

Abstract
Wilms tumor (WT) is the most common renal malignancy of childhood and accounts for 6% of all childhood malignancies. With current therapies, the 5-yr overall survival (OS) for children with unilateral favorable histology WT is greater than 85%. The prognosis is worse, however, for the roughly 15% of patients who relapse, with only 50%–80% OS reported in those with recurrence. Herein, we describe the extended and detailed clinical course of a rare case of a child with recurrent, pulmonary metastatic, favorable histology WT harboring a BRAF V600E mutation. The BRAF V600E mutation, commonly found in melanoma and other cancers, and previously undescribed in WT, has recently been reported by our group in a subset of epithelial-predominant WT. This patient, who was included in that series, presented with unilateral, stage 1, favorable histology WT and was treated with standard chemotherapy. Following the completion of therapy, the patient relapsed with pulmonary metastatic disease, that then again recurred despite an initial response to salvage chemotherapy and radiation. Next-generation sequencing (NGS) on the metastatic pulmonary nodule revealed a BRAF V600E mutation. After weighing the therapeutic options, a novel approach with dual BRAF/MEK inhibitor combination therapy was initiated. Complete radiographic response was observed following 4 months of therapy with dabrafenib and trametinib. At 12 months following the start of BRAF/MEK combination treatment, the patient continues with a complete response and has experienced minimal treatment-related side effects. This represents the first case, to our knowledge, of effective treatment with BRAF/MEK molecularly targeted therapy in a pediatric Wilms tumor patient.
Keywords: nephroblastoma (Wilms tumor)
INTRODUCTION
Wilms tumor (WT) is the most common renal malignancy of childhood, occurring in 1 in 10,000 children (Doganis et al. 2019) and accounting for 6% of childhood malignancies (Davidoff 2012). Current strategies to treat WT include chemotherapy, surgery, and radiation therapy. This multimodal approach, along with advances in diagnostic evaluation and optimal risk stratification, has led to an increase in the 5-year overall survival for children with unilateral favorable histology WT to greater than 85% (Dome et al. 2013).
Numerous somatic genetic alterations associated with WT have been identified, the most common of which involve WT1, CTNNB1 (β-catenin), DROSHA, AMER1 (WTX), and TP53 (in anaplastic histology WT). Additionally, epigenetic alterations involving IGF2 are commonly observed (Kalapurakal et al. 2004). Mutations in genes that encode elements of the RAS-BRAF-ERK signaling pathway, however, have not been previously associated with WT. BRAF V600E mutations, which are commonly found in melanoma, colon cancer, and other cancers, have been described in the majority of metanephric neoplasms, which morphologically overlap with differentiated forms of WT (Argani et al. 2016). The family of metanephric neoplasms of the kidney include metanephric adenoma (MA) and metanephric stromal tumor (MST), which harbor BRAF V600E mutations in 90% and 65% of cases, respectively (Chami et al. 2015; Udager et al. 2015). We recently described the novel finding of BRAF V600E mutations occurring in epithelial predominant WT with differentiated areas overlapping with MA in children and adults and noted briefly that one child had responded to BRAF-targeted therapy (Wobker et al. 2019). The efficacy of BRAF/MEK inhibition in BRAF V600E-mutated WT has not been reported previously. A basket trial of nonmelanoma cancers with BRAF V600E mutations demonstrated a modest response rate to inhibition of mutant BRAF in a wide range of tumors (Hyman et al. 2015). Herein, we describe the details of the clinical response to dual BRAF/MEK inhibition in a pediatric patient with metastatic epithelial-predominant WT with BRAF V600E mutation. The imaging, pathologic findings, molecular findings, and therapeutic approach are discussed.
RESULTS
Clinical Presentation
This case was previously included in our initial study of BRAF-mutated epithelial predominant WT with MA-like areas (Wobker et al. 2019; Case 8), but with only minimal clinical information. A 6-yr-old boy presented with right flank pain and hematuria. Abdominal computed tomography (CT) revealed a 7.4 × 8.4-cm mass in the right kidney (Fig. 1). No other sites of disease were identified. The patient underwent immediate right radical nephrectomy, as per North American practice, and histopathologic analysis of the specimen revealed a stage 1 WT of favorable histology. Of note, the tumor was triphasic but epithelial-predominant and contained differentiated areas that overlapped morphologically with MA (Fig. 2A–C). The patient received chemotherapy according to the National Wilms Tumor Study-5, Regimen EE-4A (vincristine, dactinomycin). Following an unremarkable 5-month treatment course, the patient was considered to have no evidence of disease. A surveillance chest CT scan performed 5 months post–completion of planned therapy revealed a solitary 3-mm left upper lobe (LUL) lung nodule of uncertain significance. Additional imaging obtained 3 months later showed an increase in size to 5 mm, without evidence of tumor recurrence in the abdomen or pelvis. Excisional biopsy of the pulmonary nodule was performed, and pathologic examination revealed metastatic WT (Fig. 2D). Next-generation sequencing (NGS), using the Johns Hopkins institutional Solid Tumor Panel version 3.0 (Dalton et al. 2017), on the pulmonary nodule demonstrated a BRAF V600E mutation, without other actionable mutations (Tables 1 and 2; Supplemental Table 1).
Figure 1.
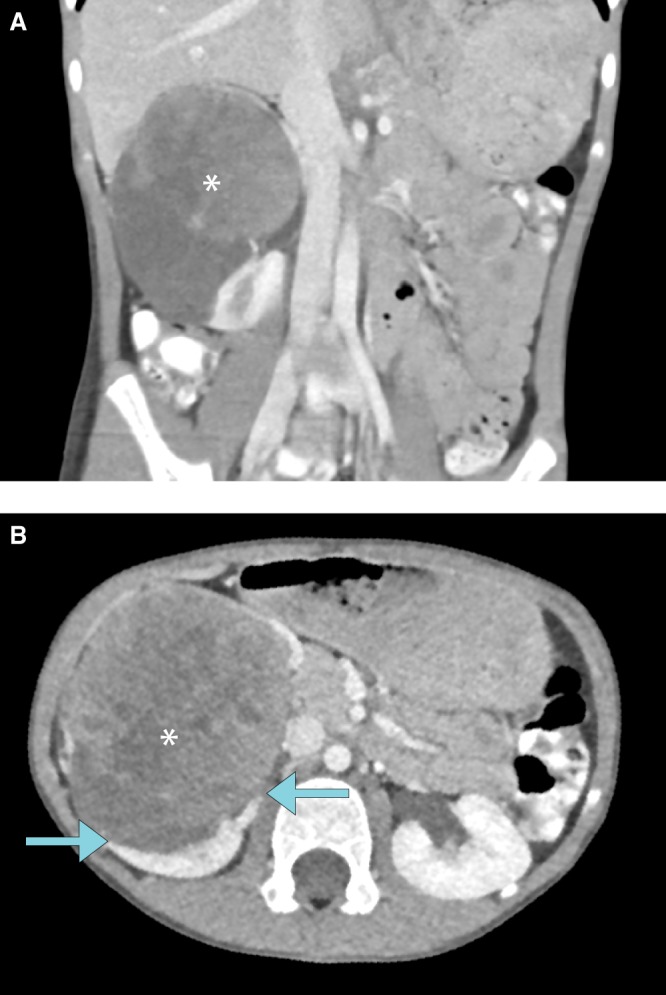
(A) Coronal contrast-enhanced computed tomography (CECT) image of the abdomen demonstrates a heterogeneously enhancing mass measuring 7.3 × 8.4 × 8.4 cm and arising from the right kidney. Calcific foci are present within the mass (not shown). (B) Origin of the mass from the right kidney, as indicated by the “claw sign” (arrows), is redemonstrated on the axial CT image.
Figure 2.

(A) The nephrectomy revealed an epithelial predominant Wilms tumor (WT). However, there was a differentiated area resembling metanephric adenoma (MA) associated with sclerosis (upper right). Normal kidney adjacent to tumor is at the upper left. (B) WT, triphasic area. Note the mitotic figures. (C) WT, differentiated area mimicking MA. Note the absence of mitotic figures and minimal cytoplasm of the neoplastic cells, which form tubules. (D) Lung metastasis of WT. (E) Cytoplasmic BRAF V600E protein immunoreactivity in the primary renal WT. (F) Cytoplasmic BRAF V600E protein immunoreactivity in the metastatic WT in the lung.
Table 1.
Genomic variants identified using the Johns Hopkins Solid Tumor Panel v. 3.0: Genomic analysis
| Chr:Pos | Base change | Reference database ID | Gene | AA_ change | VAF (%) |
|---|---|---|---|---|---|
| Chr 7:140453136 | A > T | COSM476 | BRAF | p.V600E | 45.14 |
(VAF) Variant allele frequency.
Table 2.
Variant table
| Gene | Chromosome | HGVS DNA reference | HGVS protein reference | Variant type (substitution, deletion, etc.) | Predicted effect | dbSNP/dbVar ID | Genotype (heterozygous/homozygous) |
|---|---|---|---|---|---|---|---|
| ATR | Chr 3:142178067 | NM_001184.3: c.7349 + 2T > C | NA (splicing variant) | Substitution (splicing) | VUS | rs200556378; not reported in ClinVar | Heterozygous |
| BRAF | Chr 7:140453136 | NM_004333.4: c.1799T > A | p.V600E | Substitution (missense) | Activation | rs113488022; ClinVar (29000) | Likely heterozygous |
| CDH1 | Chr 16:68835713 | NM_004360.4: c.304G > A | p.A102T | Substitution (missense) | Benign/Likely benign/VUS | rs368492235; ClinVar (152292) | Heterozygous |
| DICER1 | Chr 14:95574707 | NM_177438.2: c.2390A > G | p.D797G | Substitution (missense) | VUS | rs755375348; ClinVar (463780) | Heterozygous |
| DUSP9 | Chr X:152914768 | NM_001395.3: c.455C > T | p.P152L | Substitution (missense) | VUS | rs782504547; not reported in ClinVar | NA (X chromosome in male) |
| EP300 | Chr 22:41574829 | NM_001429.3: c.7114A > G | p.M2372V | Substitution (missense) | VUS | rs768061933; not reported in ClinVar | Likely heterozygous |
| FAS | Chr 10:90762940 | NM_000043.5: c.185C > G | p.P62R | Substitution (missense) | VUS | rs757780022; not reported in ClinVar | Heterozygous |
| GPR124 | Chr 8:37697730 | NM_032777.9: c.2603C > T | p.A868V | Substitution (missense) | VUS | rs1443346346; not reported in ClinVar | Heterozygous |
| MAFB | Chr 20:39316965 | NM_005461.4: c.526G > A | p.A176T | Substitution (missense) | VUS | rs750186410; not reported in ClinVar | Heterozygous |
| MYO18A | Chr 17:27417892 | NM_078471.3: c.5240G > A | p.R1747Q | Substitution (missense) | VUS | rs767048813; not reported in ClinVar | Heterozygous |
| PASK | Chr 2:242066165 | NM_015148.3: c.2165T > C | p.L722P | Substitution (missense) | VUS | rs201982321; not reported in ClinVar | Heterozygous |
| PCLO | Chr 7:82474598 | NM_033026.5: c.14035G > C | p.G4679R | Substitution (missense) | VUS | Not reported in ClinVar | Heterozygous |
| SYNE1 | Chr 6:152631852 | NM_182961.3: c.16867C > T | p.R5623C | Substitution (missense) | VUS | rs570556738; not reported in ClinVar | Heterozygous |
| TCF3 | Chr 19:1615488 | NM_003200.3: c.1618C > G | p.P540A | Substitution (missense) | VUS | rs778885981; not reported in ClinVar | Heterozygous |
(HGVS) Human Genome Variation Society, (dbSNP) Single Nucleotide Polymorphism Database, (dbVar) Database of Genomic Structural Variation, (VUS) variant of uncertain significance, (NA) not applicable.
Relapse therapy consisted of radiation therapy to the lungs (12 Gy) and chemotherapy per Children's Oncology Group protocol AREN0321 Regimen I (vincristine, doxorubicin, cyclophosphamide, etoposide). The patient completed 6 months of therapy and then was again considered to be without evidence of disease. Surveillance CT scan performed 3 months after completion of planned treatment, revealed at least ten 2- to 3-mm pulmonary nodules concerning for second metastatic relapse (Fig. 3A). Therapeutic options were considered, including conventional chemotherapy (including vincristine, irinotecan, carboplatin) followed by high-dose chemotherapy with autologous stem cell rescue (ASCR), as this approach has demonstrated efficacy in patients with WT in first relapse. Alternatively, a novel approach using molecularly targeted agents was considered, given the finding of BRAF V600E detected in the first metastatic tumor via NGS. The original kidney tumor and the lung metastasis were examined and confirmed to express BRAF V600E via immunohistochemistry (Fig. 2E,F). This finding lent support to the idea that BRAF V600E was an early event in the original tumor and not unique to the resected, first pulmonary metastasis. Given the expectation of a manageable toxicity profile, compared to chemotherapy followed by ASCR, combined BRAF and MEK inhibitor therapy was commenced. The patient was dosed according to recommended phase 2 doses (RP2D) determined in the phase 1 studies of these agents (McCowage et al. 2018; Kieran et al. 2019).
Figure 3.

Axial nonenhanced chest CT (NECT) images demonstrating metastatic pulmonary nodules in the same region of the right upper lobe over a period of time on treatment, including (A) baseline (week 0), with multiple (>10) 2- to 3-mm nodules, (B) after 8 weeks of treatment, with a decrease in size of several nodules and resolution of others, (C) after 16 weeks of treatment, with complete resolution of all previously visualized nodules, and (D) 6 months after initiation of treatment on combined dabrafenib/trametinib, with continued complete imaging response to treatment.
Treatment Outcome
The patient began treatment with dabrafenib, a selective inhibitor of mutant BRAF, and trametinib, a selective allosteric inhibitor of MEK1/2. Chest CT obtained 8 weeks after start of therapy demonstrated resolution of several previously identified pulmonary nodules, and decrease in size of the remaining nodules (Fig. 3A,B). Additional imaging obtained at 16 weeks on therapy revealed complete radiographic resolution of all previously identified pulmonary nodules (Fig. 3C), and no new evidence of tumor recurrence. The patient remains in a complete radiographic response, now 12 months after starting therapy (Fig. 3D). He continues to receive dabrafenib and trametinib with minimal treatment-emergent toxicities. Two episodes of pyrexia have been attributed to dabrafenib/trametinib treatment, each of several days’ duration and resolving in less than 48 h upon holding medication, at which point the patient resumed treatment at the previous dose.
DISCUSSION
WT is a malignant embryonal neoplasm originating from metanephric blastema (Argani and Beckwith 2015). Until recently, knowledge of the genetic basis for WT was restricted to aberrations of WT1, abnormalities of 11p15 methylation, and mutations that activate the WNT signaling pathway (CTNNB1, AMER1/WTX). Recent comprehensive genomic analyses of WT have identified additional mutations involved in Wilms tumorigenesis, including DROSHA, DGCR8, DICER1, SIX1, SIX2, MYCN, and MLLT1 (Gadd et al. 2017). No clinically targetable oncogene mutations, however, have been identified in population-based sets of WT cases (Dalpa et al. 2017; Gadd et al. 2017). Recently, BRAF V600E mutations have been described in a subset of renal malignancies with overlapping histologies of WT and MA (Wobker et al. 2019). MA was previously thought to be a differentiated form of epithelial WT (Argani et al. 2016), with BRAF mutations occurring in upward of 85% of cases (Chami et al. 2015; Udager et al. 2015). A case series (including this case) from our institution described the novel finding of epithelial-predominant WT with areas of MA with BRAF V600E mutations (Wobker et al. 2019). This finding raised the possibility that it could represent a targetable alteration that could yield a sustained clinical response.
BRAF (sarcoma viral oncogene homolog B) is a member of the RAF serine/threonine protein kinase family and functions to transmit signals from upstream RAS to regulate ERK activity through direct phosphorylation of its substrates MEK1 and MEK2 (Ikawa et al. 1988; Marais and Marshall 1996). BRAF is commonly mutated in cancer, with the most common mutation (80%–90% of cases) a T to A transversion at nucleotide 1799 (T1799A), which results in a substitution of valine (V) for glutamic acid (E) at codon 600 (V600E) (Davies et al. 2002). The resulting mutation results in elevated BRAF kinase activity and thereby constitutive activation of ERK signaling (Wan et al. 2004). BRAF mutations are most often seen in melanoma, thyroid and colon carcinomas, and histiocytic disorders; they occur less frequently in breast and lung carcinoma, sarcomas, gliomas, leukemias, and lymphomas (Brose et al. 2002; Davies et al. 2002; Kimura et al. 2003). BRAF mutations have been shown to induce cellular transformation and increased cell survival in many model systems (Pritchard et al. 2007).
Vemurafenib was the first selective ATP-competitive inhibitor of mutant BRAF to gain FDA approval, based on demonstration of improved progression-free survival (PFS) seen in patients with metastatic BRAF-mutated melanoma compared to dacarbazine (McArthur et al. 2014). Similarly, dabrafenib was studied in patients with BRAF V600E-mutated metastatic melanoma and resulted in significantly improved PFS compared to dacarbazine (Hauschild et al. 2012). Dabrafenib gained FDA approval for treatment of unresectable or metastatic melanoma with BRAF V600E mutation in 2013.
Since then, BRAF inhibitors have demonstrated efficacy in a number of other tumor types, including colon (limited benefit as single agent) (Falchook et al. 2012; Kopetz et al. 2015); lung cancer (Planchard et al. 2016b) (FDA-approved); and histiocytic disorders (Langerhans cell histiocytosis [LCH] and Erdheim–Chester disease [ECD]) (Haroche et al. 2015). Among a histology-agnostic population of patients enrolled in a BRAF V600E selected basket trial, an overall objective response rate of 42% was seen in patients with non-small-cell lung cancer (NSCLC) and 43% in patients with ECD or LCH, confirming the driver role of the oncogene in a diverse selection of tumor types (Hyman et al. 2015). Likewise, a pediatric histology-agnostic phase 1 trial of dabrafenib in BRAF V600E tumors has been conducted in patients with low- or high-grade gliomas, LCH, and thyroid cancer (Kieran et al. 2019).
Despite the initial exciting responses seen in patients with BRAF-mutant cancers treated with vemurafenib and dabrafenib, most patients developed acquired resistance over time. In addition, in cells with wild-type RAF, including normal tissues, RAF inhibitors induce ERK signaling through transactivation of CRAF leading to MEK activation (Hatzivassiliou et al. 2010; Heidorn et al. 2010; Joseph et al. 2010; Poulikakos et al. 2010); this phenomenon underlies the spectrum of treatment-emergent hyperproliferative toxicities that may be seen (Su et al. 2012). MEK inhibitors in combination with BRAF inhibitors were trialed as a strategy to overcome both acquired resistance as well as paradoxical MEK activation (Planchard et al. 2016a). Dual pathway inhibition (dabrafenib plus trametinib) showed superior efficacy to dabrafenib monotherapy in V600E metastatic melanoma (Long et al. 2015) and NSCLC (Planchard et al. 2016a). Recent publications report responses to dabrafenib plus trametinib in patients with high-grade gliomas (Schreck et al. 2018; Toll et al. 2019). A phase 2 study investigating the activity of dabrafenib in combination with trametinib in pediatric patients with low-grade glioma or refractory high-grade glioma with BRAF V600 mutation (NCT02684058) is ongoing (Hargrave et al. 2018).
Dabrafenib plus trametinib in combination has been well-tolerated, with rash, fatigue, joint pain, photosensitivity, and pyrexia being the most frequently reported toxicities (Grimaldi et al. 2015). High-dose chemotherapy followed by ASCR is established as effective therapy for first relapse in patients with high-risk or very-high-risk relapse, and is not required in standard-risk patients—that is, those treated initially with VCR/actinomycin (Ha et al. 2013). Its role, however, is not established in patients in second relapse. Further, high-dose chemotherapy with ASCR would have been accompanied by prolonged cytopenias, hospitalizations, and potentially life-threatening complications, with unproven benefit either in second relapse or in the unique subset of WT with BRAF mutations. Based on these factors, combination therapy with dabrafenib and trametinib was an attractive therapeutic option in the case of our patient.
This report provides clinical details of the first case, to our knowledge, of a complete response to RAF/MEK inhibition in a pediatric patient with metastatic epithelial-predominant BRAF V600E-mutated WT. This case adds to the ongoing discovery of driver mutations in WT and highlights the importance of adding molecular diagnostic studies to the histologic diagnosis. The finding of a BRAF V600E mutation allowed for the application of a targeted therapy that had not been previously reported in a child with WT. Of note, a recent single-patient case report described the rare case of an adult with metastatic epithelial-predominant WT with BRAF V600E mutation who achieved a partial response with BRAF inhibitor therapy (de Vries-Brilland et al. 2019). Together, these findings highlight the utility of histology-independent genomically selected basket studies for rare tumor types with targetable mutations. Objective responses seen across tissue types support the use of targeted therapies in tumor types which otherwise may not be studied in a tissue-specific trial.
Several questions remain unanswered, however. The optimal duration of combination therapy following a sustained CR is not established. The feasibility of maintaining the response remains to be seen, as acquired resistance has been shown to develop with dual RAF/MEK inhibition, as with single-agent RAF inhibitors. The majority of melanoma patients receiving RAF/MEK inhibitors will relapse within 1 year following an initial response (Arozarena and Wellbrock 2017). Various resistance mechanisms have been described with the majority of them converging on ERK reactivation (Nazarian et al. 2010; Villanueva et al. 2010; Poulikakos et al. 2011; Samatar and Poulikakos 2014; Wang et al. 2018). Preclinical studies investigating an optimal dosing schedule to prevent the development of resistance have suggested that intermittent dosing may forestall tumor adaptive changes (Abdel-Wahab et al. 2014). A new class of RAF dimer inhibitors is under clinical investigation and their use following the emergence of resistance to first-generation RAF inhibitors warrants continued exploration (Peng et al. 2015; Yao et al. 2015, 2019; Zhang et al. 2015). One could also consider the possibility of ASCR therapy to consolidate a response to molecularly targeted therapy, but this approach is one that has not yet been tested or validated in clinical trials.
Pediatric WT with BRAF V600E mutation has only recently been described. This is the first case of a patient with metastatic recurrence responsive to therapeutic RAF/MEK inhibition. This case supports the use of RAF/MEK inhibition as a therapeutic option in an increasing range of tumor types with BRAF V600E mutation.
METHODS
DNA was isolated from formalin-fixed paraffin-embedded tumor tissue using the automated Siemens Tissue Preparation System (Siemens Healthcare Diagnostics). NGS was also conducted using the HiSeq 2500 platform (Illumina). Briefly, libraries were prepared using the SureSelect-XT Target Enrichment System (Agilent Technologies) and an Agilent custom panel covering full coding regions of 432 cancer-related genes for clinical reporting of solid tumors. Sequencing was performed to an average 500×–1000× read depth on the HiSeq 2500 platform (Illumina). All reads were aligned to the human genome (GRCh37/hg19), using the Burrows–Wheeler alignment (BWA) algorithm. BMA files were generated using Picard Tools v1.119, and variant calling was performed using an in-house variant caller algorithm (MDLVC v5.0) and the HaplotypeCaller (Genome Analysis Tool Kit 3.3). Variants were reviewed using the Integrated Genomics Viewer (Broad Institute) and annotated using the Single Nucleotide Polymorphism Database (dbSNP) and Catalogue of Somatic Mutations in Cancer (COSMIC) database (Gondek et al. 2016; Palsgrove et al. 2018).
ADDITIONAL INFORMATION
Data Deposition and Access
Coding variants identified in the Johns Hopkins Solid Tumor Panel analysis are reported in the body of the manuscript. The patient did not provide consent for public deposition of all raw sequencing data for the 400 genes included in the Solid Tumor Panel version 3.0. The variants were submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and can be found under accession numbers SCV001147030–SCV001147043.
Ethics Statement
The minor patient's parent has provided written consent to publication of relevant clinical information presented in the manuscript and was given the opportunity to read the manuscript prior to publication. The patient's parent understands that material will be freely available on the web and can be freely redistributed and that complete anonymity cannot be guaranteed.
Acknowledgments
The authors acknowledge Ming-Tseh Lin and Christopher Gocke for assistance with NGS data. We additionally acknowledge the patient and his family.
Author Contributions
S.S., D.S.R., J.S.D., A.D.F., and C.A.P. were responsible for patient care and clinical decision-making. P.O. and C.A.P. were responsible for the concept and design. E.D. provided radiographic interpretation and figures. P.A. provided pathologic diagnosis and figures. L.J. was responsible for the next-generation sequencing assay. P.O. and C.A.P. wrote the manuscript. All authors performed a final review of the manuscript.
Funding
The authors have no funding sources to acknowledge.
Competing Interest Statement
C.A.P. is a paid consultant for Genentech/Roche (cobimetinib).
Supplementary Material
Footnotes
[Supplemental material is available for this article.]
REFERENCES
- Abdel-Wahab O, Klimek VM, Gaskell AA, Viale A, Cheng D, Kim E, Rampal R, Bluth M, Harding JJ, Callahan MK. 2014. Efficacy of intermittent combined RAF and MEK inhibition in a patient with concurrent BRAF- and NRAS-mutant malignancies. Cancer Discov 4: 538–545. 10.1158/2159-8290.CD-13-1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argani P, Beckwith JB. 2015. Renal neoplasms of childhood. In Sternberg's diagnostic surgical pathology, 6th ed. (ed. Mills SE), pp. 201–846 Wolters Kluwer Health Adis (ESP), Philadelphia. [Google Scholar]
- Argani P, Lee J, Netto GJ, Zheng G, Tseh-Lin M, Park BH. 2016. Frequent BRAF V600E mutations in metanephric stromal tumor. Am J Surg Pathol 40: 719–722. 10.1097/PAS.0000000000000603 [DOI] [PubMed] [Google Scholar]
- Arozarena I, Wellbrock C. 2017. Overcoming resistance to BRAF inhibitors. Ann Transl Med 5: 387 10.21037/atm.2017.06.09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brose MS, Volpe P, Feldman M, Kumar M, Rishi I, Gerrero R, Einhorn E, Herlyn M, Minna J, Nicholson A. 2002. BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res 62: 6997–7000. [PubMed] [Google Scholar]
- Chami R, Yin M, Marrano P, Teerapakpinyo C, Shuangshoti S, Thorner PS. 2015. BRAF mutations in pediatric metanephric tumors. Hum Pathol 46: 1153–1161. 10.1016/j.humpath.2015.03.019 [DOI] [PubMed] [Google Scholar]
- Dalpa E, Gourvas V, Soulitzis N, Spandidos DA. 2017. K-Ras, H-Ras, N-Ras and B-Raf mutation and expression analysis in Wilms tumors: association with tumor growth. Med Oncol 34: 6 10.1007/s12032-016-0862-5 [DOI] [PubMed] [Google Scholar]
- Dalton WB, Forde PM, Kang H, Connolly RM, Stearns V, Gocke CD, Eshleman JR, Axilbund J, Petry D, Geoghegan C, et al. 2017. Personalized medicine in the oncology clinic: implementation and outcomes of the Johns Hopkins molecular tumor board. JCO Precis Oncol 2017 10.1200/PO.16.00046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidoff AM. 2012. Wilms tumor. Adv Pediatr 59: 247–267. 10.1016/j.yapd.2012.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W. 2002. Mutations of the BRAF gene in human cancer. Nature 417: 949 10.1038/nature00766 [DOI] [PubMed] [Google Scholar]
- de Vries-Brilland M, Sionneau B, Dutriaux C, Blay J-Y, Ravaud A, Gross-Goupil M. 2019. Successful treatment of metastatic adult Wilms tumor with anti-BRAF treatment: a case report and a brief review of the literature. Clin Genitourin Cancer 17: e721–e723. 10.1016/j.clgc.2019.02.012 [DOI] [PubMed] [Google Scholar]
- Doganis D, Zborovskaya A, Trojanowski M, Zagar T, Bouka P, Baka M, Moschovi M, Polychronopoulou S, Papakonstantinou E, Tragiannidis A. 2019. Wilms tumour event–free and overall survival in Southern and Eastern Europe: pooled analyses of clinical data from four childhood cancer registries (1999–2017). Eur J Cancer 115: 37–46. 10.1016/j.ejca.2019.04.008 [DOI] [PubMed] [Google Scholar]
- Dome JS, Fernandez CV, Mullen EA, Kalapurakal JA, Geller JI, Huff V, Gratias EJ, Dix DB, Ehrlich PF, Khanna G. 2013. Children's Oncology Group's 2013 blueprint for research: renal tumors. Pediatr Blood Cancer 60: 994–1000. 10.1002/pbc.24419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falchook GS, Long GV, Kurzrock R, Kim KB, Arkenau TH, Brown MP, Hamid O, Infante JR, Millward M, Pavlick AC, et al. 2012. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet 379: 1893–1901. 10.1016/S0140-6736(12)60398-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadd S, Huff V, Walz AL, Ooms AH, Armstrong AE, Gerhard DS, Smith MA, Auvil JMG, Meerzaman D, Chen Q-R. 2017. A Children's Oncology Group and TARGET initiative exploring the genetic landscape of Wilms tumor. Nat Genet 49: 1487 10.1038/ng.3940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gondek LP, Zheng G, Ghiaur G, DeZern AE, Matsui W, Yegnasubramanian S, Lin MT, Levis M, Eshleman JR, Varadhan R, et al. 2016. Donor cell leukemia arising from clonal hematopoiesis after bone marrow transplantation. Leukemia 30: 1916–1920. 10.1038/leu.2016.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimaldi AM, Simeone E, Festino L, Vanella V, Palla M, Ascierto PA. 2015. Novel mechanisms and therapeutic approaches in melanoma: targeting the MAPK pathway. Discov Med 19: 455–461. [PubMed] [Google Scholar]
- Ha TC, Spreafico F, Graf N, Dallorso S, Dome JS, Malogolowkin M, Furtwängler R, Hale JP, Moroz V, Machin D, et al. 2013. An international strategy to determine the role of high dose therapy in recurrent Wilms’ tumour. Eur J Cancer 49: 194–210. 10.1016/j.ejca.2012.07.010 [DOI] [PubMed] [Google Scholar]
- Hargrave D, Witt O, Cohen K, Packer R, Lissat A, Kordes U, Laetsch T, Hoffman L, Lassaletta A, Gerber N. 2018. 407TiP Phase II open-label, global study evaluating dabrafenib in combination with trametinib in pediatric patients with BRAF V600–mutant high-grade glioma (HGG) or low-grade glioma (LGG). Ann Oncol 29: mdy273.395 10.1093/annonc/mdy273.395 [DOI] [Google Scholar]
- Haroche J, Cohen-Aubart F, Emile JF, Maksud P, Drier A, Tolédano D, Barete S, Charlotte F, Cluzel P, Donadieu J, et al. 2015. Reproducible and sustained efficacy of targeted therapy with vemurafenib in patients with BRAFV600E-mutated Erdheim–Chester disease. J Clin Oncol 33: 411–418. 10.1200/JCO.2014.57.1950 [DOI] [PubMed] [Google Scholar]
- Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, Ludlam MJ, Stokoe D, Gloor SL, Vigers G, et al. 2010. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 464: 431–435. 10.1038/nature08833 [DOI] [PubMed] [Google Scholar]
- Hauschild A, Grob J-J, Demidov LV, Jouary T, Gutzmer R, Millward M, Rutkowski P, Blank CU, Miller WH Jr, Kaempgen E. 2012. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 380: 358–365. 10.1016/S0140-6736(12)60868-X [DOI] [PubMed] [Google Scholar]
- Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, Hussain J, Reis-Filho JS, Springer CJ, Pritchard C, et al. 2010. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 140: 209–221. 10.1016/j.cell.2009.12.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman DM, Puzanov I, Subbiah V, Faris JE, Chau I, Blay J-Y, Wolf J, Raje NS, Diamond EL, Hollebecque A. 2015. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med 373: 726–736. 10.1056/NEJMoa1502309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikawa S, Fukui M, Ueyama Y, Tamaoki N, Yamamoto T, Toyoshima K. 1988. B-raf, a new member of the raf family, is activated by DNA rearrangement. Mol Cell Biol 8: 2651–2654. 10.1128/MCB.8.6.2651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph EW, Pratilas CA, Poulikakos PI, Tadi M, Wang W, Taylor BS, Halilovic E, Persaud Y, Xing F, Viale A, et al. 2010. The RAF inhibitor PLX4032 inhibits ERK signaling and tumor cell proliferation in a V600E BRAF-selective manner. Proc Natl Acad Sci 107: 14903–14908. 10.1073/pnas.1008990107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalapurakal JA, Dome JS, Perlman EJ, Malogolowkin M, Haase GM, Grundy P, Coppes MJ. 2004. Management of Wilms’ tumour: current practice and future goals. Lancet Oncol 5: 37–46. 10.1016/S1470-2045(03)01322-6 [DOI] [PubMed] [Google Scholar]
- Kieran MW, Geoerger B, Dunkel IJ, Broniscer A, Hargrave DR, Hingorani P, Aerts I, Bertozzi AI, Cohen KJ, Hummel TR, et al. 2019. A phase 1 and pharmacokinetic study of oral dabrafenib in children and adolescent patients with recurrent or refractory BRAF V600 mutation-positive solid tumors. Clin Cancer Res 25: 7294–7302. 10.1158/1078-0432.CCR-17-3572 [DOI] [PubMed] [Google Scholar]
- Kimura ET, Nikiforova MN, Zhu Z, Knauf JA, Nikiforov YE, Fagin JA. 2003. High prevalence of BRAF mutations in thyroid cancer: genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res 63: 1454–1457. [PubMed] [Google Scholar]
- Kopetz S, Desai J, Chan E, Hecht JR, O'Dwyer PJ, Maru D, Morris V, Janku F, Dasari A, Chung W, et al. 2015. Phase II pilot study of vemurafenib in patients with metastatic BRAF-mutated colorectal cancer. J Clin Oncol 33: 4032–4038. 10.1200/JCO.2015.63.2497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long GV, Stroyakovskiy D, Gogas H, Levchenko E, De Braud F, Larkin J, Garbe C, Jouary T, Hauschild A, Grob J-J. 2015. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. Lancet 386: 444–451. 10.1016/S0140-6736(15)60898-4 [DOI] [PubMed] [Google Scholar]
- Marais R, Marshall CJ. 1996. Control of the ERK MAP kinase cascade by Ras and Raf. Cancer Surv 27: 101–125. [PubMed] [Google Scholar]
- McArthur GA, Chapman PB, Robert C, Larkin J, Haanen JB, Dummer R, Ribas A, Hogg D, Hamid O, Ascierto PA. 2014. Safety and efficacy of vemurafenib in BRAFV600E and BRAFV600K mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol 15: 323–332. 10.1016/S1470-2045(14)70012-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCowage GB, Mueller S, Pratilas CA, Hargrave DR, Moertel CL, Whitlock J, Fox E, Hingorani P, Russo MW, Dasgupta K, et al. 2018. Trametinib in pediatric patients with neurofibromatosis type 1 (NF-1)–associated plexiform neurofibroma: a phase I/IIa study. J Clin Oncol 36: 10504 10.1200/JCO.2018.36.15_suppl.10504 [DOI] [Google Scholar]
- Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, et al. 2010. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 468: 973–977. 10.1038/nature09626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palsgrove DN, Brosnan-Cashman JA, Giannini C, Raghunathan A, Jentoft M, Bettegowda C, Gokden M, Lin D, Yuan M, Lin MT, et al. 2018. Subependymal giant cell astrocytoma-like astrocytoma: a neoplasm with a distinct phenotype and frequent neurofibromatosis type-1-association. Mod Pathol 31: 1787–1800. 10.1038/s41379-018-0103-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng SB, Henry JR, Kaufman MD, Lu WP, Smith BD, Vogeti S, Rutkoski TJ, Wise S, Chun L, Zhang Y, et al. 2015. Inhibition of RAF isoforms and active dimers by LY3009120 leads to anti-tumor activities in RAS or BRAF mutant cancers. Cancer Cell 28: 384–398. 10.1016/j.ccell.2015.08.002 [DOI] [PubMed] [Google Scholar]
- Planchard D, Besse B, Groen HJ, Souquet P-J, Quoix E, Baik CS, Barlesi F, Kim TM, Mazieres J, Novello S. 2016a. Dabrafenib plus trametinib in patients with previously treated BRAFV600E-mutant metastatic non–small cell lung cancer: an open-label, multicentre phase 2 trial. Lancet Oncol 17: 984–993. 10.1016/S1470-2045(16)30146-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planchard D, Kim TM, Mazieres J, Quoix E, Riely G, Barlesi F, Souquet PJ, Smit EF, Groen HJ, Kelly RJ, et al. 2016b. Dabrafenib in patients with BRAF(V600E)-positive advanced non-small-cell lung cancer: a single-arm, multicentre, open-label, phase 2 trial. Lancet Oncol 17: 642–650. 10.1016/S1470-2045(16)00077-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. 2010. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 464: 427–430. 10.1038/nature08902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, Shi H, Atefi M, Titz B, Gabay MT, et al. 2011. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 480: 387–390. 10.1038/nature10662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard C, Carragher L, Aldridge V, Giblett S, Jin H, Foster C, Andreadi C, Kamata T. 2007. Mouse models for BRAF-induced cancers. Biochem Soc Trans 35: 1329–1333. 10.1042/BST0351329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samatar AA, Poulikakos PI. 2014. Targeting RAS–ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov 13: 928 10.1038/nrd4281 [DOI] [PubMed] [Google Scholar]
- Schreck KC, Guajardo A, Lin DDM, Eberhart CG, Grossman SA. 2018. Concurrent BRAF/MEK inhibitors in BRAF V600-mutant high-grade primary brain tumors. J Natl Compr Canc Netw 16: 343–347. 10.6004/jnccn.2017.7052 [DOI] [PubMed] [Google Scholar]
- Su F, Viros A, Milagre C, Trunzer K, Bollag G, Spleiss O, Reis-Filho JS, Kong X, Koya RC, Flaherty KT, et al. 2012. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med 366: 207–215. 10.1056/NEJMoa1105358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toll SA, Tran HN, Cotter J, Judkins AR, Tamrazi B, Biegel JA, Dhall G, Robison NJ, Waters K, Patel P. 2019. Sustained response of three pediatric BRAFV600E mutated high-grade gliomas to combined BRAF and MEK inhibitor therapy. Oncotarget 10: 551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udager AM, Pan J, Magers MJ, Palapattu GS, Morgan TM, Montgomery JS, Weizer AZ, Hafez KS, Miller DC, Wolf JS Jr. 2015. Molecular and immunohistochemical characterization reveals novel BRAF mutations in metanephric adenoma. Am J Surg Pathol 39: 549 10.1097/PAS.0000000000000377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, Wubbenhorst B, Xu X, Gimotty PA, Kee D, et al. 2010. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 18: 683–695. 10.1016/j.ccr.2010.11.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ, Barford D, et al. 2004. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 116: 855–867. 10.1016/S0092-8674(04)00215-6 [DOI] [PubMed] [Google Scholar]
- Wang J, Yao Z, Jonsson P, Allen AN, R Qin AC, Uddin S, Dunkel IJ, Petriccione M, Manova K, Haque S, et al. 2018. A secondary mutation in BRAF confers resistance to RAF inhibition in a BRAF(V600E)-mutant brain tumor. Cancer Discov 8: 1130–1141. 10.1158/2159-8290.CD-17-1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wobker SE, Matoso A, Pratilas CA, Mangray S, Zheng G, Lin M-T, Debeljak M, Epstein JI, Argani P. 2019. Metanephric adenoma-epithelial Wilms tumor overlap lesions: an analysis of BRAF status. Am J Surg Pathol 43: 1157–1169. 10.1097/PAS.0000000000001240 [DOI] [PubMed] [Google Scholar]
- Yao Z, Torres NM, Tao A, Gao Y, Luo L, Li Q, de Stanchina E, Abdel-Wahab O, Solit DB, Poulikakos PI, et al. 2015. BRAF mutants evade ERK-dependent feedback by different mechanisms that determine their sensitivity to pharmacologic inhibition. Cancer Cell 28: 370–383. 10.1016/j.ccell.2015.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z, Gao Y, Su W, Yaeger R, Tao J, Na N, Zhang Y, Zhang C, Rymar A, Tao A, et al. 2019. RAF inhibitor PLX8394 selectively disrupts BRAF dimers and RAS-independent BRAF-mutant-driven signaling. Nat Med 25: 284–291. 10.1038/s41591-018-0274-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Spevak W, Zhang Y, Burton EA, Ma Y, Habets G, Zhang J, Lin J, Ewing T, Matusow B, et al. 2015. RAF inhibitors that evade paradoxical MAPK pathway activation. Nature 526: 583–586. 10.1038/nature14982 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Coding variants identified in the Johns Hopkins Solid Tumor Panel analysis are reported in the body of the manuscript. The patient did not provide consent for public deposition of all raw sequencing data for the 400 genes included in the Solid Tumor Panel version 3.0. The variants were submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and can be found under accession numbers SCV001147030–SCV001147043.