

Introduction:
IgG4-related disease (IgG4-RD) is an immune-mediated condition that can cause fibro-inflammatory lesions in nearly any organ.1 The cause remains unknown. Common manifestations include dacryoadenitis (lacrimal gland inflammation), sclerosing sialoadenitis of the major salivary glands (submandibular, parotid, and sublingual), autoimmune pancreatitis, sclerosing cholangitis, and retroperitoneal fibrosis (RPF). Though variable across cohorts, up to 35% of IgG4-RD patients2 have manifestations in the chest. These include lung nodules, pleural thickening, pleural effusions, aortitis, sclerosing pericarditis, lymphadenopathy, and paravertebral masses. Patients with thoracic disease often present with non-specific symptoms including dyspnea or cough, but asymptomatic thoracic involvement is detected incidentally in many if not the majority of IgG4-RD cases involving the chest.2 The differential diagnosis of thoracic IgG4-RD is broad and includes infections (e.g., tuberculosis), other autoimmune conditions (e.g., ANCA-associated vasculitis), and malignancy, (e.g., lymphoma, lung cancer). Here, we review IgG4-RD with an emphasis on thoracic disease, including its epidemiology, common presentations, diagnostic approach, and management.
Epidemiology:
Because of its relatively recent recognition, the incidence and prevalence of IgG4-RD are difficult to estimate. Knowledge of IgG4-RD and recognition of its manifestations has improved in recent years but persistent delays in diagnosis underscore the problem of under-recognition of this disease.3
Demographics:
Much of our understanding of the epidemiology of IgG4-RD derives from single-center case series from around the world4–7 as well as a large, international cohort assembled to derive and validate the forthcoming American College of Rheumatology/European League Against Rheumatism IgG4-RD Classification Criteria.3 Across cohorts, males tend to be affected more often than females and patients typically present in their 5th, 6th, and 7th decades of life. However, patients of all ages, including children, have been diagnosed with IgG4-RD. Though it was originally described in a cohort of Japanese patients with autoimmune pancreatitis, IgG4-RD has now been reported in patients of nearly all racial backgrounds. A recent cluster analysis suggests that Asian patients and females are at increased risk for head and neck disease but the cause of this organ predilection in these disease subsets is unclear.3
Risk Factors:
Risk factors for IgG4-RD are poorly understood. Tobacco and asbestos exposure have been found to be risk factors for idiopathic retroperitoneal fibrosis in multiple studies conducted before IgG4-RD was recognized as a common cause of RPF.8,9 In a recent case-control study including patients with diverse manifestations, tobacco exposure was found to be a risk factor for IgG4-RD but much of this association was driven by a very strong association observed among the subset of patients with RPF.10 Potential associations between tobacco exposure and the risk of IgG4-RD in the chest, particularly pulmonary manifestations, require further evaluation. Antecedent malignancy has been identified as a risk factor for the subsequent development of IgG4-RD but this observation requires further study.11 The association between malignancy and IgG4-RD is especially relevant in the chest, where the differential of IgG4-RD includes lung cancer and lymphoma. Other environmental exposures, especially occupational antigens (e.g., solvents), have also been suggested as risk factors for IgG4-RD.12
Etiopathogenesis:
Our understanding of the pathogenesis of IgG4-RD has expanded rapidly in recent years. Although genetic (e.g. HLA-DR4 in autoimmune pancreatitis) and other risk factors (e.g. cigarette smoking, as above) have been proposed, precise inciting mechanisms have not been identified.13,14
Plasmablasts:
In IgG4-RD, IgG4-expressing and oligoclonally-expanded plasmablasts (i.e., short-lived antibody-producing cells, often defined as being CD20-19+38+27+) have been described in affected tissue and found to be elevated in the circulation when compared to controls.15 Circulating plasmablast levels correlate with disease activity and the number of organs involved, regardless of serum IgG4 concentration, suggesting a pathogenic role.16,17 Moreover, B-cell depletion, which eliminates the progenitors of plasmablasts, consistently results in clinical improvement.18–23
Auto-Antibodies:
At least eleven autoantigens have been reported in IgG4-RD but their pathogenic significance remains undefined.24 Their identification supports the hypothesis that IgG4-RD is an autoimmune condition. One of the identified autoantigens, galectin-3, was shown to be an antigenic target of dominantly-expanded plasmablasts and present in 28% of a diverse, multi-organ cohort.25 Another, a human protein named laminin-511, was detected in 50% of IgG4-related pancreatitis subjects. Immunization of mice with laminin-511 resulted in antibody responses and pancreatic injury.26
T Follicular Helper Cells:
Follicular helper T cells (TFH) are essential to B cell maturation and immunoglobulin (Ig) class switching. In IgG4-RD, activated type 2 TFH cells are expanded in the circulation, accumulate in affected organs, and correlate with serum IgG4 concentrations, plasmablast levels, and the number of affected organs.27 In addition, type 2 TFH cells can induce naïve B cell differentiation into IgG4-producing plasmablasts in vitro, implicating them in a key feature of pathophysiology.27 The mechanism through which type 2 TFH cells drive naïve B cell differentiation appears to be through IL-4 secretion.
IgG4 Molecule:
The precise role of the IgG4 molecule itself in IgG4-RD remains surprisingly unclear.28 The phenomenon of IgG4 class-switching is nonspecific and occurs in the setting of any chronic antigen exposure (e.g., allergic disease). Furthermore, in contrast to other IgG subclasses, IgG4 is not very effective at activating complement or binding to Fc receptors on immune cells.29 There are a number of reasons for believing, therefore, that IgG4 is not central to the disease bearing IgG4 in its name. However, recent experimental data30–32 have suggested a possible pathologic role for IgG4. For instance, IgG4 derived from the serum of patients IgG4-RD has been found to bind C1q with enhanced affinity compared to IgG4 from healthy controls.30 Moreover, in an adoptive transfer model of purified human IgG4, the recipient mice demonstrated acute pancreatitis, with demonstrable deposition of human IgG4 within the pancreas.33
CD4+ Cytotoxic T Lymphocytes:
Even though B cells are clearly important in the pathogenesis of IgG4-RD, the tissue infiltrate is dominated by T cells34, particularly CD4+ cytotoxic T lymphocytes (CD4+CTLs).35 CD4+CTLs are clonally-expanded in the circulation of IgG4-RD patients, decline in conjunction with treatment-induced remission, and correlate with the number of affected organs.35,36 In addition to driving cytotoxicity via expression of perforin and granzymes, CD4+CTLs also secrete a collection of pro-fibrotic molecules such as IL-1β and TGF-β.35 The precise contributions of these CD4+CTLs to the fibrosis that characterizes IgG4-RD remains a focus of investigation, with potential relevance to other conditions associated with fibrosis.
Clinical Manifestations:
Overview:
IgG4-RD can affect nearly any organ (Table 1).37 Patients may present with involvement isolated to a single organ or with multiple organs involved at the same time. In untreated patients, manifestations in different organs may accumulate over time in a pattern that is termed “metachronous”. In contrast to the acute presentation that typically characterizes conditions such as infections, IgG4-RD typically presents in an indolent fashion and is sometimes discovered incidentally. When present, symptoms are typically attributable to mass effect caused by the tumefactive lesion(s) or injury caused by the inflammatory lesion(s). For instance, patients with sclerosing sialoadenitis often come to medical attention because of the appearance of a painless lump or mass. In contrast, a patient with aortitis may present with acute pain related to dissection.
Table 1:
Manifestations of IgG4-Related Disease
| Head and Neck |
| Hypertrophic pachymeningitis |
| Hypophysitis |
| Orbital pseudotumor and/or orbital myositis |
| Lacrimal gland enlargement (dacryoadenitis) |
| Sinusitis |
| Submandibular, parotid, and/or sublingual gland enlargement (sialoadenitis) |
| Fibrosing thyroiditis |
| Pulmonary |
| Airway |
| Tracheobronchial stenosis |
| Lung Parenchyma |
| Pulmonary nodules and/or infiltrates |
| Interstitial lung disease |
| Mediastinal and/or hilar lymphadenopathy |
| Pleura |
| Pleural thickening and nodules |
| Pleural effusion |
| Cardiac/Mediastinum |
| Thoracic periaortitis with or without aneurysm |
| Fibrosing pericarditis and/or mediastinitis |
| Paravertebral mass |
| Pancreato-hepato-biliary |
| Autoimmune pancreatitis (Type 1) |
| Sclerosing cholangitis |
| Hepatic pseudotumors |
| Renal |
| Tubulointerstitial nephritis |
| Membranous glomerulonephritis |
| Abdominal Aorta and Iliac Arteries |
| Retroperitoneal fibrosis |
| Periaortitis |
| Lymphadenopathy (any region) |
| Other |
| Sclerosing mesenteritis |
| Breast (mastitis) |
| Prostate (prostatitis) |
Constitutional symptoms such as fatigue and arthralgias may occur in IgG4-RD but are generally subtle and insidious rather than profound and sudden in onset.37 Substantial weight loss can occur in patients with IgG4-related autoimmune pancreatitis because of exocrine pancreatic failure. Such patients simply cannot absorb the requisite nutrition from ingested food and weight loss of up to 20–50 pounds over a period of months may be reported. Fever is atypical but has been reported in cases of thoracic IgG4-RD.2,38 The presence of prominent fevers should prompt a search for an alternative explanation, such as ascending cholangitis complicating biliary disease.
Symptoms Related to IgG4-Related Disease in the Chest
Respiratory symptoms are sometimes unimpressive even in patients with substantial thoracic involvement by IgG4-RD.2 In one study, however, approximately 25% of patients presented with what would eventually be diagnosed as IgG4-RD because of respiratory symptoms.39 Approximately 50% of IgG4-RD patients with lung involvement note at least some respiratory symptoms, but the remainder are often asymptomatic.2,38,40,41 Thoracic symptoms are nonspecific and can include cough (~65%), shortness of breath (~30%) and chest pain (~20%).38,39,42 Hemoptysis is uncommon but has been reported38; like fever, it should raise concern for an alternative etiology. Of note, cases of coronary involvement resulting in acute coronary syndrome (e.g., chest pain) have been reported.43–46 Patients with large vessel disease (e.g., aorta) are often asymptomatic except in cases in which their disease has been complicated by dissection or flow-limiting vessel narrowing. Allergy and/or asthma are common comorbidities in patients with IgG4-RD and may lead to symptoms that are not related to IgG4-RD lesions.2,40,47 The diagnosis of asthma in patients with IgG4-related lung disease may be related to airway inflammation resulting from IgG4-RD itself. The relationship between allergic conditions and IgG4-RD requires further study.
Chest Manifestations
Chest computed tomography (CT) is frequently used for the detection and characterization of intrathoracic IgG4-RD which can affect any structure in the chest, including the lungs, airways, mediastinum, and/or pleura.2,38,48–50 Radiographs are less sensitive than CT for excluding IgG4-RD involvement in the chest. We will review the manifestations of IgG4-RD in the chest by framing the discussion around imaging findings.
Multiple simultaneous locations can be found to be affected by IgG4-RD with cross-sectional imaging and the constellation of features is often more specific for IgG4-RD than any single abnormality in isolation. It is important to note that the thoracic radiological findings of IgG4-RD can resemble those associated with other conditions, most notably lymphoma, infection and other inflammatory disorders, including sarcoidosis and ANCA-associated vasculitis.
Lymphadenopathy:
The most common chest CT finding is lymphadenopathy (Figure 1). This is usually symmetrical and tends to involve multiple stations in the neck, axillae, hila and mediastinum. Nodes are mildly enlarged, well-defined, and often demonstrate enhancement following intravenous contrast administration. The nodes are often fluorodeoxyglucose (FDG)-avid on positron emission tomography (PET) scans (Figure 2).
Figure 1.
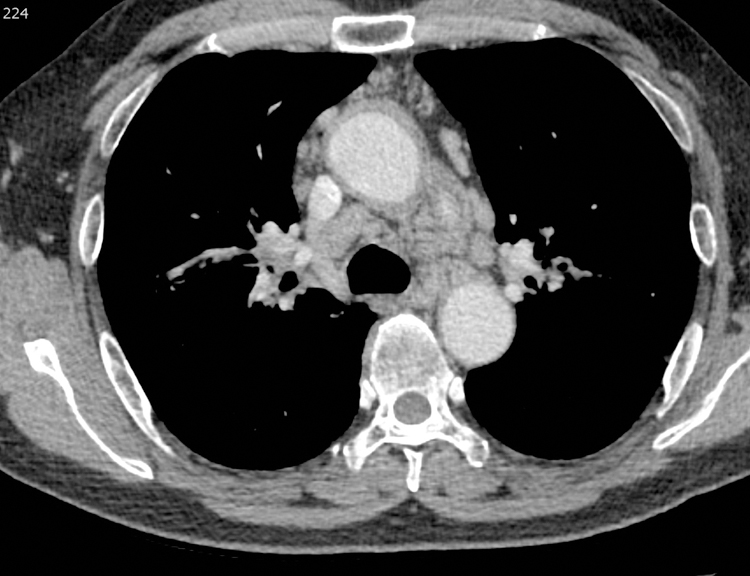
Hilar and mediastinal lymphadenopathy in a 61-year-old male with IgG4-RD. Chest CT axial scan at the level of the aortopulmonary window on soft tissue windows. There is bilateral, mild mediastinal and hilar lymphadenopathy (arrow). In addition, soft tissue surrounds the ascending aorta, consistent with peri-aortitis (arrowhead).
Figure 2.
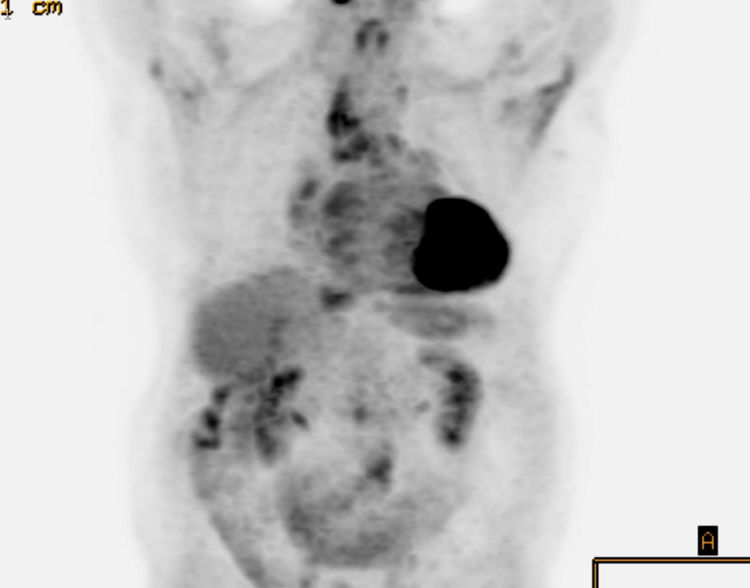
Fluorodeoxyglucose (FDG)-avid lymphadenopathy in a 77 year-old-male with IgG4-RD. Coronal PET CT demonstrates FDG-avidity in the hilar and mediastinal nodes (arrows).
Paravertebral mass:
A striking mediastinal manifestation that occurs in a minority of IgG4-RD patients is a paravertebral mass. It is most often right sided, spans multiple thoracic vertebral bodies, and does not typically encase the aorta, extend into the spine, or cause bony destruction (Figure 3). Few disease entities other than IgG4-RD lead to this thoracic disease manifestation.
Figure 3.
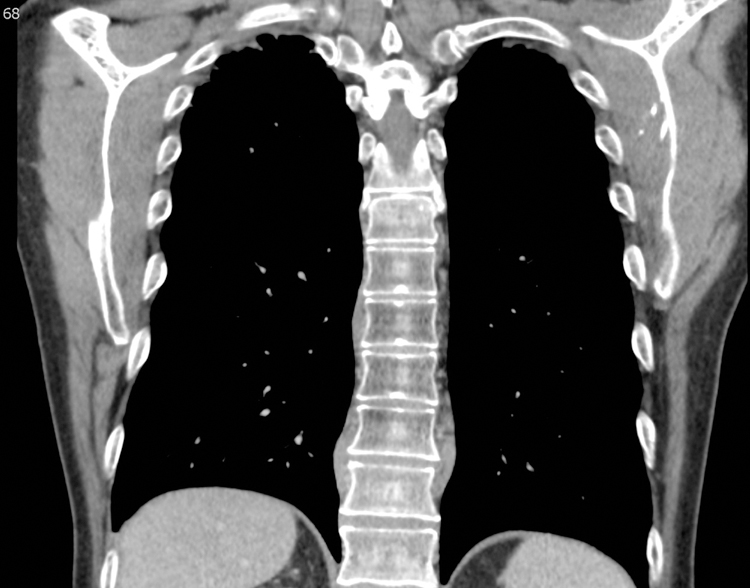
Bilateral paravertebral masses in a 61 year-old-male with IgG4-RD. Coronal reformatted image from a Chest CT scan at the level of the thoracic spine on soft tissue windows. There are bilateral paravertebral masses, seen on the right at T8 and T10/11 and on the left at T10/11 (arrows). Biopsy confirmed IgG4-RD.
Airway Involvement:
Airway involvement is seen in up to half of the IgG4-RD patients with abnormalities on chest CT and is associated with pulmonary symptoms including cough and dyspnea.51 Airway changes are characterized most frequently by smooth or nodular thickening of the airway wall (Figure 4). Small airway obstruction and subsequent air trapping related to IgG4-RD can cause mosaic attenuation of the lung parenchyma on expiratory images. Saber sheath trachea, an abnormality in which the intrathoracic trachea becomes elongated in the sagittal dimension, can also be present in IgG4-RD (Figure 5). This remodeling has been described in patients with other forms of chronic airflow obstruction and may be due to chronically raised intrathoracic pressure from airway wall thickening.
Figure 4.
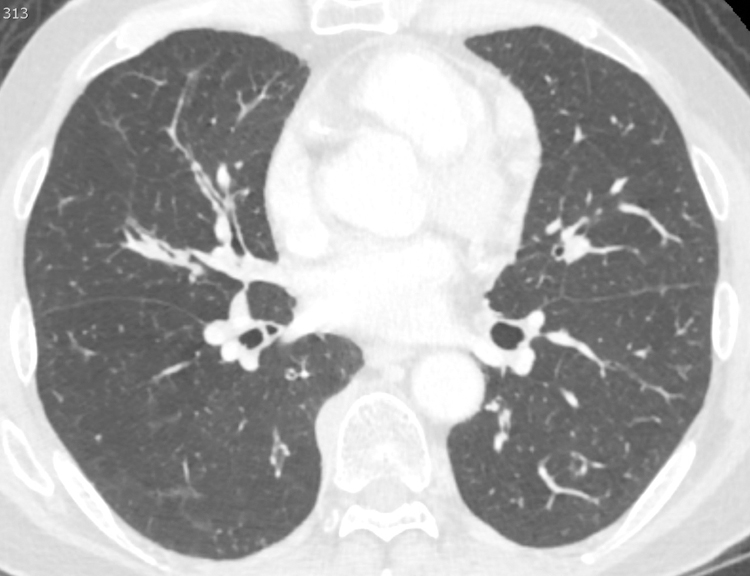
Bronchial wall thickening in 61 year-old-male with cough and IgG4-RD. Axial Chest CT scan on lung windows demonstrates diffuse, multifocal nodular thickening of the bronchial walls, best seen in the right middle lobe (arrow).
Figure 5.
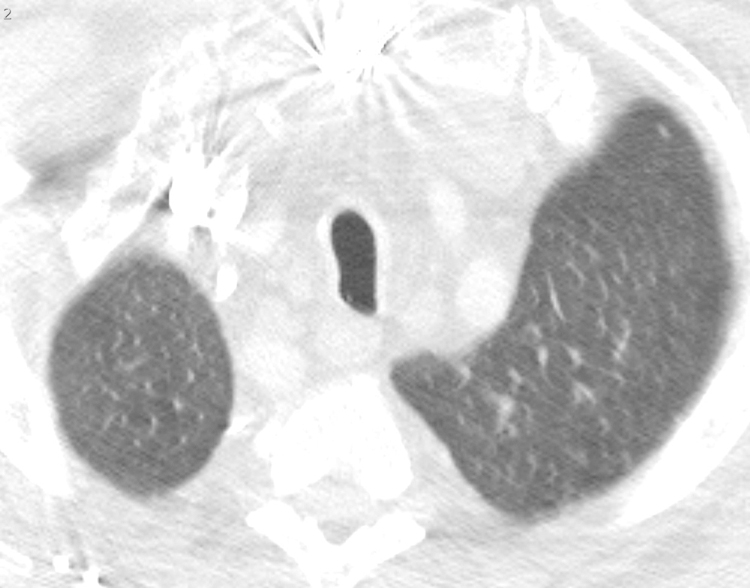
Saber sheath trachea in a 65-year-old male with IgG4-RD. There is elongation in the sagittal plane of the trachea relative to the coronal plane.
Pleuro-Pulmonary Manifestations:
A variety of pulmonary parenchymal findings can be observed in IgG4-RD. These abnormalities are typically peripheral in location, often involving the subpleural lung. Consolidation, ground glass opacities, pulmonary nodules and/or septal thickening are the most frequent intrapulmonary manifestations (Figures 6 and 7). Fibrosis has been observed but is less common; when present, it may rarely be associated with honeycombing and traction bronchiolectasis.
Figure 6.
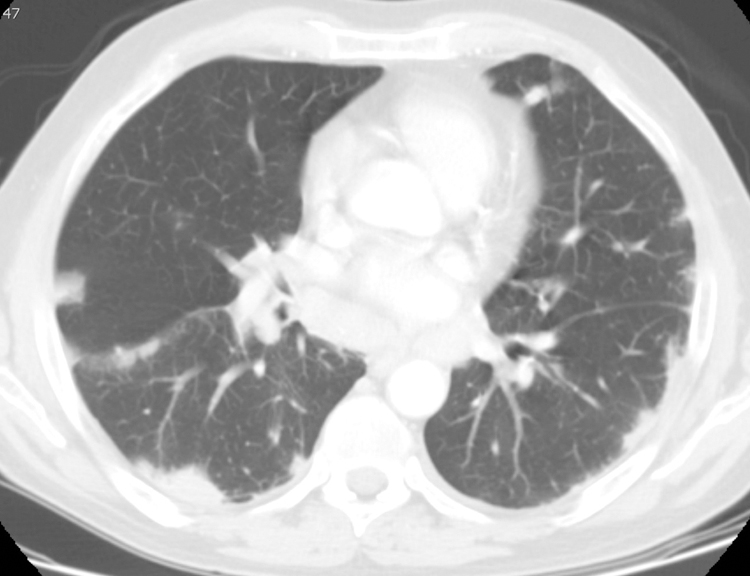
Peripheral nodules and consolidation in 69-year-old male with IgG4-RD. Chest CT axial scan at the level of the aortic root on lung windows. There are multiple, bilateral peripheral lung nodules and consolidative opacities, that also extend along the right major and minor fissures, in a perilymphatic distribution that is characteristic of IgG4-RD. The patient denied pulmonary symptoms.
Figure 7.
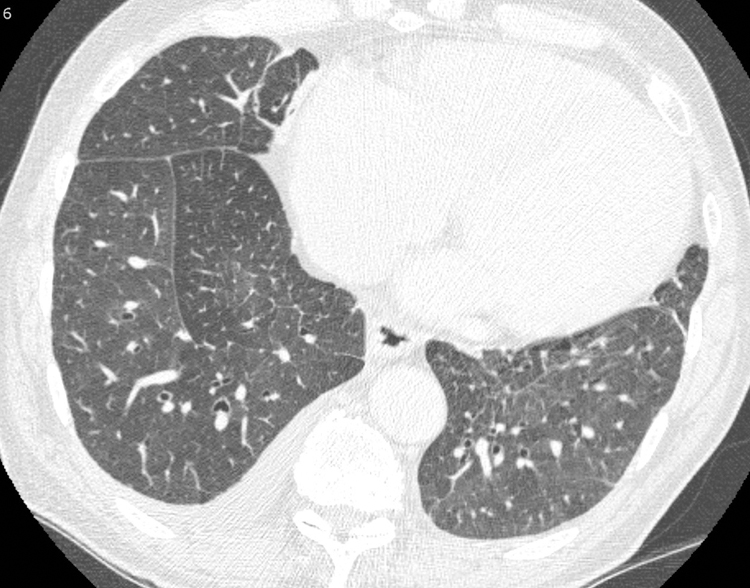
Septal thickening and right pleural effusion in a 65-year-old male with IgG4-RD. Chest CT axial scan at the level of interventricular septum on lung windows demonstrates smooth interlobular septal thickening (arrow) and a small right pleural effusion. The appearances mimic pulmonary edema. Echocardiogram showed normal cardiac function.
Pleural thickening and effusions may also be seen in IgG4-RD (Figure 7). The combination of pulmonary and pleural findings differentiates the disease from sarcoidosis or organizing pneumonia, both of which also cause subpleural, peripheral and peribronchiolar nodules and opacities but rarely involve the pleura.
Heart and Great Vessel Involvement:
Chest CT also allows assessment of the heart and great vessels in IgG4-RD.52 Cardiac features include coronary artery pseudotumors and aneurysms as well as pericardial thickening (Figure 8). Periaortitis due to IgG4-RD typically manifests as a soft-tissue formation around the vessel with or without vessel wall thickening and enhancement53; this can be found affecting the aorta, pulmonary artery and/or great vessels (Figure 1).
Figure 8.
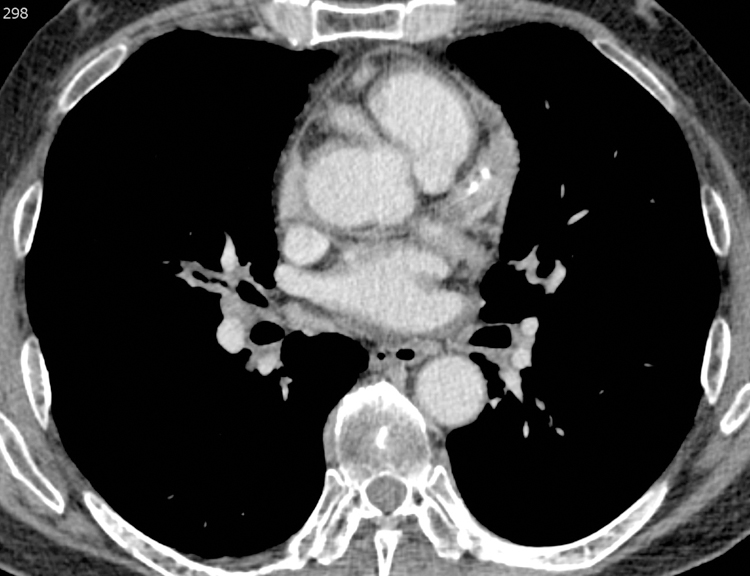
Pseudotumors of the coronary arteries in a 61-year-old male with IgG4-RD. Chest CT axial scan at the level of the coronary arteries on mediastinal windows. There is soft tissue surrounding the right coronary artery, left anterior descending and circumflex arteries (arrows).
Features Suggestive of Alternative Diagnoses:
Certain manifestations of disease in the chest are uncommon in IgG4-RD. These include diffuse alveolar hemorrhage, large pleural or pericardial effusions, pulmonary hypertension, and airway stenosis or collapse. When present, clinicians should consider other explanations more commonly associated with these findings such as vasculitis, systemic lupus erythematosus, systemic sclerosis, or relapsing polychondritis.
Diagnosing IgG4-RD:
Overview:
There is no single diagnostic test for IgG4-RD. The diagnosis of IgG4-RD requires a careful consideration of all available data, including the history, physical examination, laboratory results, imaging findings, and pathology.
Serum IgG4 concentration elevations are neither sensitive nor specific for the diagnosis54–56, though they can certainly support the diagnosis in the proper clinical setting. Moreover, while IgG4+ plasma cell infiltrates in the tissue are the sine qua non of the condition, these too are not specific for IgG4-RD and may be encountered in other conditions, including ANCA-associated vasculitis and malignancy.57–59 Diagnostic criteria have previously been proposed by different groups around the world, some for specific organ manifestations and others for any manifestation of IgG4-RD.60–62 These criteria often place excessive weight on histopathologic findings and serum IgG4 concentration elevations, approaches that likely maximize specificity at the expense of sensitivity. Although they can help inform the approach to diagnosis, we do not strictly use these criteria in the day-to-day clinical care of patients with suspected IgG4-RD.
Considering Other Organ Involvement:
The first step in diagnosing IgG4-RD is considering the full extent of potential organ involvement. When referred a patient because of abnormal findings in the lung, for instance, it is important to consider whether there is involvement of organs that may reveal key physical examination findings. The eyes and major salivary glands must be examined carefully. Lacrimal gland enlargement, readily detected on examination (Figure 9A and 9B), may heighten the diagnostic suspicion for IgG4-RD but can also be found in IgG4-RD mimickers (e.g., sarcoidosis, granulomatosis with polyangiitis, and Sjögren’s syndrome). Proptosis may be caused by IgG4-RD infiltrating and enlarging extra-ocular muscles. Isolated submandibular gland enlargement (Figure 9C) can be a major clue to IgG4-RD, but the parotid (Figure 9D) and sublingual glands may also be strikingly enlarged.
Figure 9A.
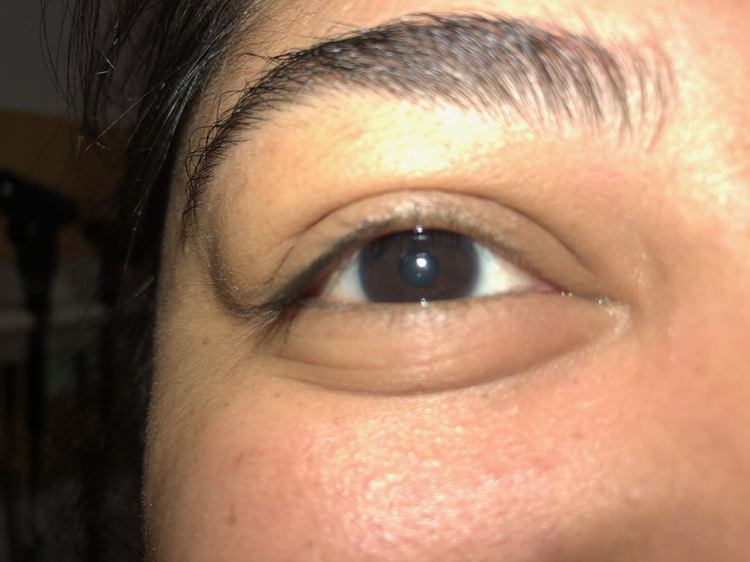
Right lacrimal gland enlargement evidenced by a bulge over the patient’s upper lateral right eye.
Figure 9B.
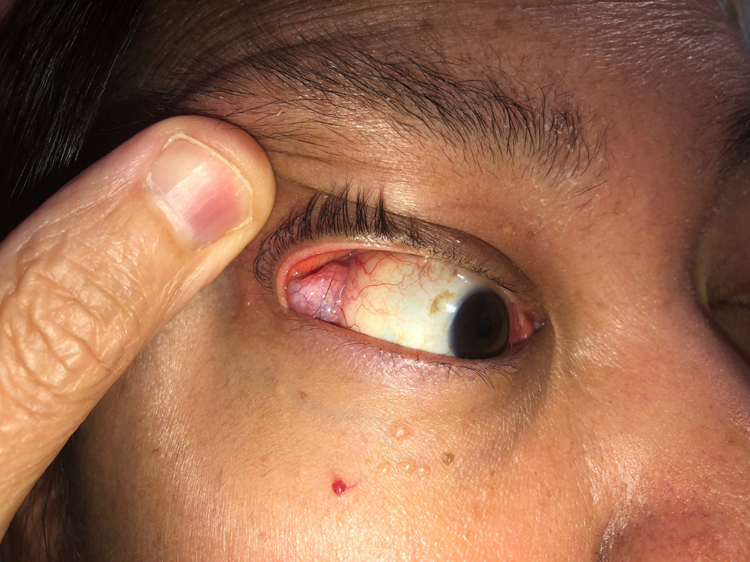
Right lacrimal gland enlargement revealed by simple retraction of the eyelid.
Figure 9C.
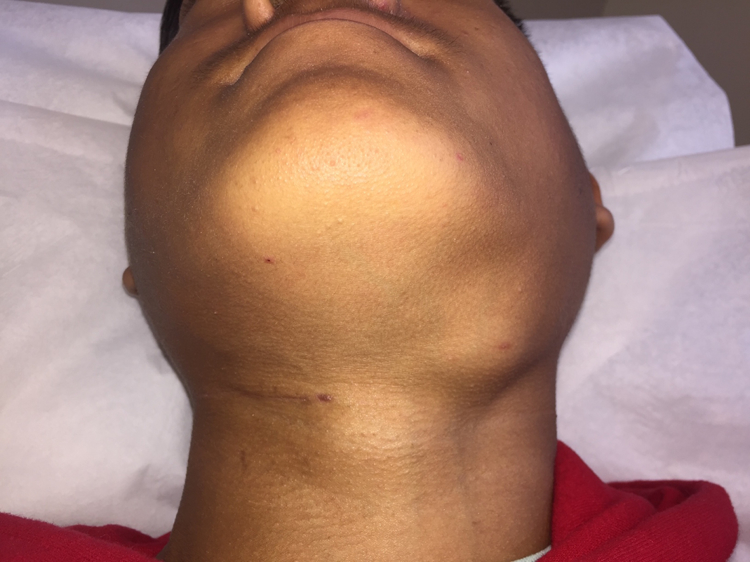
Left submandibular gland enlargement in a 16-year-old boy with IgG4-related disease affecting the major salivary glands, lungs, kidneys, pancreas, and biliary tree. Note that the right submandibular gland has been resected - removed for diagnostic purposes.
Figure 9D.
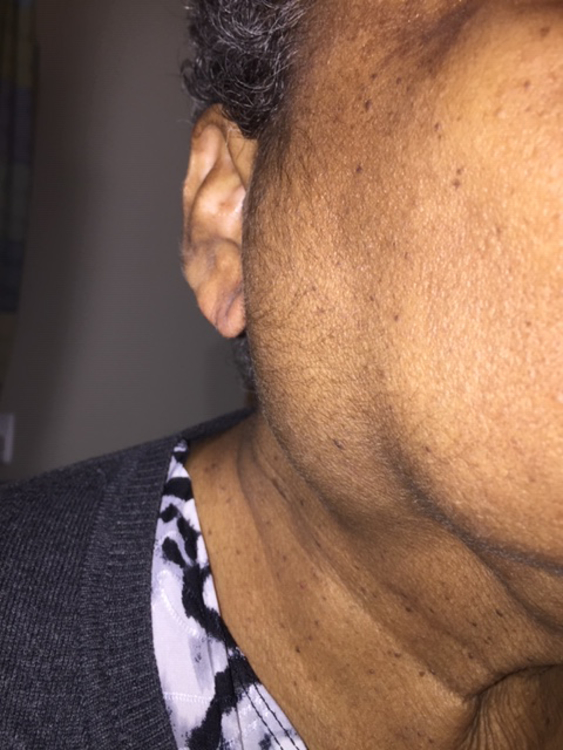
Right parotid enlargement in a 75-year-old woman with IgG4-related disease affecting the major salivary glands, kidneys (interstitial nephritis), and pleura.
Moreover, a careful history-taking might reveal symptoms suggestive of organ involvement elsewhere. For instance, new-onset diabetes or loose and pale colored stool may suggest pancreatic disease. New lower back and/or groin pain may be suggestive of retroperitoneal fibrosis. The patient’s medical history can also be useful when it suggests previous complications (e.g., autoimmune pancreatitis, tubulointerstitial nephritis) likely related to what was unrecognized IgG4-RD at the time.
Cross-sectional imaging of the chest and the abdomen/pelvis can reveal asymptomatic disease in a distribution that supports a diagnosis of IgG4-RD. In the case of suspected pancreato-hepatobiliary disease, magnetic resonance cholangiopancreatography (MRCP) may be useful for diagnosing IgG4-RD and ruling out alternative etiologies. Patients with significantly elevated serum IgG4 concentrations (e.g., > 3x the upper limit of normal) are more likely to have multi-organ disease and cross-sectional imaging may be higher yield in these patients.3,39
Considering Alternative Diagnoses:
The second step is to consider the common mimickers of IgG4-RD and whether any of those conditions might better explain the patient’s presentation or if additional workup is indicated to exonerate those conditions (Table 2). In the chest, the differential includes sarcoidosis, ANCA-associated vasculitis, Sjögren’s syndrome63, lymphoma and primary/metastatic lung cancer, Erdheim-Chester Disease64, infections (including bacterial infections as well as atypical mycobacterial or fungal infections), and myofibroblastic tumors65. To exonerate these conditions, a new biopsy, a review of pathological tissue available from earlier procedures, or additional laboratory testing can be helpful (Table 2).
Table 2:
Common Mimickers of IgG4-Related Disease in the Chest
| Disease | Differentiating factors |
|---|---|
| Sjogren’s Syndrome | • Parotid gland involvement more common • Positive Anti-SSA (Ro) and/or -SSB (La) exclude IgG4-RD |
| Sarcoidosis | • Cutaneous disease more common • ACE may be elevated, not expected in IgG4-RD • Splenomegaly can be seen but would be atypical in IgG4-RD • Granulomas exclude IgG4-RD |
| ANCA-associated vasculitis (AAV, e.g. granulomatosis with polyangiitis, microscopic polyangiitis, eosinophilic granulomatosis with polyangiitis (EGPA)) | • Fever and very high CRP may be present and atypical for IgG4-RD • A positive MPO- or PR3-ANCA generally excludes IgG4-RD; only present in ~50% of EGPA cases • High-grade eosinophilia >3,000/mm3, as seen in EGPA, generally excludes IgG4-RD • Necrotizing vasculitis and/or granulomas exclude IgG4-RD |
| Giant Cell Arteritis | • Fever and very high CRP may be present and atypical in IgG4-RD • Cranial symptoms (headache, scalp tenderness, jaw claudication, vision change) not typical in IgG4-RD |
| Lymphoma | • Fever may be present • Malignant pathology excludes IgG4-RD • Rapid progression across tissue planes not seen in IgG4-RD |
| Infection | • Fevers and very high CRP common, atypical in IgG4-RD • Rapid progression across tissue planes not seen in IgG4-RD |
| Multicentric Castleman’s Disease (MCD) | • Fever and very high CRP often present and atypical in IgG4-RD • MCD pathology is distinct from IgG4-RD though both may have IgG4+ plasma cells infiltrating tissue |
| Erdheim-Chester Disease | • Classic long-bone abnormalities (e.g., sclerosis) • Distinct pathology which includes foamy histiocytes excludes IgG4-RD • May have a BRAF mutation detectable in tissue and/or circulating blood |
| Inflammatory Myofibroblastic Tumor | • Much more common in pediatric patients than adult patients • Distinct pathology reveals spindle cells • ALK (anaplastic lymphoma kinase), ROS-1, and other gene rearrangements detectable in ~50% of cases |
Biopsy Specimens to Diagnose IgG4-RD:
In many cases, a biopsy of an affected lesion can help support a diagnosis of IgG4-RD and/or rule out alternative etiologies. In fact, histopathologic confirmation was considered essential to diagnosing IgG4-RD in the years following its initial recognition. However, the full panoply of histopathologic findings associated with IgG4-RD may be difficult to detect in more limited biopsy specimens that are often preferred now because they are less invasive and can help exonerate infection and malignancy. Review of an archived pathology sample from a biopsy of another affected site can be diagnostic, even if the sample is many years old. Fresh re-cuts of paraffin-embedded specimens can be stained for IgG4+ plasma cells even if immunostains were not performed originally. Lymphadenopathy is the most common manifestation of IgG4-RD in the chest but it is important to note that histology of involved lymph nodes tends to show non-specific hyperplastic or reactive histologic changes rather than the typical features of IgG4-RD66–68; fibrosis may be variably encountered in lymph node biopsies. Caution should be used when diagnosing IgG4-RD using only lymph node biopsies.
In 2012, an international group of experts published a consensus statement on the pathology of IgG4-RD.69 Intended to act as diagnostic guidelines for practicing pathologists, this statement provides clear emphasis on two aspects of the pathology findings in IgG4-RD: a characteristic histologic appearance and a prominent infiltrate of IgG4-expressing plasma cells identified using immunostaining.
The characteristic histologic features include a dense lymphoplasmacytic infiltrate, fibrosis (often in a storiform pattern), and phlebitis (obliterative or non-obliterative) (Figure 10). The collection of at least 2 of these features has classically formed the diagnostic foundation of IgG4-RD, but any single feature in isolation lacks specificity. Tumefactive pulmonary lesions often extend along the bronchovascular tree (Figure 10A) and can be subpleural (Figure 10B). The polyclonal lymphoplasmacytic infiltrate is diffusely distributed with lymphocytes, especially T lymphocytes, dominating over plasma cells. CD20+ B cells are often present in extra-nodal germinal centers. A modest eosinophilic tissue infiltration can be seen but IgG4-RD is not considered to be a hypereosinophilic disorder. Consequently, a dominant eosinophilic infiltrate suggests an alternative etiology. Macrophages may be a prominent feature, as in any inflammatory lesion, but discrete giant cells, granulomas and predominant granulomatous inflammation should not be present in biopsies of IgG4-RD. Similarly, monoclonal cell populations, necrosis, and prominent histiocytic infiltrates are atypical of IgG4-RD.69
Figure 10.
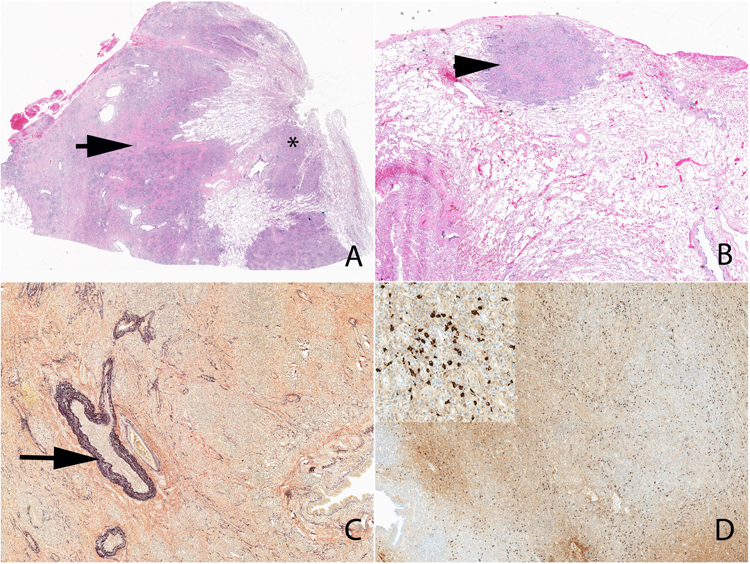
Low power view of IgG4-related pulmonary disease. Note the tumefactive lesion (arrow) (A), extension along the bronchovascular tree (A) (*) and subpleural involvement (B) (arrowhead). An elastic stain highlights the focus of obliterative phlebitis (C) (arrow). An immunohistochemical stain shows a diffuse increase in IgG4+ plasma cells (D and inset).
Fibrosis is a core feature of IgG4-RD lesions and, at least focally, should have a storiform or spiraling pattern (Figure 10A). In cases of limited tissue sampling such as with retroperitoneal fibrosis, the fibrotic component may predominate. Phlebitis (obliterative or non-obliterative) represents destruction of the venous wall and filling of the lumen by infiltrating immune cells (Figure 10C). Pulmonary lesions related to IgG4-RD are unique in that pathology can reveal obliterative arteritis as well as obliterative phlebitis – neither of which should be confused with necrotizing vasculitis. As in the vein, obliterative arteritis is characterized by a non-necrotizing lymphoplasmacytic infiltrate with or without obliteration of the lumen. Elastin stains are often required to visualize obliterative phlebitis or arteritis because of the extensive vessel wall destruction.
Immunostaining for both IgG4+ and IgG+ plasma cells is essential to confirm a suspected histopathologic diagnosis (Figure 10D). Interpretation of IgG4 immunostaining relies on both the quantification of IgG4+ plasma cells per high power field and the ratio of IgG4+/IgG+ plasma cells. The diagnostic cutoff for the former is organ-specific while a ratio greater than or equal to 40% is a general rule of thumb used to confirm the diagnosis. However, ratios of less than 40% are often encountered in cases of IgG4-RD, as much depends on adequate sampling and its converse: sampling error. Nevertheless, a true lack of IgG4+ plasma cells or a low proportion of IgG4+ plasma cells infiltrating tissue suggests that one consider an alternative diagnosis. It is also critical to recognize that many important mimickers of IgG4-RD can be associated with a prominent IgG4+ plasma cell infiltrate.57–59 Major examples include primary sclerosing cholangitis, ANCA-associated vasculitis, multicentric Castleman’s disease, B-cell lymphomas, and pancreatic cancer.
Although histopathology and immunostaining can be useful in suggesting or confirming an IgG4-RD diagnosis, clinicopathological correlation remains critical in making this diagnosis. This is important to keep in mind because the pathology of IgG4-RD in the lung may lack the typical storiform fibrosis and/or obliterative phlebitis often seen in biopsies from other organs affected by IgG4-RD.40,69
Laboratory Testing in IgG4-RD:
Certain laboratory tests can be useful to screen for specific organ involvement by IgG4-RD (Table 3). Elevations in the erythrocyte sedimentation rate (ESR) are common because of the hypergammaglobulinemia that typically accompanies untreated disease. ESR measurements over 100 mm/hr, however, are unusual. C-reactive protein (CRP) elevations can be observed in IgG4-RD but these are less common than ESR elevations. There is often an acute phase reactant discordance characterized by a high ESR and low CRP.
Table 3:
Useful Lab Tests When Evaluating a Patient for IgG4-Related Disease
| Lab Test | Reason for Test |
|---|---|
| General | |
| IgG Subclasses | IgG4 elevated in ~70% of patients with IgG4-RD |
| IgE and Peripheral Eosinophilia | May be elevated in patients with IgG4-RD; may predict risk of flare after treatment |
| Renal Disease | |
| CH50, C3, and C4 | Often low in IgG4-related renal disease |
| Creatinine and Glomerular Filtration Rate | To evaluate for potential IgG4-related renal disease |
| Urinalysis | Proteinuria may be present in IgG4-related renal disease |
| Pancreatic Disease | |
| Amylase and Lipase | To evaluate for potential IgG4-related pancreatitis |
| Bilirubin | To evaluate for potential IgG4-related pancreatitis |
| A1c | To evaluate for potential IgG4-related pancreatitis causing endocrine insufficiency |
| Stool elastase | To evaluate for potential IgG4-related pancreatitis causing exocrine insufficiency |
Other laboratory findings that make IgG4-RD less likely include pancytopenia (especially involving multiple lines) and the presence of disease-specific auto-antibodies. While patients can be screened for many possible disease-specific auto-antibodies (Table 2), these are not meant to be checked in all patients in whom the diagnosis of IgG4-RD is being considered; rather, they should be checked in patients with presentations that are suggestive of those specific diseases (e.g., lupus, ANCA-associated vasculitis, Sjögren’s syndrome). Considering these diagnoses and interpreting lab results may require consultation with other specialists such as a rheumatologist.
Perhaps the most commonly used laboratory test to evaluate for IgG4-RD is the serum IgG4 concentration. Though IgG4-RD was initially described in a cohort of patients with autoimmune pancreatitis and uniform elevations in serum IgG4 concentrations, it is now recognized that approximately 30% of patients with biopsy-proven IgG4-RD actually have normal serum IgG4 concentrations.4 Moreover, elevated serum IgG4 concentrations have been described in other conditions, including patients with interstitial lung disease, ANCA-associated vasculitis, and allergic conditions undergoing immunotherapy.57–59 In sum, serum IgG4 concentrations have an approximate specificity for IgG4-RD of 83%.70
In the past, some patients with IgG4-RD and multi-organ disease were found to have surprisingly normal serum IgG4 concentrations that did not fit clinically. Ultimately, it was discovered that in patients with very high serum IgG4 concentrations, a prozone effect caused falsely low measurements because of excess antigen that overwhelmed the assays.71 To overcome this limitation, most commercial labs now use serial dilution of samples to avoid the prozone effect, but providers should be aware of this phenomenon when interpreting IgG4 concentrations.
Other laboratory anomalies commonly observed in IgG4 are elevations of other IgG subclasses as well as elevations of IgE.20 Hypocomplementemia is often observed in patients with IgG4-related tubulointerstitial nephritis but is sometimes detected in patients without overt renal disease. The pattern of hypocomplementemia in IgG4-RD frequently calls to mind the hypocomplementemia observed in SLE or mixed cryoglobulinemia; namely, a reduced C3 and an extremely low or even unmeasureable C4.
Pulmonary Function Tests:
There is scant literature describing the frequencies of pulmonary function test (PFT) abnormalities in patients with IgG4-RD.50,72 We suspect that findings on PFTs would lack sensitivity and specificity to distinguish IgG4-RD from other conditions with thoracic involvement. PFTs may have a role in monitoring disease progression in certain patients but this requires further study.
Initial Management:
Nearly all patients with IgG4-RD should be treated with immunosuppression to prevent accrual of additional organ involvement and damage in affected organs.73,74 Exceptions to this include those in whom lesions have been fully resected or in those with asymptomatic and mild lymphadenopathy or salivary gland enlargement.73,74 In those cases, “watchful waiting” may be considered. Glucocorticoids are the most frequently utilized first-line treatment but other strategies that incorporate glucocorticoid-sparing agents are often used. The goal of initial management is to achieve a complete remission. It is important to note that in those with elevated pre-treatment serum IgG4 concentration, there is typically a decrease in the lab measurement following treatment, but levels may not normalize even in those achieving clinical remission.18,20
Glucocorticoids
Glucocorticoids have been the mainstay of treatment for IgG4-RD because of their efficacy as well as affordability. Failure to respond to glucocorticoid therapy suggests that the diagnosis is incorrect. In rare circumstances, a highly fibrotic lesion (e.g., orbital lesion, retroperitoneal fibrosis) may not respond to glucocorticoid therapy but this should be interpreted cautiously.
A single arm clinical trial75 found a response rate of greater than 93% to glucocorticoids and a complete remission rate of 66%, consistent with our general experience using glucocorticoids. It should be noted, however, that patients in that study were maintained on significant doses of prednisone (≥7.5mg/day) throughout the one-year follow-up period.
Glucocorticoids are typically used first-line with equivalent prednisone doses ranging from 20mg to 60mg/day depending on the severity of the presentation. The general practice is to begin a slow taper of glucocorticoids after two to four weeks of therapy with the goal of discontinuation by three months, sometimes sooner. During this time, the patient should be monitored for flares of disease which are common, especially at lower doses and following discontinuation.76,77 In the previously mentioned single-arm clinical trial, 15% of patients flared during follow-up.
The risks of glucocorticoid therapy must be weighed against these benefits, especially in light of the demographics of IgG4-RD. This is a group of patients at increased risk for glucocorticoid-related toxicity (e.g., diabetes mellitus, osteoporosis, infection) because of their age and comorbidities, especially those with pancreatic insufficiency. For instance, in the clinical trial of glucocorticoid therapy in IgG4-RD, over 40% of patients experienced glucose intolerance and nearly half of these patients were newly diagnosed with diabetes mellitus. Because of the high incidence of glucocorticoid complications in this patient population, it is usually our practice to move quickly to treatment regimens that minimize glucocorticoids for long-term disease control.
Conventional Disease Modifying Anti-Rheumatic Drugs (DMARDs)
Much of our understanding of the efficacy of conventional DMARDs (e.g., methotrexate, azathioprine, mycophenolate mofetil) in IgG4-RD is extrapolated from the autoimmune pancreatitis literature.21 Results from these uncontrolled observational studies have not suggested good efficacy of conventional DMARDs such as azathioprine for maintaining disease remission better than glucocorticoid monotherapy.73 However, a recent open-label randomized clinical trial of IgG4-RD patients with diverse manifestations suggests that mycophenolate mofetil added to glucocorticoids may be efficacious, especially for maintaining remission when compared to maintenance therapy with glucocorticoid monotherapy.78 Future studies are necessary to clarify the potential role of conventional DMARDs in IgG4-RD.
Rituximab
A small pilot clinical trial18 as well as several cases series suggest that rituximab is effective for the treatment of IgG4-RD.21–23 A unique benefit of rituximab in IgG4-RD is that it may be effective when used as monotherapy. When used, the dosing regimen is typically 1,000mg administered twice over two weeks. A response may be observed over the first eight weeks following treatment but the maximum benefit from rituximab may not be observed for approximately twelve weeks after treatment. If necessary, treatment may be repeated approximately six months after the initial doses but many patients do not require retreatment at that time and can be monitored prospectively for evidence of a flare.20,22 Retreatment at six months, however, may reduce the risk of subsequent flare.23 One benefit of prospectively monitoring without preemptive retreatment at six month is a reduced risk of infection.23
General Approach
Most often, therapy is initiated with glucocorticoids and the decision of whether to combine glucocorticoids with an additional steroid-sparing immunosuppressant is guided by the severity of the presentation as well as the patient’s comorbidities.73 As discussed, some experts prefer to treat without glucocorticoids which may be reasonable if the patient’s presentation is not organ-(e.g., renal failure, aortitis, cholangitis) or life-threatening. When organ- or life-threatening disease is present, upfront glucocorticoids can be important for obtaining quick control of the disease. Regardless of the initial treatment strategy in patients with multi-organ disease, the various manifestations respond similarly to therapy but some reports suggest that the response of thoracic IgG4-RD to treatment may not be synchronous with response elsewhere in the body.2,40,79 Additional prospective studies are necessary to define optimal treatment strategies and to better characterize whether certain manifestations respond differently to treatment.
Summary:
IgG4-RD is an immune-mediated disease that causes fibro-inflammatory lesions in nearly any organ, including structures in the chest. An accurate diagnosis requires careful clinicopathologic correlation. Neither elevated serum IgG4 concentrations nor IgG4+ plasma cell infiltrates are diagnostic of IgG4-RD. Mimickers of IgG4-RD in the chest include malignancy, infection, and other immune-mediated diseases such as vasculitis and sarcoidosis. Glucocorticoids and/or steroid-sparing agents are the cornerstone of treatment.
Key Points:
Thoracic manifestations of IgG4-related disease (IgG4-RD) include lung nodules, pleural thickening, aortitis, and lymphadenopathy.
Thoracic manifestations of IgG4-RD are often detected incidentally on imaging but can present with nonspecific symptoms such as dyspnea or cough.
Biopsies of pulmonary lesions can distinguish IgG4-RD from common mimickers, including sarcoidosis, ANCA-associated vasculitis, and malignancy.
Synopsis:
IgG4-related disease (IgG4-RD) can cause fibroinflammatory lesions in nearly any organ and lead to organ dysfunction and irreversible damage. In addition to frequent involvement of the salivary glands, lacrimal glands, and/or pancreas, IgG4-RD often affects the chest. Thoracic manifestations include lung nodules and consolidations, pleural thickening, aortitis, and lymphadenopathy. The diagnosis is made after careful clinicopathologic correlation because there is no single diagnostic test with excellent sensitivity or specificity, even serum IgG4 concentrations. Biopsy of pulmonary lesions can be useful for distinguishing IgG4-RD from common mimickers such as ANCA-associated vasculitis and sarcoidosis. Immunosuppressive regimens, such as glucocorticoids and/or glucocorticoid-sparing agents like rituximab, form the cornerstone of treatment.
Acknowledgments
Disclosure Statement: Dr. Wallace received grant support through a Scientist Development Award from the Rheumatology Research Foundation and from the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS/NIH; Loan Repayment Award and K23 AR073334).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Commercial or Financial Conflicts of Interest: None
References
- 1.Kamisawa T, Zen Y, Pillai S, Stone JH. IgG4-related disease. Lancet 2015;385(9976):1460–1471. [DOI] [PubMed] [Google Scholar]
- 2.Fei Y, Shi J, Lin W, et al. Intrathoracic Involvements of Immunoglobulin G4-Related Sclerosing Disease. Medicine (Baltimore) 2015;94(50):e2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wallace ZS, Zhang Y, Perugino CA, et al. Clinical phenotypes of IgG4-related disease: an analysis of two international cross-sectional cohorts. Ann Rheum Dis 2019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wallace ZS, Deshpande V, Mattoo H, et al. IgG4-Related Disease: Clinical and Laboratory Features in One Hundred Twenty-Five Patients. Arthritis Rheumatol 2015;67(9):2466–2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Inoue D, Yoshida K, Yoneda N, et al. IgG4-Related Disease: Dataset of 235 Consecutive Patients. Medicine (Baltimore) 2015;94(15):e680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huggett MT, Culver EL, Kumar M, et al. Type 1 autoimmune pancreatitis and IgG4-related sclerosing cholangitis is associated with extrapancreatic organ failure, malignancy, and mortality in a prospective UK cohort. Am J Gastroenterol 2014;109(10):1675–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin W, Lu S, Chen H, et al. Clinical characteristics of immunoglobulin G4-related disease: a prospective study of 118 Chinese patients. Rheumatology (Oxford) 2015;54(11):1982–1990. [DOI] [PubMed] [Google Scholar]
- 8.Goldoni M, Bonini S, Urban ML, et al. Asbestos and smoking as risk factors for idiopathic retroperitoneal fibrosis: a case-control study. Ann Intern Med 2014;161(3):181–188. [DOI] [PubMed] [Google Scholar]
- 9.Uibu T, Oksa P, Auvinen A, et al. Asbestos exposure as a risk factor for retroperitoneal fibrosis. Lancet 2004;363(9419):1422–1426. [DOI] [PubMed] [Google Scholar]
- 10.Wallwork R, Choi HK, Perugino CA, Zhang Y, Stone JH, Wallace Z. Cigarette Smoking Is a Risk Factor for IgG4-Related Disease [abstract]. Arthritis Rheumatol 2018;70(suppl 10). [Google Scholar]
- 11.Wallace ZS, Wallace CJ, Lu N, Choi HK, Stone JH. Association of IgG4-Related Disease With History of Malignancy. Arthritis Rheumatol 2016;68(9):2283–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Buy Wenniger LJ, Culver EL, Beuers U. Exposure to occupational antigens might predispose to IgG4-related disease. Hepatology 2014;60(4):1453–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kawa S, Ota M, Yoshizawa K, et al. HLA DRB10405-DQB10401 haplotype is associated with autoimmune pancreatitis in the Japanese population. Gastroenterology 2002;122(5):1264–1269. [DOI] [PubMed] [Google Scholar]
- 14.Brandt AS, Kamper L, Kukuk S, Haage P, Roth S. Associated findings and complications of retroperitoneal fibrosis in 204 patients: results of a urological registry. J Urol 2011;185(2):526–531. [DOI] [PubMed] [Google Scholar]
- 15.Mattoo H, Mahajan VS, Della-Torre E, et al. De novo oligoclonal expansions of circulating plasmablasts in active and relapsing IgG-related disease. J Allergy Clin Immunol 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wallace ZS, Mattoo H, Carruthers M, et al. Plasmablasts as a biomarker for IgG4-related disease, independent of serum IgG4 concentrations. Ann Rheum Dis 2015;74(1):190–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin W, Zhang P, Chen H, et al. Circulating plasmablasts/plasma cells: a potential biomarker for IgG4-related disease. Arthritis Res Ther 2017;19(1):25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carruthers MN, Topazian MD, Khosroshahi A, et al. Rituximab for IgG4-related disease: a prospective, open-label trial. Ann Rheum Dis 2015;74(6):1171–1177. [DOI] [PubMed] [Google Scholar]
- 19.Khosroshahi A, Carruthers MN, Deshpande V, Unizony S, Bloch DB, Stone JH. Rituximab for the treatment of IgG4-related disease: lessons from 10 consecutive patients. Medicine (Baltimore) 2012;91(1):57–66. [DOI] [PubMed] [Google Scholar]
- 20.Wallace ZS, Mattoo H, Mahajan VS, et al. Predictors of disease relapse in IgG4-related disease following rituximab. Rheumatology (Oxford) 2016;55(6):1000–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hart PA, Topazian MD, Witzig TE, et al. Treatment of relapsing autoimmune pancreatitis with immunomodulators and rituximab: the Mayo Clinic experience. Gut 2013;62(11):1607–1615. [DOI] [PubMed] [Google Scholar]
- 22.Ebbo M, Grados A, Samson M, et al. Long-term efficacy and safety of rituximab in IgG4-related disease: Data from a French nationwide study of thirty-three patients. PLoS One 2017;12(9):e0183844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Majumder S, Mohapatra S, Lennon RJ, et al. Rituximab Maintenance Therapy Reduces Rate of Relapse of Pancreaticobiliary Immunoglobulin G4-related Disease. Clin Gastroenterol Hepatol 2018;16(12):1947–1953. [DOI] [PubMed] [Google Scholar]
- 24.Du H, Shi L, Chen P, et al. Prohibitin Is Involved in Patients with IgG4 Related Disease. PLoS One 2015;10(5):e0125331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perugino CA, AlSalem SB, Mattoo H, et al. Identification of galectin-3 as an autoantigen in patients with IgG4-related disease. J Allergy Clin Immunol 2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shiokawa M, Kodama Y, Sekiguchi H, et al. Laminin 511 is a target antigen in autoimmune pancreatitis. Sci Transl Med 2018;10(453). [DOI] [PubMed] [Google Scholar]
- 27.Akiyama M, Yasuoka H, Yamaoka K, et al. Enhanced IgG4 production by follicular helper 2 T cells and the involvement of follicular helper 1 T cells in the pathogenesis of IgG4-related disease. Arthritis Res Ther 2016;18:167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nirula A, Glaser SM, Kalled SL, Taylor FR. What is IgG4? A review of the biology of a unique immunoglobulin subtype. Curr Opin Rheumatol 2011;23(1):119–124. [DOI] [PubMed] [Google Scholar]
- 29.Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol 2014;5:520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sugimoto M, Watanabe H, Asano T, et al. Possible participation of IgG4 in the activation of complement in IgG4-related disease with hypocomplementemia. Mod Rheumatol 2016;26(2):251–258. [DOI] [PubMed] [Google Scholar]
- 31.Culver EL, van de Bovenkamp FS, Derksen NI, et al. Unique patterns of glycosylation in immunoglobulin subclass G4-related disease and primary sclerosing cholangitis. J Gastroenterol Hepatol 2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shiokawa M, Kodama Y, Kuriyama K, et al. Pathogenicity of IgG in patients with IgG4-related disease. Gut 2016;65(8):1322–1332. [DOI] [PubMed] [Google Scholar]
- 33.Shiokawa M, Kodama Y, Yoshimura K, et al. Risk of cancer in patients with autoimmune pancreatitis. Am J Gastroenterol 2013;108(4):610–617. [DOI] [PubMed] [Google Scholar]
- 34.Mahajan VS, Mattoo H, Deshpande V, Pillai SS, Stone JH. IgG4-Related Disease. Annu Rev Pathol 2014;9:315–347. [DOI] [PubMed] [Google Scholar]
- 35.Mattoo H, Mahajan VS, Maehara T, et al. Clonal expansion of CD4(+) cytotoxic T lymphocytes in patients with IgG4-related disease. J Allergy Clin Immunol 2016;138(3):825–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maehara T, Mattoo H, Ohta M, et al. Lesional CD4+ IFN-gamma+ cytotoxic T lymphocytes in IgG4-related dacryoadenitis and sialoadenitis. Ann Rheum Dis 2017;76(2):377–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med 2012;366(6):539–551. [DOI] [PubMed] [Google Scholar]
- 38.Sun X, Liu H, Feng R, et al. Biopsy-proven IgG4-related lung disease. BMC Pulm Med 2016;16:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Corcoran JP, Culver EL, Anstey RM, et al. Thoracic involvement in IgG4-related disease in a UK-based patient cohort. Respir Med 2017;132:117–121. [DOI] [PubMed] [Google Scholar]
- 40.Zen Y, Inoue D, Kitao A, et al. IgG4-related lung and pleural disease: a clinicopathologic study of 21 cases. Am J Surg Pathol 2009;33(12):1886–1893. [DOI] [PubMed] [Google Scholar]
- 41.Matsui S, Hebisawa A, Sakai F, et al. Immunoglobulin G4-related lung disease: clinicoradiological and pathological features. Respirology 2013;18(3):480–487. [DOI] [PubMed] [Google Scholar]
- 42.Hui P, Mattman A, Wilcox PG, Wright JL, Sin DD. Immunoglobulin G4-related lung disease: a disease with many different faces. Can Respir J 2013;20(5):335–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ruggio A, Iaconelli A, Panaioli E, et al. Coronary Artery Aneurysms Presenting as Acute Coronary Syndrome: An Unusual Case of IgG4-Related Disease Vascular Involvement. Can J Cardiol 2018;34(8):1088.e1087–1088.e1010. [DOI] [PubMed] [Google Scholar]
- 44.Barbu M, Lindstrom U, Nordborg C, Martinsson A, Dworeck C, Jeppsson A. Sclerosing Aortic and Coronary Arteritis Due to IgG4-Related Disease. Ann Thorac Surg 2017;103(6):e487–e489. [DOI] [PubMed] [Google Scholar]
- 45.Keraliya AR, Murphy DJ, Aghayev A, Steigner ML. IgG4-Related Disease With Coronary Arteritis. Circ Cardiovasc Imaging 2016;9(3):e004583. [DOI] [PubMed] [Google Scholar]
- 46.Bito Y, Sasaki Y, Hirai H, et al. A surgical case of expanding bilateral coronary aneurysms regarded as immunoglobulin G4-related disease. Circulation 2014;129(16):e453–456. [DOI] [PubMed] [Google Scholar]
- 47.Della Torre E, Mattoo H, Mahajan VS, Carruthers M, Pillai S, Stone JH. Prevalence of atopy, eosinophilia, and IgE elevation in IgG4-related disease. Allergy 2014;69(2):269–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Inoue D, Zen Y, Abo H, et al. Immunoglobulin G4-related lung disease: CT findings with pathologic correlations. Radiology 2009;251(1):260–270. [DOI] [PubMed] [Google Scholar]
- 49.Keenan JC, Miller E, Jessurun J, Allen T, Kim HJ. IgG4-related lung disease: a case series of 6 patients and review of the literature. Sarcoidosis Vasc Diffuse Lung Dis 2016;32(4):360–367. [PubMed] [Google Scholar]
- 50.Saraya T, Ohkuma K, Fujiwara M, et al. Clinical characterization of 52 patients with immunoglobulin G4-related disease in a single tertiary center in Japan: Special reference to lung disease in thoracic high-resolution computed tomography. Respir Med 2017;132(62–67). [DOI] [PubMed] [Google Scholar]
- 51.Lim SY, McInnis M, Wallace Z, Deshpande V, Sharma A, Stone JH. Intrathoracic Manifestations of IgG4-Related Disease: Findings in a Cohort Study from North America [abstract]. Arthritis Rheumatol 2016;68(suppl 10). [Google Scholar]
- 52.Oyama-Manabe N, Yabusaki S, Manabe O, Kato F, Kanno-Okada H, Kudo K. IgG4-related Cardiovascular Disease from the Aorta to the Coronary Arteries: Multidetector CT and PET/CT. Radiographics 2018;38(7):1934–1948. [DOI] [PubMed] [Google Scholar]
- 53.Perugino CA, Wallace ZS, Meyersohn N, Oliveira G, Stone JR, Stone JH. Large vessel involvement by IgG4-related disease. Medicine (Baltimore) 2016;95(28):e3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Carruthers MN, Khosroshahi A, Augustin T, Deshpande V, Stone JH. The diagnostic utility of serum IgG4 concentrations in IgG4-related disease. Ann Rheum Dis 2015;74(1):14–18. [DOI] [PubMed] [Google Scholar]
- 55.Ryu JH, Horie R, Sekiguchi H, Peikert T, Yi ES. Spectrum of Disorders Associated with Elevated Serum IgG4 Levels Encountered in Clinical Practice. Int J Rheumatol 2012;2012:232960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yamamoto M, Tabeya T, Naishiro Y, et al. Value of serum IgG4 in the diagnosis of IgG4-related disease and in differentiation from rheumatic diseases and other diseases. Mod Rheumatol 2012;22(3):419–425. [DOI] [PubMed] [Google Scholar]
- 57.Chang SY, Keogh KA, Lewis JE, et al. IgG4-positive plasma cells in granulomatosis with polyangiitis (Wegener’s): a clinicopathologic and immunohistochemical study on 43 granulomatosis with polyangiitis and 20 control cases. Hum Pathol 2013;44(11):2432–2437. [DOI] [PubMed] [Google Scholar]
- 58.Bledsoe J, Wallace Z, Deshpande V, et al. Atypical IgG4+ plasmacytic proliferations and lymphomas: characterization of 11 cases. Am J Clin Pathol 2017;148(3):215–235. [DOI] [PubMed] [Google Scholar]
- 59.Kiil K, Bein J, Schuhmacher B, et al. A high number of IgG4-positive plasma cells rules out nodular lymphocyte predominant Hodgkin lymphoma. Virchows Arch 2018;473(6):759–764. [DOI] [PubMed] [Google Scholar]
- 60.Chari ST, Smyrk TC, Levy MJ, et al. Diagnosis of autoimmune pancreatitis: the Mayo Clinic experience. Clin Gastroenterol Hepatol 2006;4(8):1010–1016; quiz 1934. [DOI] [PubMed] [Google Scholar]
- 61.Otsuki M, Chung JB, Okazaki K, et al. Asian diagnostic criteria for autoimmune pancreatitis: consensus of the Japan-Korea Symposium on Autoimmune Pancreatitis. J Gastroenterol 2008;43(6):403–408. [DOI] [PubMed] [Google Scholar]
- 62.Shimosegawa T, Chari ST, Frulloni L, et al. International consensus diagnostic criteria for autoimmune pancreatitis: guidelines of the International Association of Pancreatology. Pancreas 2011;40(3):352–358. [DOI] [PubMed] [Google Scholar]
- 63.Ahuja J, Arora D, Kanne JP, Henry TS, Godwin JD. Imaging of Pulmonary Manifestations of Connective Tissue Diseases. Radiol Clin North Am 2016;54(6):1015–1031. [DOI] [PubMed] [Google Scholar]
- 64.Mirmomen SM, Sirajuddin A, Nikpanah M, et al. Thoracic involvement in Erdheim-Chester disease: computed tomography imaging findings and their association with the BRAFV600E mutation. Eur Radiol 2018;. [DOI] [PubMed] [Google Scholar]
- 65.Surabhi VR, Chua S, Patel RP, Takahashi N, Lalwani N, Prasad SR. Inflammatory Myofibroblastic Tumors: Current Update. Radiol Clin North Am 2016;54(3):553–563. [DOI] [PubMed] [Google Scholar]
- 66.Cheuk W, Chan JK. Lymphadenopathy of IgG4-related disease: an underdiagnosed and overdiagnosed entity. Semin Diagn Pathol 2012;29(4):226–234. [DOI] [PubMed] [Google Scholar]
- 67.Sato Y, Notohara K, Kojima M, Takata K, Masaki Y, Yoshino T. IgG4-related disease: historical overview and pathology of hematological disorders. Pathol Int 2010;60(4):247–258. [DOI] [PubMed] [Google Scholar]
- 68.Cheuk W, Yuen HK, Chu SY, Chiu EK, Lam LK, Chan JK. Lymphadenopathy of IgG4-related sclerosing disease. Am J Surg Pathol 2008;32(5):671–681. [DOI] [PubMed] [Google Scholar]
- 69.Deshpande V The pathology of IgG4-related disease: critical issues and challenges. Semin Diagn Pathol 2012;29(4):191–196. [DOI] [PubMed] [Google Scholar]
- 70.Hao M, Liu M, Fan G, Yang X, Li J. Diagnostic Value of Serum IgG4 for IgG4-Related Disease: A PRISMA-compliant Systematic Review and Meta-analysis. Medicine (Baltimore) 2016;95(21):e3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Khosroshahi A, Cheryk LA, Carruthers MN, Edwards JA, Bloch DB, Stone JH. Brief Report: spuriously low serum IgG4 concentrations caused by the prozone phenomenon in patients with IgG4-related disease. Arthritis Rheumatol 2014;66(1):213–217. [DOI] [PubMed] [Google Scholar]
- 72.Cao L, Chen YB, Zhao DH, Shi WF, Meng S, Xie LX. Pulmonary function tests findings and their diagnostic value in patients with IgG4-related disease. J Thorac Dis 2017;9(3):547–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Khosroshahi A, Wallace ZS, Crowe JL, et al. International consensus guidance statement on the management and treatment of IgG4-related disease. Arthritis Rheumatol 2015;67(7):1688–1699. [DOI] [PubMed] [Google Scholar]
- 74.Shimizu Y, Yamamoto M, Naishiro Y, et al. Necessity of early intervention for IgG4-related disease--delayed treatment induces fibrosis progression. Rheumatology (Oxford) 2013;52(4):679–683. [DOI] [PubMed] [Google Scholar]
- 75.Masaki Y, Matsui S, Saeki T, et al. A multicenter phase II prospective clinical trial of glucocorticoid for patients with untreated IgG4-related disease. Mod Rheumatol 2017;27(5):849–854. [DOI] [PubMed] [Google Scholar]
- 76.Hart PA, Kamisawa T, Brugge WR, et al. Long-term outcomes of autoimmune pancreatitis: a multicentre, international analysis. Gut 2013;62(12):1771–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Masamune A, Nishimori I, Kikuta K, et al. Randomised controlled trial of long-term maintenance corticosteroid therapy in patients with autoimmune pancreatitis. Gut 2017;66(3):487–494. [DOI] [PubMed] [Google Scholar]
- 78.Yunyun F, Yu P, Panpan Z, et al. Efficacy and safety of low dose Mycophenolate mofetil treatment for immunoglobulin G4-related disease: a randomized clinical trial. Rheumatology (Oxford) 2019;58(1):52–60. [DOI] [PubMed] [Google Scholar]
- 79.Wang L, Zhang P, Wang M, et al. Failure of remission induction by glucocorticoids alone or in combination with immunosuppressive agents in IgG4-related disease: a prospective study of 215 patients. Arthritis Res Ther 2018;20(1):65. [DOI] [PMC free article] [PubMed] [Google Scholar]