-25, CX3CR1 antibody was applied to treat CX3CR1&IL-25-LV-MSCs before their injection. Alternatively, IL-25 antibody was intraperitoneally administrated to mice with DSS-induced colitis after MSC tail vein injection. Survival rate (Figure 7A) and body weight (Figure 7B) were monitored over 16 days. At day 16, mice were sacrificed, and colons were excised for macroscopic observation (Figure 7C) and DAI score (Figure 7D). Colon length (Figure 7E) and MPO activity (Figure 7F) were measured, and tissues were fixed in formalin and prepared for histology (Figures 7G and 7H). Compared with treatment with CX3CR1&IL-25-LV-MSCs, either CX3CR1 antibody incubation or IL-25 antibody injection impaired the anti-inflammation efficacy of CX3CR1&IL-25-LV-MSCs in mice with DSS-induced colitis. Therefore, the improved anti-colitis effects of engineered MSCs in mice with DSS-induced colitis were based on the function of CX3CR1 and IL-25, rather than the paracrine factors basically expressed by MSCs.
Figure 7.

Anti-colitis Effect of Engineered MSC Depends on CX3CR1 and IL-25 Function
CX3CR1&IL-25-LV-MSCs pre-incubated with CX3CR1 antibody (10 μg/mL) or mouse IgG were administered into mice with DSS colitis by tail vein injection on days 4, 6, and 8. Mice were treated with dual-functionalized MSCs (intravenous injection) and IL-25 antibody (intraperitoneal injection) on days 4, 6, and 8. (A and B) Survival analysis was performed (A), and body weight was measured daily to monitor colitis severity (B). (C) Colons excised from mice with DSS-induced colitis were photographed. (D and E) The DAI was determined (D), and the colon length was measured (E). (F) Colonic MPO activity was examined. (G and H) Colon sections from mice that had undergone different treatments were examined by H&E staining (G), and histopathological scoring was analyzed (H). Scale bars, 100 μm. Values are expressed as the mean ± SEM (n = 7 mice per group). *p < 0.05; **p < 0.01.
Discussion
For therapies based on MSCs, the most essential aspects are maximizing efficiency of MSC delivery to the diseased sites and ensuring that these MSCs produce as many immunomodulatory factors as possible in a short period of time.17 Therefore, several approaches have been developed to modify MSCs, including biomaterial-assisted methods,18, 19, 20 enzymatic modification of cell-surface proteins,21 anti-addressin antibody coating,8 genetic modification,22 and “painting” MSCs with complement factor H.23 In regard to MSC-mediated IBD treatment, recent reports have demonstrated that preconditioning with poly(I:C), a ligand of Toll-like receptor 3 (TLR3), or MSCs transfected with a let-7a inhibitor improve therapeutic functionality and immunosuppressive activity of MSCs in experimental colitis.24,25 All these methodologies have been proven to effectively enhance the therapeutic potential of MSCs. In the present study, we applied LV vectors to equip MSCs with improved targeting molecules and potent therapeutic agents. However, there are concerns about the safety of MSCs genetically modified with stable DNA in clinical investigations.26,27 Thus, phenotypic characterization analysis and immunogenicity investigation were performed to evaluate the safety of engineered MSCs in the present study. Our results indicated that LV infection showed no influences on the phenotype or cell proliferation rate of MSCs. In vitro immunogenicity analysis and in vivo immunorejection testing indicated that the engineered MSCs were hypoimmunogenic and could not induce detectable immune rejection responses in mice with colitis. Biodistribution assay indicated that xenogeneic MSCs could be detected in the colon tissues 8 d after MSCs injection through the fluorescent signal. Because the fluorophore could be released from MSCs, and the fluorescence signal could persist long after MSCs were dead, other markers such as the level of IL-25 should be presented to better reflect the function of MSCs. As shown in Figure S4B, the IL-25 level in the colon tissues from mice with colitis treated with CX3CR1&IL-25-LV-MSCs was significantly higher than that in colitis mice without any treatment or with unmodified MSC treatment. The results implied that the engineered MSCs in the colon tissues were still alive 8 days after the last injection and could effectively secrete IL-25. Overall, the engraftment of engineered MSCs in this way may not pose a biological safety problem, and the dual functionalized MSCs could stay in the colon tissues long enough to exert anti-colitis activity.
The CX3CL1-CX3CR1 axis was selected to recruit MSCs to the inflamed colon for the following reasons. First, it has been reported that CX3CL1 expression is significantly increased in the colonic epithelium and vascular endothelium in CD patients.12 Consistent with previous reports, this study observed that the CX3CL1 level was upregulated in the colon tissues from DSS mice compared with that from healthy mice. Moreover, we also found that the colonic concentration of CX3CL1 was higher than that in other organs in mice with DSS-induced colitis. Second, unlike other chemokines, CX3CL1 has two forms: the membrane-bound form and the soluble form.28 These different structural forms allow CX3CL1 to function as an adhesion molecule and a chemoattractant, respectively.29 Therefore, CX3CL1 can maintain a potent concentration gradient in the blood, which is efficient for recruiting circulating CX3CR1-positive cells into the blood vessels of the inflamed colon.30 Then, the upregulated CX3CL1 level on the inflamed endothelial cells can bind with CX3CR1, which generates rolling of the CX3CR1-positive cells along inflamed blood vessel wall.31 The rolling movement along the endothelium can slow down the CX3CR1-positive cells in the blood flow, which is beneficial for the subsequent firm adhesion of the CX3CR1-positive cells to the blood vessel and the infiltration of the CX3CR1-positive cells into the inflamed colon tissues.32 Indeed, the experimental data indicated that this process is feasible. An in vitro migration assay indicated that CX3CR1-positive MSCs exhibited enhanced chemotactic activity toward medium containing CX3CL1. We also observed increased accumulation of engineered MSCs in the colon in DSS-challenged mice at three indicated time points after cell injection. Results of immunofluorescence staining further indicated that the engineered MSCs were not simply trapped within the blood vessel but could move into the extravascular space via trans-endothelial migration. Therefore, we believe that the application of CX3CR1-LV to upregulate CX3CR1 expression in MSCs can be useful as a strategy for enhancing the homing properties of MSCs.
As an important immunomodulatory cytokine, IL-25 exhibits significantly downregulated expression in the colon tissues from IBD patients and a variety of murine colitis models.33 Functional studies have revealed that IL-25 can impair mucosal Th1/Th17 responses by suppressing IL-12/IL-23 production in colonic APCs.34 Although endogenous IL-25 seems to act as a proinflammatory factor in DSS-induced colitis,35 it has been indicated that the exogenous administration of high-dose IL-25 protects against colitis in different murine models, including DSS-induced colitis,36 2,4,6-trinitrobenzenesulfonic acid (TNBS)-induced colitis,37 and IL-10-knockout (KO) spontaneous colitis.38 Therefore, we speculate that infecting MSCs with an LV encoding IL-25 can significantly upregulate the local concentration of IL-25 in the colon and enhance the anti-inflammatory activity of these cells. Our experimental data further supported this speculation. When compared with untreated MSCs, MSCs encoding IL-25 showed an enhanced ability to suppress Th1/Th17 responses. Additionally, anti-IL-25 mAb neutralization abolished the anti-Th1/Th17 response ability of engineered MSCs. These results prove that IL-25 plays a key role in the engineered MSC-mediated immunomodulatory effect. Our findings that IL-25 antibody could reduce the therapeutic effect of engineered MSCs agreed with those of previous studies on TNBS, IL-10 KO, and azoxymethane (AOM)/DSS mice (Th1/Th17 cell involved) but contradicted the report involving the oxazolone model (Th2 cell dominated).38, 39, 40 The different therapeutic effect of IL-25 antibody on a variety of colitis models may be caused by the different pathogenesis of colitis models.
MSCs themselves exert immunomodulatory effects by recruiting regulatory T cells (Tregs) and secreting anti-inflammatory mediators that can inhibit innate immune responses and Th1/Th17 responses and promote tissue regeneration.41 Consistent with previous studies, this study found that blank MSCs had certain therapeutic effects.42 Moreover, CX3CR1&IL-25-LV-MSCs showed increased therapeutic efficacy compared with blank MSCs and MSCs infected with an LV encoding CX3CR1 or IL-25 alone. Therefore, the possible reasons for the enhanced therapeutic effects mediated by the CX3CR1&IL-25-LV-MSCs include the following two points. Initially, CX3CR1-LV infection improves the homing ability of MSCs and promotes the influx of more engineered MSCs into inflamed sites. Moreover, IL-25-LV treatment enhances the anti-inflammatory activity of MSCs, and IL-25 secreted by these engineered MSCs might be more effective at suppressing mucosal Th1/Th17-cell-mediated inflammatory responses. Antibody-blocking studies indicated that the improved therapeutic effects of engineered MSCs were based on CX3CR1 and IL-25 function. Therefore, equipment of MSCs with homing and therapeutic molecules leads to an increased number of MSCs with improved immunomodulatory activities in the colitis colon, which is beneficial for achieving an improved therapeutic effect. Certainly, there is still much room to improve our work. Further efforts can be made on how to increase therapeutic efficacy of the engineered MSCs in colitis by avoiding the nonspecific retention of MSCs in lung, liver, and spleen.
Overall, the protective effects of engineered MSCs expressing CX3CR1 and IL-25 on experimental colitis were demonstrated in the present study. This strategy to impair mucosal inflammatory responses through engineered MSCs represents a potential therapeutic approach that may be valuable for IBD treatment.
Materials and Methods
Animals and Reagents
Four to five-week-old female Sprague-Dawley rats and 6- to 8-week-old female C57BL/6 mice were purchased from the Laboratory Animal Center of Nanjing University (Nanjing, China). All animals received humane care in accordance with the guidelines approved by the Animal Care Ethics Committee of Nanjing University. Throughout acclimatization and study periods, all animals had access to food and water ad libitum and were maintained on a 12-h/12-h light/dark cycle (23°C ± 2°C, with a relative humidity of 50% ± 10%) in a specific pathogen-free (SPF) animal facility. DSS (molecular weight, 36–50 kDa) was obtained from MP Biomedicals (Irvine, CA, USA). An MPO activity assay kit was obtained from Jiancheng Biotech (Nanjing, China). A DiI cell membrane-labeling solution was obtained from Beyotime Biotechnology (Nantong, China). Other chemical reagents were purchased from Sigma (St. Louis, MO, USA).
Isolation and Culture of MSCs
MSCs were isolated from Sprague-Dawley rats as previously described.43 Briefly, rat femurs and tibias were excised from the hindlimbs and carefully cleaned to remove adherent tissue under sterile conditions. Then, the dissected femurs and tibias were placed in 75% alcohol for 30 s and transferred to sterile precooled phosphate-buffered saline (PBS). Bone marrow plugs were extracted from the bones by flushing the bone marrow cavity repeatedly with complete culture medium. Then, the homogeneous cell suspension was filtered through a 70-μm nylon strainer (BD Biosciences, Bedford, MA, USA), centrifuged at 1,000 rpm for 5 min at 4°C, and resuspended in low-glucose Dulbecco’s modified eagle medium (DMEM; Life Technologies, Grand Island, New York, USA) containing 10% fetal bovine serum (FBS; low endotoxin ⩽ 10 endotoxin units (EU)/mL, #1091148, Life Technologies), 1% HEPES (Life Technologies), 1% nonessential amino acids (Life Technologies), and an antibiotics mixture (100 U/mL penicillin and 100 μg/mL streptomycin; Beyotime Biotechnology). Subsequently, MSCs were plated in 25-cm2 cell-culture flasks (Corning, Corning, NY, USA) and incubated at 37°C in a humidified atmosphere with 5% CO2 for 24 h before the first medium change. After that, the medium was replaced every 3 days until the MSCs reached 80% confluence. Then, the MSCs were passaged at 1:3 in triple flasks under the same conditions. All experiments were carried out with MSCs passaged no more than five times.
Construction and Phenotypic Analysis of Engineered MSCs
LV vectors with CMV promoter encoding mouse IL-25 (GenBank: NM_080729.3), CX3CR1 (GenBank: NM_009987.4), and CX3CR1&IL-25 were generated by Genecopoeia (Guangzhou, China). An empty negative vector was used as a control. MSCs at passage 2 were seeded into a 6-well plate 24 h prior to viral infection. When the cells reached 70%–80% confluence, the appropriate LV was added into each well at a multiplicity of infection (MOI) of 5:1 and incubated with the cells at 37°C with 5% CO2 overnight. The supernatants were collected, and IL-25 was detected by using an ELISA kit (Thermo Fisher Scientific, Rockford, IL, USA), and the cells were harvested for western blotting, immunofluorescence staining, and flow cytometry analysis. For a cell proliferation assay, MSCs infected with different LVs were seeded into 96-well plates (103 cells per well). A growth curve was constructed via a CCK-8 (Dojindo Laboratory, Kumamoto, Japan) assay. Absorbance was measured at 450 nm using a microplate spectrometer (Tecan Group, Männedorf, Switzerland). Specifically, the levels of paracrine factors in the supernatant from MSCs with different treatments were examined with ELISA kits (rat IGF-1, #70-EK9131-24, MultiSciences, Hangzhou, China; rat IL-10, #70-EK310/2-24, MultiSciences; rat PGE2, #JEB-13530-1, JinYibai Biological Technology, Nanjing, China).
Chemotaxis Assay
For an in vitro chemotaxis assay, MSCs that received different treatments (2 × 105 cells per 200 μL complete medium) were cultured in the upper chamber of an 8-μm-pore polycarbonate membrane (Transwell, Corning). The lower chamber contained 0.8 mL low-glucose DMEM supplemented with 1% FBS and 100 μg/mL CX3CL1 (#300-31, PeproTech, Rocky Hill, NJ, USA). After a 24-h incubation, the nonmigrating cells were gently removed. The cells attached to the lower chamber were fixed with 90% ethanol and stained using 0.1% crystal violet. A Nikon microscope (TE2000U, Tokyo, Japan) was used to analyze 10 different randomly selected fields.
Immunosuppressive Ability Assay for MSCs
APCs were isolated from the spleen by anti-MHC-II antibody-coated paramagnetic beads (Miltenyi Biotech, Cologne, Germany). The purity of the APCs (>95%) was confirmed by a flow cytometer using a fluorescein isothiocyanate (FITC)-labeled anti-MHC-II antibody (#ab24882, Abcam). The supernatants from MSCs that received different treatments were collected at 96 h post-LV infection and mixed with fresh culture medium at a ratio of 1:1. Under some circumstances, 2 μg/mL LEAF-purified anti-mouse IL-25 mAb (#514403, BioLegend, San Diego, CA, USA) was added. Then, mixed medium supplemented with 100 ng/mL LPS (Escherichia coli, L2630, serotype O111:B4, Sigma) was added to stimulate the APCs for 12 h, and the supernatants were collected to determine IL-12p70 and IL-23 levels for ELISA and further functional analyses.
To examine the influence of activated APCs on T cell differentiation, naive CD4+ T cells were first purified from the splenocytes of 6- to 8-week-old C57BL/6 mice using the MagniSort Mouse CD4 Naive T Cell Enrichment Kit (eBioscience, San Diego, CA) according to the manufacturer’s instructions. Purified naive CD4+ T cells were seeded in 48-well plates precoated with an anti-CD3ε antibody (5 μg/mL) (#557306, BD Biosciences, East Rutherford, NJ, USA) and cultured in the aforementioned collected medium from APCs containing 1 μg/mL soluble anti-CD28 antibody (#557398, BD Biosciences). After 96 h of culture, differentiated T cells were harvested for intracellular staining and flow cytometry analysis.
Mixed-Lymphocyte Reactions In Vitro
In order to assess the immunogenicity of MSCs, cell co-culture experiments were performed. Lymphocytes isolated from the spleen of mice with DSS-induced colitis according to a previous report were used as effector cells.44 As stimulator cells, MSCs were mitotically inactivated by the addition of mitomycin C (50 μg/mL; #HY-13316, MedChem Express, Princeton, NJ, USA) in the dark at 37°C for 30 min. Then, 200 μL culture medium (RPMI 1640 supplemented with 10% FBS) containing 1 × 104 mitotically inactivated MSCs and 1 × 105 lymphocytes were seeded in 96-well flat-bottom plates. Lymphocytes treated with concanavalin A (Con A) (5 μg/mL; #C0412, Sigma) were used as positive controls. Lymphocytes without any treatment were used as negative controls. Mitotically inactivated MSCs infected with corresponding types of LVs were used as basal controls. After co-culture for 3 days, the proliferation of lymphocytes was assayed with CCK-8 (Dojindo). The following formula was used to calculate the stimulation index (SI): for co-culture groups, SI = (sample OD − basal OD)/negative control OD; for lymphocytes and lymphocytes + Con A groups, SI = sample OD/negative control OD, where OD is optical density.
Western Blotting
Protein extracts from MSCs were prepared using RIPA lysis buffer containing a protease inhibitor cocktail (Sigma), and protein concentrations were determined via a bicinchoninic acid (BCA) protein assay kit (Beyotime Biotechnology). Samples were incubated with a protein loading buffer at 100°C for 5 min, centrifuged at 12,000 rpm for 5 min at 4°C, separated on a 10% SDS-polyacrylamide gel, and blotted onto polyvinylidene fluoride (PVDF) membranes. Bands were detected immunologically using an anti-CX3CR1 antibody (#ab8021, Abcam, MA, USA, 1:1,000). Subsequently, the corresponding horseradish peroxidase (HRP)-conjugated anti-rabbit immunoglobulin G (IgG) secondary antibody (#111-035-003, Jackson ImmunoResearch, West Grove, PA, USA) was applied. GAPDH (#5174, Cell Signaling Technology, 1:1,000) was used as the internal control. The bands were detected using HRP Substrate Luminol Reagent (Millipore, Billerica, MA, USA).
Establishment of Experimental Colitis Models and Treatment
C57BL/6 mice received either regular drinking water (control) or 5% (w/v) DSS drinking water (model) for 7 days. Body weight and survival rate were monitored over 16 days, and at day 16, mice were sacrificed, colon length and weight were measured, and tissues were fixed in formalin and prepared for histology.
To examine the biodistribution of engineered MSCs in vivo, CX3CR1&IL-25-LV-MSCs and empty vector-infected MSCs were labeled with DiI prior to tail vein injection. After the C57BL/6 mice were treated with 5% DSS on day 4, prepared MSCs (2.5 × 106 cells per mouse) were administered via tail vein injection. The heart, liver, spleen, lung, kidney, small intestine, and colon were harvested from the experimental colitic mice at 2 h, 24 h, and 8 days after injection, imaged by an IVIS Lumina XR system (PerkinElmer, Waltham, MA, USA), and then embedded in optimum cutting temperature compound (OCT) for frozen sections. Mice in group A were treated with empty-LV-infected MSCs without labeling DiI red fluorescent probes, and this group was used to eliminate the background fluorescence signal. For fluorescent MSC localization, cryosectioned tissue was counterstained with 4,6-diamidino-2-phenylindole (DAPI; Sigma) and imaged by a Nikon confocal microscope (C2+, Nikon, Tokyo, Japan).
For a therapeutic study, mice were randomly assigned to control, DSS-treated; DSS+MSC-treated; DSS+empty-LV-infected MSC-treated; DSS+IL-25-LV-infected MSC-treated; DSS+CX3CR1-LV-infected MSC-treated; and DSS+CX3CR1&IL-25-LV-MSC-treated groups. The different populations of treated MSCs (2.5 × 106 cells per mouse) were administered by tail vein injection on days 4, 6, and 8. All measurements and data analysis was performed through double-blinded methods. Animals were weighed daily and sacrificed on day 16, and the colon was excised for macroscopic observation, histopathological examination, MPO activity measurement, T cell isolation, and cytokine (IL-25, IFN-γ, IL-12p70, IL-17A, IL-23, IL-1β, IL-6, TNF-α, and CX3CL1) analysis with ELISA kits (Beijing 4A Biotech, Beijing, China). A DAI based on a previously published grading system was used to evaluate body weight loss, stool consistency, and rectal bleeding at day 16.45 Colon tissues near the rectum (approximately 3–5 cm) were used for hematoxylin and eosin (H&E) staining and scored according to the previously reported approach.46 The combined scores were averaged to obtain the final DAI score and the histological score.
To investigate whether the enhanced anti-colitis effect of the dual-functionalized MSCs was dependent on CX3CR1 and IL-25 function, 2.5 × 106 CX3CR1&IL-25-LV-MSCs were pre-incubated with CX3CR1 antibody (10 μg/mL; #153702, BioLegend) or mouse IgG at 4°C for 30 min and then administered into mice with DSS-induced colitis by tail vein injection. Additionally, mice were treated with dual-functionalized MSCs (2.5 × 106 cells per mouse, tail vein injection) and IL-25 antibody (100 μg/20 g mouse weight; BioLegend, #514418, intraperitoneal injection) on days 4, 6, and 8 in mice with DSS-induced colitis. Body weight and survival rate were monitored over 16 days, and at day 16, mice were sacrificed. Colon tissues were collected for macroscopic observation, histopathological examination, and MPO activity measurement.
Flow Cytometry Analysis
MSCs that received different treatments were collected at day 4 after LV infection for flow cytometry analysis. The MSCs were stained with fluorescence-conjugated mAbs against rat CD11b, CD45, CD90, CD29, CD80, CD86, and MHC-I (#210817, #202207, #205903, #102207, #200205, #200307, and #205208, BioLegend) and CD40 and MHC-II (#48-0402-80, #12-5980-81, eBioscience); corresponding isotype controls were applied for phenotypic characterization. To examine the expression level of CX3CR1, the MSCs were stained with a rabbit anti-mouse/rat CX3CR1 antibody (#ab8021, Abcam, 1:100) followed by an Alexa Fluor 546-conjugated donkey anti-rabbit secondary antibody (#A-10040, Life Technologies).
For intracellular IFN-γ and IL-17A staining, in vitro differentiated T cells were stimulated with phorbol 12-myristate 13-acetate (PMA; 50 ng/mL), ionomycin (500 ng/mL) and brefeldin A (10 μg/mL) (all from Sigma) for 4 h. Then, the cell surface was stained with a FITC-conjugated anti-mouse CD4 antibody (#100405, BioLegend), and the cells were fixed in 1 × Intracellular Fixation Buffer (eBioscience, San Diego, CA, USA). The fixed cells were permeabilized with 1 × Permeabilization Buffer (eBioscience), followed by intracellular staining with an Alexa Fluor 647-conjugated anti-mouse IFN-γ antibody (#505814, BioLegend) or Alexa Fluor 647-conjugated anti-mouse IL-17A antibody (#506912, BioLegend). Then, the cells were examined with a flow cytometer (BD FACSCalibur, BD Biosciences).
To measure the proportion of T and B cells in peripheral blood, mice with DSS-induced colitis that were intravenously injected with LV-infected MSCs (2.5 × 106 cells per mouse) were sacrificed at 2 h, 24 h, and 8 days. Organs were harvested for ELISA assay (Beijing 4A Biotech, Beijing, China). Blood was collected, and erythrocytes were lysed to generate the leukocyte suspension. Then, leukocytes were incubated with the following antibodies: CD45, CD3, CD4, CD8, CD19 (#103114, #100204, #100408, # 100712, and #115512, BioLegend) or corresponding IgG controls. After that, the cells were examined with a flow cytometer (BD FACSCalibur).
Immunofluorescence Analysis
For IL-25 and CX3CR1 expression studies, empty-vector-infected MSCs and CX3CR1&IL-25-LV-MSCs seeded on coverslips were fixed in 4% paraformaldehyde (PFA), permeabilized with 0.1% Triton X-100 (Beyotime Biotechnology) for 5 min, and blocked with 3% bovine serum albumin (BSA) (Sun Shine Biotechnology, Nanjing, China) for 1 h. Different MSCs were incubated with a rat anti-mouse IL-25 antibody (#514403, BioLegend, 1:100) and a rabbit anti-mouse/rat CX3CR1 antibody (#ab8021, Abcam, 1:100) at 4°C overnight and then stained with an Alexa Fluor 488-conjugated goat anti-rat secondary antibody (#A-11006, Life Technologies) and an Alexa Fluor 546-conjugated donkey anti-rabbit secondary antibody (#A-10040, Life Technologies) at room temperature for 1 h, followed by nuclear staining with DAPI. The cells were imagined by a Nikon confocal microscope and analyzed using Nis-Elements Advanced Research software (Nikon). Cells stained with matched isotype controls were used as negative controls to eliminate nonspecific binding by the antibodies (data not shown).
To investigate whether dual-functionalized (DiI-labeled) MSCs could move to extravascular space through the trans-endothelial migration, cryosections of colons tissue were stained with anti-mouse CD31 mouse monoclonal antibody (#sc-376764, Santa Cruz Biotechnology, CA, USA) at 4°C overnight. Then, an Alexa Fluor 488-conjugated donkey anti-mouse secondary antibody (#A-21202, Life Technologies) was applied for 45 min at room temperature, followed by nuclear staining with DAPI. The colonic tissue sections were imaged by using a Nikon confocal microscope. Colonic tissue sections stained with matched isotype controls were used as negative controls to eliminate nonspecific binding by the antibodies (data not shown).
To detect the infiltration of Th1 or Th17 cells in the inflamed colon, cryosections of colon tissue were stained with a rabbit anti-mouse CD4 antibody (#sc-7219, Santa Cruz Biotechnology, CA, USA) and a hamster anti-mouse IFN-γ or a rat anti-mouse IL-17A antibody (#513202 and #506901; BioLegend), respectively, at 4°C overnight. Then, an Alexa Fluor 488-conjugated donkey anti-rabbit secondary antibody (#A-21206, Life Technologies) and an Alexa Fluor 594-conjugated goat anti-hamster or donkey anti-rat secondary antibody (#A-21113 and #A-21209; Life Technologies), respectively, were applied for 45 min at room temperature, followed by nuclear staining with DAPI. The colonic tissue sections were imaged by using a Nikon confocal microscope. Colonic tissue sections stained with matched isotype controls were used as negative controls to eliminate nonspecific binding by the antibodies (data not shown).
mRNA Quantification
Lamina propria mononuclear cells (LPMCs) were isolated according to the protocol described in a previous report.47 Colonic CD4+ T cells were separated from the LPMCs by CD4 microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany). Total RNA was isolated from colonic T cells by using TRIzol reagent (Life Technology). Quantitative real-time PCR was performed by using the One Step SYBR PrimeScript RT-PCR Kit (Takara, Shiga, Japan) according to the manufacturer’s protocol. β-actin was used as an internal control. The following primers were used: T-bet sense, 5′-TGCCAGGGAACCGCTTATAT-3′; T-bet antisense, 5′-GTTGGAAGCCCCCTTGTTGT-3′; RORγt sense, 5′-CCAGGAG CAATGGAAGTCG-3′; RORγt antisense, 5′-CCGTGTAGAGGGCAATCTCA-3′; β-actin sense, 5′-GGTGTGATGGTGGGAATGGG-3′; β-actin antisense, 5′-ACG GTTGGCCTTAGGGTTCAG-3′.
Data Statistics
Results are expressed as the means ± standard error of the mean (mean ± SEM). Data were statistically analyzed using GraphPad Prism software (v.5.01, GraphPad Software, La Jolla, CA, USA). Data were checked for distribution normality. Differences among multiple groups were compared using one-way ANOVA with a post hoc Bonferroni correction. Differences between two groups were evaluated using a two-tailed Student’s t test. Survival curves were analyzed by the Kaplan-Meier log-rank test. A value of p < 0.05 was considered significant.
Author Contributions
Y.F. conducted the experiments and wrote the paper. J.N., Jiahui Chen, G.M., M.Z., S.Z., T.S., and J. Zhu conducted the experiments. J. Zhang designed the experiments. Jiangning Chen and Z.H. designed the experiments and wrote the paper.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (31571458, 31870821, 31771550, 81972267, 81673380, and 81973273), the National Key Research and Development Program of China (2017YFC0909700), and the Fundamental Research Funds for the Central Universities (020814380122).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.01.020.
Contributor Information
Zhen Huang, Email: zhenhuang@nju.edu.cn.
Junfeng Zhang, Email: jfzhang@nju.edu.cn.
Jiangning Chen, Email: jnchen@nju.edu.cn.
Supplemental Information
References
- 1.Khor B., Gardet A., Xavier R.J. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011;474:307–317. doi: 10.1038/nature10209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abraham C., Cho J.H. Inflammatory bowel disease. N. Engl. J. Med. 2009;361:2066–2078. doi: 10.1056/NEJMra0804647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang Z., Shi T., Zhou Q., Shi S., Zhao R., Shi H., Dong L., Zhang C., Zeng K., Chen J., Zhang J. miR-141 regulates colonic leukocytic trafficking by targeting CXCL12β during murine colitis and human Crohn’s disease. Gut. 2014;63:1247–1257. doi: 10.1136/gutjnl-2012-304213. [DOI] [PubMed] [Google Scholar]
- 4.Papadakis K.A., Targan S.R. Role of cytokines in the pathogenesis of inflammatory bowel disease. Annu. Rev. Med. 2000;51:289–298. doi: 10.1146/annurev.med.51.1.289. [DOI] [PubMed] [Google Scholar]
- 5.Nancey S., Holvöet S., Graber I., Joubert G., Philippe D., Martin S., Nicolas J.F., Desreumaux P., Flourié B., Kaiserlian D. CD8+ cytotoxic T cells induce relapsing colitis in normal mice. Gastroenterology. 2006;131:485–496. doi: 10.1053/j.gastro.2006.05.018. [DOI] [PubMed] [Google Scholar]
- 6.Present D.H., Rutgeerts P., Targan S., Hanauer S.B., Mayer L., van Hogezand R.A., Podolsky D.K., Sands B.E., Braakman T., DeWoody K.L. Infliximab for the treatment of fistulas in patients with Crohn’s disease. N. Engl. J. Med. 1999;340:1398–1405. doi: 10.1056/NEJM199905063401804. [DOI] [PubMed] [Google Scholar]
- 7.Plevy S.E., Targan S.R. Future therapeutic approaches for inflammatory bowel diseases. Gastroenterology. 2011;140:1838–1846. doi: 10.1053/j.gastro.2011.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ko I.K., Kim B.-G., Awadallah A., Mikulan J., Lin P., Letterio J.J., Dennis J.E. Targeting improves MSC treatment of inflammatory bowel disease. Mol. Ther. 2010;18:1365–1372. doi: 10.1038/mt.2010.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levy O., Zhao W., Mortensen L.J., Leblanc S., Tsang K., Fu M., Phillips J.A., Sagar V., Anandakumaran P., Ngai J. mRNA-engineered mesenchymal stem cells for targeted delivery of interleukin-10 to sites of inflammation. Blood. 2013;122:e23–e32. doi: 10.1182/blood-2013-04-495119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sarkar D., Spencer J.A., Phillips J.A., Zhao W., Schafer S., Spelke D.P., Mortensen L.J., Ruiz J.P., Vemula P.K., Sridharan R. Engineered cell homing. Blood. 2011;118:e184–e191. doi: 10.1182/blood-2010-10-311464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zaph C., Du Y., Saenz S.A., Nair M.G., Perrigoue J.G., Taylor B.C., Troy A.E., Kobuley D.E., Kastelein R.A., Cua D.J. Commensal-dependent expression of IL-25 regulates the IL-23-IL-17 axis in the intestine. J. Exp. Med. 2008;205:2191–2198. doi: 10.1084/jem.20080720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brand S., Hofbauer K., Dambacher J., Schnitzler F., Staudinger T., Pfennig S., Seiderer J., Tillack C., Konrad A., Göke B. Increased expression of the chemokine fractalkine in Crohn’s disease and association of the fractalkine receptor T280M polymorphism with a fibrostenosing disease phenotype. Am. J. Gastroenterol. 2006;101:99–106. doi: 10.1111/j.1572-0241.2005.00361.x. [DOI] [PubMed] [Google Scholar]
- 13.Bruno G., Saracino A., Monno L., Angarano G. The revival of an “old” marker: CD4/CD8 ratio. AIDS Rev. 2017;19:81–88. [PubMed] [Google Scholar]
- 14.An J.H., Song W.J., Li Q., Kim S.M., Yang J.I., Ryu M.O., Nam A.R., Bhang D.H., Jung Y.C., Youn H.Y. Prostaglandin E2 secreted from feline adipose tissue-derived mesenchymal stem cells alleviate DSS-induced colitis by increasing regulatory T cells in mice. BMC Vet. Res. 2018;14:354. doi: 10.1186/s12917-018-1684-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao D., Xie J., Zhang J., Feng C., Yao B., Ma K., Li J., Wu X., Huang S., Fu X. MSC attenuate diabetes-induced functional impairment in adipocytes via secretion of insulin-like growth factor-1. Biochem. Biophys. Res. Commun. 2014;452:99–105. doi: 10.1016/j.bbrc.2014.08.060. [DOI] [PubMed] [Google Scholar]
- 16.Li J., Ezzelarab M.B., Cooper D.K.C. Do mesenchymal stem cells function across species barriers? Relevance for xenotransplantation. Xenotransplantation. 2012;19:273–285. doi: 10.1111/xen.12000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oggu G.S., Sasikumar S., Reddy N., Ella K.K.R., Rao C.M., Bokara K.K. Gene delivery approaches for mesenchymal stem cell therapy: strategies to increase efficiency and specificity. Stem Cell Rev. Rep. 2017;13:725–740. doi: 10.1007/s12015-017-9760-2. [DOI] [PubMed] [Google Scholar]
- 18.Moussa L., Pattappa G., Doix B., Benselama S.-L., Demarquay C., Benderitter M., Sémont A., Tamarat R., Guicheux J., Weiss P. A biomaterial-assisted mesenchymal stromal cell therapy alleviates colonic radiation-induced damage. Biomaterials. 2017;115:40–52. doi: 10.1016/j.biomaterials.2016.11.017. [DOI] [PubMed] [Google Scholar]
- 19.Ricles L.M., Hsieh P.-L., Dana N., Rybalko V., Kraynak C., Farrar R.P., Suggs L.J. Therapeutic assessment of mesenchymal stem cells delivered within a PEGylated fibrin gel following an ischemic injury. Biomaterials. 2016;102:9–19. doi: 10.1016/j.biomaterials.2016.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen J., Chen H., Li P., Diao H., Zhu S., Dong L., Wang R., Guo T., Zhao J., Zhang J. Simultaneous regeneration of articular cartilage and subchondral bone in vivo using MSCs induced by a spatially controlled gene delivery system in bilayered integrated scaffolds. Biomaterials. 2011;32:4793–4805. doi: 10.1016/j.biomaterials.2011.03.041. [DOI] [PubMed] [Google Scholar]
- 21.Sackstein R., Merzaban J.S., Cain D.W., Dagia N.M., Spencer J.A., Lin C.P., Wohlgemuth R. Ex vivo glycan engineering of CD44 programs human multipotent mesenchymal stromal cell trafficking to bone. Nat. Med. 2008;14:181–187. doi: 10.1038/nm1703. [DOI] [PubMed] [Google Scholar]
- 22.Cheng Z., Ou L., Zhou X., Li F., Jia X., Zhang Y., Liu X., Li Y., Ward C.A., Melo L.G., Kong D. Targeted migration of mesenchymal stem cells modified with CXCR4 gene to infarcted myocardium improves cardiac performance. Mol. Ther. 2008;16:571–579. doi: 10.1038/sj.mt.6300374. [DOI] [PubMed] [Google Scholar]
- 23.Lombardi M.S., Gilliéron C., Dietrich D., Gabay C. SIK inhibition in human myeloid cells modulates TLR and IL-1R signaling and induces an anti-inflammatory phenotype. J. Leukoc. Biol. 2016;99:711–721. doi: 10.1189/jlb.2A0715-307R. [DOI] [PubMed] [Google Scholar]
- 24.Yu Y., Liao L., Shao B., Su X., Shuai Y., Wang H., Shang F., Zhou Z., Yang D., Jin Y. Knockdown of microRNA Let-7a improves the functionality of bone marrow-derived mesenchymal stem cells in immunotherapy. Mol. Ther. 2017;25:480–493. doi: 10.1016/j.ymthe.2016.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qiu Y., Guo J., Mao R., Chao K., Chen B.L., He Y., Zeng Z.R., Zhang S.H., Chen M.H. TLR3 preconditioning enhances the therapeutic efficacy of umbilical cord mesenchymal stem cells in TNBS-induced colitis via the TLR3-Jagged-1-Notch-1 pathway. Mucosal Immunol. 2017;10:727–742. doi: 10.1038/mi.2016.78. [DOI] [PubMed] [Google Scholar]
- 26.Sage E.K., Thakrar R.M., Janes S.M. Genetically modified mesenchymal stromal cells in cancer therapy. Cytotherapy. 2016;18:1435–1445. doi: 10.1016/j.jcyt.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hammer K., Kazcorowski A., Liu L., Behr M., Schemmer P., Herr I., Nettelbeck D.M. Engineered adenoviruses combine enhanced oncolysis with improved virus production by mesenchymal stromal carrier cells. Int. J. Cancer. 2015;137:978–990. doi: 10.1002/ijc.29442. [DOI] [PubMed] [Google Scholar]
- 28.Vitale S., Cambien B., Karimdjee B.F., Barthel R., Staccini P., Luci C., Breittmayer V., Anjuère F., Schmid-Alliana A., Schmid-Antomarchi H. Tissue-specific differential antitumour effect of molecular forms of fractalkine in a mouse model of metastatic colon cancer. Gut. 2007;56:365–372. doi: 10.1136/gut.2005.088989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Imai T., Hieshima K., Haskell C., Baba M., Nagira M., Nishimura M., Kakizaki M., Takagi S., Nomiyama H., Schall T.J., Yoshie O. Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell. 1997;91:521–530. doi: 10.1016/s0092-8674(00)80438-9. [DOI] [PubMed] [Google Scholar]
- 30.Jamieson W.L., Shimizu S., D’Ambrosio J.A., Meucci O., Fatatis A. CX3CR1 is expressed by prostate epithelial cells and androgens regulate the levels of CX3CL1/fractalkine in the bone marrow: potential role in prostate cancer bone tropism. Cancer Res. 2008;68:1715–1722. doi: 10.1158/0008-5472.CAN-07-1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fong A.M., Robinson L.A., Steeber D.A., Tedder T.F., Yoshie O., Imai T., Patel D.D. Fractalkine and CX3CR1 mediate a novel mechanism of leukocyte capture, firm adhesion, and activation under physiologic flow. J. Exp. Med. 1998;188:1413–1419. doi: 10.1084/jem.188.8.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gittens B.R., Bodkin J.V., Nourshargh S., Perretti M., Cooper D. Galectin-3: a positive regulator of leukocyte recruitment in the inflamed microcirculation. J. Immunol. 2017;198:4458–4469. doi: 10.4049/jimmunol.1600709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fina D., Franzè E., Rovedatti L., Corazza G.R., Biancone L., Sileri P.P., Sica G., MacDonald T.T., Pallone F., Di Sabatino A., Monteleone G. Interleukin-25 production is differently regulated by TNF-α and TGF-β1 in the human gut. Mucosal Immunol. 2011;4:239–244. doi: 10.1038/mi.2010.68. [DOI] [PubMed] [Google Scholar]
- 34.Monteleone G., Pallone F., Macdonald T.T. Interleukin-25: a two-edged sword in the control of immune-inflammatory responses. Cytokine Growth Factor Rev. 2010;21:471–475. doi: 10.1016/j.cytogfr.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 35.Reynolds J.M., Lee Y.H., Shi Y., Wang X., Angkasekwinai P., Nallaparaju K.C., Flaherty S., Chang S.H., Watarai H., Dong C. Interleukin-17B antagonizes interleukin-25-mediated mucosal inflammation. Immunity. 2015;42:692–703. doi: 10.1016/j.immuni.2015.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mchenga S.S.S., Wang D., Li C., Shan F., Lu C. Inhibitory effect of recombinant IL-25 on the development of dextran sulfate sodium-induced experimental colitis in mice. Cell. Mol. Immunol. 2008;5:425–431. doi: 10.1038/cmi.2008.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Caruso R., Sarra M., Stolfi C., Rizzo A., Fina D., Fantini M.C., Pallone F., MacDonald T.T., Monteleone G. Interleukin-25 inhibits interleukin-12 production and Th1 cell-driven inflammation in the gut. Gastroenterology. 2009;136:2270–2279. doi: 10.1053/j.gastro.2009.02.049. [DOI] [PubMed] [Google Scholar]
- 38.Shi T., Xie Y., Fu Y., Zhou Q., Ma Z., Ma J., Huang Z., Zhang J., Chen J. The signaling axis of microRNA-31/interleukin-25 regulates Th1/Th17-mediated inflammation response in colitis. Mucosal Immunol. 2017;10:983–995. doi: 10.1038/mi.2016.102. [DOI] [PubMed] [Google Scholar]
- 39.Thelen T.D., Green R.M., Ziegler S.F. Acute blockade of IL-25 in a colitis associated colon cancer model leads to increased tumor burden. Sci. Rep. 2016;6:25643. doi: 10.1038/srep25643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Camelo A., Barlow J.L., Drynan L.F., Neill D.R., Ballantyne S.J., Wong S.H., Pannell R., Gao W., Wrigley K., Sprenkle J., McKenzie A.N. Blocking IL-25 signalling protects against gut inflammation in a type-2 model of colitis by suppressing nuocyte and NKT derived IL-13. J. Gastroenterol. 2012;47:1198–1211. doi: 10.1007/s00535-012-0591-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luz-Crawford P., Kurte M., Bravo-Alegría J., Contreras R., Nova-Lamperti E., Tejedor G., Noël D., Jorgensen C., Figueroa F., Djouad F., Carrión F. Mesenchymal stem cells generate a CD4+CD25+Foxp3+ regulatory T cell population during the differentiation process of Th1 and Th17 cells. Stem Cell Res. Ther. 2013;4:65. doi: 10.1186/scrt216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grégoire C., Lechanteur C., Briquet A., Baudoux É., Baron F., Louis E., Beguin Y. Review article: mesenchymal stromal cell therapy for inflammatory bowel diseases. Aliment. Pharmacol. Ther. 2017;45:205–221. doi: 10.1111/apt.13864. [DOI] [PubMed] [Google Scholar]
- 43.Soleimani M., Nadri S. A protocol for isolation and culture of mesenchymal stem cells from mouse bone marrow. Nat. Protoc. 2009;4:102–106. doi: 10.1038/nprot.2008.221. [DOI] [PubMed] [Google Scholar]
- 44.Malliaras K., Li T.S., Luthringer D., Terrovitis J., Cheng K., Chakravarty T., Galang G., Zhang Y., Schoenhoff F., Van Eyk J. Safety and efficacy of allogeneic cell therapy in infarcted rats transplanted with mismatched cardiosphere-derived cells. Circulation. 2012;125:100–112. doi: 10.1161/CIRCULATIONAHA.111.042598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alex P., Zachos N.C., Nguyen T., Gonzales L., Chen T.E., Conklin L.S., Centola M., Li X. Distinct cytokine patterns identified from multiplex profiles of murine DSS and TNBS-induced colitis. Inflamm. Bowel Dis. 2009;15:341–352. doi: 10.1002/ibd.20753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alex P., Zachos N.C., Nguyen T., Gonzales L., Chen T.E., Conklin L.S., Centola M., Li X. Distinct cytokine patterns identified from multiplex profiles of murine DSS and TNBS-induced colitis. Inflamm. Bowel Dis. 2009;15:341–352. doi: 10.1002/ibd.20753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weigmann B., Tubbe I., Seidel D., Nicolaev A., Becker C., Neurath M.F. Isolation and subsequent analysis of murine lamina propria mononuclear cells from colonic tissue. Nat. Protoc. 2007;2:2307–2311. doi: 10.1038/nprot.2007.315. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.