

Abstract
In various real-world problems, we are presented with classification problems with positive and unlabeled data, referred to as presence-only responses. In this article we study variable selection in the context of presence only responses where the number of features or covariates p is large. The combination of presence-only responses and high dimensionality presents both statistical and computational challenges. In this article, we develop the PUlasso algorithm for variable selection and classification with positive and unlabeled responses. Our algorithm involves using the majorization-minimization framework which is a generalization of the well-known expectation-maximization (EM) algorithm. In particular to make our algorithm scalable, we provide two computational speed-ups to the standard EM algorithm. We provide a theoretical guarantee where we first show that our algorithm converges to a stationary point, and then prove that any stationary point within a local neighborhood of the true parameter achieves the minimax optimal mean-squared error under both strict sparsity and group sparsity assumptions. We also demonstrate through simulations that our algorithm outperforms state-of-the-art algorithms in the moderate p settings in terms of classification performance. Finally, we demonstrate that our PUlasso algorithm performs well on a biochemistry example. Supplementary materials for this article are available online.
Keywords: Majorization-minimization; Nonconvexity, PU-learning; Regularization
1. Introduction
In many classification problems, we are presented with the problem where it is either prohibitively expensive or impossible to obtain negative responses and we only have positive and unlabeled presence-only responses (see, e.g., Ward et al. 2009). For example, presence-only data are prevalent in geographic species distribution modeling in ecology where presences of species in specific locations are easily observed but absences are difficult to track (see, e.g., Ward et al. 2009), text mining (see, e.g., Liu et al. 2006), bioinformatics (see, e.g., Elkan and Noto 2008), and many other settings. Classification with presence-only data is sometimes referred to as PU-learning (see, e.g., Liu et al. 2006; Elkan and Noto 2008). In this article, we address the problem of variable selection with presence-only responses.
1.1. Motivating Application: Biotechnology
Although the theory and methodology we develop apply generally, a concrete application that motivates this work arises from biological systems engineering. In particular, recent high-throughput technologies generate millions of biological sequences from a library for a protein or enzyme of interest (see, e.g., Fowler and Fields 2014; Hietpas, Jensen, and Bolon 2011). In Section 5, the enzyme of interest is beta-glucosidase (BGL) which is used to decompose disaccharides into glucose which is an important step in the process of converting plant matter to biofuels (Romero, Tran, and Abate 2015). The performance of the BGL enzyme is measured by the concentration of glucose that is produced and a positive response arises when the disaccharide is decomposed to glucose and a negative response arises otherwise. Hence, there are two scientific goals: firstly to determine how the sequence structure influences the biochemical functionality; secondly, using this relationship to engineer and design BGL sequences with improved functionality.
Given these two scientific goals, we are interested in both the variable selection and classification problem since we want to determine which positions in the sequence most influence positive responses as well as classify which protein sequences are functional. Furthermore, the number of variables here is large since we need to model long and complex biological sequences. Hence, our variable selection problem is high-dimensional. In Section 5, we demonstrate the success of our algorithm in this application context.
1.2. Problem Setup
To state the problem formally, let be a p-dimensional covariate such that , y ∈ {0, 1} an associated response, and z ϵ {0, 1} an associated label. If a sample is labeled (z = 1), its associated outcome is positive (y = 1). On the other hand, if a sample is unlabeled (z = 0), it is assumed to be randomly drawn from the population with only covariates x not the response y being observed. Given nℓ labeled and nu unlabeled samples, the goal is to draw inferences about the relationship between y and x. We model the relationship between the probability of a response y being positive and (x, θ) using the standard logistic regression model
| (1) |
and where refers to the unknown true parameter. Also, we assume the label z is assigned only based on the latent response y independent from x. Viewing z as a noisy observation of latent y, this assumption corresponds to a missing at random assumption, a classical assumption in latent variable problems.
Given such z, we select nl labeled and nu unlabeled samples from samples with z = 1 and z = 0, respectively. An important issue is how positive and unlabeled samples are selected. In this article, we adopt a case-control approach (e.g., McCullagh and Nelder 2006)which is suitable for our biotechnology application and many others. In particular, we introduce another binary random variable s ∈ {0, 1} representing whether a sample is selected (s = 1) or not (s = 0) to model different sampling rates in selecting labeled and unlabeled samples. Since there are nℓ labeled and nu unlabeled samples, we have
and we see only selected samples, . It is further assumed that the selection is only based on the label z, independent of x and y. We note that this case-control scheme (Lancaster and Imbens 1996;Wardet al. 2009), opposed to the single training sampling scheme (Elkan and Noto 2008) is needed to model the case where unlabeled samples are random draws from the original population, since positive samples have to be overrepresented in the dataset to satisfy such model assumption.
In our biotechnology application the case-control setting is appropriate since the high-throughput technology leads to the unlabeled samples being drawn randomly from the original population (see Romero, Tran, and Abate 2015 for details). As is displayed in Figure 1, sequences are selected randomly from a library and positive samples are generated through a screening step. Hence, the positive sequences are sampled randomly from the positive sequences while the unlabeled sequences are based on random sampling from the original sequence library. This experiment corresponds exactly to the case-control sampling scheme discussed.
Figure 1.
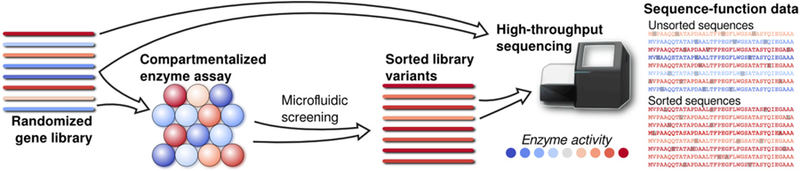
High-throughput sequencing diagram.
Furthermore, the true positive (TP) prevalence is
| ? | (2) |
and π is assumed known. In our biotechnology application, π is estimated precisely using an alternative experiment (Romero, Tran, and Abate 2015).
In the biological sequence engineering example, correspond to binary covariates of biological sequences. In the BGL example, for each of the d positions, there are M possible categories of amino acids. Therefore, the covariates correspond to the indicator of an amino acid appearing in a given position (p = O(dM)) as well as pairs of amino acids (p = O(d2M2)), and so on. Here d = O(1000) and M ≈ 20 make the problem high-dimensional.
High-dimensional PU-learning presents computational challenges since the standard logistic regression objective leads to a nonconvex likelihood when we have positive and unlabeled data. To address this challenge, we build on the expectation maximization (EM) procedure developed in Ward et al. (2009) and provide two computational speed-ups. In particular, we introduce the PUlasso for high-dimensional variable selection with positive and unlabeled data. Prior work that involves the EM algorithm in the low-dimensional setting in Ward et al. (2009) involves solving a logistic regression model at the M-step. To adapt to the high-dimensional setting and make the problem scalable, we include an ℓ1-sparsity or ℓ1/ℓ2-group sparsity penalty and provide two speed-ups. First, we use a quadratic majorizer of the logistic regression objective, and secondly, we use techniques in linear algebra to exploit sparsity of the design matrix X which commonly arises in the applications we are dealing with. Our PUlasso algorithm fits into the majorization minimization (MM) framework (see, e.g., Lange, Hunter, and Yang 2000; Ortega and Rheinboldt 2000) for which the EM algorithm is a special case.
1.3. Our Contributions
In this article we make the following major contributions:
Develop the PUlasso algorithm for doing variable selection and classification with presence-only data. In particular, we build on the existing EM algorithm developed in Ward et al. (2009) and add two computational speed-ups, quadratic majorization and exploiting sparse matrices. These two speed-ups improve speed by several orders of magnitude and allows our algorithm to scale to datasets with millions of samples and covariates.
Provide theoretical guarantees for our algorithm. First we show that our algorithm converges to a stationary point of the nonconvex objective, and then show that any stationary point within a local neighborhood of θ* achieves the minimax optimal mean-squared error for sparse vectors. To provide statistical guarantees we extend the existing results of generalized linear model with a canonical link function (Negahban et al. 2012; Loh and Wainwright 2006) to a noncanonical link function and show optimality of stationary points of nonconvex objectives in high-dimensional statistics. To the best of our knowledge the PUlasso is the first algorithm where PUlearning is provably optimal in the high-dimensional setting.
Demonstrate through a simulation study that our algorithm performs well in terms of classification compared to stateof-the-art PU-learning methods in Du Marthinus, Niu, and Sugiyama (2015), Elkan and Noto (2008), and Liu et al. (2006), both for low-dimensional and high-dimensional problems.
Demonstrate that our PUlasso algorithmallows us to develop improved protein-engineering approaches. In particular, we applyour PUlassoalgorithmto sequencesofBGL enzymes to determine which sequences are functional. We demonstrate that sequences selected by our algorithm have a good predictive accuracy and we also provide a scientific experiment which shows that the variables selected lead to BGL proteins that are engineered with improved functionality.
The remainder of the article is organized as follows: in Section 2 we provide the background and introduce the PUlasso algorithm, including our two computational speed-ups and provide an algorithmic guarantee that our algorithm converges to a stationary point; in Section 3 we provide statistical meansquared error guarantees which show that our PUlasso algorithm achieves the minimax rate; Section 4 provides a comparison in terms of classification performance of our PUlasso algorithm to state-of-the-art PU-learning algorithms; finally in Section 5, we apply our PUlasso algorithm to the BGL data application and provide both a statistical validation and simple scientific validation for our selected variables.
Notation: For scalars a, , we denote a ∧ b = min{a, b}, a ∨ b = max{a, b}. Also, we denote a ≳ b if there exists a universal constant c > 0 such that a ≥ cb. For , we denote ℓ1, ℓ2, and ℓ∞ norm as , , and and use to denote Hadamard product (entry-wise product) of v,w. For a set S, we use |S| to denote the cardinality of S. For any subset , denotes the subvector of the vector v by selecting the components with indices in S. Likewise for matrix , denotes a submatrix by selecting columns with indices in S. For a group ℓ1/ℓ2 norm, the norm is characterized by a partition of {1, … , p} and associated weights . We let and define the ℓ1/ℓ2 norm as . We often need a dual norm of . We use to denote and write . Finally, we write for an ℓq ball with radius r centered at , and denote as if v = 0.
For a convex function , we use ∂f (x) to denote the set of subgradients at the point x and ∇f (x) to denote an element of ∂f (x). Also for a function f + g such that f is differentiable (but not necessarily convex) and g is convex, we define with a slight abuse of notation. Also, we say f (n) = O(g(n)), f (n) = Ω(g(n)), and f (n) = Θ(g(n)) if |f | is asymptotically bounded above, bounded below, and bounded above and below by g.
For a random variable , we say x is a sub-Gaussian random variable with sub-Gaussian parameter σx > 0 if for all and we denote as with a slight abuse of notation. Similarly, we say x is a sub-exponential random variable with sub-exponential parameter (ν, b) if E[exp(t(x − E[x]))] ≤ exp(t2ν2/2) for all |t| ≤ 1/b and we denote as x ∼ subExp(ν, b). A collection of random variables (x1,…, xn) is referred to as .
2. PUlasso Algorithm
In this section, we introduce our PUlasso algorithm. First, we discuss the prior EM algorithm approach developed in Ward et al. (2009) and apply a simple regularization scheme. We then discuss our two computational speed-ups, the quadratic majorization for the M-step and exploiting sparse matrices. We prove that our algorithm has the descending property and converges to a stationary point, and show that our two speedups increase speed by several orders of magnitude.
2.1. Prior Approach: EM Algorithm With Regularization
First we use the prior result in Ward et al. (2009) to determine the observed log-likelihood (in terms of the zis) and the full log-likelihood (in terms of the unobserved yis and zis). The following lemma, derived in Ward et al. (2009), gives the form of the observed and the full log-likelihood in the case-control sampling scheme.
Lemma 2.1 (Ward et al. 2009). The observed log-likelihood for our presence-only model in terms of is
| (3) |
The full log-likelihood in terms of is
| (4) |
where nℓ, nu are the number of positive and unlabeled observations, n = nℓ + nu and π is defined in (2).
The proof can be found in Ward et al. (2009). Our goal is to estimate the parameter , which we assume to be unique. In the setting where p is large, we add a regularization term. We are interested in cases when there exists or does not exist a group structure within covariates. To be general we use the group ℓ1/ℓ2-penalty for which ℓ1 is a special case. Hence, our overall optimization problem is
| (5) |
where log L(θ; xi, zi) is the observed log-likelihood. For a penalty term, we use the group sparsity regularizer
| (6) |
with , such that is a partition of (1,…, p) and wj > 0.We note that if J = p, gj = {j} and wj = 1, ∀j. For notational convenience we denote the overall objective as
| (7) |
where we define the loss function as and .
In the original proposal of the group lasso, Yuan and Lin (2006) recommended to use (6) for orthonormal group matrices , that is,. If group matrices are not orthonormal, however, it is unclear whether we should orthonormalize group matrices prior to application of the group lasso. This question was addressed in Simon and Tibshirani (2012), and the authors provide a compelling argument that prior orthonormalization has both theoretical and computational advantages. In particular, Simon and Tibshirani (2012) demonstrated that the following orthonormalization procedure is intimately connected with the uniformly most powerful invariant testing for inclusion of a group. To describe this orthonormalization explicitly, we obtain standardized group matrices and scale matrices for j ≥ 2 using the QR-decomposition such that
| (8) |
where is the projection matrix onto the orthogonal space of . Letting , the original optimization problem (5) can be expressed in terms of qis and becomes
| (9) |
where we use the transformation θ to ν
| (10) |
We note that this corresponds to the standard centering and scaling of the predictors in the case of standard lasso. For more discussion about group lasso and standardization (see, e.g., Huang, Breheny, and Ma 2012).
A standard approach to performing this minimization is to use the EM-algorithm approach developed in Ward et al. (2009). In particular, we treat as hidden variables and estimate them in the E-step. Then use estimated to obtain the full log-likelihood in the M-step.

The E-step follows from since zi = 1 implies yi = 1 and when zi = 0, observations in the unlabeled data are random draws from the population. An initialization θ0 can be any vector such that where θnull is the parameter corresponding to the intercept-only model. If we are provided with no additional information, we may use θnull for the initialization. We use θ0 = θnull as the initialization for the remainder of the article. For the M-step, it was originally proposed to use a logistic regression solver. We can use a regularized logistic regression solver such as the glmnet R package to solve (12). We discuss a computationally more efficient way of solving (12) in the subsequent section.
2.2. PUlasso: A Quadratic Majorization for the M-Step
Now we develop our PUlasso algorithm which is a faster algorithm for solving (5) by using quadratic majorization for the M-step. The main computational bottleneck in algorithm 1 is the M-step which requires minimizing a regularized logistic regression loss at each step. This subproblem does not have a closed-form solution and needs to be solved iteratively, causing inefficiency in the algorithm. However, the most important property of the objective function in the M-step is that it is a surrogate function of the likelihood which ensures the descending property (see, e.g., Lange, Hunter, and Yang 2000). Hence, we replace a logistic loss function with a computationally faster quadratic surrogate function. In this aspect, our approach is an example of the more general MM framework (see, e.g., Lange, Hunter, and Yang 2000; Ortega and Rheinboldt 2000).
On the other hand, our loss function itself belongs to a generalized linear model family, as we will discuss in more detail in the subsequent section. A number of works have developed methods for efficiently solving regularized generalized linear model problems. A standard approach is to make a quadratic approximation of the log-likelihood and use solvers for a regularized least-square problem. Works include using an exact Hessian (Lee et al. 2006; Friedman, Hastie, and Tibshirani 2010), an approximate Hessian (Meier, Van De Geer, and Bühlmann 2008) or a Hessian bound (Krishnapuramet al. 2005; Simon and Tibshirani 2012; Breheny and Huang 2013) for the second-order term. Solving a second-order approximation problem amounts to taking a Newton step, thus convergence is not guaranteed without a step-size optimization (Lee et al. 2006;Meier, Van De Geer, and Bühlmann 2008), unless a global bound of the Hessian matrix is used. Our work can be viewed as in the line of these works where a quadratic approximation of the loss function is made and then an upper bound of the Hessian matrix is used to preserve a majorization property.
A coordinate descent (CD) algorithm (Wu and Lange 2008; Friedman, Hastie, and Tibshirani 2010) or a block coordinate descent (BCD) algorithm (Yuan and Lin 2006; Puig et al. 2011; Simon and Tibshirani 2012; Breheny and Huang 2013) has been a very efficient and standard way to solve a quadratic problem with ℓ1 penalty or ℓ1/ℓ2 penalty and we also take this approach. When a feature matrix is sparse, we can set up the algorithm to exploit such sparsity through a sparse linear algebra calculation. We discuss this implementation strategy in Section 2.2.1.
Now we discuss the PUlasso algorithm and the construction of quadratic surrogate functions in more details. Using the MM framework, we construct the set of majorization functions with the following two properties
| (13) |
where our goal is to minimize −Q where .
Using the Taylor expansion of Q(θ; θm) at θ = θm, we obtain Q(θ; θm)
where we define , , and is a diagonal matrix with . The inequality follows from , ∀θ. Thus, setting as follows
satisfies both conditions in (13). Also with some algebra, it follows that
for some c(θm) which does not depend on θ. Hence, acts as a quadratic surrogate function of−Q which replaces our M-step for the original EM algorithm. Therefore, our PUlasso algorithm can be represented as follows.

Now we state the following proposition to show that both the regularized EM and PUlasso algorithms have the desirable descending property and converge to a stationary point. For convenience we define the feasible region , which contains all θ whose objective function value is better than that of the intercept-only model, defined as
| (17) |
where , an estimate corresponding to the intercept-only model. We let be the set of stationary points satisfying the first-order optimality condition, that is,
| (18) |
One of the important conditions is to ensure that all iterates of our algorithm lie in which is trivially satisfied if θ0 = θnull.
Proposition 2.1. The sequence of estimates (θm) obtained by Algorithms 1 or 2 satisfies
, and if .
All limit points of are elements of the set , and converges monotonically to for some .
The sequence (θm) has at least one limit point, which must be a stationary point of by (ii).
Proposition 2.1 shows that we obtain a stationary point of the objective (7) as an output of both the regularized EM algorithm and our PUlasso algorithm. The proof uses the standard arguments based on Jensen’s inequality, convergence of EM algorithm and MM algorithms and is deferred to the supplement S1.1.
2.2.1. Block Coordinate Descent Algorithm for M-Step and Sparse Calculation
In this section, we discuss the specifics of finding a minimizer for the M-step (16) for each iteration of our PUlasso algorithm. After preprocessing the design matrix as described in (9) and (10), we solve the following optimization problem using a standard block-wise coordinate descent algorithm.
| (19) |

S(., λ) is the soft thresholding operator defined as follows
Note that we do not need to keep updating the intercept ν1 since Qgj, j ≥ 2 are orthogonal to . For more details (see, e.g., Breheny and Huang 2013).
For our biochemistry example and many other examples, X is a sparse matrix since each entry is an indicator of whether an amino acid is in a position. In Algorithm 3, we do not exploit this sparsity since Q will not be sparse even when X is sparse. If we want to exploit sparse X we use the following algorithm.

To explain the changes to this algorithm, we modify (20) and (22) so that we directly use X rather than Q to exploit the sparsity of X. Using (8), we first substitute with to obtain
| (28) |
| (29) |
However, carrying out (28) – (29) instead of (20) – (22) incurs a greater computational cost. Calculating requires n|gj| operations. On the contrary, the minimal number of operations required to do a matrix multiplication of is , when it is parenthesized as . Inmany cases |gj| is small (for standard lasso, |gj| = 1, ∀j and for our biochemistry example, |gj| is at most 20), but the additional increase in n can be very costly (especially in our example where n is over 4 million).
For a more efficient calculation, we first exploit the structure of when multiplying P0 with a vector, which reduces the cost from n2 operations to 2n operations. Also, we carry out calculations using instead of when calculating residuals and do the corrections all at once.
Before going into detail about (23)–(26), we first discuss the computational complexity. Comparing (23) with (20), the first term only requires an additional |gj|2 operations. The second term can be stored during the initial QR decomposition; thus the only potentially expensive operation is calculating an average of r which requires n operations. Comparing (25) with (22), only |gj|2 additional operations are needed when we parenthesize as . Note that if we had kept P0, there would have been an additional 2n operations even though we had used the structure of P0. In the calculation of (27), we note that n operations are involved in subtracting from r because aj are scalars. In summary, we essentially reduce additional computational cost from O(n2) to nJ per cycle by carrying out (23)–(26) instead of (28)–(29).
Now we derive/explain the formulas in Algorithm 4. To make quantities more explicit, we use rj and to denote a residual vector before/after update at j using Algorithm 3 and and using Algorithm 4. By definition, and . Also we note that in the beginning of the cycle . Equation (23) can be obtained from (28) by replacing P0 with . Now we show that modified residuals still correctly update coefficient. Starting from j = 2, a calculated is a constant vector off from a correct residual , as we see below
| (30) |
| (31) |
| (32) |
where we recall that . We note because . Then the next zj+1, thus new , are still correctly calculated since
| (33) |
The next residual is again off by a constant from the correct residual . To see this, by (29). Going through (30) – (32) with j being replaced by j + 1, we obtain
Inductively, we have correct zj, thus for all j ≥ 2. At the end of the cycle, we correct the residual vector all at once by letting .
2.3. R Package Details
We provide a publicly available R implementation of our algorithm in the PUlasso package. For a fast and efficient implementation, all underlying computation is implemented in C++. The package uses warm start and strong rule (Friedman et al. 2007; Tibshirani et al. 2012), and a cross-validation function is provided as well for the selection of the regularization parameter λ. Our package supports a parallel computation through the R package parallel.
2.4. Run-Time Improvement
Now we illustrate the run-time improvements for our two speed-ups. Note that we only include p up to 100 so that we can compare to the original regularized EM algorithm. For our biochemistry application p = O(104) and n = O(106) which means the regularized EM algorithm is too slow to run efficiently. Hence, we use smaller values of n and p in our run-time comparison. It is clear from our results that the quadratic majorization step is several orders of magnitude faster than the original EM algorithm, and exploiting the sparsity of X provides a further 30% speed-up.
3. Statistical Guarantee
We now turn our attention to statistical guarantees for our PUlasso algorithm under the statistical model (1). In particular, we provide error bounds for any stationary point of the nonconvex optimization problem (5). Proposition 2.1 guarantees that we obtain a stationary point from our PUlasso algorithm.
We first note that the observed likelihood (3) is a generalized linear model (GLM) with a noncanonical link function. To see this, we rewrite the observed likelihood (3) as
| (34) |
after some algebraic manipulations, where we define and . Also, we let µ(ηi) := A′(ηi), which is the conditional mean of zi given xi, by the property of exponential families. For the convenience of the reader, we include the derivation from (3) to (34) in the supplementary materials (S2.1). The mean of zi is related with θTxi via the link function g through g(µ(ηi)) = θTxi, where g satisfies . Because (g ○ µ)−1 is not the identity function, the likelihood is not convex anymore. For a more detailed discussion about the GLM with noncanonical link (see, e.g., McCullagh and Nelder 2006; Fahrmeir and Kaufmann 1985).
A number of works have been devoted to sparse estimation for generalized linear models. A large number of previous works have focused on generalized linear models with convex loss functions (negative log-likelihood with a canonical link) plus ℓ1 or ℓ1/ℓ2 penalties. Results with the ℓ1 penalty include a risk consistency result (van de Geer 2008) and estimation consistency in ℓ2 or ℓ1 norms (Kakade et al. 2010). For a group-structured penalty, a probabilistic bound for the prediction error was given in Meier, Van De Geer, and Bühlmann (2008). An ℓ2 estimation error bound in the case of the group lasso was given in Blazère, Loubes, and Gamboa (2014).
Negahban et al. (2012) rederived an ℓ2 error bound of an ℓ1-penalized GLM estimator under the unified framework for M-estimators with a convex loss function. This result about the regularized GLM was generalized in Loh and Wainwright (2006) where penalty functions are allowed to be nonconvex, while the same convex loss function was used. Since the overall objective function is nonconvex, authors discuss error bounds obtained for any stationary point, not a global minimum. In this aspect, our work closely follows this idea. However, our setting differs from Loh and Wainwright (2006) in two aspects: first, the loss function in our setting is nonconvex, in contrast with a convex loss function (a negative log-likelihood with a canonical link)with nonconvex regularizer in Loh andWainwright (2006). Also, an additive penalty function was used in the work of Loh and Wainwright (2006), but we consider a group-structured penalty.
After the initial draft of this article was written, we became aware of two recent papers (Elsener and van de Geer 2018; Mei, Bai, and Montanari 2018) which studied nonconvex M-estimation problems in various settings including binary linear classification, where the goal is to learn θ* such that for a known σ(·). The proposed estimators are stationary points of the optimization problem: in both papers. As the focus of our article is to learn a model with a structural contamination in responses, our choice of mean and loss functions differ from both papers. In particular, our choice of mean function is different from the sigmoid function, which was the representative example of σ(·) in both papers, and we use the negative log-likelihood loss in contrast to the squared loss. We establish error bounds by proving a modified restricted strong convexity condition, which will be discussed shortly, while error bounds of the same rates were established in Elsener and van de Geer (2018) through a sharp oracle inequality, and a uniform convergence result over population risk in Mei, Bai, and Montanari (2018).
Due to the nonconvexity in the observed log-likelihood, we limit the feasible region Θ0 to
| (35) |
for theoretical convenience. Here r0, Rn > 0 must be chosen appropriately and we discuss these choices later. Similar restriction is also assumed in Loh andWainwright (2006).
3.1. Assumptions
We impose the following assumptions. First, we define a sub-Gaussian tail condition for a random vector ; we say x has a sub-Gaussian tail with parameter , if for any fixed , there exists σx > 0 such that for any . We recall that θ* is the true parameter vector, which minimizes the population loss.
Assumption 1. The rows , i = 1,2, … , n of the design matrix are iid samples from a mean-zero distribution with sub-Gaussian tails with parameter . Moreover, is a positive definite and with minimum eigenvalue λmin(∑x) ≥ K0 where K0 is a constant bounded away from 0.We further assume that are independent for all j ∈ gj and .
Similar assumptions appear in, for example, Negahban et al. (2012). This restricted minimum eigenvalue condition (see, e.g., Raskutti, Wainwright, and Yu 2010 for details) is satisfied for weakly correlated design matrices. We further assume independence across covariates within groups since sub-Gaussian concentration bound assuming independence within groups is required.
Assumption 2. For any r > 0, there exists such that a.s. for all θ in the set .
Assumption 2 ensures that is bounded a.s., which guarantees that the underlying probability is between 0 and 1, and is also bounded within a compact sparse neighborhood of θ* which ensures concentration to the population loss. Comparable assumptions are made in Elsener and van de Geer (2018); Mei, Bai, and Montanari (2018) where similar nonconvex M-estimation problems are investigated.
Assumption 3. The ratio of the number of labeled to unlabeled data, that is, nℓ/nu is lower bounded away from 0 and upper bounded for all n = nℓ + nu, as n → ∞. Equivalently, there is a constant K2 such that Assumption 3 ensures that the number of labeled samples nℓ is not too small or large relative to n. The reason why nℓ cannot be too large is that the labeled samples are only positives and we need a reasonable number of negative samples which are a part of the unlabeled samples.
Assumption 4 (Rate conditions). We assume a high-dimensional regime where both (n, p) → ∞ log p = o(n). For and , we assume J = Ω(nβ) for some β > 0, m = o(n ∧ J), minj wj = Ω(1), and maxj wj = o(n ∧ J).
Assumption 4 states standard rate conditions in a high-dimensional setting. In terms of the group structure, we assume that growth of p is not totally attributed to the expansion of a few groups; the number of groups J increases with n, and the maximum group size m is of small order of both n and J. Also we note that a typical choice of satisfies Assumption 4 because minj wj ≥ 1, and ,.
Finally, we define the restricted strong convexity assumption for a loss function following the definition in Loh and Wain-wright (2006).
Definition 3.1 (Restricted strong convexity). We say satisfies a restricted strong convexity (RSC) condition with respect to θ* with curvature α > 0 and tolerance function τ over Θ0 if the following inequality is satisfied for all θ ∈ Θ0
| (36) |
where Δ:= θ – θ* and .
In the special case where and hence , similar RSC conditions were discussed in Negahban et al. (2012) and Loh and Wainwright (2006) with different τ and Θ0. One of the important steps in our proof is to prove that RSC holds for the objective function .
3.2. Guarantee
Under Assumptions 1 – 4, we will show in Theorem 3.2 that the RSC condition holds with high probability over and therefore over Θ0, for Θ0 defined in (35). Under the RSC assumption, the following proposition, which is a modification of Theorem1 in Loh and Wainwright (2006), provides ℓ1/ℓ2 and ℓ2 bounds of an error vector . Recall that m = maxj |gj| (the size of the largest group) and J is the number of groups.
Proposition 3.1. Suppose the empirical loss satisfies the RSC condition (36) with over Θ0 where Θ0 is feasible region for the objective (5), as defined in (35), and the true parameter vector θ* is feasible, that is, θ* ∈ Θ0. Consider λ such that
| (37) |
Let be a stationary point of (5). Then the following error bounds
| (38) |
hold where and s :=|S|.
The proof for Proposition 3.1 is deferred to the supplementary materials (S2.3). From (38), we note the squared ℓ2-error to grow proportionally with s and λ2. If θ* ∈ Θ0 and the choice of satisfies the inequality (37), we obtain squared ℓ2 error which scales as , provided that the RSC condition holds over Θ0. In the case of lasso we recover parametric optimal rate since J = p, m = 1.
With the choice of and 1, we ensure θ* is feasible and satisfies the inequality (37) with high probability. Clearly is of the order with the choice of , and following Lemma 3.1, we have with high probability. Thus, inequality (37) is satisfied with w.h.p. as well.
Lemma 3.1. Under Assumptions 1 – 4, for any given ϵ > 0, there is a positive constant c such that
given a sample size .
The proof for Lemma 3.1 is provided in the supplement S2.4. Now we state them a in theorem of this section which shows that RSC condition holds uniformly over a neighborhood of the true parameter.
Theorem 3.2. For any given r > 0 and ϵ > 0, there exist strictly positive constants α, τ1, and τ2 depending on σx, K0, , and K2 such that
| (39) |
holds for all θ such that with probability at least 1 – ϵ, given (n, p) satisfying .
The proof of Theorem3.2 is deferred to the supplement S2.5. There are a couple of notable remarks about Theorem 3.2 and Proposition 3.1.
The application of the Proposition 3.1 requires for a RSC condition to hold over a feasible regionΘ0. Setting r = 2r0 in Theorem3.2, inequality (39) holds over w.h.p., therefore, over .
We discuss how underlying parameters r0, σx, and constants K0–K2 in Assumptions 1–3 are related to the ℓ2-error bound. From Proposition 3.1, we see that ℓ2-error is proportional to τ1/α and τ2/α. The proof of Theorem3.2 reveals that and , where L0 and K3 are also constants defined as and . As L0 is inversely related to K2 and r0, ℓ2-error is proportional to the r0, σx, and K2 in Assumptions 2 and 3, but inversely related to the minimum eigenvalue bound K0 in Assumption 1.
The mean-squared error in the case of J = p is verified below in Figure 2 and both the mean-squared error and ℓ1 errors are minimax optimal for high-dimensional linear regression (Raskutti,Wainwright, and Yu 2011).
Figure 2.
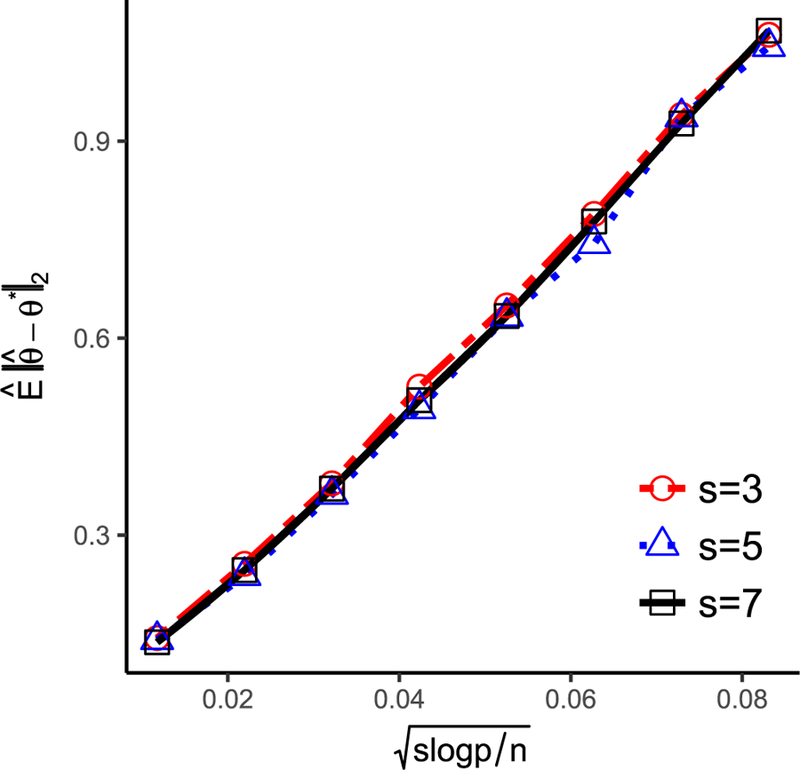
plotted against with fixed p = 500 and varying s and n.
To validate the mean-squared error upper bound of in Section 3, a synthetic dataset was generated according to the logistic model (1) with p = 500 covariates and X ∼ N(0, I500×500). Varying s and n were considered to study the rate of convergence of . The ratio nℓ/nu was fixed to be 1. For each dataset, was obtained by applying PUlasso algorithm with a lambda sequence for a suitably chosen cs for each s. We repeated the experiment 100 times and average ℓ2-error was calculated. In Figure 2, we illustrate the rate of convergence of . In particular, against is plotted with varying s and n. The error appears to be linear in , and thus we also empirically conclude that our algorithm achieves the optimal rate.
4. Simulation Study: Classification Performance
In this section, we provide a simulation study which validates the classification performance for PUlasso. In particular, we provide a comparison in terms of classification performance to state-of-the-art methods developed in Du Marthinus, Niu, and Sugiyama (2015), Elkan and Noto (2008), and Liu et al. (2006). The focus of this section is classification rather than variable selection since many of the state-of-the-artmethods we compare to are developed mainly for classification and are not developed for variable selection.
4.1. Comparison Methods
Our experiments compare six algorithms: (i) logistic regression model assuming we know the true responses (oracle estimator); (ii) our PUlasso algorithm; (iii) a bias-corrected logistic regression algorithm in Elkan and Noto (2008); (iv) a second algorithm from Elkan and Noto (2008) that is effectively a one step EM algorithm; (v) the biased SVM algorithm from Liu et al. (2006); and (vi) the PU-classification algorithm based on an asymmetric loss from Du Marthinus, Niu, and Sugiyama (2015).
The biased SVM from Liu et al. (2006) is based on the supported vector machine (SVM) classifier with two tuning parameters which parameterize misclassification costs of each kind. The first algorithm from Elkan and Noto (2008) estimates label probabilities and corrects the bias in the classifier via the estimation of under the assumption of a disjoint support between and . Their secondmethod is amodification of the first method; a unit weight is assigned to each labeled sample, and each unlabeled example is treated as a combination of a positive and negative example with weight and , respectively. Du Marthinus, Niu, and Sugiyama (2015) suggested using asymmetric loss functions with ℓ2-penalty. Asymmetric loss function is considered to cancel the bias induced by separating positive and unlabeled samples rather than positive and negative samples. Any convex surrogate of 0–1 loss function can be used for the algorithm. There is a publicly available matlab implementation of the algorithm when a surrogate is the squared loss on the author’s webpage2 and since we use their code and implementation, the squared loss is considered.
4.2. Setup
We consider a number of different simulation settings: (i) small and large p to distinguish the low and high-dimensional setting; (ii) weakly and strongly separated populations; (iii) weakly and highly correlated features; and (iv) correctly specified (logistic) or mis-specified model. Given dimensions (n, p), sparsity level s, predictor autocorrelation ρ, separation distance d, and model specification scheme (logistic, misspecified), our setup is the following
Choose the active covariate set S ⊆ {1, 2,…, p} by taking s elements uniformly at random from(1, 2,…, p). We let true such that .
-
Draw samples , iid from where ,. More concretely, firstly draw u ∼ Ber(0.5). If u = 1, draw x from and draw x from otherwise.
Mean vectors µ1, are chosen so that they are s-sparse, that is, , and variance of μi does not depend on d. Specifically, we sample μ1,μ2 such that for j ∈ S, we let , μ2j = −μ1j, and for j ∉ S, μij = 0 for i ∈ (1, 2).
A covariance matrix is taken to be where Kρ is chosen so that . This scaling of Σρ is made to ensure that the signal strength stays the same across ρ.
Draw responses y ∈ {0, 1}. If scheme = logistic, we draw y such that where . In contrast, if scheme = mis-specified, we let y = 1 if x was drawn from , and zero otherwise; that is,.
To compare performances both in low and high dimensional setting, we consider (p = 10, s = 5) and (p = 5000, s = 5). We set the sample size nℓ = nu = 500 in both cases. Autocorrelation level ρ takes values in (0, 0.2, 0.4, 0.6, 0.8). In the high dimensional setting, we excluded algorithm (v), since (v) requires a grid search over two dimensions, which makes the computational cost prohibitive. For algorithms (i)–(iv), tuning parameters λ are chosen based on the 10-fold cross-validation.
4.3. Classification Comparison
We use two criteria, misclassification rate and F1 score, to evaluate performances. F1 is the harmonic mean of the precision and recall, which is calculated as . The F1 score ranges from 0 to 1, where 1 corresponds to perfect precision and recall. Experiments are repeated 50 times and the average score and SEs are reported. The result for the misclassification rate under correct model specification is displayed in Figure 3.
Figure 3.
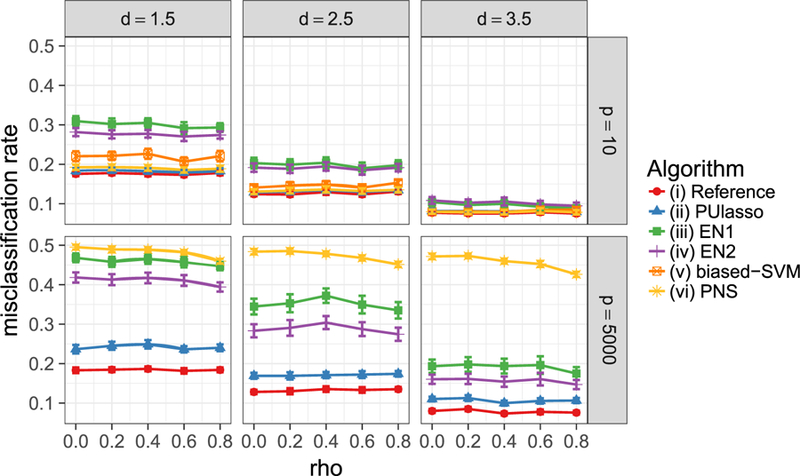
Misclassification rates of algorithms (i)–(vi) under correct (logistic) model specification. Each error bar represents two SEs of the mean.
Not surprisingly the oracle estimator has the best accuracy in all cases. PUlasso and algorithm (vi) performs almost as well as the oracle in the low-dimensional setting and better than remaining methods in most cases. It must be pointed out that both PUlasso and algorithm (vi) use additional knowledge π of the true prevalence in the unlabeled samples. PUlasso performs best in the high-dimensional setting while the performance of algorithm (vi) becomes significantly worse because estimation errors can be greatly reduced by imposing many 0s on the estimates in PUlasso due to the ℓ1-penalty (compared to ℓ2-penalty in algorithm (vi)). The performance of (iii)–(iv) is greatly improved when positive and negative samples are more separated (large d), because algorithms (iii)–(iv) assume disjoint support between two distributions. The algorithms showsimilar performancewhen evaluated with the F1 scoremetric and in the mis-specified setting. Due to space constraints, we defer the full set of remaining results in the supplementarymaterials (Section S3).
5. Analysis of Beta-Glucosidase Sequence Data
Our original motivation for developing the PUlasso algorithm was to analyze a large-scale dataset with positive and unlabeled responses developed by the lab of Dr. Philip Romero (Romero, Tran, and Abate 2015). The prior EM algorithm approach of Ward et al. (2009) did not scale to the size of this dataset. In this section, we discuss the performance of our PUlasso algorithm on a dataset involving mutations of a natural BGL enzyme. To provide context, BGL is a hydrolytic enzyme involved in the deconstruction of biomass into fermentable sugars for biofuel production. Functionality of the BGL enzyme is measured in terms of whether the enzyme deconstructs disaccharides into glucose or not. Dr. Romero used a microfluidic screen to generate a BGL dataset containing millions of sequences (Romero, Tran, and Abate 2015).3
Main effects and two-way interaction models are fitted using our PUlasso algorithm with ℓ1 and ℓ1/ℓ2 penalties (we discuss how the groups are chosen shortly) over a grid of λ values. We test stability of feature selection and classification performance using a modified ROC and AUC approach. Finally a scientific validation is performed based on a follow-up experiment conducted by the Romero lab. The variables selected by PUlasso were used to design a new BGL enzyme and the performance is compared to the original BGL enzyme.
5.1. Data Description
The dataset consists of nℓ = 2,647,877 labeled and functional sequences and nu = 1,567,203 unlabeled sequences where each of the observation σ = (σ1,…, σ500) is a sequence of amino acids of length d = 500. Each of the position σj ∈ (A, R,…,V, *) takes one of M = 21 discrete values, which correspond to the 20 amino acids in the DNA code and an extra to include the possibility of a gap(*).
Another important aspect of the millions of sequences generated is that a “base wild-type BGL sequence” was considered and known to be functional (y = 1), and the millions of sequences were generated by mutating the base sequence. Single mutations (changing one position from the base sequence) and double mutations (changing two positions) from the base sequence were common but higher-order mutations were not prevalent using the deep mutational scanning approach in Romero, Tran, and Abate (2015). Hence, the sequences generated were not random samples across the entire enzyme sequence space, but rather very local sequences around the wild-type sequence. Hence, the number of possible mutations in each position and consequently the total number of observed sequences is also reduced dramatically. With this dataset, we want to determine which mutations should be applied to the wild-type BGL sequence.
Categorical variables σ are converted into indicator variables: where 1 ≤ j ≤ 500, for the main-effects model, where 1 ≤ j, k≤ 500, for the pairwise interaction models, where represents the amino acid of the wild-type sequence at the lth position. In other words, each variable corresponds to an indicator of mutation from the base sequence or interaction between mutations. Although there are in principle p ≈ d(M − 1) variables for a main-effects model and p ≈ d2(M − 1)2 if we include main-effects and two-way interactions, there are many amino acids that never appear in any position or appear only a small number of times. For features corresponding to the main-effects , those sparse features are aggregated within each position until the number of mutations of the aggregated column reaches 100 or 1% of the total number of mutations in each position; accordingly, each aggregated column is an indicator of any mutations to those sparse amino acids. For two-way interactions features , sparse features (≤ 25 out of 4,215,080 samples) are simply removed from the feature space. Using this basic preprocessing we obtained only 3075 corresponding to single mutations and 930 binary variables corresponding to double mutations. They correspond to 500 unique positions and 820 two-way interactions between positions, respectively. As mentioned earlier, we consider both ℓ1 and group ℓ1/ℓ2 penalties. We use the ℓ1-penalty for the main-effects model and the ℓ1/ℓ2 for the two-way interaction models. For the two-way interaction model each group gj corresponds to a different position (500 total) and pair of positions (820 total) where mutations occur in the preprocessed design matrix and the group size |gj| corresponds to the number of different observed mutations in each position or pair of mutations in pair of positions (for this dataset m = maxj |gj| = 8). Higher-order interactions were not modeled as they did not frequently arise. Hence, the main-effects and two-way interaction model we consider have p = 3076 (1 + 3075) and p = 4006 (1 + 3075 + 930) and J = 1320 (500 + 820) groups, respectively. In summary, we consider the following two models and corresponding design matrices
and the response vector z = [1, …, 1, 0,…, 0]T ∈ {0, 1}4,215,080.
5.2. Classification Validation and Model Stability
Next we validate the classification performance for both the main-effect and two-way interaction models. We fit models using 90% of the randomly selected samples both from the positive and unlabeled set and use area under the ROC curve (AUC) to evaluate the classification performance on the 10% of the hold-out set. Since positive and negative samples are mixed in the unlabeled test dataset this is a nontrivial task with presence-only responses. A naive approach is to treat unlabeled samples as negative and estimate AUC, but if we do so, the AUC is inevitably downward-biased because of the inflated false positive (FP) rate. We note that a TP rate can be estimated in an unbiased manner using positive samples. To adjust such bias, we follow the methodology suggested in Jain, White, and Radivojac (2017) and adjust FP rate and AUC value using the following equation
where π is the prevalence of positive samples.
As Figure 4 shows, we have a significant improvement in AUC over random assignment (AUC = 0.5) in both the main effect (AUC = 0.7933) and two-way interaction (AUC = 0.7938) models. The performances of the two models in terms of AUC values are very similar at their best λ values chosen by 10-fold cross-validation. This is not very surprising as only a small number of two-way interactions are observed in the experiments.
Figure 4.
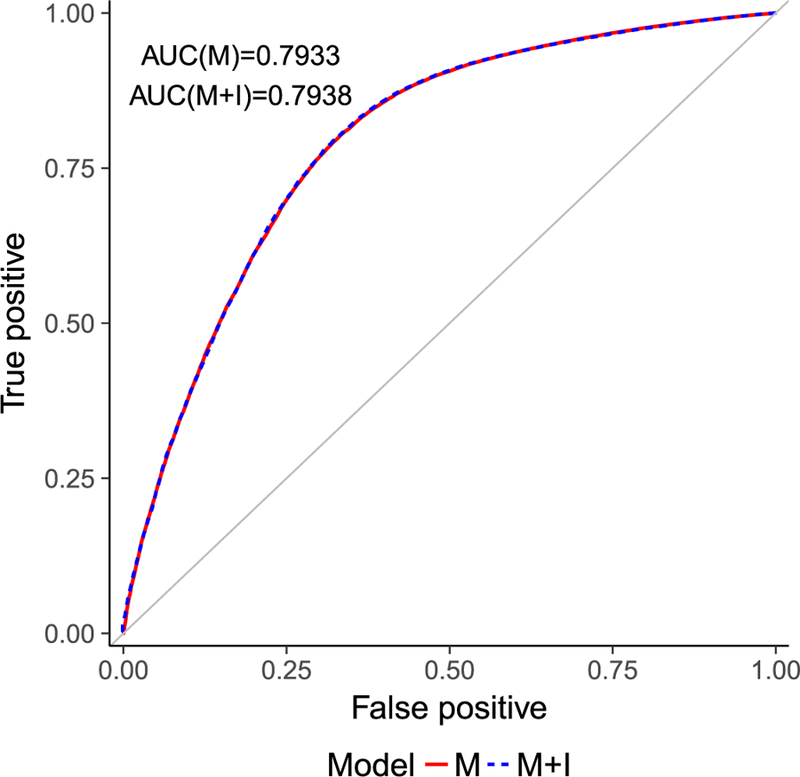
ROC curves of main effects (M) and two-way interaction model (M+I) with λ chosen based on 10-fold cross-validation.
We also examined the stability of the selected features for both models as the training data changes. Following the methodology of Kalousis, Prados, and Hilario (2007), we measure similarity between two subsets of features s, s′ using SS(s, s′) defined as . SS takes values in [0, 1], where 0 means that there is no overlap between the two sets, and 1 that the two sets are identical. Ss is computed for each pair of two training folds (i.e., we have pairs) using selected features and computed values are finally averaged over all pairs. Feature selection turned out to be very stable across all tuning parameter λ values: on average we had about 95% overlap of selection in main effect model (M) and about 98% overlap in main effect+interaction model (M+I). Stability score is higher in the latter model since we do a feature selection on groups, whose number is much less than individual variables (1320 groups vs. 3076 individual variables).
5.3. Scientific Validation: Designed BGL Sequence
Finally, we provide a scientific validation of the mutations estimated by our PUlasso algorithm. In particular, we fit the model with the PUlasso algorithm and selected the best λ = 0.0001 based on the 10-fold cross-validation. We use the top 10 mutations based on the largest size of coefficients with positive signs from our PUlasso algorithm because we are interested in mutations that enhance the performance of the sequence. Dr. Romero’s lab designed the BGL sequence with the 10 positive mutations from Table 4. This sequence was synthesized, expressed, and assayed for its hydrolytic activity. Hence, the designed sequence has 10 mutations compared to the wild-type (base) BGL sequence.
Table 4.
Ten positive mutations.
| Base/position/mutated | ||
|---|---|---|
| T197P | E495G | |
| K300P | A38G | |
| G327A | S486P | |
| A150D | T478S | |
| D164E | D481N |
Figure 5 shows firstly that the designed protein sequence folds which in itself is remarkable given that 10 positions are mutated. Secondly, Figure 5 shows that the designed sequence decomposes disaccharides into glucose more quickly than the wild-type sequence. These promising results suggest that our variable selection method is able to identify positions of the wild-type sequences with improved functionality.
Figure 5.
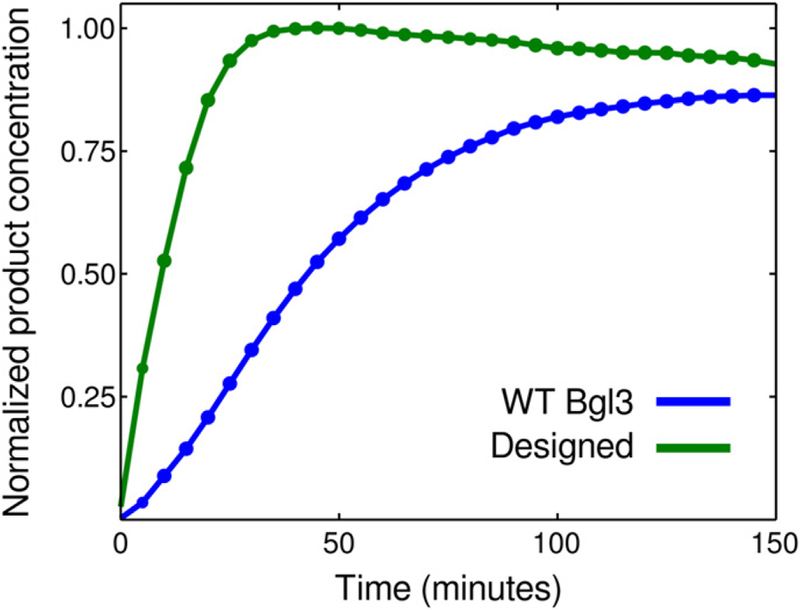
Kinetics 10 positive mutations used in the lab(base state/position/mutated state) and kinetics of designed BGL enzyme versus wild-type (WT) BGL sequence. The designed BGL enzyme based on mutations from Table 4 displays faster kinetics than the WT BGL sequence.
6. Conclusion
In this article, we developed the PUlasso algorithm for both variable selection and classification for high-dimensional classification with presence-only responses. Theoretically, we showed that our algorithm converges to a stationary point and every stationary point within a local neighborhood of θ* achieves an optimal mean squared error (up to constant). We also demonstrated that our algorithm performs well on both simulated and real data. In particular, our algorithm produces more accurate results than the existing techniques in simulations and performs well on a real biochemistry application.
Supplementary Material
Table 1.
Timings (in seconds).
| (n, p) | PUlasso | EM | Time reduction (%) | |
|---|---|---|---|---|
| Dense matrix | n = 1000, p = 10 | 0.94 | 443.72 | 99.79 |
| n = 5000, p = 50 | 2.52 | 1844.98 | 99.86 | |
| n = 10,000, p = 100 | 9.45 | 5066.86 | 99.81 | |
| Sparse matrix | n = 1000, p = 10 | 0.40 | 196.86 | 99.80 |
| n = 5000, p = 50 | 2.01 | 614.65 | 99.67 | |
| n = 10,000, p = 100 | 4.29 | 1201.09 | 99.64 |
NOTE: Sparsity level in X = 0.95, nℓ/nu = 0.5. Total time for 100 λ values, averaged over 3 runs.
Table 2.
Timings (in seconds) using sparse and dense calculation for fitting the same simulated data.
| (n, p) | Sparse calculation | Dense calculation | Time reduction (%) |
|---|---|---|---|
| n = 10,000, p = 100 | 12.91 | 19.24 | 32.89 |
| n = 30,000, p = 100 | 25.64 | 38.73 | 33.79 |
| n = 50,000, p = 100 | 39.47 | 57.18 | 30.97 |
NOTE: Sparsity level in X = 0.95, nℓ/nu = 0.5. Total time for 100 λ values, averaged over 3 runs.
Table 3.
Summary of stability scores across all tuning parameter λ values.
| 1st Qu. | Median | Mean | 3rd Qu. | |
|---|---|---|---|---|
| M | 93.3% | 94.9% | 94.9% | 96.8% |
| M+I | 97.9% | 98.8% | 98.4% | 99.3% |
Acknowledgments
Funding
Both HS and GR were partially supported by NSF-DMS 1407028. GR was also partially supported by ARO W911NF-17–1-0357.
Footnotes
Color versions of one or more of the figures in the article can be found online at www.tandfonline.com/r/JASA.
Supplementary Materials
In the supplementary material, we provide proofs of results in the Sections 2 and 3 of the main article. In addition, extra simulation results are included.
we note that the group ℓ1 constraint is active only if . If , by the ℓ1-ℓ2 inequality, that is, if ,. The other direction is trivial, and thus Θ0 is reduced to .
Available at http://www.ms.k.u-tokyo.ac.jp/software.html
The raw data is available in https://github.com/RomeroLab/seq-fcn-data.git
Supplementary materials for this article are available online. Please go to www.tandfonline.com/r/JASA.
References
- Blazère M, Loubes JM, and Gamboa F (2014), “Oracle Inequalities for a Group Lasso Procedure Applied to Generalized Linear Models in High Dimension,” IEEE Transactions on Information Theory, 60, 2303–2318. [Google Scholar]
- Breheny P, and Huang J (2013), “Group Descent Algorithms for Non-convex Penalized Linear and Logistic Regression Models With Grouped Predictors,” Statistics and Computing, 25, 173–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Marthinus P, Niu G, and Sugiyama M (2015), “Convex Formulation for Learning From Positive and Unlabeled Data,” in Proceedings of the 32nd International Conference on Machine Learning, pp. 1386–1394. [Google Scholar]
- Elkan C, and Noto K (2008), “Learning Classifiers From Only Positive and Unlabeled Data,” in Proceedings of the 14th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, KDD ‘08, New York, NY, USA: ACM, pp. 213–220. [Google Scholar]
- Elsener A, and van de Geer S (2018), “Sharp Oracle Inequalities for Stationary Points of Nonconvex Penalized M-Estimators,” arXiv no. 1802.09733.
- Fahrmeir L, and Kaufmann H (1985), “Consistency and Asymptotic Normality of the Maximum Likelihood Estimator in Generalized Linear Models,” The Annals of Statistics, 13, 342–368. [Google Scholar]
- Fowler DM, and Fields S (2014), “Deep Mutational Scanning: A New Style of Protein Science,” Nature Methods, 11, 801–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman J, Hastie T, Höfling H, and Tibshirani R (2007), “Pathwise Coordinate Optimization,” The Annals of Applied Statistics, 1, 302–332. [Google Scholar]
- Friedman J, Hastie T, and Tibshirani R (2010), “Regularization Paths for Generalized Linear Models via Coordinate Descent,” Journal of Statistical Software, 33, 1–22. [PMC free article] [PubMed] [Google Scholar]
- Hietpas RT, Jensen JD, and Bolon DNA (2011), “Experimental Illumination of a Fitness Landscape,” Proceedings of the National Academy of Sciences of the USA, 108, 7896–7901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Breheny P, and Ma S (2012), “A Selective Review of Group Selection in High-Dimensional Models,” Statistical Science, 27, 481–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S, White M, and Radivojac P (2017), “Recovering True Classifier Performance in Positive-Unlabeled Learning,” in Proceedings of the Thirty-First AAAI Conference on Artificial Intelligence, February 4–9, 2017, San Francisco, California, USA, pp. 2066–2072. [Google Scholar]
- Kakade S, Shamir O, Sindharan K, and Tewari A (2010), “Learning Exponential Families in High-Dimensions: Strong Convexity and Sparsity,” in Proceedings of the Thirteenth International Conference on Artificial Intelligence and Statistics, pp. 381–388. [Google Scholar]
- Kalousis A, Prados J, and Hilario M (2007), “Stability of Feature Selection Algorithms: A Study on High-Dimensional Spaces,” Knowledge and Information Systems, 12, 95–116. [Google Scholar]
- Krishnapuram B, Carin L, Figueiredo MAT, and Hartemink AJ (2005), “Sparse Multinomial Logistic Regression: Fast Algorithms and Generalization Bounds,” IEEE Transactions on Pattern Analysis and Machine Intelligence, 27, 957–968. [DOI] [PubMed] [Google Scholar]
- Lancaster T, and Imbens G (1996), “Case-Control Studies With Contaminated Controls,” Journal of Econometrics, 71, 145–160. [Google Scholar]
- Lange K, Hunter DR, and Yang I (2000), “Optimization Transfer Using Surrogate Objective Functions,” Journal of Computational and Graphical Statistics, 9, 1–20. [Google Scholar]
- Lee S, Lee H, Abbeel P, and Ng AY (2006), “Efficient L1 Regularized Logistic Regression,” in Proceedings of the Twenty-First National Conference on Artificial Intelligence (AAAI-06), pp. 1–9. [Google Scholar]
- Liu B, Dai Y, Li X, Lee WS, and Yu P (2006), “Building Text Classifiers Using Positive and Unlabeled Examples,” in Proceedings of the Third IEEE International Conference on Data Mining (ICDM’03). [Google Scholar]
- Loh P-L, and Wainwright MJ (2006), “Regularized M-Estimators With Nonconvexity: Statistical and Algorithmic Theory for Local Optima,” Journal of Machine Learning Research, 1, 1–9. [Google Scholar]
- McCullagh P, and Nelder JA (2006), Generalized Linear Models (Vol. 28), Boca Raton, FL: Chapman and Hall/CRC. [Google Scholar]
- Mei S, Bai Y, and Montanari A (2018), “The Landscape of Empirical Risk for Nonconvex Losses,” Annals of Statistics, 46, 2747–2774. [Google Scholar]
- Meier L, Van De Geer S, and Bühlmann P (2008), “The Group Lasso for Logistic Regression,” Journal of the Royal Statistical Society, Series B, 70, 53–71. [Google Scholar]
- Negahban SN, Pradeep R, Yu B, and Wainwright MJ (2012), “A Unified Framework for High-Dimensional Analysis of M-Estimators With Decomposable Regularizers,” Statistica Sinica, 27, 538–557. [Google Scholar]
- Ortega JM, and Rheinboldt WC (2000), Iterative Solution of Nonlinear Equations in Several Variables, Classics in Applied Mathematics, New York: SIAM. [Google Scholar]
- Puig AT, Wiesel A, Fleury G, and Hero AO (2011), “Multidimensional Shrinkage-Thresholding Operator and Group LASSO Penalties,” IEEE Signal Processing Letters, 18, 363–366. [Google Scholar]
- Raskutti G, Wainwright MJ, and Yu B (2010), “Restricted Eigenvalue Conditions for Correlated Gaussian Designs,” Journal of Machine Learning Research, 11, 2241–2259. [Google Scholar]
- Raskutti G, Wainwright MJ, and Yu B (2011), “Minimax Rates of Estimation for High-Dimensional Linear Regression Over 4q-Balls,” IEEE Transactions on Information Theory, 57, 6976–6994. [Google Scholar]
- Romero PA, Tran TM, and Abate AR (2015), “Dissecting Enzyme Function With Microfluidic-Based Deep Mutational Scanning,” Proceedings of the National Academy of Sciences of the USA, 112, 7159–7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon N, and Tibshirani R (2012), “Standardization and the Group Lasso Penalty,” Statistica Sinica, 22, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibshirani R, Bien J, Friedman J, Hastie T, Simon N, Taylor J, and Tibshirani RJ (2012), “Strong Rules for Discarding Predictors in Lasso- Type Problems,” Journal of the Royal Statistical Society, Series B, 74, 245–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Geer SA (2008), “High-Dimensional Generalized Linear Models and the Lasso,” The Annals of Statistics, 36, 614–645. [Google Scholar]
- Ward G, Hastie T, Barry S, Elith J, and Leathwick JR (2009), “Presence-Only Data and the EM Algorithm,” Biometrics, 65, 554–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TT, and Lange K (2008), “Coordinate Descent Algorithms for Lasso Penalized Regression,” The Annals of Applied Statistics, 2, 224–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan M, and Lin Y (2006), “Model Selection and Estimation in Regression With Grouped Variables,” Journal of the Royal Statistical Society, Series B, 68, 49–67. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.