

ABSTRACT
One major cause of endoplasmic reticulum (ER) stress is homeostatic imbalance between biosynthetic protein folding and protein folding capacity. Cells utilize mechanisms such as the unfolded protein response (UPR) to cope with ER stress. Nevertheless, when ER stress is prolonged or severe, cell death may occur, accompanied by production of mitochondrial reactive oxygen species (ROS). Using a yeast model (Saccharomyces cerevisiae), we describe an innate, adaptive response to ER stress to increase select mitochondrial proteins, O2 consumption and cell survival. The mitochondrial response allows cells to resist additional ER stress. The ER stress-induced mitochondrial response is mediated by activation of retrograde (RTG) signaling to enhance anapleurotic reactions of the tricarboxylic acid cycle. Mitochondrial response to ER stress is accompanied by inactivation of the conserved TORC1 pathway, and activation of Snf1/AMPK, the conserved energy sensor and regulator of metabolism. Our results provide new insight into the role of respiration in cell survival in the face of ER stress, and should help in developing therapeutic strategies to limit cell death in disorders linked to ER stress.
This article has an associated First Person interview with the first author of the paper.
KEY WORDS: Mitochondria, Endoplasmic reticulum, ER stress, Yeast
Highlighted Article: Increased respiration is an innate, adaptive response to endoplasmic reticulum stress that is mediated by retrograde signaling and is independent of the unfolded protein response.
INTRODUCTION
Major activities in the endoplasmic reticulum (ER) include folding, modification and assembly of newly synthesized proteins destined either for residence within the endomembrane system or secretion from the cell. When demand for protein folding at the ER is increased (e.g. in pancreatic beta cells during hyperglycemic conditions) or when protein misfolding occurs, cells can experience ER stress. Under these conditions, cells activate a transcriptional program called the unfolded protein response (UPR), which helps to maintain ER homeostasis by enhancing capacity for protein folding and promoting destruction of misfolded proteins (Walter and Ron, 2011). In yeast (herein Saccharomyces cerevisiae), UPR signaling in response to protein misfolding occurs via the Ire1 sensor, whereas in mammalian cells, canonical UPR signaling includes not only the Ire1 branch but also engages two additional distinct branches mediated by PERK and ATF6. Failure to satisfactorily resolve chronic or severe ER stress can lead to cell death; moreover, ER stress and cell death contribute to the pathogenesis of many disorders, including metabolic and neurodegenerative diseases (Oakes and Papa, 2015; Wang and Kaufman, 2016).
There is accumulating evidence that beyond the UPR, cells exhibit further responses to ER stress (Appenzeller-Herzog and Hall, 2012; Darling and Cook, 2014; Knupp et al., 2018). The TOR signaling network, a master cellular regulator of anabolic activities, has been suggested to interconnect with ER stress response signaling (Appenzeller-Herzog and Hall, 2012; Bachar-Wikstrom et al., 2013). The multiprotein kinase complex TORC1 is a central component of a signaling network sensing nutrient (amino acid) availability for a myriad of anabolic activities, such as ribosomal biogenesis and protein translation. A role for activation of AMP-activated protein kinase (AMPK) has also been suggested in alleviating ER stress (Jung and Choi, 2016). AMPK and its yeast ortholog Snf1 act as energy sensors, regulators of metabolism, and promote mitochondrial biogenesis and autophagy (Hardie et al., 2012). The TORC1 and Snf1/AMPK signaling networks have broadly opposing effects on metabolism. Although the points of interplay between the two pathways are numerous, ER stress signaling is most often associated with induction of catabolic activities, such as attenuation of protein synthesis and autophagy, which accompanies inactivation of the TORC1 signaling pathway (Bravo et al., 2013; Kapahi et al., 2010).
Ongoing work has elucidated a complex and interdependent relationship between mitochondria and the ER. Physical contact sites connect mitochondria and ER for lipid and calcium exchange (Phillips and Voeltz, 2016), proper targeting of mitochondrial proteins is assisted by the ER (Hansen et al., 2018), and there is accruing evidence to suggest that mitochondria participate in the ER stress response (Knupp et al., 2018; Malhotra and Kaufman, 2011). Mitochondria are involved in cell death decisions upon unresolved ER stress, and the mitochondrial electron transport chain (ETC) is a major source of reactive oxygen species (ROS) that are damaging to cells (Malhotra and Kaufman, 2011). Mitochondria also have a protective role against ER stress by providing ATP to fuel chaperone activity and playing a critical role in cellular calcium homeostasis. Because mitochondrial DNA encodes components of the oxidative phosphorylation machinery, regulation of mitochondrial respiratory activity involves coordination of nuclear and mitochondrial gene expression (Nunnari and Suomalainen, 2012).
Recently, we reported on genetic strategies that limit ROS accumulation by increasing the rate and efficiency of electron transport; these approaches were effective in promoting cell survival and resistance to ER stress (Knupp et al., 2018; Turrens, 2003). In yeast growing in the presence of glucose, respiration is repressed in favor of glycolysis (Broach, 2012); however, we now show that despite abundant glucose, yeast responds to ER stress with increased O2 consumption, increased mitochondrial membrane potential, and up-regulation of levels of select mitochondrial proteins. Enhanced respiration and adaptation to ER stress is mediated by retrograde (RTG) signaling to promote anapleurotic reactions of the tricarboxylic acid (TCA) cycle. Activation of RTG signaling in response to ER stress is accompanied by inactivation of TORC1-mediated signaling, and activation of Snf1/AMPK.
RESULTS
Adaptation to ER stress is accompanied by increased electron transport chain activity
When exposed to prolonged ER stress, yeast cells exponentially growing in abundant glucose (in which respiration is ordinarily repressed) responded with an increase in specific mitochondrial proteins such as Cox2 [a mitochondrially encoded component of cytochrome c oxidase (COX)], the outer membrane protein Por1/VDAC, and a slight increase in the Hsp70 protein and component of the inner membrane import motor, Ssc1 (Craig, 2018) (Fig. 1A). A time course shows that Cox2 protein levels were progressively increased after addition of tunicamycin [which promotes ER protein misfolding by inhibiting N-linked glycosylation; Cox2 protein levels were constitutively increased upon constitutive expression of misfolded CPY* (Ng et al., 2000) (Fig. 1A)].
Fig. 1.

Time course of mitochondrial respiratory response to ER stress. Cells were exponentially growing in SC medium. (A) Left panel: western blot sequentially blotted for Cox2, Por1 and Ssc1, showing that these proteins were progressively increased at various times after tunicamycin (tun) addition (0.5 µg/ml). Lysates were normalized to total protein. Pgk1, the loading control and the alpha subunit of ATP synthase remained much the same. Right panel: western blot showing that Cox2, Por1 and Ssc1, but not ATP synthase alpha subunit protein levels, were elevated in wild-type (WT) cells constitutively expressing misfolded CPY*. Cox2 induction by ER stress (0.5 µg/ml tunicamycin for 5 h) was dependent on Rtg1 (right panel) but independent of Ire1 (left panel). (B) Relative mitochondrial numbers as measured by COX2 mitochondrial DNA content. Semi-quantitative PCR was used to determine COX2 DNA level normalized to ACT1. Cells growing exponentially in SC, treated with 0.5 µg/ml tunicamycin (Tun) for 5 h, were compared with untreated cells (−), cells over-expressing (OE) SAK1 or HAP4 grown in SC, and cells grown overnight in SC with the nonfermentable carbon source glycerol (Gly). Error bars indicate s.e.m.; n=3. (C) Cellular O2 consumption was progressively increased after various times of tunicamycin (0.5 µg/ml) treatment, as measured by high resolution respirometry. Oxygen consumption decreased to 0 upon addition of antimycin (2 µM). Oxygen consumption was increased to a maximal rate upon addition of the protonophore CCCP (4 µM). O2 consumption was also increased by CPY* expression (compared with untreated control), P=0.0028. (D) Adaptation to ER stress is dependent on RTG signaling. Cell survival was assayed by colony formation assay. Exposure of wild-type cells to low (‘lo’) ER stress (0.5 µg/ml tunicamycin or 1 mM DTT for 4 h) rendered the surviving cells more resistant to a subsequent high dose (‘hi’) of an ER stressor not previously experienced (10 mM DTT and 10 µg/ml tunicamycin, respectively, for 4 h). Error bars indicate s.e.m.; n=3; P<0.05. (E) ER stress-induced ROS accumulation, assayed by DHE staining. Exponentially growing cells were exposed to tunicamycin (5 µg/ml), antimycin (AmA; 4 µM), or both for 5 h. Cells were then resuspended in PBS with 5 µg/ml DHE for 15 min before imaging cells by fluorescence microscopy. Fraction of fluorescent cells of total was determined by counting >100 cells. Error bars indicate s.e.m.; n=3.
Significantly, Cox2 protein induction was not dependent on canonical signaling of ER stress by Ire1 (Fig. 1A, bottom-left panel). However, increased Cox2 protein induced by ER stress was dependent on Rtg1, a transcriptional activator of the retrograde (RTG) signaling pathway that conveys mitochondrial needs to changes in nuclear gene expression, in particular, to replenish TCA cycle intermediates (Liu and Butow, 2006) (Fig. 1A, bottom-right panel). Cox2 protein induction was dependent on translation by mitochondrial ribosomes, as it was inhibited by the specific inhibitor pentamidine (Zhang et al., 2000); by contrast, Por1, encoded by a nuclear gene, was induced by tunicamycin, unaffected by pentamidine (Fig. S1A). Furthermore, Cox2 translation required its translational activator Pet111, which is encoded by nuclear DNA (Green-Willms et al., 2001); in response to ER stress, PET111 undergoes Rtg1-dependent transcriptional induction (Fig. S1B). Because nuclear-encoded proteins are dependent on mitochondrial membrane potential for import (Green-Willms et al., 2001), ER stress-mediated induction of both Cox2 and Por1 was abrogated in rho0 cells, deficient in mtDNA and mitochondrial membrane potential (Fig. S1A).
To determine whether increased mitochondrial protein levels reflect increased numbers of mitochondria in response to ER stress, COX2 DNA levels (encoded by the mitochondrial genome) were measured by PCR and normalized to actin (nuclear) DNA levels (Fig. 1B). When cells growing exponentially in glucose were incubated for 5 h with tunicamycin, COX2 DNA levels were not increased beyond that in unstressed control cells, indicating that mitochondrial numbers remain constant while select proteins per mitochondrion increased (Fig. 1B). By contrast, COX2 DNA levels were increased in cells growing in the non-fermentable carbon source glycerol (Fig. 1B), indicating de novo mitochondrial biogenesis (in excess of turnover) under conditions in which respiration is de-repressed. Similarly, mitochondrial numbers were also increased (Fig. 1B) upon de-repression of respiration by over-expression of SAK1, encoding a regulatory component of the glucose repression machinery (Knupp et al., 2018), or HAP4, encoding a transcriptional activator of respiratory gene expression (Lascaris et al., 2002; Lin et al., 2004).
Consistent with the idea that mitochondrial response to ER stress does not involve a change in mitochondrial numbers, Cox2 protein was still induced by tunicamycin in dnm1Δ cells in which mitochondrial fission is prevented (Bleazard et al., 1999) (Fig. S2A,B). By contrast, in fzo1Δ cells defective in mitochondrial fusion in which mitochondria are fragmented and mtDNA is lost (Sesaki et al., 2003), ER stress-induced Cox2 protein was not observed (Fig. S2A,B). In dnm1Δ fzo1Δ double mutants, mitochondrial morphology and mtDNA stability are recovered, but fusion remains impaired (Sesaki et al., 2003); nevertheless, induction of Cox2 and Por1 proteins by ER stress was detectable (Fig. S2A). Without an increase in mitochondrial numbers, the observed increase in Cox2 protein in response to ER stress was matched by elevated cellular respiration (O2 consumption rate measured by high resolution respirometry; Fig. 1C). Increased O2 consumption was induced by tunicamycin as well as CPY* expression (Fig. 1C), strongly suggesting that ER stress triggers increased respiratory response.
To determine whether mitochondrial response to ER stress is an adaptive response, cell viability was measured by colony formation assay. Wild-type cells were fairly resistant to death from low-dose tunicamycin or DTT (Fig. 1D). A larger fraction of cells die with a 20-fold higher tunicamycin or 10-fold higher DTT dose (Fig. 1D). However, when cells had prior exposure to a low dose ER stressor, they acquired resistance to cell death when subsequently challenged with a higher dose of a different ER stress agent (Fig. 1D). In rtg1Δ cells deficient in RTG signaling, mitochondrial Cox2 protein no longer responded to ER stress (Fig. 1A, right panel), and the cells could not adapt with increased survivability (Fig. 1D). By contrast, UPR response to tunicamycin was similar in both wild-type and rtg1Δ cells (Fig. S3A). These results suggest that mitochondrial response is a critical contributor to ER stress survival that requires RTG signaling.
The involvement of the ETC in ROS accumulation during ER stress was examined by staining cells with dihydroethidium (DHE). Upon oxidation by superoxide, DHE becomes fluorescent (Dikalov and Harrison, 2014). In Fig. 1E, wild-type cells were treated with tunicamycin for 5 h in the presence or absence of antimycin A, an inhibitor of Complex III of the ETC, and then stained with DHE. A low level of ROS was detected by counting fluorescent cells treated with tunicamycin or antimycin A alone for 5 h. When combined, the ER stressor and oxidative phosphorylation inhibitor induced a synergistic effect on ROS accumulation (Fig. 1E). These results support an ameliorative effect of respiration on ER stress, and are in agreement with our previous results showing that ER stress-induced cell death is linked to mitochondrial ROS production (Knupp et al., 2018).
Activation of retrograde signaling during mitochondrial response to ER stress
Because Rtg1 is required for adaptation to ER stress, we assayed for activation of RTG signaling by tunicamycin treatment. CIT2, encoding a citrate synthase isozyme, is a prototypical target of RTG regulation; induction of CIT2 leads to enhanced anapleurotic reactions of the TCA cycle for biosynthetic and oxidative phosphorylation processes (Chen et al., 2017; Liao et al., 1991). A CIT2-lacZ reporter was assayed after cells were treated with ER stressors for 5 h, including tunicamycin, DTT and CPY*. As a positive control, the activity of CIT2-lacZ was assayed after rapamycin addition, as it has been well established that RTG signaling is induced when TORC1 is inhibited (Butow and Avadhani, 2004). Indeed, Fig. 2A shows that CIT2-lacZ activity was increased in response to rapamycin, and this induction was blocked in rtg1Δ cells. RTG signaling was increased during growth in the absence of glutamate (Fig. 2A, yeast nitrogen base minimal medium), reflecting the role of glutamate depletion in promoting amino acid biosynthesis from TCA cycle intermediates, as previously reported (Liu and Butow, 2006). Although CIT2-lacZ activity was already high in the absence of glutamate, tunicamycin addition further increased RTG signaling (Fig. 2A). Even in the presence of glutamate in synthetic complete (SC) medium with abundant amino acids, CIT2-lacZ activity was increased upon tunicamycin addition, and induction was dependent on Rtg1 (Fig. 2A). Moreover, tunicamycin induced RTG signaling in ire1Δ cells, and was thus independent of the UPR. These findings support a role for RTG signaling in communicating protein misfolding in the ER to promote mitochondrial response.
Fig. 2.

Activation of retrograde signaling promotes mitochondrial response to ER stress. Cells exponentially growing in SC were analyzed +/− ER stress. (A) Effect of ER stress on CIT2 expression, as assayed with a CIT2-lacZ reporter. Wild-type (WT) cells were exponentially growing in SC-uracil, supplemented YNB, or supplemented YNB medium with 0.02% glutamate. Cells constitutively expressing CPY* were analyzed, or cells were treated with or without tunicamycin (tun; 0.5 μg/ml) or 1 mM DTT for 5 h. As a positive control, wild-type cells were treated with 200 nM rapamycin (rap) for 5 h. β-Galactosidase activity was measured in cell lysates, and is expressed as μmol/min/mg protein. Error bars indicate s.e.m.; n≥3. In CPY*-expressing cells, enzyme activity is significantly higher (P<0.05) in tunicamycin-treated versus untreated cells. (B) Mitochondrial protein levels in cells upon activation of RTG signaling. Cells exponentially growing in SC medium were treated with tunicamycin (T; 0.5 μg/ml) or washed with water and then shifted to minimal medium without glutamate (−E) for 5 h (arrow). Cells growing overnight to mid-log phase in YNB medium (without glutamate) were treated with tunicamycin (0.5 μg/ml) for 5 h. Lysates were normalized to protein and examined by western blot sequentially blotted for Cox2, Por1, Ssc1 and ATP1; Pgk1 was blotted as a loading control. (C) Cellular O2 consumption rate is increased in rtg1Δ cells. In wild-type cells, O2 consumption rate was decreased to 0 upon addition of antimycin A (AMA, 2 μM), and O2 consumption rate was increased to a maximal rate upon addition of the protonophore CCCP (4 μM). The constitutive O2 consumption rate in rtg1Δ cells was increased significantly in comparison with wild-type cells, but remained unaffected after tunicamycin treatment. (D) Mitochondrial membrane potential is increased by ER stress, and impaired in rtg1Δ cells. Cells were untreated or treated with 0.5 μg/ml tunicamycin for 5 h, and then stained with 5 nM TMRM (nonquenching mode) for 30 min before visualizing by fluorescence microscopy.
In Fig. 2B, western blots show that Cox2 and Por1 proteins levels are significantly increased when RTG signaling is activated by shifting cells into glutamate-free medium for 5 h (Fig. 2B, arrow). Strikingly, addition of tunicamycin to cells growing without glutamate further increased Cox2 protein levels, in agreement with further RTG activation shown in Fig. 2A.
To examine further how loss of Rtg1 affects ER stress-induced mitochondrial response, O2 consumption was measured in wild-type and rtg1Δ cells. In wild-type cells, basal O2 consumption was completely inhibited by addition of antimycin A, an inhibitor of respiratory chain Complex III (Liu and Barrientos, 2013) (Fig. 2C), indicating that O2 consumption is entirely attributable to mitochondria. Upon addition of the protonophore CCCP, O2 consumption increased to a maximal level (Fig. 2C), as expected upon collapse of the membrane potential and uncoupling of ATP production. After 5 h of ER stress, O2 consumption rate in wild-type cells was increased to approximately half of maximal capacity (Figs 2C and 1C). Surprisingly, basal O2 consumption rate in rtg1Δ cells exceeded maximal capacity of wild-type cells by ∼5-fold, and was near or at its maximal capacity, i.e. CCCP addition did not produce a significant further increase in O2 consumption (Fig. 2C).
To better understand high O2 consumption in rtg1Δ cells, mitochondrial membrane potential was assessed by staining cells with the fluorescent dye TMRM, whose accumulation in mitochondria is dependent on membrane potential (Perry et al., 2011). As shown in Fig. 2D, fluorescent TMRM staining was increased in wild-type cells after 5 h incubation with tunicamycin, indicating increased mitochondrial membrane potential, as previously reported (Bravo et al., 2011; Knupp et al., 2018). By contrast, TMRM staining of rtg1Δ cells was barely detectable, and after ER stress, TMRM was not significantly increased, in agreement with loss of Cox2 induction by tunicamycin (Fig. 1A). These findings suggest that TCA cycle function is impaired in rtg1Δ cells (Butow and Avadhani, 2004; Liu and Butow, 1999) during growth in glucose, resulting in a defective electron transport chain and loss of a mitochondrial membrane potential.
ER stress induces inactivation of TORC1 signaling without eliciting a nutrient deprivation response
The RTG signaling pathway is under negative regulation by TORC1 (Liu and Butow, 2006; see also Fig. 3A, right panel). We therefore asked whether mitochondrial response to ER stress requires inactivation of TORC1. Tor1 is a component of the TORC1 kinase complex, and Tor1 loss results in inactivation of TORC1 signaling (Loewith and Hall, 2011). As shown in Fig. 3A (left panel), Cox2 was constitutively increased in tor1Δ cells, and became more abundant after tunicamycin addition. Consistent with a role for retrograde signaling downstream of TORC1 (Liu and Butow, 2006; see also Fig. 3A, right panel), Cox2 induction was abrogated in a tor1Δ rtg1Δ double mutant (Fig. 3A, left panel).
Fig. 3.

Inactivation of TORC1 signaling by ER stress. Cells were exponentially growing in SC medium. (A) Western blot showing that Cox2 and Por1 proteins were constitutively increased by TORC1 inactivation in tor1Δ cells; these protein levels were further increased by ER stress. In npr2Δ cells, Cox2 increase in response to tunicamyin (tun; 0.5 μg/ml) was impaired; in snf1Δ cells, tunicamycin-induced Cox2 increase was slightly impaired. Blotting with anti-Pgk1 is shown as a loading control. (B) Tunicamycin does not induce an amino acid starvation response. Cells bearing pGCN4-lacZ exponentially growing in SC-uracil were treated with tunicamycin (0.5 μg/ml) or rapamycin (rap; 200 nM) for 4 h. Control cells were washed with water and resuspended in nitrogen-free medium for 4 h. Cells were harvested by freezing with liquid nitrogen. β-Galactosidase activity was assayed in cell lysates and expressed as μmol/mg/min. (C) TORC1 activity as revealed by Rps6 phosphorylation. Top panel: time course of TORC1 inactivation after tunicamycin (0.5 μg/ml) addition, as measured by Rps6 phosphorylation (S6-P). Bottom panel: phosphorylation of Rps6 was analyzed after tunicamycin (0.5 μg/ml) or rapamycin (200 nM) were added for 5 h to wild-type (WT) and npr2Δ cells. eIF2α protein is shown as a loading control. The vertical line indicates removal of unrelated lanes. (D) RTG signaling was activated after 5 h incubation with tunicamycin (0.5 μg/ml) or rapamycin (200 nM). Tunicamycin-induced CIT2-lacZ expression was further augmented in tor1Δ cells, but was prevented in npr2Δ cells. β-Galactosidase activity was assayed in cell lysates, and expressed as μmol/min/mg protein. Error bars indicate s.e.m.; n=3. (E) Mitochondrial activity as reflected by O2 consumption rate. Cellular O2 consumption rate after 5 h with tunicamycin (0.5 μg/ml) was significantly decreased in npr2Δ cells by contrast with that in wild-type cells (P=0.0345). In tor1Δ cells, cellular O2 consumption was constitutively increased and then further elevated by tunicamycin. Error bars indicate s.e.m.; n=3. (F) Sensitivity and adaptation to ER stress, assayed as described in the legend to Fig. 1D. Adaptation to ER stress after prior exposure to low-dose stressor was abrogated in npr2Δ cells, but tor1Δ cells displayed high viability after exposure to high-dose ER stressor.
We addressed the possibility that mitochondrial response to ER stress is an indirect consequence of nutrient depletion because amino acid deprivation and glutamate depletion are known to trigger TORC inactivation (González and Hall, 2017) and RTG signaling (Liu and Butow, 1999), respectively. When cells experience amino acid deprivation, the transcription factor Gcn4 is transcriptionally activated to promote an adaptation response by increasing amino acid biosynthesis (Hinnebusch, 2005). When cells were treated under control conditions such as rapamycin treatment or nitrogen deprivation, a GCN4-lacZ reporter was induced (Fig. 3B). However, transcriptional activation of GCN4 was not induced by tunicamycin treatment (Fig. 3B). Moreover, increased Cox2 protein level in response to tunicamycin was not affected in gcn4Δ cells or prototrophic cells (Fig. S3B,C), suggesting that the ER stress-mediated response is not due to amino acid deprivation.
To show that TORC1 signaling is inactivated by ER stress, phosphorylation of ribosomal S6 (Rps6) was assayed as it reflects one branch of the TORC1 signaling network (Urban et al., 2007). As expected, Rps6 was dephosphorylated in wild-type cells by rapamycin addition, but also upon tunicamycin treatment (Fig. 3C, bottom panel). A time course of tunicamycin treatment shows significant Rps6 dephosphorylation by 90 min (Fig. 3C, top panel). Inactivation of TORC1 activity appears to underlie increased mitochondrial membrane potential in response to tunicamycin as increased TMRM staining was observed after rapamycin treatment (Fig. S4).
Cells impaired in inactivation of TORC1 activity were then examined to determine whether TORC1 inactivation is a requisite component of ER stress-mediated mitochondrial respiratory response. TORC1 signaling is regulated by a conserved upstream Rag GTPase complex that is modulated by guanine nucleotide exchange factors (GEFs) and GTP activating protein complexes (GAPs) (Hatakeyama and De Virgilio, 2016). Npr2 is a component of the yeast SEACIT/GATOR1 complex whose GAP activity regulates the Gtr1/2 GTPase upstream of TORC1 (Hatakeyama and De Virgilio, 2016; Neklesa and Davis, 2009). It has been reported that TORC1 activity is increased in npr2Δ cells (Panchaud et al., 2013). Fig. 3C (bottom panel) shows that, in npr2Δ cells, tunicamycin was far less effective in its ability to induce Rps6 dephosphorylation, while rapamycin (acting downstream of Npr2) remained effective in decreasing Rps6 phosphorylation (Fig. 3C, bottom panel). Significantly, in npr2Δ cells, mitochondrial response during ER stress was impaired as Cox2 and Por1 proteins were not increased in response to tunicamycin (Fig. 3A). Together, the data in Fig. 3A and C underscore inactivation of TORC1 activity in response to ER stress, and this inactivation is necessary for an optimum mitochondrial response.
To confirm the impact of TORC1 inactivation on ER stress-mediated RTG signaling, CIT2-lacZ activity was assayed in tor1Δ cells and npr2Δ cells. As shown in Fig. 3D, CIT2-lacZ was increased by tunicamycin to a greater extent in tor1Δ cells than in wild-type cells, whereas induction of CIT2-lacZ activity by ER stress was impaired in npr2Δ cells. Consistent with these effects, elevation of O2 consumption by ER stress was considerably higher in tor1Δ cells than in wild-type cells, whereas npr2Δ cells displayed no detectable change in O2 consumption in response to tunicamycin (Fig. 3E). Together, these results suggest that ER stress-induced TORC1 inactivation potentiates RTG signaling.
The effect of TORC1 inactivation on adaptation to ER stress was tested in tor1Δ and npr2Δ cells. As shown in Fig. 3F, tor1Δ cells were resistant to high dose ER stress whereas npr2Δ cells with activated TORC1 were unable to acquire resistance after exposure to a low dose ER stressor followed by subsequent challenge with a high dose ER stress agent. Although npr2Δ cells were unable to adapt to ER stress, their ability to mount a UPR response was not impaired (Fig. S3A), consistent with the idea that a mitochondrial response participates in stress adaptation.
A role for Snf1/AMPK signaling in mitochondrial response to ER stress
Snf1/AMPK signaling is a key regulator of respiratory metabolism (Broach, 2012), and inactivation of TORC1 has been reported to activate Snf1 (Orlova et al., 2006). Therefore, we tested whether Snf1/AMPK plays a role in ER stress-induced mitochondrial response. As shown in Fig. 3A, Cox2 increase in response to tunicamycin was slightly diminished in snf1Δ cells by comparison with wild-type cells. To examine further the role of Snf1/AMPK in the ER stress response, Snf1/AMPK activation was assayed with an antibody to phospho-AMPK Thr172 of the catalytic (alpha) subunit (Orlova et al., 2006). As a positive control, exponentially growing cells were shifted from SC medium with 2% glucose into low glucose (0.05%) medium for 1 h, resulting in Snf1 activation (Fig. 4A, arrow; see also Hedbacker and Carlson, 2009). Strikingly, Snf1 activation was also detectable within ∼1.5 h after tunicamycin addition (Fig. 4A), supporting Snf1 involvement in the ER stress response.
Fig. 4.
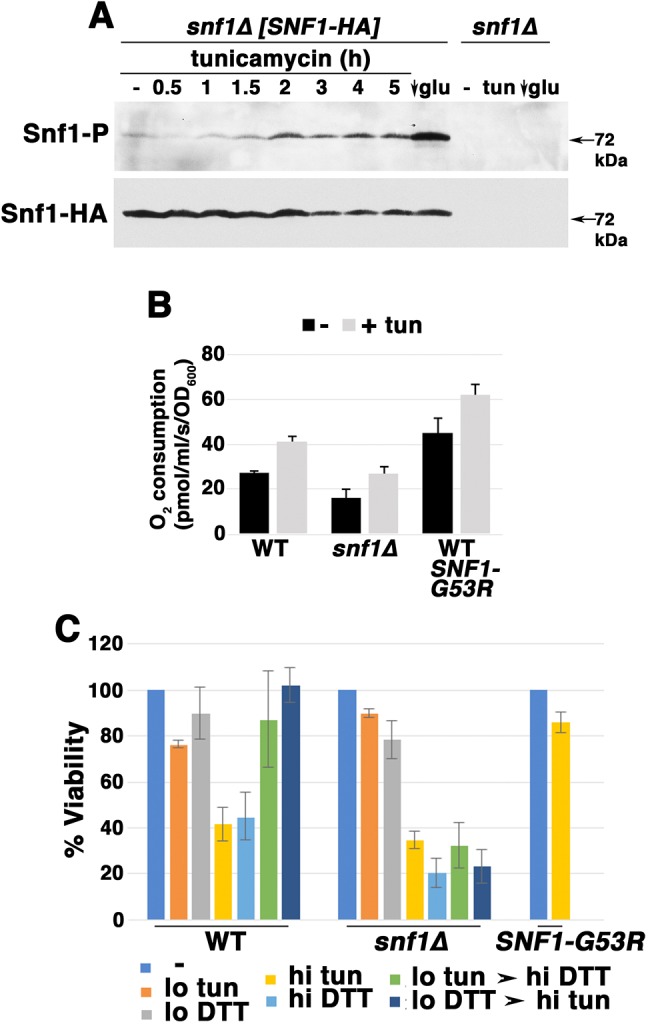
Snf1 activation during ER stress. (A) Time course of Snf1 activation, as assayed by western blot with anti-phospho-Snf1. As a positive control, cells were shifted to low glucose (0.05%) SC medium for 1 h, leading to Snf1 activation. Snf1 was phosphorylated by addition of tunicamycin (0.5 μg/ml). Snf1-HA levels were measured by blotting with anti-HA. (B) O2 consumption rate in snf1Δ cells and in cells expressing constitutively active Snf1-G53R, as measured by high resolution respirometry. Exponentially growing cells were assayed before and after treating with tunicamycin (tun) for 5 h. (C) Adaptation to ER stress, assayed as described in the legend to Fig. 1D.
CIT2-lacZ activity was assayed to measure RTG signaling in snf1Δ cells. Consistent with a role for Snf1/AMPK in inducing mitochondrial response, RTG signaling was diminished in snf1Δ cells under both basal and ER stress conditions (Fig. 2A). Moreover, in snf1Δ cells, respiration was constitutively decreased, while in response to ER stress, O2 consumption rose only to the level of that in untreated wild-type cells (Fig. 4B). By contrast, in wild-type cells expressing constitutively active Snf1-G53R, O2 consumption was constitutively higher than that seen in wild-type cells, with further elevation upon ER stress (Fig. 4B). Finally, adaptive response to ER stress was impaired in snf1Δ cells while constitutively active Snf1-G53R conferred immediate resistance to high-dose ER stress, even in cells that were not previously adapted to low-dose ER stress (Fig. 4C). These results support the idea that Snf1 activation contributes to an optimal response to ER stress to promote cell survival.
The relationship between TORC1 inactivation and Snf1/AMPK activation during ER stress was examined. Upon rapamycin-induced TORC1 inactivation, Snf1/AMPK was activated (Fig. 5A, lane 3). Consistently, Snf1 was constitutively activated in tor1Δ cells (Fig. 5B, lane 4). In npr2Δ cells with constitutively activated TORC1 (Panchaud et al., 2013), Snf1 was still phosphorylated after tunicamycin addition, although the extent of phosphorylation was somewhat diminished by comparison with that of wild-type control cells (Fig. 5C, top panel). Interestingly, Snf1 activation in response to low glucose was not different in npr2Δ and wild-type cells (Fig. 5C, arrows in top panel). These results suggest that optimum Snf1 activation in response to ER stress requires TORC1 inactivation.
Fig. 5.

Relationship between Snf1 activation and TORC1 inactivation during ER stress. (A) Activation of Snf1 by inactivation of TORC1. Lysate was prepared from cells incubated without or with rapamycin (rap; 200 nM), tunicamycin (tun; 0.5 μg/ml), or 1 mM DTT for 5 h. As a positive control, cells were shifted to SC medium with low (0.05%) glucose (glu) for 1 h (arrow). Lysates were normalized to protein content and analyzed by western blot with anti-phospho-Snf1 antibody and anti-phospho-Rps6 antibodies. (B) Western blot showing phosphorylation of Snf1 in tor1Δ cells (lane 4). (C) Top panels: western blot showing Snf1 phosphorylation in response to tunicamycin (0.5 μg/ml) in wild-type (WT) and npr2Δ cells. As a positive control, cells were shifted to low 0.05% glucose for 1 h (arrow). Pgk1 is shown as a loading control. Bottom panel: western blot showing Rps6 phosphorylation after treatment with tunicamycin for 1 and 2 h in wild-type and snf1Δ cells. Pgk1 is shown as a loading control. (D) Proposed model for ER stress-induced mitochondrial biogenesis (detailed in Discussion). ER stress leads to inactivation of TORC1 signaling; subsequently, activation of retrograde signaling leads to mitochondrial response. Snf1 activation is induced by TORC1 inactivation, and contributes to ER stress response by mitochondria.
ER stress-induced dephosphorylation of Rps6 appeared unaffected by absence of Snf1 (Fig. 5C, bottom panel), suggesting that response of TORC1 to ER stress is essentially independent of Snf1. Rps6 dephosphorylation in the presence of tunicamycin was also unaffected by constitutively active Snf1-G53R (Fig. S5A). Furthermore, Cox2 protein induction by rapamycin was not affected in snf1Δ cells (Fig. S5B). These data suggest that TORC1 inactivation and Snf1 activation during ER stress are required for optimal mitochondrial response. A model summarizing these results is shown in Fig. 5D, and further detailed in the Discussion.
DISCUSSION
ER stress promotes accumulation of mitochondrial ROS and susceptibility to cell death (Knupp et al., 2018; Yoboue et al., 2018); previously, we found that ROS accumulation and cell death are mitigated by genetic strategies that bypass glucose-mediated repression of respiration (Knupp et al., 2018). We now report that prolonged ER stress elicits an innate cellular survival response, comprising increased respiration accompanied by changes in select mitochondrial proteins without an increase in mitochondrial numbers (Fig. 1B). We show that respiratory activity is required during ER stress even in the presence of abundant glucose when the cells have a glycolytic/fermentative metabolism. ROS accumulation during ER stress is exacerbated when ETC function is inhibited by antimycin A (Fig. 1E). These results support a model in which respiratory response restricts ROS production during ER stress. Although it is also possible that enhanced oxidative phosphorylation fuels an increased demand for ATP for protein folding, we currently favor a role for mitochondrial response in ROS repression as ER stress-induced death is rescued by the protonophore CCCP (which dissipates the mitochondrial membrane potential necessary for ATP synthase function) (Knupp et al., 2018).
Multiple pathways have been described that elicit changes in nuclear gene expression in response to mitochondrial needs, such as response to mitochondrial protein misfolding (UPRmt) (Melber and Haynes, 2018), response to impaired mitochondrial import (Haynes, 2015), and mitoCPR (Weidberg and Amon, 2018). Of these pathways, retrograde signaling via the RTG pathway was the first discovered response to loss of a functional ETC (Parikh et al., 1987). The RTG pathway serves to replenish metabolic intermediates of the TCA cycle upon impairment of the ETC or glutamate deprivation (Epstein et al., 2001). We report that disruption of retrograde signaling in rtg1Δ cells results in loss of mitochondrial membrane potential (Fig. 2D), consistent with defects in the TCA cycle in rtg1Δ cells in the presence of glucose (Velot et al., 1996).
A novel finding of this study is that the RTG pathway is activated in response to ER stress. Consistent with negative regulation of the RTG pathway by TORC1, ER stress-induced mitochondrial response is linked to inactivation of TORC1 signaling (Fig. 3). Inactivation of TORC1 signaling by rapamycin or in tor1Δ cells results in increased RTG signaling, levels of select mitochondrial proteins, O2 consumption, and mitochondrial membrane potential; in tor1Δ cells or cells deprived of glutamate to activate RTG signaling, mitochondrial response is further enhanced by tunicamycin, suggesting that TORC1 inactivation and RTG activation potentiate response to ER stress (Fig. 3A,D,E; see also Bonawitz et al., 2007); these effects are linked to resistance to ER stress (Fig. 3F). By contrast, in npr2Δ cells, constitutive activation of TORC1 activity impairs mitochondrial response to ER stress, although induction of the UPR is unimpeded (Fig. 3; Fig. S3). These results suggest that TORC1 inactivation is necessary for mitochondrial response to ER stress. Importantly, Pan and Shadel reported previously that increased O2 consumption in tor1Δ cells is not due to increased mitochondrial biogenesis (numbers), but by increased mitochondrial translation of mtDNA-encoded oxidative phosphorylation subunits (Pan and Shadel, 2009). Underscoring the importance of mitochondrial translation in response to ER stress, a regulator of mitochondrial translation, MRM1, was previously identified in an over-expression genetic screen for rescue from ER stress-mediated toxicity (Knupp et al., 2018). Moreover, the cofactor heme is a regulator of mitochondrial translation (Dennerlein et al., 2017), and increased heme synthesis has been shown to mitigate ER stress-induced cytotoxicity in yeast and mammalian cells (Knupp et al., 2018).
The working model in Fig. 5D proposes that retrograde signaling for mitochondrial response occurs downstream of TORC1, as supported by loss of ER stress-induced Cox2 induction in rtg1Δ tor1Δ cells (Fig. 3A), and in agreement with previous reports that the Rtg1–Rtg3 transcription factor complex acts downstream of TORC1 (Dilova et al., 2002).
In addition to TORC1 inactivation and RTG signaling, there is a lesser role for Snf1/AMPK activation in mitochondrial response to ER stress. Our evidence suggests that Snf1 is activated upon TORC1 inhibition (Fig. 5A), consistent with crosstalk between Snf1 and TORC1 pathways that has been described previously (Shashkova et al., 2015). Although AMPK has been shown to inhibit TORC1 in yeast and mammalian cells (Hughes Hallett et al., 2015; Inoki et al., 2003), ER stress-induced TORC1 inactivation in yeast is not impacted by loss of Snf1 or constitutively active Snf1 (Fig. S5). Because loss of Snf1 has only a small negative impact (Figs 3A and 2A), the model in Fig. 5D places Snf1 signaling as a secondary pathway in mitochondrial reaction to ER stress.
At present, it is unclear how ER stress is signaled to mitochondria, or how ER stress leads to TORC1 inactivation, activation of RTG signaling, and Snf1/AMPK activation. It is plausible that activation of retrograde signaling and/or inactivation of TORC1 signaling are triggered by an ER stress-elicited mitochondrial change. At present there is no clear consensus on a mitochondrial signal that elicits the RTG response, although numerous diverse signals have been proposed, including ROS and calcium dynamics (da Cunha et al., 2015).
It has been well established that respiration is repressed in favor of glycolysis in yeast exponentially growing in glucose (Broach, 2012). We show here, however, that respiratory activity is inducible despite glucose-repressing conditions. During ER stress, glucose-mediated repression of respiration is over-ridden to activate RTG signaling and drive the TCA cycle to contribute electrons to the ETC. Similarly, the UPR itself may promote respiration because it transcriptionally activates heme biosynthetic genes (Travers et al., 2000), and heme enhances metabolic flux in the TCA cycle and the ETC (Knupp et al., 2018; Zhang et al., 2017).
How our findings translate to mammalian systems awaits further study; however, increased O2 consumption is also associated with resistance to ER stress in mammalian cells (Knupp et al., 2018), and a recent study reports up-regulation of mitochondrial components during ER stress response in human cervical cancer cells (Rendleman et al., 2018). Our findings may help to devise therapeutic strategies to limit cell death in disorders linked to ER stress.
MATERIALS AND METHODS
Strains and media
Strains used in this study were in the BY4742/BY4741 background, and except as noted, strains were analyzed during exponential growth at 30°C in standard synthetic complete (SC) medium with 2% glucose, or yeast nitrogen base (YNB) supplemented with auxotrophic requirements and 2% glucose, as described in Sherman et al. (1986). Yeast transformations were by the lithium acetate method. Deletion strains were confirmed by PCR. A tor1Δ::clonNAT (ACY112) strain was constructed by transformation of BY4742 with primers (sequences available upon request) amplified using pAG25 as the template (Goldstein and McCusker, 1999). The diploid ACX433 was constructed by cross of rtg1Δ::G418r with ACY112; dissection of a tetratype tetrad yielded wild-type, single and double rtg1Δ tor1Δ mutants. A dnm1Δ fzo1Δ double mutant was obtained by tetrad dissection. The prototroph is a parent of the BY4742/BY4741 strains, a gift from Fred Winston (Harvard University, Boston, MA).
Molecular biology
URA3-marked centromeric plasmids expressing HA-Snf1 and HA-Snf1-G53R (pIT517) were a gift from S. Kutchin (University of Wisconsin, Milwaukee, WI). pDN436 is a LEU2-marked centromeric plasmid expressing CPY* driven by the native promoter (Ng et al., 2000), kindly provided by Davis Ng (National University of Singapore). A LEU2-marked plasmid for over-expressing HAP4 was cut with Pac1 for integration at the ADH1 promoter, and was a gift from Su-Ju Lin (University of California, Davis, CA) (Lin et al., 2004). SAK1 on a URA3-marked 2 μm plasmid was from Martin Schmidt (University of Pittsburgh, PA). pCIT2-lacZ, a URA3-marked centromeric plasmid, was a gift from Zhengchang Liu (University of New Orleans, LA) (Liu and Butow, 1999). pJC104, a 2 µm URA3-marked plasmid bearing UPRE-lacZ (Cox et al., 1997) was a gift from Peter Walter (University of California, San Francisco, CA). pGCN4-lacZ (Hinnebusch, 1985), a URA3-marked centromeric plasmid was a gift from Alan Hinnebusch (NIH, Bethesda, MD).
To assay mitochondrial DNA content after ER stress, COX2 content was determined by PCR and normalized to ACT1 content using genomic DNA. Primers were designed to amplify a small region of each gene; sequences are available upon request.
Western blots and enzyme assays
Cells were harvested by freezing in liquid nitrogen. Cell lysates were made by vortexing cells with glass beads in sorbitol buffer (0.3 M sorbitol, 0.1 M NaCl, 5 mM MgCl2, 10 mM Tris; pH 7.4) with a protease inhibitor cocktail, including PMSF, as described previously (Chang and Slayman, 1991). Cell lysate prepared in this way was used for assaying β-galactosidase activity, as described previously (Rose et al., 1990). Protein content was determined by Bradford assay (Bradford, 1976). Western blots were visualized by incubating with primary antibody, followed by peroxidase-conjugated secondary antibody and detection by chemiluminescence.
To assay Rps6 phosphorylation, exponentially growing cells were harvested and frozen in liquid nitrogen and trichloroacetic acid, as described previously (Liu et al., 2012). Lysate was produced by vortexing with glass beads, and protein content was determined by BCA (Pierce) assay.
Snf1 activation was assayed by western blot to detect Snf1 phosphorylation at the activation loop Thr210. For these experiments, snf1Δ cells were transformed with centromeric plasmids bearing HA-SNF1 in order to assess total Snf1 levels. Cell cultures were boiled prior to protein extraction to prevent spurious Snf1 activation, as described by Orlova and colleagues (Orlova et al., 2008).
Anti-Cox2 (ab110271) and anti-Por1 (ab110326) monoclonal antibodies were from Abcam, Inc. (Cambridge, UK). Anti-ATP synthase subunit alpha (ATP1) monoclonal antibody (459240) was from MitoSciences, Inc. (Eugene, OR). Anti-Pgk1 antibody (#459250) was from Thermo Fisher Scientific. Anti-HA monoclonal antibody (MMS101P) was from CovanceI (Princeton, NJ). Anti-phospho-AMPKα (Thr172) rabbit antibody and anti-phospho-S6 ribosomal protein (Ser235/236) antibody (#2211) were from Cell Signaling Technology (Danvers, MA). Antibody to yeast eIF2α was a gift from Tom Dever (NIH). Rabbit anti-Ssc1 was a kind gift from Kai Hell (Ludwig-Maximilians-Universität München, Munich, Germany).
Cell viability, ER stress adaptation, membrane potential assay and ROS staining
For viability assay, cells exponentially growing in SC medium (2% glucose) were diluted to ∼0.15 OD600/ml for treatment with 0.5 µg/ml tunicamycin or 1 mM DTT. After 4 h incubation, cells were normalized to 0.1 OD600/ml, and then further serially diluted onto YPD plates. For adaptation assay, cells were treated with low dose tunicamycin or DTT for 4 h, followed by addition of high dose DTT (10 mM) or tunicamycin (10 µg/ml), respectively, for 4 h. Cells were then normalized and serially diluted for viability assay. After 2 days incubation at 30°C, colonies were counted and expressed as a percentage of colony numbers of untreated cells. Controls from all strains ranged from 167 to 411 colonies.
For TMRM staining in nonquenching mode, live cells were stained with TMRM (5 nM) for 30 min, and visualized with an Olympus fluorescent microscope, and images were collected with a Hamamatsu CCD camera.
For detection of ROS, mid-log cells were treated (or not) with tunicamycin (0.5 µg/ml), antimycin (4 µM) and both for 5 h. Cells were then resuspended in PBS at 2 OD600/ml with 5 mg/ml DHE for 15 min at 30°C; cells were washed once before fluorescence microscopy.
High-resolution respirometry
For whole-cell oxygen consumption, exponentially growing cells were pelleted and resuspended in SC medium at 20 OD600/ml. Cells were then added to a high-resolution Orobos Oxygraph 2K at 25°C at a concentration of 2 OD600/ml. O2 flux was determined by measuring the fall in O2 concentration in the sealed oxygraph.
Real-time quantitative polymerase chain reaction
Real-time PCR was performed as described (Yang et al., 2014). In brief, RNA samples were extracted with TRIzol/choloroform reagent (Invitrogen) and purified using a PureLink RNA mini kit (Invitrogen). After treatment of total RNA with PureLink Dnase (Invitrogen), approximately 6 μg of purified RNA was used for first-strand complementary DNA synthesis using PrimeScript Reverse Transcriptase (TaKaRa) with oligo dT primers. RT-PCR was performed using specific PET111 primers (sequences on request) and Power SYBR Green PCR Master Mix in a StepOnePlus Real-time PCR System (Thermo Fisher). Relative transcript levels were determined by the comparative threshold method, and normalized to that of ACT1. qPCR for each gene was done with at least five biological replicates.
Supplementary Material
Acknowledgements
We thank Sergei Kuchin, Zhengchang Liu, Davis Ng, Su-Ju Lin, Peter Walter, Alan Hinnebusch and Martin Schmidt for plasmids; Tom Dever, Kai Hell and Doron Rapaport for antibodies; Dan Beard's laboratory for sharing their expertise and oxygraphy; and Peter Arvan for helpful discussions.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: A.C.; Investigation: I.H., J.K., A.C.; Writing - review & editing: I.H., J.K., A.C.; Supervision: A.C.; Funding acquisition: A.C.
Funding
This work was supported by funds from the University of Michigan Protein Folding Disease Initiative (A.C.) and the National Institutes of Health [R21 AG058862 to A.C.]. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.241539.supplemental
Peer review history
The peer review history is available online at https://jcs.biologists.org/lookup/doi/10.1242/jcs.241539.reviewer-comments.pdf
References
- Appenzeller-Herzog C. and Hall M. N. (2012). Bidirectional crosstalk between endoplasmic reticulum stress and mTOR signaling. Trends Cell Biol. 22, 274-282. 10.1016/j.tcb.2012.02.006 [DOI] [PubMed] [Google Scholar]
- Bachar-Wikstrom E., Wikstrom J. D., Kaiser N., Cerasi E. and Leibowitz G. (2013). Improvement of ER stress-induced diabetes by stimulating autophagy. Autophagy 9, 626-628. 10.4161/auto.23642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleazard W., McCaffery J. M., King E. J., Bale S., Mozdy A., Tieu Q., Nunnari J. and Shaw J. M. (1999). The dynamin-related GTPase Dnm1 regulates mitochondrial fission in yeast. Nat. Cell Biol. 1, 298-304. 10.1038/13014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonawitz N. D., Chatenay-Lapointe M., Pan Y. and Shadel G. S. (2007). Reduced TOR signaling extends chronological life span via increased respiration and upregulation of mitochondrial gene expression. Cell Metab. 5, 265-277. 10.1016/j.cmet.2007.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248-254. 10.1016/0003-2697(76)90527-3 [DOI] [PubMed] [Google Scholar]
- Bravo R., Vicencio J. M., Parra V., Troncoso R., Munoz J. P., Bui M., Quiroga C., Rodriguez A. E., Verdejo H. E., Ferreira J. et al. (2011). Increased ER-mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J. Cell Sci. 124, 2143-2152. 10.1242/jcs.080762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo R., Parra V., Gatica D., Rodriguez A. E., Torrealba N., Paredes F., Wang Z. V., Zorzano A., Hill J. A., Jaimovich E. et al. (2013). Endoplasmic reticulum and the unfolded protein response: dynamics and metabolic integration. Int. Rev. Cell Mol. Biol. 301, 215-290. 10.1016/B978-0-12-407704-1.00005-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broach J. R. (2012). Nutritional control of growth and development in yeast. Genetics 192, 73-105. 10.1534/genetics.111.135731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butow R. A. and Avadhani N. G. (2004). Mitochondrial signaling: the retrograde response. Mol. Cell 14, 1-15. 10.1016/S1097-2765(04)00179-0 [DOI] [PubMed] [Google Scholar]
- Chang A. and Slayman C. W. (1991). Maturation of the yeast plasma membrane [H+]ATPase involves phosphorylation during intracellular transport. J. Cell Biol. 115, 289-295. 10.1083/jcb.115.2.289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Sutter B. M., Shi L. and Tu B. P. (2017). GATOR1 regulates nitrogenic cataplerotic reactions of the mitochondrial TCA cycle. Nat. Chem. Biol. 13, 1179-1186. 10.1038/nchembio.2478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J. S., Chapman R. E. and Walter P. (1997). The unfolded protein response coordinates the production of endoplasmic reticulum protein and endoplasmic reticulum membrane. Mol. Biol. Cell 8, 1805-1814. 10.1091/mbc.8.9.1805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig E. A. (2018). Hsp70 at the membrane: driving protein translocation. BMC Biol. 16, 11 10.1186/s12915-017-0474-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Cunha F. M., Torelli N. Q. and Kowaltowski A. J. (2015). Mitochondrial retrograde signaling: triggers, pathways, and outcomes. Oxidative Med. Cell. Longevity 482582, 1-10. 10.1155/2015/482582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling N. J. and Cook S. J. (2014). The role of MAPK signalling pathways in the response to endoplasmic reticulum stress. Biochim. Biophys. Acta 1843, 2150-2163. 10.1016/j.bbamcr.2014.01.009 [DOI] [PubMed] [Google Scholar]
- Dennerlein S., Wang C. and Rehling P. (2017). Plasticity of mitochondrial translation. Trends Cell Biol. 27, 712-721. 10.1016/j.tcb.2017.05.004 [DOI] [PubMed] [Google Scholar]
- Dikalov S. I. and Harrison D. G. (2014). Methods for detection of mitochondrial and cellular reactive oxygen species. Antioxid. Redox Signal. 20, 372-382. 10.1089/ars.2012.4886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilova I., Chen C.-Y. and Powers T. (2002). Mks1 in concert with TOR signaling negatively regulates RTG target gene expression in S. cerevisiae. Curr. Biol. 12, 389-395. 10.1016/S0960-9822(02)00677-2 [DOI] [PubMed] [Google Scholar]
- Epstein C. B., Waddle J. A., Hale W. T., Davé V., Thornton J., Macatee T. L., Garner H. R. and Butow R. A. (2001). Genome-wide responses to mitochondrial dysfunction. Mol. Biol. Cell 12, 297-308. 10.1091/mbc.12.2.297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein A. L. and McCusker J. H. (1999). Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast 15, 1541-1553. [DOI] [PubMed] [Google Scholar]
- González A. and Hall M. N. (2017). Nutrient sensing and TOR signaling in yeast and mammals. EMBO J. 36, 397-408. 10.15252/embj.201696010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green-Willms N. S., Butler C. A., Dunstan H. M. and Fox T. D. (2001). Pet111p, an inner membrane-bound translational activator that limits expression of the Saccharomyces cerevisiae mitochondrial gene COX2. J. Biol. Chem. 276, 6392-6397. 10.1074/jbc.M009856200 [DOI] [PubMed] [Google Scholar]
- Hansen K. G., Aviram N., Laborenz J., Bibi C., Meyer M., Spang A., Schuldiner M. and Herrmann J. M. (2018). An ER surface retrieval pathway safeguards the import of mitochondrial membrane proteins in yeast. Science 361, 1118-1122. 10.1126/science.aar8174 [DOI] [PubMed] [Google Scholar]
- Hardie D. G., Ross F. A. and Hawley S. A. (2012). AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 13, 251-262. 10.1038/nrm3311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatakeyama R. and De Virgilio C. (2016). Unsolved mysteries of Rag GTPase signaling in yeast. Small GTPases 7, 239-246. 10.1080/21541248.2016.1211070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes C. M. (2015). Surviving import failure. Nature 524, 419-420. 10.1038/nature14644 [DOI] [PubMed] [Google Scholar]
- Hedbacker K. and Carlson M. (2009). Snf1/AMPK pathways in yeast. Front. Biosci. 13, 2408-2420. 10.2741/2854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnebusch A. G. (1985). A hierarchy of trans-acting factors modulates translation of an activator of amino acid biosynthetic genes in Saccharomyces cerevisiae. Mol. Cell. Biol. 5, 2349-2360. 10.1128/MCB.5.9.2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnebusch A. G. (2005). Translational regulation of GCN4 and the general amino acid control of yeast. Annu. Rev. Microbiol. 59, 407-450. 10.1146/annurev.micro.59.031805.133833 [DOI] [PubMed] [Google Scholar]
- Hughes Hallett J. E., Luo X. and Capaldi A. P. (2015). Snf1/AMPK promotes the formation of Kog1/Raptor-bodies to increase the activation threshold of TORC1 in budding yeast. eLife 4, e09181 10.7554/eLife.09181.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K., Zhu T. and Guan K.-L. (2003). TSC2 mediates cellular energy response to control cell growth and survival. Cell 115, 577-590. 10.1016/S0092-8674(03)00929-2 [DOI] [PubMed] [Google Scholar]
- Jung T. W. and Choi K. M. (2016). Pharmacological modulators of endoplasmic reticulum stress in metabolic diseases. Int. J. Mol. Sci. 17, 192 10.3390/ijms17020192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapahi P., Chen D., Rogers A. N., Katewa S. D., Li P. W.-L., Thomas E. L. and Kockel L. (2010). With TOR, less is more: a key role for the conserved nutrient-sensing TOR pathway in aging. Cell Metab. 11, 453-465. 10.1016/j.cmet.2010.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knupp J., Arvan P. and Chang A. (2019). Increased mitochondrial respiration promotes survival from endoplasmic reticulum stress. Cell Death Differ. 26, 487-501. 10.1038/s41418-018-0133-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lascaris R., Bussemaker H. J., Boorsma A., Piper M., van der Spek H., Grivell L. and Blom J. (2002). Hap4p overexpression in glucose-grown Saccharomyces cerevisiae induces cells to enter a novel metabolic state. Genome Biol. 4, R3 10.1186/gb-2002-4-1-r3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao X. S., Small W. C., Srere P. A. and Butow R. A. (1991). Intramitochondrial functions regulate nonmitochondrial citrate synthase (CIT2) expression in Saccharomyces cerevisiae. Mol. Cell. Biol. 11, 38-46. 10.1128/MCB.11.1.38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S.-J., Ford E., Haigis M., Liszt G. and Guarente L. (2004). Calorie restriction extends yeast life span by lowering the level of NADH. Genes Dev. 18, 12-16. 10.1101/gad.1164804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J. and Barrientos A. (2013). Transcriptional regulation of yeast oxidative phosphorylation hypoxic genes by oxidative stress. Antioxid Redox Signal. 19, 1916-1927. 10.1089/ars.2012.4589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z. and Butow R. A. (1999). A transcriptional switch in the expression of yeast tricarboxylic acid cycle genes in response to a reduction or loss of respiratory function. Mol. Cell. Biol. 19, 6720-6728. 10.1128/MCB.19.10.6720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z. and Butow R. A. (2006). Mitochondrial retrograde signaling. Annu. Rev. Genet. 40, 159-185. 10.1146/annurev.genet.40.110405.090613 [DOI] [PubMed] [Google Scholar]
- Liu M., Huang C. J., Polu S. R., Schneiter R. and Chang A. (2012). Regulation of sphingolipid synthesis through Orm1 and Orm2 in yeast. J. Cell Sci. 125, 2428-2435. 10.1242/jcs.100578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewith R. and Hall M. N. (2011). Target of rapamycin (TOR) in nutrient signaling and growth control. Genetics 189, 1177-1201. 10.1534/genetics.111.133363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra J. D. and Kaufman R. J. (2011). ER stress and its functional link to mitochondria: role in cell survival and death. Cold Spring Harbor Perspect. Biol. 3, a004424 10.1101/cshperspect.a004424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melber A. and Haynes C. M. (2018). UPRmt regulation and output: a stress response mediated by mitochondrial-nuclear communication. Cell Res. 28, 281-295. 10.1038/cr.2018.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neklesa T. K. and Davis R. W. (2009). A genome-wide screen for regulators of TORC1 in response to amino acid starvation reveals a conserved Npr2/3 complex. PLoS Genet. 5, e1000515 10.1371/journal.pgen.1000515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng D. T. W., Spear E. D. and Walter P. (2000). The unfolded protein response regulates multiple aspects of secretory and membrane protein biogenesis and endoplasmic reticulum quality control. J. Cell Biol. 150, 77-88. 10.1083/jcb.150.1.77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunnari J. and Suomalainen A. (2012). Mitochondria: in sickness and in health. Cell 148, 1145-1159. 10.1016/j.cell.2012.02.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakes S. A. and Papa F. R. (2015). The role of endoplasmic reticulum stress in human pathology. Annu. Rev. Pathol. Mech. Dis. 10, 173-194. 10.1146/annurev-pathol-012513-104649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlova M., Kanter E., Krakovich D. and Kuchin S. (2006). Nitrogen availability and TOR regulate the Snf1 protein kinase in Saccharomyces cerevisiae. Eukaryot. Cell 5, 1831-1837. 10.1128/EC.00110-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlova M., Barrett L. K. and Kuchin S. (2008). Detection of endogenous Snf1 and its activation state: application to Saccharomyces and Candida species. Yeast 25, 745-754. 10.1002/yea.1628 [DOI] [PubMed] [Google Scholar]
- Pan Y. and Shadel G. S. (2009). Extension of chronological life span by reduced TOR signaling requires down-regulation of Sch9p and involves increased mitochondrial OXPHOS complex density. Aging 1, 131-143. 10.18632/aging.100016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panchaud N., Peli-Gulli M.-P. and De Virgilio C. (2013). Amino acid deprivation inhibits TORC1 through a GTPase-activating protein complex for the Rag family GTPase Gtr1. Sci. Signal. 6, ra42 10.1126/scisignal.2004112 [DOI] [PubMed] [Google Scholar]
- Parikh V. S., Morgan M. M., Scott R., Clements L. S. and Butow R. A. (1987). The mitochondrial genotype can influence nuclear gene expression in yeast. Science 235, 576-580. 10.1126/science.3027892 [DOI] [PubMed] [Google Scholar]
- Perry S. W., Norman J. P., Barbieri J., Brown E. B. and Gelbard H. A. (2011). Mitochondrial membrane potential probes and the proton gradient: a practical usage guide. BioTechniques 50, 98-115. 10.2144/000113610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips M. J. and Voeltz G. K. (2016). Structure and function of ER membrane contact sites with other organelles. Nat. Rev. Mol. Cell Biol. 17, 69-82. 10.1038/nrm.2015.8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rendleman J., Cheng Z., Maity S., Kastelic N., Munschauer M., Allgoewer K., Teo G., Zhang Y. B. M., Lei A., Parker B. et al. (2018). New insights into the cellular temporal response to proteostatic stress. eLife 7, e39054 10.7554/eLife.39054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose M. D., Winston F. and Hieter P. (1990). Methods in Yeast Genetics: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Sesaki H., Southard S. M., Yaffe M. P. and Jensen R. E. (2003). Mgm1p, a dynamin-related GTPase, is essential for fusion of the mitochondrial outer membrane. Mol. Biol. Cell 14, 2342-2356. 10.1091/mbc.e02-12-0788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shashkova S., Welkenhuysen N. and Hohmann S. (2015). Molecular communication: crosstalk between the Snf1 and other signaling pathways. FEMS Yeast Res. 15, 1-10. 10.1093/femsyr/fov026 [DOI] [PubMed] [Google Scholar]
- Sherman F., Hicks J. B. and Fink G. R. (1986). Methods in Yeast Genetics: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Travers K. J., Patil C. K., Wodicka L., Lockhart D. J., Weissman J. S. and Walter P. (2000). Functional and genomic analyses reveal an essential coordination between the unfolded protein response and endoplasmic reticulum-associated degradation. Cell 101, 249-258. 10.1016/S0092-8674(00)80835-1 [DOI] [PubMed] [Google Scholar]
- Turrens J. F. (2003). Mitochondrial formation of reactive oxygen species. J. Physiol. 552, 335-344. 10.1113/jphysiol.2003.049478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban J., Soulard A., Huber A., Lippman S., Mukhopadhyay D., Deloche O., Wanke V., Anrather D., Ammerer G., Riezman H. et al. (2007). Sch9 is a major target of TORC1 in Saccharomyces cerevisiae. Mol. Cell 26, 663-674. 10.1016/j.molcel.2007.04.020 [DOI] [PubMed] [Google Scholar]
- Velot C., Haviernik P. and Lauquin G. J.-M. (1996). The Saccharomyces cerevisiae RTG2 gene is a regulator of aconitase expression under catabolite repression conditions. Genetics 144, 893-903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P. and Ron D. (2011). The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081-1086. 10.1126/science.1209038 [DOI] [PubMed] [Google Scholar]
- Wang M. and Kaufman R. J. (2016). Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 529, 326-335. 10.1038/nature17041 [DOI] [PubMed] [Google Scholar]
- Weidberg H. and Amon A. (2018). MitoCPR-a surveillance pathway that protects mitochondria in response to protein import stress. Science 360, eaan4146 10.1126/science.aan4146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X., Ren W., Zhao Q., Zhang P., Wu F. and He Y. (2014). Homodimerization of HYL1 ensures the correct selection of cleavage sites in primary miRNA. Nucleic Acids Res. 42, 12224-12236. 10.1093/nar/gku907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoboue E. D., Sitia R. and Simmen T. (2018). Redox crosstalk at endoplasmic reticulum (ER) membrane contact sites (MCS) uses toxic waste to deliver messages. Cell Death Dis. 9, 331-345. 10.1038/s41419-017-0033-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Bell A., Perlman P. S. and Leibowitz M. J. (2000). Pentamidine inhibits mitochondrial intron splicing and translation in Saccharomyces cerevisiae. RNA 6, 937-951. 10.1017/S1355838200991726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T., Bu P., Zeng J. and Vancura A. (2017). Increased heme synthesis in yeast induces a metabolic switch from fermentation to respiration even under conditions of glucose repression. J. Biol. Chem. 292, 16942-16954. 10.1074/jbc.M117.790923 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.