

Abstract
Rhabdomyosarcoma (RMS) is the most common childhood soft-tissue sarcoma. The largest subtype of RMS is embryonal rhabdomyosarcoma (ERMS) and accounts for 53% of all RMS. ERMS typically occurs in the head and neck region, bladder, or reproductive organs and portends a promising prognosis when localized; however, when metastatic the 5-yr overall survival rate is ∼43%. The genomic landscape of ERMS demonstrates a range of putative driver mutations, and thus the recognition of the pathological mechanisms driving tumor maintenance should be critical for identifying effective targeted treatments at the level of the individual patients. Here, we report genomic, phenotypic, and bioinformatic analyses for a case of a 3-yr-old male who presented with bladder ERMS. Additionally, we use an unsupervised agglomerative clustering analysis of RNA and whole-exome sequencing data across ERMS and undifferentiated pleomorphic sarcoma (UPS) tumor samples to determine several major endotypes inferring potential targeted treatments for a spectrum of pediatric ERMS patient cases.
Keywords: embryonal rhabdomyosarcoma
INTRODUCTION
Rhabdomyosarcoma (RMS) is a highly malignant tumor of mesenchymal origin that accounts for ∼5%–8% of all childhood soft-tissue sarcomas. RMS is most common in children under the age of 10 with male to female predominance ratio of 1.5:1 (Ruymann and Grovas 2000). RMS is divided into two major subtypes: alveolar (ARMS) and embryonal (ERMS). The molecular characteristics of ARMS and ERMS vary greatly. ARMS is defined by either the t(2;13) or t(1;13) chromosomal translocation, resulting in a fusion gene of PAX3:FOXO1 or PAX7:FOXO1 and a limited number of secondary genomic alterations. Further, diagnostic criteria from the International Classification of Rhabdomyosarcoma (ICR) classifies FOXO1 fusion-negative RMS with only focal alveolar histology to be FOXO1 fusion-negative ARMS (Barr et al. 2006). In contrast, ERMS has several implied causative mutations with p53 loss (Taylor et al. 2000; Pérot et al. 2010), RAS pathway activation (Stratton et al. 1989), and MYOD1 mutation (Kohsaka et al. 2014) being among the frequent molecular features of this disease. The MYOD1 mutation and VGLL2 gene fusions define an aggressive and rare subtype with distinct morphological features apart from ERMS called sclerosing and spindle cell rhabdomyosarcoma (Mentzel and Katenkamp 2000; Mentzel and Kuhnen 2006; Mentzel 2010; Agaram et al. 2019). Botryoid RMS, on the other hand, with a morphologic appearance resembling grapes (“botryoid”), is considered to be a subtype of ERMS by the fourth edition of the World Health Organization (WHO) Classification of Tumors of Soft Tissue and Bone. One report suggests that the RAS pathway alone may be mutationally activated in ∼45% of ERMS tumors (Shern et al. 2014), but this observation is yet to be borne out with clinical responses to MEK inhibitors. Because of the complex and crowded genetic landscape, one might surmise that ERMS has several subtypes and intragenic (nonexonic) or epigenetic features.
Clinical features of ERMS and ARMS also differ. ERMS has a more favorable prognosis, whereas ARMS has a higher rate of metastasis and experiences a poorer outcome (Rudzinski et al. 2017). The 5-yr event-free survival (EFS) for ERMS is 43%, whereas the 5-yr EFS for PAX3:FOXO1, PAX7:FOXO1, unknown fusion status, and fusion-negative ARMS are 8%, 17%, 17%, and 29%, respectively (Rudzinski et al. 2017). This outcome has not seen improvement over several decades (Breneman et al. 2003; Williams et al. 2004; Davis and Keller 2012; Malempati and Hawkins 2012; Rudzinski et al. 2017). Moreover, intensified treatments historically carry a 3%–4% treatment-related mortality rate (Crist et al. 2001). Thus, new treatment options that provide long-term survival benefits are needed.
To define these ERMS endotypes (subgroups of disease originating from distinct pathobiological mechanisms), we have used a hierarchical agglomerative clustering approach to organize a wide range of ERMS samples. To cover the full breadth of ERMS, we have aggregated RNA and DNA sequencing data from genetically engineered mouse models (GEMMs), patient-derived xenografts (PDXs), canine soft-tissue sarcoma biopsies, patient surgical biopsies, patient surgical autopsies, and cell lines. Using these surrogates, we sought to match targeted treatments to each endotype.
Here, we present a case report of a 3-yr-old male with the botryoid type of ERMS arising in the urinary bladder. Using whole-exome sequencing we have identified notable variants, including the ERMS-associated genes BUB1B, DICER1, and FGFR1.
RESULTS
Clinical Presentation
A 19-mo-old boy presented with blood spots in his diaper. The spots were later uncovered to be hematuria. The patient underwent two ultrasounds as the first ultrasound was inconclusive. The second ultrasound, 2 weeks later, demonstrated a large mass thought to be malignant. Magnetic resonance imaging (MRI) was performed, showing a 3 cm × 2 cm × 2 cm mass arising from the posterior bladder base (Fig. 1).
Figure 1.

MRI of the abdomen and pelvis. A multilobulated heterogeneous T2 hyperintense and T1 isointense intraluminal mass (red asterisk) was found arising from the posterior bladder base with the following sequences: axial T1 radial VIBE with fat saturation +C on the pelvis (A) and sagittal radial VIBE with fat saturation (B). A T1 hypointense and T2 hyperintense lesion ventral to the anterior urethra was found with the following sequences: axial T2 with fat saturation on the pelvis (C) and sagittal IR triggered (D).
At week 4, a cystoscopy and urinary bladder biopsy confirmed the patient's diagnosis to be ERMS. Operative findings included a circumferential bladder neck and prostate extension. Tumor cells were found to be diffusely immunoreactive for desmin (cytoplasmic), and a subset of tumor cells were positive for myogenin (nuclear) (Fig. 2).
Figure 2.

Histology sections. Hematoxylin and eosin (H&E) staining sections from a small biopsy revealing a small round blue cell tumor favor embryonal rhabdomyosarcoma (top) and a surgical resection revealing a botryoid pattern (bottom). Scale bars, (top) (left) 10 µM; (middle) 100 µM; (right) 100 µM; (bottom) (left) 10 µM; (right) 100 µM.
Thereafter, the patient underwent resection of the bladder, prostate, and urethra. A cystoprostatectomy revealed focal tumor invasion into the bladder muscularis. The tumor arose within the urinary bladder and extended by suburothelial spread into the prostatic and penile urethra and into the left ureteral orifice. The tumor was present 0.1 cm from the anterior bladder soft-tissue margin and 0.6 cm from the posterior bladder soft-tissue margin. Distal urethral margin, bilateral ureteral margins, and seminal vesicles were negative for tumor. The patient's final diagnosis following surgery was stage III embryonal rhabdomyosarcoma (botryoid type) with T2b N0 M0 pathological staging.
Sections from the patient's surgery revealed neoplastic cells present in single-file clusters or as infiltrative single cells undermining the bladder and prostate urothelium focally invading the bladder muscularis mucosa. The neoplastic cells were small and round with hyperchromatic nuclei with indistinct chromatin with either scant cytoplasm in the more poorly differentiated population or with rhabdomyoblastic features in the more well-differentiated cells. The patient's treatment plan was a 43-wk protocol with vincristine, actinomycin-D, and cyclophosphamide (VAC). The patient has remained disease-free for 41 mo from the time of diagnosis.
With respect to past medical history, the patient was born at 5 pounds 7 ounces (average weight between 5 and 8 pounds) at 36 wk gestation. The patient had jejunal atresia on day 3 of life, requiring immediate corrective surgery. At age 3, the patient weighed 12.5 kg, was 91 cm tall, and had a head circumference of 48 cm (41, 62, and 31 percentiles, respectively). There were no signs of growth delay before birth or after birth nor was microcephaly present in the patient's history. At 7 mo of age, the patient began early intervention services after discovering right-sided weakness, a potential consequence of unrecognized neonatal stroke. The patient has been under ongoing care for developmental delays in gross motor function and expressive language.
Genomic Analyses
Previous Analysis
Molecular assessment of the resected tumor and peripheral blood was performed prior to our analysis. Of note, a somatic variant of unknown significance was identified in the TP53 gene (c. 91G > A p.V31I). TP53 is a well-studied tumor-suppressor gene, whereby loss of function of germline TP53 has been found to be associated with Li–Fraumeni cancer predisposition (LFS). LFS can increase the risk of developing rhabdomyosarcoma in children (Diller et al. 1995). The variant found in the patient, TP53 p.V31I, has been reported with varying degrees of interpretation, such as benign, uncertain significance, and pathogenic, in ClinVar with respect to LFS (Landrum et al. 2014). Additionally, somatic mutations in the BUB1B (c.1649G > A p.R550Q) and CSF1R (c.2737dupC p.Q913Pfs*29) genes were identified using the OncoPanel assay. The OncoPanel detects mutations in exonic DNA sequences of 300 cancer genes and 113 introns across 35 genes for rearrangement detection (Supplemental Information; Methods). Loss of BUB1B, a critical component of the mitotic checkpoint, has been reported to be associated with chromosomal instability in ERMS (Shern et al. 2014). More specifically, reports of the patient's somatic variant R550Q have been found to cause mosaic-variegated aneuploidy (MVA) (Hanks et al. 2004), along with mutations in CEP57 (Snape et al. 2011) or TRIP13 (Yost et al. 2017). Moreover, missense mutations in BUB1B have been found in five families with MVA. Two of these five families have children who have developed ERMS (Hanks et al. 2004). Clinical manifestations of MVA are microcephaly, prenatal growth failure, eye anomalies, dysmorphism, and developmental delays. Because of the history of at least one congenital anomaly (esophageal atresia), ERMS diagnosis, and discovery of the somatic mutation in the BUB1B gene, the patient was simultaneously screened for MVA and evaluated for germline mutations in BUB1B. Furthermore, two tests were subsequently explored: the first sequenced the patient's peripheral blood for deletion/duplication analysis of the BUB1B gene, and the second test examined germline chromosomal mosaicism. The deletion/duplication analysis was negative, and there was no evidence of aneuploidy from the mosaicism karyotype analysis. Based on these results, the patient's congenital anomaly and ERMS diagnosis were reported to not be a result of germline mutations in the BUB1B gene or MVA.
Whole-Exome Sequencing Analysis
Whole-exome sequencing from fixed-formalin, paraffin-embedded (FFPE) tumor tissue and buccal swab was performed for the detection of somatic mutations, insertion/deletion (indel) events, and/or copy-number alterations, as well as potential germline mutations (Table 1; Methods). After filtering for somatic mutations bearing high or moderate impact, 6280 nonsynonymous somatic variants were identified. In addition, somatic mutations were identified in BUB1B, DCC, DICER1, KRAS, NF1, PAX3, PIK3AP1, PIK3C2G, PIK3CA, PIK3CB, PIK3CD, PIK3IP1, PIK3R1, PIK3R2, PIK3R3, PIK3R5, PIK3R6, and PTEN (Table 2). After filtering further for mutations that also showed increased copy number (log2 tumor/normal read ratio > 0.4) and TPM > 100, we identified 137 mutations of interest (Fig. 3; Supplemental Table S1), including mutations in FGFR1.
Table 1.
Sequencing coverage using HiSeq 4000 (paired-end, 100-bp)
| Sequencing | Data output |
|---|---|
| Normal DNA exome | 800× |
| Tumor DNA exome | 100× |
| Tumor RNA | 30 million reads |
Table 2.
DICER1, BUB1B, APC, DCC, KRAS, NF1, ALK, PAX3, PIK3AP1, PIK3C2G, PIK3CA, PIK3CB, PIK3CD, PIK3IP1, PIK3R1, PIK3R2, PIK3R3, PIK3R5, PIK3R6, and TP53 mutations
| Gene | Germline | Somatic | Variant | HGVS DNA reference | HGVS protein reference | Genotype | Variant allele fractions |
|---|---|---|---|---|---|---|---|
| DICER1 | X | Stop gained | c.2782C > T | p.Gln928* | Het | 2.7% of 187 reads | |
| DICER1 | X | Missense variant | c.2018C > T | p.Ser673Leu | Het | 2.8% of 143 reads | |
| DICER1 | X | Frameshift variant | c.1111_1112insTAATAA TAGAAATCAGGAT | p.Lys371fs | Het | 6.9% of 102 reads | |
| BUB1B | X | Stop gained | c.508A > T | p.Lys170* | Het | 3.6% of 194 reads | |
| BUB1B | X | Stop gained | c.550A > T | p.Lys184* | Het | 3.6% of 194 reads | |
| BUB1B | X | Missense variant | c.1733G > T | p.Cys578Phe | Het | 3.7% of 107 reads | |
| BUB1B | X | Missense variant | c.1775G > T | p.Cys592Phe | Het | 3.7% of 107 reads | |
| BUB1B | X | X | Missense variant | c.1046G > A | p.Arg349Gln | Het | 46% of 609 reads |
| BUB1B | X | X | Missense variant | c.1088G > A | p.Arg363Gln | Het | 46% of 609 reads |
| BUB1B | X | X | Missense variant | c.1649G > A | p.Arg550Gln | Het | 50% of 664 reads |
| BUB1B | X | X | Missense variant | c.1691G > A | p.Arg564Gln | Het | 50% of 664 reads |
| APC | X | X | Missense variant | c.5465T > A | p.Val1822Asp | Het | 100% of 305 reads |
| DCC | X | Frameshift variant | c.1735_1736delCC | p.Pro579fs | Het | 4.8% of 83 reads | |
| DCC | X | Frameshift variant | c.1867_1868delCC | p.Pro623fs | Het | 4.8% of 83 reads | |
| DCC | X | Frameshift variant | c.1936_1937delCCb | p.Pro646fs | Het | 4.8% of 83 reads | |
| DCC | X | Frameshift variant | c.901_902delCC | p.Pro301fs | Het | 4.8% of 83 reads | |
| DCC | X | X | Missense variant | c.67T > C | p.Phe23Leu | Het | 100% of 2,079 reads |
| KRAS | X | Splice acceptor variant & intron variant | c.-11-2A > T | None | Het | 5.6% of 89 reads | |
| NF1 | X | Missense variant | c.419G > A | p.Gly140Glu | Het | 3% of 230 reads | |
| NF1 | X | Missense variant | c.218G > A | p.Gly73Glu | Het | 3% of 230 reads | |
| NF1 | X | Missense variant | c.299G > A | p.Gly100Glu | Het | 3% of 230 reads | |
| ALK | X | X | Stop gained | c.218G > A | p.Trp73* | Het | 100% of 345 reads |
| PAX3 | X | Missense variant | c.332C > T | p.Thr111Met | Het | 3% of 97 reads | |
| PIK3AP1 | X | Sequence feature | c.1376-4555C > T | None | Het | 11% of 36 reads | |
| PIK3AP1 | X | X | Missense variant | c.1913A > G | p.Lys638Arg | Het | 23% of 66 reads (TU) |
| PIK3AP1 | X | X | Missense variant | c.710A > G | p.Lys237Arg | Het | 23% of 66 reads |
| PIK3AP1 | X | X | Missense variant | c.1379A > G | p.Lys460Arg | Het | 23% of 66 reads |
| PIK3C2G | X | Missense variant | c.1952C > A | p.Pro651Gln | Het | 4% of 123 reads | |
| PIK3C2G | X | Missense variant | c.1829C > A | p.Pro610Gln | Het | 4% of 123 reads | |
| PIK3C2G | X | X | Conservative in-frame deletion | c.385_387delCCC | p.Pro129del | Het | 37% of 505 reads |
| PIK3C2G | X | X | Missense variant | c.437C > T | p.Pro146Leu | Het | 37% of 453 reads |
| PIK3CA | X | Stop gained | c.418C > T | p.Arg140* | Het | 1.9% of 208 reads | |
| PIK3CA | X | Structural interaction variant | c.418C > T | None | Het | 1.9% of 208 reads | |
| PIK3CA | X | X | Structural interaction variant | c.3075C > T | None | Het | 43% of 639 reads |
| PIK3CB | X | Missense variant | c.1836C > A | p.Phe612Leu | Het | 4.9% of 103 reads | |
| PIK3CB | X | Missense variant | c.432C > A | p.Phe144Leu | Het | 4.9% of 103 reads | |
| PIK3CB | X | Missense variant | c.729C > A | p.Phe243Leu | Het | 4.9% of 103 reads | |
| PIK3CB | X | Missense variant | c.174C > A | p.Phe58Leu | Het | 4.9% of 103 reads | |
| PIK3CD | X | Stop gained | c.2492C > A | p.Ser831* | Het | 3.0% of 203 reads | |
| PIK3CD | X | Stop gained | c.2564C > A | p.Ser855* | Het | 3.0% of 203 reads | |
| PIK3CD | X | Missense variant | c.2604G > A | p.Met868Ile | Het | 3.0% of 198 reads | |
| PIK3CD | X | Missense variant | c.2532G > A | p.Met844Ile | Het | 3.0% of 198 reads | |
| PIK3CD | X | Missense variant | c.2604G > A | p.Met868Ile | Het | 3.0% of 198 reads | |
| PIK3IP1 | X | Missense variant | c.752C > G | p.Thr251Ser | Het | ||
| PIK3R1 | X | Missense variant | c.218C > T | p.Ser73Phe | Het | 2.5% of 198 reads | |
| PIK3R1 | X | Missense | c.512C > T | p.Ser171Phe | Het | 2.5% of 198 reads | |
| PIK3R1 | X | X | Structural interaction variant | c.219C > T | None | Het | 53% of 618 reads |
| PIK3R1 | X | X | Protein–protein contact | c.1176C > T | None | Het | 50% of 576 reads |
| PIK3R2 | X | Missense variant | c.700A > C | p.Ser234Arg | Het | 100% of 39 reads | |
| PIK3R2 | X | X | Missense variant | c.937T > C | p.Ser313Pro | Het | 100% of 58 reads |
| PIK3R3 | X | Missense variant | c.743G > A | p.Gly248Glu | Het | 4.1% of 147 reads | |
| PIK3R3 | X | Missense variant | c.121_122delCCinsTT | p.Pro41Leu | Het | 3.3% of 120 reads | |
| PIK3R3 | X | X | Missense variant | c.1031A > G | p.Asn344Ser | Het | 50% of 125 reads |
| PIK3R3 | X | X | Missense variant | c.854A > G | p.Asn285Ser | Het | 50% of 125 reads |
| PIK3R3 | X | X | Missense variant | c.849T > A | p.Asn283Lys | Het | 54% of 107 reads |
| PIK3R5 | X | Frameshift variant | c.222_225delCTAC | p.Tyr75fs | Het | 5.0% of 59 reads | |
| PIK3C2G | X | X | Conservative in-frame deletion | c.385_387delCCC | p.Pro129del | Het | |
| PIK3R5 | X | Missense variant | c.221C > G | p.Thr74Ser | Het | 5.3% of 57 reads | |
| PIK3R6 | X | X | Missense | c.1870G > A | p.Asp624Asn | Het | 35% of 201 reads |
| PIK3R6 | X | X | Missense | c.785C > T | p.Ala262Val | Het | 43% of 92 reads |
| TP53 | X | X | Missense variant | c.91G > A | p.Val31Ile | Het | 47% of 574 reads |
Figure 3.

Circos plot. Circos plots were generated using tumor DNA exome, normal DNA exome, and tumor RNA sequencing data. Exome data was analyzed for somatic point mutation, indel, and copy-number variation data, as denoted by the inner and middle rings, respectively. Gene expression from RNA sequencing data was plotted on the outer ring.
Expression Analysis
Gene expression from the FFPE tumor tissue was quantified as TPM (transcripts per million). To note, the sample was deemed nonoptimal after RNA extraction on account of sample degradation and size (DV200 [23%] < 30%, RIN [1.4] < 2.0, total mass [0.0375 µg] < 0.2 µg, concentration [1.25 ng/µL] < 70 ng/µL), which is common for FFPE-derived RNA. Several mitochondrial genes were found to have the highest expression of all the genes (Supplemental Table S2). PABPC1 (3,507 TPM), RPL26 (3,323 TPM), RAB7A (3,213 TPM), and EEF1A1 (2,291 TPM) were also among the most highly expressed genes and have been shown in previous studies to be involved in tumor proliferation (Zhu et al. 2015; Zhang et al. 2016; Xie et al. 2019). For comparative analysis, we used the median TPM values from the population of normal of skeletal muscle tissues (n = 564) from the Genotype-Tissue Expression (GTEx) project (GTEx Consortium 2013). We found twofold higher expression of 6520 genes comparative to the normal skeletal muscle cohort. Several small nuclear RNAs (snoRNAs) were among the highest expressed genes compared to the population of skeletal muscle tissue (Supplemental Table S3). INS-IGF2 and RN7SL751P were also highly expressed to normal skeletal muscle tissue. PABPC1, RPL26, RAB7A, and EEF1A1 all had twofold higher expression.
Fusion Genes
STAR-fusion was used to identify gene fusion events from RNA isolated from FFPE tumor tissue (Fig. 3; Supplemental Table S4; Dobin et al. 2013). A novel interchromosomal fusion was found between Chromosome 5 and Chromosome 3 (NIPBL-TM4SF18). Additionally, a high-impact frameshift mutation was found in NIPBL (c.3360_3376delTGACAGAAGAAGCTCTG p.Asp1121fs). A second novel interchromosomal fusion was found between Chromosomes 17 and 21 (BTBD1–CSTB). The other fusions detected were found to be local inversions.
Hierarchical Cluster Analysis
Because genomic analysis did not define a high-value target for therapeutic intervention, we turned to functional studies. Because a fresh tumor sample was not provided and thus a patient-derived cell model could not be developed for downstream experimental analysis, we used an unsupervised clustering approach to determine a genetically similar experimental model. To add diversity, we included canine soft-tissue sarcoma/ERMS biopsy samples as well. Hierarchical clustering of RNA sequencing and whole-exome sequencing from canine samples (n = 14), fusion-negative GEMMs (n = 8), PDX models (n = 6), patient surgical samples (n = 53), and cell lines (n = 25) revealed several endotypes, or subtypes defined by distinct pathobiological mechanisms (Fig. 4). Large clusters were formed by samples harboring mutations in TP53, FGFR4, NRAS, HRAS, and the PAX7:FOXO1 fusion. Cell lines also formed their own cluster with the patient's case with 42 mutated genes in common. SMS-CTR, RD, and Hs729T were found to be the closest in the cluster with the patient. Both the patient and the cell lines were found to have mutations in TP53, BUB1B, APC, ATRX, HRAS, ATR, CPS1, AURKA, LAG3, MUC16, AURKB, SYNE1, PKHD1, FOS, DCC, CHD9, ALK, and CENPF. Hierarchical clustering of the top 1000 genes with highest expression level in the patient's tumor sample (Supplemental Fig. S1) clusters the patient's tumor sample away from the other samples, perhaps indicating that the genes that are most highly expressed in the patient's tumor have uniquely high expression levels compared to the cohort. As surrogate cell models for the patient's endotype, we then performed chemical screens on ERMS human cell lines RD, SMS-CTR, RMS559, and a genetically engineered mouse (GEM) cell culture U57810 (Fig. 5). Each of the human cell lines clustered closely with the patient's sample. Because none of the mouse cell lines clustered closely with the patient's sample, a mouse ERMS cell line was chosen that grew well in culture. Cell lines were validated through short tandem repeat (STR) profiling (Supplemental Table S5). Cell cultures were treated with BYL-719 (PI3Kα inhibitor), GSK1059615 (PI3K α/β/δ/γ and mTOR inhibitor), TC-KHNS11 (PI3K δ inhibitor), BEZ-235 (PI3K α/β/δ/γ and mTOR inhibitor), CUDC-907 (PI3Kα and HDAC 1/2/3/10 inhibitor), entinostat (HDAC 1/3), and BKM-120 (PI3K α/β/δ/γ inhibitor). The cell cultures were most sensitive to the dual HDAC and PI3Kα inhibitor CUDC-907 (IC50 range 2 to 123 nM) and the dual PI3K and mTOR inhibitor BEZ-235 (IC50 range 6 to 616 nM). Both of these drugs have been evaluated clinically and were found to have unusual pharmacokinetics, complicating comparison of the observed IC50 values to clinically achievable Cmax values (Bendell et al. 2015; Oki et al. 2017). For CUDC-907, the compound is readily metabolized and the metabolites have reduced PI3K activity and no HDAC activity (Oki et al. 2017). On the other hand, BEZ-235 has a reported Cmax of 1.4 µM (range 494 nM to 5.7 µM [Bendell et al. 2015]).
Figure 4.
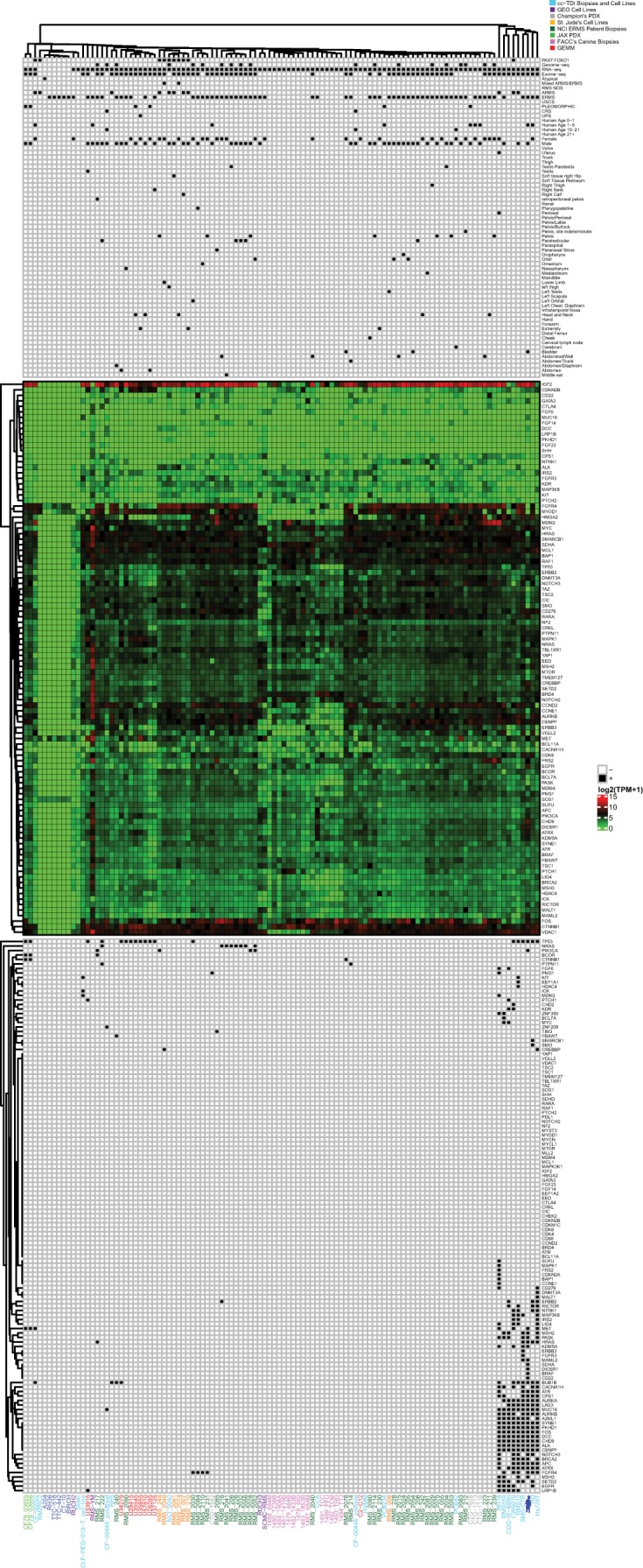
Index case endotype unsupervised clustering of embryonal rhabdomyosarcoma (ERMS) and non-rhabdomyosarcoma soft-tissue sarcoma (NRSTS) samples with alveolar rhabdomyosarcoma (ARMS) samples harboring the PAX7:FOXO1 fusion gene as a control determined several endotypes. Clustering was performed using DNA and RNA sequencing from tumor samples from our IRB-approved CuReFAST initiative and cell lines (light blue), cell lines from Gene Expression Omnibus (GEO) (purple), PDX mouse models from Champions Oncology (light gray), cell lines from St. Jude Children's Research Hospital (blue), ARM patient's biopsies from National Cancer Institute (orange), ERMS patient's biopsies from NCI (dark green), PDX mouse models from the Jackson Laboratory (light green), canine samples from Flint Animal Cancer Center (pink), genetically engineered mouse models (red), and the patient's tumor sample (dark blue avatar). The legend below marks samples with known sex, age, primary tumor site, somatic mutations, and diagnosis. Gene expression (log2 (TPM +1)) is shown below in a heatmap on a scale of 15 (red) to 0 (green). Samples without matched RNA-sequencing data were given a value 0 for all genes. Below the gene expression heatmap, samples with somatic mutations in our genes of interest are indicated by a black box. Unsupervised clustering was performed separately on the expression and somatic data within the legend (vertical dendrogram). Because we suspected a germline predisposition/syndrome for this patient, we kept germline mutations in the dendrogram but did not do the same for the other tumors as they were presumably somatic, noninherited cases.
Figure 5.

Drug validation for rhabdomyosarcoma cell lines. BYL-719, GSK1059615, CUDC-907, TC-KHNS11, BEZ-235, BKM-120, and entinostat were tested on RMS cell lines RD (blue), SMS-CTR (red), RMS559 (green), and U57810 (purple) at varying concentrations.
Because somatic mutations were found in the PI3K pathway, and we found sensitivity of cell models to the PI3K inhibitors, we explored whether PI3K signaling was enriched within the patient's endotype compared to the other endotypes. Samples counted as within the patient's endotype were SMS-CTR, Hs729T, RD, RMS13, CW9019, COG-R-486h, and RMS559. Several mutations in the PI3K pathway were found in common with the samples within the same endotype (Supplemental Table S6). Further, the case sample shares mutations in PIK3C2G, PIK3IP1, and PIK3R3 with SMS-CTR, RD, and Hs729. Some samples outside the patient's endotype were found to have PI3KCA and PI3KC2A mutations, and conversely, not all samples within the patient's endotype were found to have all of the same PI3K mutations. However, the majority of samples with whole-exome sequencing data outside of the patient's endotype did not harbor a mutation within the PI3K genes of interest, whereas all of the samples within the patient's endotype did harbor at least one PI3K mutation. Finally, the patient shares several other mutations in common with samples within the patient's endotype, further elucidating that PI3K may not be the only pathway involved.
DISCUSSION
Despite several advances in understanding the dominant molecular pathways of ERMS, our data show that only 40% of ERMS tumors have known driving mutations, leaving the remaining 60% of ERMS with a completely unknown biology. For this patient, few pathognomonic mutations were identified. We did, however, note widespread amplifications in Chromosome 8. Gene variants identified by next-generation deep sequencing, but not by ancillary tests, included BUB1B (an ERMS-associated germline variant) and a somatic variant of DICER1.
FOXM1/BUB1B signaling pathway is considered to be crucial for the growth and progression of rhabdomyosarcoma (Wan et al. 2012). BUB1B acts by regulating the spindle-assembly checkpoint and is a transcriptional target of Forkhead Box M1 (FoxM1) (Rio Frio et al. 2010; Wan et al. 2012). Furthermore, when FOXM1 is suppressed by either shRNA or FOXM1 inhibitor siomycin A, BUB1B levels decrease, leading to chromosomal loss and apoptosis (Wan et al. 2012). Overexpression of BUB1B is also related to poor prognosis and progression of other types of cancer, such as breast, gastric, colorectal, and prostate (Fu et al. 2016; Hahn et al. 2016; Hudler et al. 2016; Mansouri et al. 2016).
DICER1 is a miRNA processing gene associated with familial and sporadic ERMS. Decreased expression of DICER1 correlates with the concentration of miRNA during organ development, suggesting that abnormal miRNA regulation plays a role in the pathogenesis of ERMS (Hill et al. 2009). Further, germline mutations in DICER1 can cause DICER1 syndrome, a rare pediatric tumor predisposition syndrome. In DICER1-associated tumors, one allele is inactivated by a germline mutation in DICER1 and the other allele has a somatic missense mutation that affects one of the RNase IIIb metal ion-binding sites (Doros et al. 2014). Several publications in the literature report sarcomas arising from germline and somatic mutations in DICER1, including urogenital ERMS cases (Doros et al. 2012; de Kock et al. 2017). In this case, however, no germline mutations were found in DICER1, and the expression of DICER1 (198.53 TPM) was not decreased compared to the normal cohort of skeletal muscles from the GTEx project (median TPM, 14.03). Additionally, none of the somatic mutations found harbored the characteristic RNase IIIb hotspot variant consistent in previous sarcomas (Doros et al. 2014). The patient's frameshift variant in this case has not been reported in the International Pleuropulmonary Blastoma Registry, the majority of which are frameshift or nonsense mutations (A Hill, pers. comm.). Hence, a causal effect of DICER1 in this cases’ ERMS cannot be ruled out, but would require biochemical investigation to determine if the expressed protein had a dominant negative effect.
The PI3K pathway has been studied extensively in the context of soft-tissue sarcomas (Barretina et al. 2010; Shukla et al. 2012; Shern et al. 2014; Li et al. 2015). Shukla et al. has reported PIK3CA to be mutated in 4.9% of ERMS. A large-scale study focusing on the mutational landscape of rhabdomyosarcoma found mutations in PIK3CA and PIK3CD, as well as coexisting mutations in PIK3CA and a RAS family gene (Shern et al. 2014). On a smaller scale, a co-occurrence of two missense mutations in PIK3CA (35 G > A, G12D) and KRAS (1636C > A, Q546K) were observed in an undifferentiated pleomorphic sarcoma case (Li et al. 2015). Functionally, PIK3CA mutations have mainly been reported to cluster on helical (exon 9) and kinase (exon 20) domain (Barretina et al. 2010). In accordance with several previous genomic studies, a mutation in PIK3CA was found leading to a premature termination codon (a stop gained) at codon 140. Several other effectors of the PI3K pathway were found to be mutated, such as PIK3AP1, PIK3C2G, PIK3CB, PIK3CD, PIK3R1, PIK3R3, and PIK3R5, suggesting a role in the PI3K pathway in the oncogenesis of ERMS in this patient. Although a protein change was not found for the KRAS mutant found in this case, our data reveals a potential role of the RAS/PIK3CA genetic axis as described in previous studies (Shern et al. 2014). Further validation is required to render the genetic function of these mutations.
As a basis for surrogate functional studies, hierarchical agglomerative clustering identified SMS-CTR, RD, and Hs729T to be the most genetically similar cell line to the patient's samples. However, a significant fraction of other ERMS samples have unidentified molecular features warranting further investigation into potential driving mutations. As a caveat, we lacked matched normal (germline) controls used for the cell lines included in this study, and previous studies have shown tumor-only pipelines to be potentially unreliable with 69% of somatic mutations being false positives (Shi et al. 2018). Future studies might consider using a cohort-normal pipeline for samples that do not have a matched normal.
Notwithstanding the caveats of DNA features, the hierarchical agglomerative clustering of DNA and RNA features identified an ERMS endotype (subgroup) demonstrating consistent sensitivity to the dual HDAC-PI3K inhibitor CUDC-907 and the dual PI3K-mTOR inhibitor BEZ0235. These drugs represent candidates for further preclinical/clinical investigation in this ERMS endotype.
METHODS
Cell Lines
RMS559, CW9019, CCA, RD, Hs729T, COG-R-486h, and SMS-CTR were grown in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. COG-R-486h was maintained in hypoxic conditions. HSMM, SkMc, RH5, Rh18, CLF-PED-015T, and RMS13 were grown in Roswell Park Memorial Institute (RPMI)1640 medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. All cell lines were validated through short tandem repeat (STR) profiling (Supplemental Table S5).
Whole-Exome and RNA Sequencing
Previous analysis using the OncoPanel assay isolated DNA from FFPE tumor tissue containing 60% neoplastic cells and analyzed by massively parallel sequencing using a solution-phase Agilent SureSelect hybrid capture kit and an Illumina HiSeq 2500 sequencer. A commercial sequencing-based assay for germline TP53 mutations was performed by Ambry Genetics from peripheral mononuclear cell DNA.
Within the scope of this study, material for the generation of exome and RNA sequencing data was isolated from FFPE tumor tissue. Comparison was made with DNA from a buccal swab as the matched normal exome sample. Sequencing of the tumor tissue was performed at BGI Genomics using Agilent SureSelect capture kit v5 and HiSeq 4000 technology, and sequencing of the buccal swab was also performed at BGI Genomics using the BGI exome array and BGISEQ-500RS sequencer.
Variant Detection
Somatic and germline mutations (point mutations, insertions/deletions [indels], and functional and structural variants), as well as copy-number variation, were analyzed using tumor and matched normal exome sequencing data. Sequence reads were aligned to the GRCh38 human reference genome. Mutations and indels were called with Genome Analysis Toolkit (GATK) Version 4.0 with tumor logarithm of odds (TLOD) scores of >6.3 (McKenna et al. 2010; DePristo et al. 2011; Van der Auwera et al. 2013). Copy-number variations were identified using VarScan2, where regions with a log ratio > 0.40 were called as gained, and regions with a log ratio < −0.40 were called as lost (Koboldt et al. 2012).
Gene Expression and Fusion Detection
Gene expression and gene fusion events were analyzed using RNA sequencing data from the tumor tissue. Transcriptome data was aligned to the GRCh38 human reference genome. Normalized gene expression was quantified using STAR aligner with RSEM, and gene fusion events were identified using STAR-Fusion (Li and Dewey 2011; Dobin et al. 2013). Region-specific unmatched skeletal muscle tissue gene expression data from the GTEx project served as a population normal to identify underexpressed and overexpressed genes.
Hierarchical Clustering Analysis
DNA and RNA sequencing of canine samples, GEMMs, cell lines, PDX mouse models, and the patient's biopsy samples were collected from numerous sources including Flint Animal Cancer Center (FACC), St. Jude Children's Research Hospital, the Jackson Laboratory, Champions Oncology (COG), Gene Expression Omnibus (GEO), and the National Cancer Institute (NCI) and through our IRB-approved CuReFAST initiative. We used unsupervised hierarchical clustering analysis to determine the major ERMS endotypes or samples representative of distinct pathobiological mechanisms. Average-linkage clustering and the Euclidean distance methods were used in RStudio Version 3.6.1 to develop the dendrogram shown in Figure 4. Average-linkage clustering and the Euclidean distance methods were also used to create the vertical dendrograms.
Chemical Screens
Human cell lines RMS559, SMS-CTR, and RD and the mouse cell line U57810 were grown to ∼70% confluency in appropriate media before being trypsinized and plated into white-walled 384-well plates. BYK-719, GSK-1059615, TC-KHNS11, BEZ-235, CUDC-907, entinostat, and BKM-120 were resuspended in DMSO and added to plates at concentrations of 0.01 µM, 0.0268 µM, 0.072 µM, 0.193 µM, 0.518 µM, 1.39 µM, 3.73 µM, and 10 µM using the Tecan D300e drug printer (Tecan Life Sciences). After 72 h of incubating the cells with drugs at 37°C, 25 µL of CellTiter-Glo (G9243, Promega) was added to each well using the MultiFlo Dispenser. Cells then incubated in the dark for 15 min at room temperature while rocking. Luminescence was captured using the BioTek Synergy HT plate reader. Absolute IC50 values were found using GraphPad Prism (GraphPad).
ADDITIONAL INFORMATION
Data Deposition and Access
All raw data was deposited to our CuReFAST database. RNA sequencing data of our biopsy samples and cell lines were submitted to Gene Expression Omnibus (GEO) (accession numbers: GSE138269). DNA sequencing data of our biopsy samples and cell lines were submitted to European Genome-phenome Archive (EGA) (accession number: EGAS00001003981). Sequencing data from the laboratory of Dr. E. Alejandro Sweet-Cordero (UCSF) will be submitted separately to EGA. DNA and RNA sequencing for the UPS cell line, CLF-PED-015-T, were downloaded from dbGaP (accession number: phs001121.v1.p1). RNA sequencing data were collected from GEO for the cell lines, SkMc (accession number: GSM984615), and HSMM (accession number: GSM758578). Sequencing data from cell lines, RUCH2, RUCH3, SCMC-RMS, and RMS-YM, along with the cell lines from St. Jude Children's Hospital, are available on the OncoGenomics Database (https://pob.abcc.ncifcrf.gov/cgi-bin/JK). Somatic and gene expression data from Champions Oncology were received from the Champions TumorGraft database. National Cancer Institute (NCI) data was provided within the Supplemental Materials of Shern et al. (2014). Data from PDX models from Jackson Laboratory are available through the Mouse Tumor Biology Database (http://tumor.informatics.jax.org/mtbwi/pdxSearch.do). RNA sequencing of GEMMs and canine samples were deposited to GEO (GSE142775) and DNA sequencing of GEMMs were deposited to SRA (PRJNA613152).
Ethics Statement
The parents of this minor patient gave written consent for this study. The Institutional Review Board (IRB) approved of the collection of the patient's tumor sample and the studies that followed.
Acknowledgments
We are tremendously grateful for all the patients who kindly donated their tumor tissue as part of our CuReFAST initiative, as well as Marielle Yohe, Annette Werger, Suzanne Shesterman, Katherine Janeway, Patrick Reynolds, Lorena Landuzzi, and Jonathan Fletcher, who contributed material and data. We also would like to acknowledge those that have contributed to our sample collection: the Jackson Laboratory for giving us WES and RNA sequencing data from PDX models; Marcus Breese and E. Alejandro Sweet-Cordero for sharing sarcoma patient sample sequencing data; and the Gene Expression Omnibus (GEO) for sharing WES and RNA sequencing data from cell lines. This work was supported through Building Blocks to a Cure (1:1 match) crowdfunding campaign on Consano (https://consano.org/projects/building-blocks-to-a-cure-11-match/) as well as Rub Out Rhabdo Fund and financial contributions of Braver Stronger Smarter Inc.
Author Contributions
C.A.R., N.E.B., and C.K. participated in the design of these studies. C.A.R., K.C., K.M., M.L., E.R.R., N.E.B., and C.K. performed data collection and analysis. B.S. contributed materials for these studies. C.A.R., N.E.B., and C.K. wrote the manuscript.
Competing Interest Statement
The authors have declared no competing interest.
Supplementary Material
Footnotes
[Supplemental material is available for this article.]
REFERENCES
- Agaram NP, LaQuaglia MP, Alaggio R, Zhang L, Fujisawa Y, Ladanyi M, Wexler LH, Antonescu CR. 2019. MYOD1-mutant spindle cell and sclerosing rhabdomyosarcoma: an aggressive subtype irrespective of age. A reappraisal for molecular classification and risk stratification. Mod Pathol 32: 27–36. 10.1038/s41379-018-0120-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr FG, Smith LM, Lynch JC, Strzelecki D, Parham DM, Qualman SJ, Breitfeld PP. 2006. Examination of gene fusion status in archival samples of alveolar rhabdomyosarcoma entered on the Intergroup Rhabdomyosarcoma Study-III trial: a report from the Children's Oncology Group. J Mol Diagn 8: 202–208. 10.2353/jmoldx.2006.050124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J, Taylor BS, Banerji S, Ramos AH, Lagos-Quintana M, Decarolis PL, Shah K, Socci ND, Weir BA, Ho A, et al. 2010. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat Genet 42: 715–721. 10.1038/ng.619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendell JC, Patel MR, Infante JR, Kurkjian CD, Jones SF, Pant S, Burris HA 3rd, Moreno O, Esquibe V, Levin W, Moore KN. 2015. Phase 1, open-label, dose escalation, safety, and pharmacokinetics study of ME-344 as a single agent in patients with refractory solid tumors. Cancer 121: 1056–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breneman JC, Lyden E, Pappo AS, Link MP, Anderson JR, Parham DM, Qualman SJ, Wharam MD, Donaldson SS, Maurer HM, et al. 2003. Prognostic factors and clinical outcomes in children and adolescents with metastatic rhabdomyosarcoma—a report from the Intergroup Rhabdomyosarcoma Study IV. J Clin Oncol 21: 78–84. 10.1200/JCO.2003.06.129 [DOI] [PubMed] [Google Scholar]
- Crist WM, Anderson JR, Meza JL, Fryer C, Raney RB, Ruymann FB, Breneman J, Qualman SJ, Wiener E, Wharam M, et al. 2001. Intergroup Rhabdomyosarcoma Study-IV: results for patients with nonmetastatic disease. J Clin Oncol 19: 3091–3102. 10.1200/JCO.2001.19.12.3091 [DOI] [PubMed] [Google Scholar]
- Davis LE, Keller C. 2012. Integrative biology of rhabdomyosarcoma using genetic murine models. AACR 2012 Annual Meeting Educational Book. [Google Scholar]
- de Kock L, Rivera B, Revil T, Thorner P, Goudie C, Bouron-Dal Soglio D, Choong CS, Priest JR, van Diest PJ, Tanboon J, et al. 2017. Sequencing of DICER1 in sarcomas identifies biallelic somatic DICER1 mutations in an adult-onset embryonal rhabdomyosarcoma. Br J Cancer 116: 1621–1626. 10.1038/bjc.2017.147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43: 491–498. 10.1038/ng.806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diller L, Sexsmith E, Gottlieb A, Li FP, Malkin D. 1995. Germline p53 mutations are frequently detected in young children with rhabdomyosarcoma. J Clin Invest 95: 1606–1611. 10.1172/JCI117834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doros L, Yang J, Dehner L, Rossi CT, Skiver K, Jarzembowski JA, Messinger Y, Schultz KA, Williams G, André N, et al. 2012. DICER1 mutations in embryonal rhabdomyosarcomas from children with and without familial PPB-tumor predisposition syndrome. Pediatr Blood Cancer 59: 558–560. 10.1002/pbc.24020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doros LA, Rossi CT, Yang J, Field A, Williams GM, Messinger Y, Cajaiba MM, Perlman EJ, H ASK, Cathro P, et al. 2014. DICER1 mutations in childhood cystic nephroma and its relationship to DICER1-renal sarcoma. Mod Pathol 27: 1267–1280. 10.1038/modpathol.2013.242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X, Chen G, Cai ZD, Wang C, Liu ZZ, Lin ZY, Wu YD, Liang YX, Han ZD, Liu JC, et al. 2016. Overexpression of BUB1B contributes to progression of prostate cancer and predicts poor outcome in patients with prostate cancer. Onco Targets Ther 9: 2211–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GTEx Consortium. 2013. The Genotype-Tissue Expression (GTEx) project. Nat Genet 45: 580–585. 10.1038/ng.2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn MM, Vreede L, Bemelmans SA, van der Looij E, van Kessel AG, Schackert HK, Ligtenberg MJ, Hoogerbrugge N, Kuiper RP, de Voer RM. 2016. Prevalence of germline mutations in the spindle assembly checkpoint gene BUB1B in individuals with early-onset colorectal cancer. Genes Chromosomes Cancer 55: 855–863. 10.1002/gcc.22385 [DOI] [PubMed] [Google Scholar]
- Hanks S, Coleman K, Reid S, Plaja A, Firth H, Fitzpatrick D, Kidd A, Mehes K, Nash R, Robin N, et al. 2004. Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in BUB1B. Nat Genet 36: 1159–1161. 10.1038/ng1449 [DOI] [PubMed] [Google Scholar]
- Hill DA, Ivanovich J, Priest JR, Gurnett CA, Dehner LP, Desruisseau D, Jarzembowski JA, Wikenheiser-Brokamp KA, Suarez BK, Whelan AJ, et al. 2009. DICER1 mutations in familial pleuropulmonary blastoma. Science 325: 965 10.1126/science.1174334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudler P, Britovsek NK, Grazio SF, Komel R. 2016. Association between polymorphisms in segregation genes BUB1B and TTK and gastric cancer risk. Radiol Oncol 50: 297–307. 10.1515/raon-2015-0047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. 2012. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22: 568–576. 10.1101/gr.129684.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohsaka S, Shukla N, Ameur N, Ito T, Ng CK, Wang L, Lim D, Marchetti A, Viale A, Pirun M, et al. 2014. A recurrent neomorphic mutation in MYOD1 defines a clinically aggressive subset of embryonal rhabdomyosarcoma associated with PI3K-AKT pathway mutations. Nat Genet 46: 595–600. 10.1038/ng.2969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, Maglott DR. 2014. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 42: D980–D985. 10.1093/nar/gkt1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Dewey CN. 2011. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12: 323 10.1186/1471-2105-12-323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Li L, Li X, Wang Y, Xie Y, Liu C, Li F. 2015. Undifferentiated pleomorphic sarcoma with co-existence of KRAS/PIK3CA mutations. Int J Clin Exp Pathol 8: 8563–8567. [PMC free article] [PubMed] [Google Scholar]
- Malempati S, Hawkins DS. 2012. Rhabdomyosarcoma: review of the Children's Oncology Group (COG) Soft-Tissue Sarcoma Committee experience and rationale for current COG studies. Pediatr Blood Cancer 59: 5–10. 10.1002/pbc.24118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansouri N, Movafagh A, Sayad A, Heidary Pour A, Taheri M, Soleimani S, Mirzaei HR, Alizadeh Shargh S, Azargashb E, Bazmi H, et al. 2016. Targeting of BUB1b gene expression in sentinel lymph node biopsies of invasive breast cancer in Iranian female patients. Asian Pac J Cancer Prev 17: 317–321. 10.7314/APJCP.2016.17.S3.317 [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297–1303. 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentzel T. 2010. [Spindle cell rhabdomyosarcoma in adults: a new entity in the spectrum of malignant mesenchymal tumors of soft tissues]. Pathologe 31: 91–96. 10.1007/s00292-009-1249-6 [DOI] [PubMed] [Google Scholar]
- Mentzel T, Katenkamp D. 2000. Sclerosing, pseudovascular rhabdomyosarcoma in adults. Clinicopathological and immunohistochemical analysis of three cases. Virchows Arch 436: 305–311. 10.1007/s004280050451 [DOI] [PubMed] [Google Scholar]
- Mentzel T, Kuhnen C. 2006. Spindle cell rhabdomyosarcoma in adults: clinicopathological and immunohistochemical analysis of seven new cases. Virchows Arch 449: 554–560. 10.1007/s00428-006-0284-4 [DOI] [PubMed] [Google Scholar]
- Oki Y, Kelly KR, Flinn I, Patel MR, Gharavi R, Ma A, Parker J, Hafeez A, Tuck D, Younes A. 2017. CUDC-907 in relapsed/refractory diffuse large B-cell lymphoma, including patients with MYC-alterations: results from an expanded phase I trial. Haematologica 102: 1923–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérot G, Chibon F, Montero A, Lagarde P, de Thé H, Terrier P, Guillou L, Ranchère D, Coindre JM, Aurias A. 2010. Constant p53 pathway inactivation in a large series of soft tissue sarcomas with complex genetics. Am J Pathol 177: 2080–2090. 10.2353/ajpath.2010.100104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rio Frio T, Lavoie J, Hamel N, Geyer FC, Kushner YB, Novak DJ, Wark L, Capelli C, Reis-Filho JS, Mai S, et al. 2010. Homozygous BUB1B mutation and susceptibility to gastrointestinal neoplasia. N Engl J Med 363: 2628–2637. 10.1056/NEJMoa1006565 [DOI] [PubMed] [Google Scholar]
- Rudzinski ER, Anderson JR, Chi YY, Gastier-Foster JM, Astbury C, Barr FG, Skapek SX, Hawkins DS, Weigel BJ, Pappo A, et al. 2017. Histology, fusion status, and outcome in metastatic rhabdomyosarcoma: a report from the Children's Oncology Group. Pediatr Blood Cancer 64: e26645 10.1002/pbc.26645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruymann FB, Grovas AC. 2000. Progress in the diagnosis and treatment of rhabdomyosarcoma and related soft tissue sarcomas. Cancer Invest 18: 223–241. 10.3109/07357900009031827 [DOI] [PubMed] [Google Scholar]
- Shern JF, Chen L, Chmielecki J, Wei JS, Patidar R, Rosenberg M, Ambrogio L, Auclair D, Wang J, Song YK, et al. 2014. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov 4: 216–231. 10.1158/2159-8290.CD-13-0639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W, Ng CKY, Lim RS, Jiang T, Kumar S, Li X, Wali VB, Piscuoglio S, Gerstein MB, Chagpar AB, et al. 2018. Reliability of whole-exome sequencing for assessing intratumor genetic heterogeneity. Cell Rep 25: 1446–1457. 10.1016/j.celrep.2018.10.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla N, Ameur N, Yilmaz I, Nafa K, Lau CY, Marchetti A, Borsu L, Barr FG, Ladanyi M. 2012. Oncogene mutation profiling of pediatric solid tumors reveals significant subsets of embryonal rhabdomyosarcoma and neuroblastoma with mutated genes in growth signaling pathways. Clin Cancer Res 18: 748–757. 10.1158/1078-0432.CCR-11-2056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snape K, Hanks S, Ruark E, Barros-Núñez P, Elliott A, Murray A, Lane AH, Shannon N, Callier P, Chitayat D, et al. 2011. Mutations in CEP57 cause mosaic variegated aneuploidy syndrome. Nat Genet 43: 527–529. 10.1038/ng.822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratton MR, Fisher C, Gusterson BA, Cooper CS. 1989. Detection of point mutations in N-ras and K-ras genes of human embryonal rhabdomyosarcomas using oligonucleotide probes and the polymerase chain reaction. Cancer Res 49: 6324–6327. [PubMed] [Google Scholar]
- Taylor AC, Shu L, Danks MK, Poquette CA, Shetty S, Thayer MJ, Houghton PJ, Harris LC. 2000. P53 mutation and MDM2 amplification frequency in pediatric rhabdomyosarcoma tumors and cell lines. Med Pediatr Oncol 35: 96–103. [DOI] [PubMed] [Google Scholar]
- Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, et al. 2013. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 43: 11.10.11–11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan X, Yeung C, Kim SY, Dolan JG, Ngo VN, Burkett S, Khan J, Staudt LM, Helman LJ. 2012. Identification of FoxM1/Bub1b signaling pathway as a required component for growth and survival of rhabdomyosarcoma. Cancer Res 72: 5889–5899. 10.1158/0008-5472.CAN-12-1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams BA, Williams KM, Doyle J, Stephens D, Greenberg M, Malkin D, Pappo AS. 2004. Metastatic rhabdomyosarcoma: a retrospective review of patients treated at the hospital for sick children between 1989 and 1999. J Pediatr Hematol Oncol 26: 243–247. 10.1097/00043426-200404000-00006 [DOI] [PubMed] [Google Scholar]
- Xie J, Yan Y, Liu F, Kang H, Xu F, Xiao W, Wang H, Wang Y. 2019. Knockdown of Rab7a suppresses the proliferation, migration, and xenograft tumor growth of breast cancer cells. Biosci Rep 39: BSR20180480 10.1042/BSR20180480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yost S, de Wolf B, Hanks S, Zachariou A, Marcozzi C, Clarke M, de Voer R, Etemad B, Uijttewaal E, Ramsay E, et al. 2017. Biallelic TRIP13 mutations predispose to Wilms tumor and chromosome missegregation. Nat Genet 49: 1148–1151. 10.1038/ng.3883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Zhang J, Yan W, Chen X. 2016. p73 expression is regulated by ribosomal protein RPL26 through mRNA translation and protein stability. Oncotarget 7: 78255–78268. 10.18632/oncotarget.13126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Ding H, Wang X, Lu Q. 2015. PABPC1 exerts carcinogenesis in gastric carcinoma by targeting miR-34c. Int J Clin Exp Pathol 8: 3794–3802. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw data was deposited to our CuReFAST database. RNA sequencing data of our biopsy samples and cell lines were submitted to Gene Expression Omnibus (GEO) (accession numbers: GSE138269). DNA sequencing data of our biopsy samples and cell lines were submitted to European Genome-phenome Archive (EGA) (accession number: EGAS00001003981). Sequencing data from the laboratory of Dr. E. Alejandro Sweet-Cordero (UCSF) will be submitted separately to EGA. DNA and RNA sequencing for the UPS cell line, CLF-PED-015-T, were downloaded from dbGaP (accession number: phs001121.v1.p1). RNA sequencing data were collected from GEO for the cell lines, SkMc (accession number: GSM984615), and HSMM (accession number: GSM758578). Sequencing data from cell lines, RUCH2, RUCH3, SCMC-RMS, and RMS-YM, along with the cell lines from St. Jude Children's Hospital, are available on the OncoGenomics Database (https://pob.abcc.ncifcrf.gov/cgi-bin/JK). Somatic and gene expression data from Champions Oncology were received from the Champions TumorGraft database. National Cancer Institute (NCI) data was provided within the Supplemental Materials of Shern et al. (2014). Data from PDX models from Jackson Laboratory are available through the Mouse Tumor Biology Database (http://tumor.informatics.jax.org/mtbwi/pdxSearch.do). RNA sequencing of GEMMs and canine samples were deposited to GEO (GSE142775) and DNA sequencing of GEMMs were deposited to SRA (PRJNA613152).