

Abstract
Ergothioneine is a thiohistidine derivative with potential benefits on many aging-related diseases. The central step of aerobic ergothioneine biosynthesis is the oxidative C–S bond formation reaction catalyzed by mononuclear nonheme iron sulfoxide synthases (EgtB and Egt1). Thus far, only the Mycobacterium thermoresistibile EgtB (EgtBMth) crystal structure is available, while the structural information for the more industrially attractive Egt1 enzyme is not. Herein, we reported the crystal structure of the ergothioneine sulfoxide synthase (EgtBCth) from Candidatus Chloracidobacterium thermophilum. EgtBCth has both EgtB- and Egt1-type of activities. Guided by the structural information, we conducted Rosetta Enzyme Design calculations, and we biochemically demonstrated that EgtBCth can be engineered more toward Egt1-type of activity. This study provides information regarding the factors governing the substrate selectivity in Egt1- and EgtB-catalysis and lays the groundwork for future sulfoxide synthase engineering toward the development of an effective ergothioneine process through a synthetic biology approach.
Keywords: ergothioneine, nonheme iron enzyme, enzyme engineering, Rosetta Enzyme Design, sulfur-containing natural product
Graphical Abstract
Ergothioneine is a thiohistidine derivative. Through an ergothioneine-specific transporter, human and animals absorb ergothioneine from foods, and it accumulates in concentrations as high as 2 mM in erythrocytes, livers, kidneys, lenses, and corneas of eyes.1–4 In addition, a combination of the two most abundant natural thiols ( V5–7 for ergothioneine and V8 for glutathione) protect cells against reactive oxygen species (ROS) and reactive nitrogen species (RNS) under varying conditions.9 Ergothioneine has also been suggested to serve as a protecting agent against several diseases, including cardiovascular disorders,10 rheumatoid arthritis,11,12 Crohn’s disease,13,14 neurodegenerative diseases,15–18 and diabetes.19 Because of its potential health benefit, ergothioneine biosynthetic studies have received considerable interest in recent years.
Two aerobic biosynthetic pathways of ergothioneine have been reported (Scheme 1A): the Mycobacterium smegmatis pathway (EgtA-EgtE) and the Neurospora crassa pathway (Egt1/Egt2).20–30 In these two pathways, the crucial steps are the oxidative C–S bond formation mediated by nonheme iron sulfoxide synthases (EgtBMsm /Egt1) and the reductive C–S cleavage reaction catalyzed by PLP-dependent lyases (EgtE/Egt2), which differ from other reported sulfur-transfer strategies.31–34 Recently, the ovothiol biosynthetic pathway has also been reconstituted in vitro (Scheme 1B).27,35,36 These three sulfoxide synthases (Egt1, EgtBMsm, and OvoA) differ in at least two aspects: their substrate selectivity and their regioselectivity. First, Egt1 and EgtBMsm selectively use hercynine as the substrate, while they differ in the sulfur sources [γ-glutamyl-cysteine (γ-Glu-Cys) for EgtBMsm and L-Cys for Egt1]. On the other hand, OvoA selectively uses L-His and L-Cys as the substrates. Although OvoA can also use of hercynine and γ-Glu-Cys as substrates, sulfoxide 4 is not the major product under this condition.37 Second, for EgtB- and Egt1-catalysis, sulfur was inserted into the ε-position of imidazole side-chain (Scheme 1A), while in OvoA-catalysis, the C–S bond is formed at the imidazole δ-carbon (Scheme 1B).
Scheme 1. Ergothioneine, Ovothiol Biosynthesis, and Distinct Properties of Sulfoxide Synthase EgtBCtha.

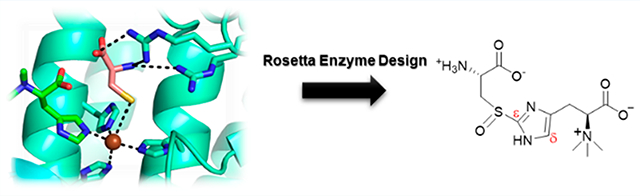
a(A) Two aerobic ergothioneine biosynthetic pathways. (B) Ovothiol biosynthetic pathway. (C) Sequence similarity network analysis of ergothioneine sulfoxide synthases and the link between the sulfoxide synthase EgtBCth (in red), Egt1 (in blue), and EgtB (in green). (D) EgtBCth exhibits Egt1- and EgtB-type activities.
Given ergothioneine’s many potential health benefits, there is an increasing demand for developing efficient industrial-scale ergothioneine production methods. The pathway involving Egt1-catalysis is preferred because the use of l-Cys as the sulfur source alleviates the competition between ergothioneine and glutathione biosynthesis, in which γ-Glu-Cys is a key biosynthetic intermediate. Among these sulfoxide synthases in ergothioneine and ovothiol biosynthesis, EgtBMth from M. thermoresistibile is the only reported structure.22 In this study, a thermophilic Candidatus Chloracidobacterium thermophilum sulfoxide synthase, Cabther_A1318 (EgtBCth, Scheme 1C), was identified from a cluster bridging between that of EgtB and Egt1 through sequence similarity network analysis. The separation of cluster of EgtBCth from that of EgtB and Egt1 is suggestive of different biochemical properties of EgtBCth. Indeed, biochemical characterization shows that EgtBCth exhibits both EgtB- and Egt1-type of activities (Scheme 1D). Encouraged by this discovery, based on the EgtBCth and EgtBMth structural information, we attempted EgtBCth activity engineering using Rosetta Enzyme Design calculation.38,39 We then selected three mutants for biochemical characterization and demonstrated that we can tune the activity of EgtBCth toward Egt1-catalysis.
Egt1 from N. crassa exhibited different substrate preference from both EgtB and OvoA (Scheme 1A,B).28,40 This discovery immediately raised questions as to which factors governed these selectivities (substrate selectivity and product regioselectivity, Scheme 1). Among these enzymes, only EgtBMth structure from a thermophilic M. thermoresistibile is available. Our attempts to crystallize N. crassa Egt1 failed. To search for a proper sulfoxide synthase to study factors governing substrate selectivities, we retrieved 21 475 protein sequences containing either DinB_2 domain (Pfam ID: PF12867) or FGE sulfatase domain (Pfam ID: PF03781) from the Pfam protein family database.41 3000 sequences out of 21 476 sequences were randomly selected for protein similarity network analysis (Scheme 1C) at an E-value cut off of 10−50. The protein similarity network was visualized by Cytoscape, which showed some sequences located between EgtB and Egt1 nodes (Scheme 1C and Table S1), implying the possibility of finding different biochemical properties. Among these sequences, Cabther_A1318 (EgtBCth) from thermophilic C. thermophilum was chosen for further studies.
The EgtBCth gene was overexpressed and purified following a reported procedure.28,40 The purified EgtBCth protein contained ~0.92 iron per monomer (Figure S1). The substrates, L-Cys and γ-Glu-Cys, were characterized before use in reactions (Figure S1). 1H NMR analysis showed that EgtBCth exhibited both EgtB- and Egt1-type activities and accepted both L-Cys and γ-Glu-Cys as the sulfur donor (Scheme 1D, Figures S2 and S3). In addition, the chemical shift (~ 7.11 ppm, the imidazole hydrogen signal of compound 3, Figure 1) is consistent with our previous results on EgtBMsm and Egt1 studies, suggesting that C–S bond is formed at the imidazole ε-position, instead of the OvoA-type in which the C–S bond is formed at the δ-position.28,40 EgtBCth was characterized kinetically using the oxygen consumption assays as previously reported in Egt1 and OvoA studies (Table 1 and Figures S4 and S5).28,40 Using hercynine and γ-Glu-Cys as substrates, the kcat of the reaction was ~18 min−1 with KM for hercynine of 41.4 ± 3.5 μM and for γ-Glu-Cys of 5.9 ± 0.9 mM. The catalytic efficiency of EgtBCth increased by ~42-fold when hercynine and L-Cys were used as the substrates exhibiting a kcat of ~26 min−1. In this reaction, EgtBCth has a KM of 87.7 ± 7.6 μM and 205 ± 18 μM, for hercynine and L-Cys, respectively (Table 1). The biochemical studies show that EgtBCth resembles the activity of EgtB and Egt1 in term of their C–S bond regioselectivity but differ in their substrate specificity as EgtBCth shows both Egt1- and EgtB-type of activities (Scheme 1D). The distinct substrate specificity might account for the cluster of EgtBCth locating between that of EgtB and Egt1 nodes in the sequence similarity analysis (Scheme 1C).
Figure 1.

1H NMR analysis of EgtBCth reactions. (A) EgtBCth reaction under EgtB reaction conditions. The two hydrogens of compound 2 imidazole side chain are labeled as 2, and the hydrogen of compound 3 imidazole hydrogen is labeled as 3. (B) EgtBCth reaction under Egt1-conditions. Compound 4 imidazole hydrogen is labeled 4. (C) Ratios of sulfoxide synthase and cysteine dioxygenase activity of EgtBCth under EgtB- and Egt1-type of reaction conditions. These two competing pathways are present in all three sulfoxide synthases (Egt1, EgtB, and OvoA). Notably, a significant level of cysteine dioxygenase activity was observed in EgtBCth-catalysis.
Table 1.
Kinetic Parameter Characterizations of EgtBCth Wild-Type and Variants
| enzyme | substrates | kcat (min−1) | KM, hercynine(μM) | kcat/ KM, hercynine(min−1μM−1) | KM, γ‑Glu‑Cys/Cys (μM) | kcat/ KM, γGluCys/Cys(min−1μM−1) | % of coupling product |
|---|---|---|---|---|---|---|---|
| wild-type | 2 + γ-Glu-Cys | 17.5 ± 0.4 | 41.4 ± 3.5 | 0.42 ± 0.04 | (5.9 ± 0.9)E3 | (3.0 ± 0.5)E-3 | 75% |
| 2 + L-Cys | 26.6 ± 0.7 | 87.7 ± 7.6 | 0.30 ± 0.03 | 205 ± 18 | 0.13 ± 0.01 | 72% | |
| A420Y | 2 + γ-Glu-Cys | 17.4 ± 0.3 | 13.2 ± 1.3 | 1.3 ± 0.1 | (3.1 ± 0.3)E3 | (5.6 ± 0.6)E-3 | 61% |
| 2 + L-Cys | 27.6 ± 0.5 | 39.2 ± 1.6 | 0.70 ± 0.03 | 28.1 ± 1.8 | 0.98 ± 0.06 | 76% | |
| D52L | 2 + γ-Glu-Cys | 12.9 ± 0.2 | 19.6 ± 1.4 | 0.66 ± 0.05 | (3.9 ± 0.3)E3 | (3.3 ± 0.2)E-3 | 50% |
| 2 + L-Cys | 27.6 ± 0.5 | 13.2 ± 1.3 | 2.1 ± 0.2 | 47.3 ± 2.4 | 0.58 ± 0.03 | 69% | |
| D52L/A420Y | 2 + γ-Glu-Cys | 18.6 ± 0.4 | 15.1 ± 1.3 | 1.2 ± 0.1 | (4.2 ± 0.5)E3 | (4.4 ± 0.5)E-3 | 52% |
| 2 + L-Cys | 32.7 ± 0.3 | 23.3 ± 1.2 | 1.4 ± 0.07 | 48.7 ± 3.5 | 0.67 ± 0.04 | 75% |
In previous studies of EgtB, Egt1, and OvoA, these enzymes also show some cysteine dioxygenase activity.25,28,37,42,43 Thus, we analyzed EgtBCth reaction for cysteine dioxygenase activity under both Egt1- and EgtB-type of catalytic conditions. We have characterized all the products, and their 1H NMR signals have been reported.28,37,42,43 The ratio between sulfoxide and cysteine oxidation activity was analyzed by 1H NMR (Figures S6 and S7).25,28 Our analysis indicated that when hercynine and γ-Glu-Cys were the substrates, sulfoxide 3 accounted for ~75% of the total products, and if hercynine and L-Cys were the substrates, sulfoxide 4 accounted for ~72% of the products (Table 1 and Figures S6 and S7). The remaining 25% and 28% were γ-Glu-Cys sulfinic acid 9 or cysteine sulfinic acid 10, respectively (Figures S6 and S7).
With this interesting biochemical information, we also initiated its crystallization studies of EgtBCth. Apo-EgtBCth crystallized in a cubic form with a space group of P21 and diffracted to 2.5 Å in a synchrotron beam source (PDB ID: 6O6M). The single-wavelength anomalous dispersion (SAD) technique was used for de novo phase determination and structure solution using selenomethionine incorporated EgtBCth crystals (Table S2). The crystallographic asymmetric unit is composed of four protein molecules (Figure 2A). The overall structure of EgtBCth is a tetramer, consistent with its oligomerization state in solution as detected in gel filtration profile. For each monomer, electron densities were resolved for residues 17 to 433, except an interdomain loop of 10 residues (184 to 193) which is missing because of its high flexibility (Figure 2A). Each monomer is composed of an N-terminal helical domain (residue 17 to 183) and a C-terminal domain that consists of an α–ββ–α fold (residue 194 to 433) (Figure 2A). A Dali44 search revealed that the N-terminal domain most closely resembles the damage-inducible protein Din-B (PDB ID: 5WK045) and the C-terminal domain is structurally similar to the formylglycine generating enzyme (PDB ID: 5NXL).46 The C-terminal domain shares a high structural similarity with the catalytic domain of EgtBMth.22
Figure 2.

Structures of EgtBCth and EgtBCth·hercynine binary complex. (A) Overall structure of EgtBCth in the tetrameric configuration with each monomer labeled. In Monomer A, the N-terminal domain (residue 17 to 183) is shown in blue and the C-terminal domain (residue 194 to 433) is shown in pink. The iron cofactor present at the active site of each monomer is shown as a brown sphere. (B) The 2mFo-DFc map of iron coordination site of EgtBCth contoured at 1.5σ (blue mesh); the metal ion is shown as a brown sphere and the coordinating residues are represented in sticks. Ordered water molecules coordinating the iron are shown as red spheres. (C) The mFo-DFc omit map of the active site of EgtBCth cocrystallized with hercynine contoured at 3σ (green mesh). The chemical structure of the substrate hercynine (shown as yellow sticks) was modeled into the positive density. (D) The interaction network between hercynine and EgtBCth active-site residues. Residues interacting with the substrate hercynine are shown in sticks with the potential interactions shown in black dash lines. (E) The previously reported structure of EgtBMth· dimethyl histidine·γ-Glu-Cys complex (PDB ID 4X8D) superimposed on the EgtBCth·herycine complex (shown in green). The side chains of the EgtBMth residues interacting with the γ-Glu-Cys are shown in sticks (white) and numbered with a superscript (′), the corresponding residues in the EgtBCth structure are shown as blue sticks. (F) The putative γ-Glu-Cys binding mode to EgtBCth (shown as yellow sticks). The potential interactions between γ-Glu-Cys and EgtBCth active-site residues were depicted as black dashed lines, and the side chains of the interacting residues are shown as blue sticks
The active site of EgtBCth is located at the interface between the N- and C-terminal domains for each monomer where a mononuclear nonheme iron is coordinated by His62, His153, His157 and three water molecules in an octahedral arrangement (Figure 2B). Upon soaking or cocrystallizing EgtBCth crystals with hercynine, close to the metal ion, strong positive density was observed replacing one of the coordinating water molecules (Figure 2C). Comparison between structures of EgtBCth and EgtBCth·hercynine binary complex, no significant conformation change was observed (Figure S8). In the EgtBCth· hercynine binary complex (PDB ID: 6O6L), hercynine coordinates to the iron center through its imidazole ε-nitrogen (Figure 2D). The hercynine imidazole δ-nitrogen forms hydrogen bonds with Gln156 (3.4 Å) and the hydroxyl group of Tyr93 (3.6 Å). In addition to the dipolar contact with Asn414, the trimethylated amino group of hercynine has cation-π interactions with the side chains of Phe415 (4.7 Å) and Phe416 (5.1 Å).
Despite the lack of overall sequence similarity between EgtBCth and EgtBMth, a comparison of active sites of the two homologous enzymes reveals a high degree of conservation of residues, suggesting a similar substrate recognition network and catalytic mechanism. The residues used to coordinate the iron center are faithfully conserved in EgtBCth and EgtBMth (Figure 2E). Additionally, the residues involved in hercynine binding between these two structures are conserved (Gln137, Asn414, and Trp415 in EgtBMth vs Gln156, Asn414, and Phe415 in EgtBCth, Figure 2E and S9). The binding pocket for the cosubstrate γ-Glu-Cys/L-Cys is located close to the iron center in EgtBMth. A similar pocket also exists in EgtBCth. As EgtBCth·hercynine·Cys or EgtBCth·hercynine·γ-Glu-Cys tertiary complexes were resistant to crystallization efforts, we used the structure of M. thermoresistibile EgtBMth in complex with dimethyl histidine and γ-Glu-Cys (PDB ID: 4X8D, Figure 2E) to guide the creation of a model of EgtBCth·hercynine·γ-Glu-Cys complex (Figure 2F). 22 Upon superimposing, the residues involved in the binding of cysteinyl portion of γ-Glu-Cys are identical for both EgtBMth and EgtBCth (Figure 2E). They share two conserved Arg residues involving in the binding of 1-carboxylate group of γ-Glu-Cys (Arg87 and Arg90 in EgtBMth vs Arg103 and Arg106 in EgtBCth (Figure 2E). These pairs of arginine residues also form salt bridges with the carboxylate of L-Cys. The thiol groups of γ-Glu-Cys and L-Cys replace one of the iron-center water ligands.
Based on the structural model in Figure 2F, the regions of active site in EgtBCth anchoring the glutamyl portion of γ-Glu-Cys vary between EgtBMth and EgtBCth. The EgtBMth Asp416 and Arg420 residues interacting with γ-Glu-Cys are replaced with Ala420 and Phe416 in EgtBCth (Figure 2F). Therefore, some hydrogen bonding and salt bridge interactions present in EgtBMth·dimethylhistidine·γ-Glu-Cys do not seem to be present in the modeled EgtBCth·hercynine·γ-Glu-Cys complex. However, favorable hydrogen-bond interaction between the γ-Glu-Cys glutamyl groups and EgtBCth Gln393 and Asp52 residues led to the binding of γ-Glu-Cys in EgtBCth as an alternative rotamer (Figure 2F) relative to that in EgtBMth. Overall, EgtBCth has a more open active site relative to that of M. thermoresistibile EgtBMth22 for cosubstrate recognition, which might account for both Egt1- and EgtB-types of activities in EgtBCth.
Based on EgtBMth and EgtBCth structures, we generated the Egt1 model using the I-TASSER (Figure S9).47 In this Egt1 structural model, the iron center histidine ligands (His370, His463, and His467) and the binding pocket for hercynine were conserved (Figure S9). On the contrary, because EgtBCth is flexible in making use of either γ-Glu-Cys or L-Cys as the sulfur donor and the fact that EgtBCth’s active-site pocket is more open than EgtBMth implies that residues next to the γ-Glu-Cys may be turned to modulate EgtBCth selectivity (Figure S9). Structural comparison of these three enzymes identified three nonconserved residues (Asp52, Phe416, and Ala420) in the EgtBCth active site relative to that of EgtBMth and Egt1 (Figure S9). We decided to test this hypothesis by engineering these residues with the goal of tuning the EgtBCth activity toward the Egt1-type to facilitate ergothioneine production.
To guide our engineering effort, we employed Rosetta Enzyme Design to optimize the active site environment for substrate binding.38 By using Rosetta energy function and conformational sampling of side chain rotamers, 8000 possible variants were ranked on the basis of the relative energy levels when hercynine and L-Cys are used as the substrates. The top 20 variants listed in Table S3 were further evaluated through Pymol to assess the potential interaction between the variants and L-Cys substrate. Additionally, the variants were further compared with Egt1 sequences retrieved from the protein sequence similarity network analysis in Scheme 1C (Figure S9). The sequence alignment shows conserved Leu360 and Tyr820 among Egt1 homologues; however, these two residues are not conserved in EgtBCth (Asp52 and Ala420). Surprisingly, the D52L and A420Y variants were listed among the top 20 variants predicted from Rosetta Enzyme Design, which warrants further examination of the role of these residues. Taking all these factors into account, we started EgtBCth engineering with two single mutations: EgtBCth A420Y and D52L.
Using hercynine and γ-Glu-Cys as the substrates for EgtBCth A420Y variant, the number of turnovers remained similar to that of wild-type EgtBCth (kcat of 17.4 ± 0.3 min−1) with lower Michaelis constant (KM of 13.2 ± 1.3 μM for hercynine, and a KM of 3.1 ± 0.3 mM for γ-Glu-Cys, Table 1 and Figure S10). Additionally, 1H NMR analysis of A420Y mutant shows that the C–S bond is formed at the imidazole side chain ε-position similar to that of wild-type EgtBCth (Figure S11). However, when hercynine and L-Cys were the substrates, the KM for L-Cys was lowered by 10-fold (205 ± 18 μM for wild-type vs 28.1 ± 1.8 μM for the A420Y variant), clearly indicating that this mutation altered substrate selectivity more toward the Egt1-type (Table 1 and Figure S12). Besides this kinetic information, 1H NMR characterization of EgtBCth A420Y variant supports the importance of this position in controlling the reaction selectivity. When hercynine and γ-Glu-Cys were used as the substrates, the amount of sulfoxide 3 decreased from 75% in wild-type EgtBCth (Table 1) to 61% in EgtBCth A420Y variant (Table 1 and Figure S13), indicating an increased amount of side-reaction (cysteine dioxygenase activity). Interestingly, when hercynine and L-Cys were used as the substrates, the amount of sulfoxide 4 was 76% in EgtBCth A420Y, which is slightly improved relative to that of the wildtype EgtBCth (Table 1 and Figure S14). Notably, structural analysis shows that this residue does not directly interact with the substrate. However, as A420Y mutant altered the substrate selectivity, this suggested that this mutation may change in the interaction network of the substrate binding site to favor L-Cys binding, which might explain its conservancy among Egt1 homologues. The mutation reduces the active-site pocket, favoring the smaller cysteine.
These studies were also repeated using EgtBCth D52L mutant, and the results are shown in Table 1 and Figures S15–S17. In D52L variant, when γ-Glu-Cys was used as the substrate, the amount of sulfoxide 3 decreased to 50% of the product mixture (Figure S18). On the contrary, when L-Cys was used as the substrate, the level of the coupling product 4 was 69%, which was comparable to the wild-type activity (Figure S19). The D52L mutation can possibly disrupt the hydrogen bond between Asp52 and glutamyl group of γ-Glu-Cys, which in turn alters the substrate selectivity of EgtBCth. Therefore, EgtBCth D52 can play some roles in controlling the partition between Egt1- and EgtB-type of activities.
The promising results from these two variants led us to further characterized the activity of EgtBCth D52L/A420Y double mutant (Figure S20). When hercynine and γ-Glu-Cys were the substrates, the kinetic parameters were not significantly altered; however, only 52% of sulfoxide product was observed (Table 1 and Figures S21 and S22). In contrast, using hercynine and L-Cys as the substrates, EgtBCth D52L/A420Y exhibits the highest turnover among the wild-type and other mutants (kcat of 32.7 ± 0.3 min−1) with 75% of sulfoxide product formation (Table 1 and Figures S23 and S24). Therefore, the double mutation also tuned EgtBCth activity toward Egt1-type.
In summary, EgtBCth exhibited distinct biochemical activities showing both Egt1- and EgtB-type of reactions (Scheme 1). Thus far, only the crystal structure of M. thermoresistibile EgtBMth sulfoxide synthase was available. In this study, we have successfully crystallized EgtBCth. The EgtBCth and EgtBMth structural information allows us the opportunity to examine the factors responsible for differentiating the substrate selectivity among these sulfoxide synthases. Guided by computational results using the Rosetta Enzyme Design and information from evolutionary-related sequences (the EgtB-node and the Egt-1 node, Scheme 1C), we selected three mutants for characterizations (EgtBCth D52L, A420Y, and D52L/A420Y variants). Indeed, even with a single mutation, we could tune the EgtBCth activity more toward Egt1-type of catalysis. For these two mutants, the catalytic proficiency (kcat/Km) changes from ~40-fold more favoring Egt1 type in wildtype EgtBCth to ~180-fold more favoring Egt1-type.
Because of difficulties faced in chemical synthetic processes in industrial ergothioneine production,48,49 there is an increasing interest of developing ergothioneine biosynthetic processes. For fermentation-based ergothioneine production, Egt1-type of pathway is preferred relative to that of the EgtB-type of pathway to alleviate the competition between ergothioneine and glutathione biosynthesis. Guided by EgtBCth structural information and predictions from Rosetta Enzyme Design calculation, we have successfully demonstrated that EgtBCth activities could be tuned toward Egt1-type (D52L, A420Y, and D52L/A420Y). These promising results open up the opportunity of engineering EgtBCth enzyme toward Egt1-type, with significantly better thermo-stability, which might benefit the ergothioneine industrial production processes.
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank Art Monzingo for his guidance during the denovo-phasing of the EgtBCth structure. This work is supported in part by a grant from the National Science Foundation (CHE-1309148 to P.L.), the Boston University Nano Center seeding grant, National Institute for Health (R01 GM104896 and 125882 to Y.J.Z), and Welch Foundation (F-1778 to Y.J.Z). W.H. and L. Z. are supported by fellowship from China Scholarship Council.
Footnotes
The authors declare no competing financial interest.
Related Articles
While this manuscript was under review, another manuscript was published on this enzyme.50
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.9b02054.
Experimental procedures, protein characterizations, crystallographic studies, NMR of reactions and products, and kinetic study results (PDF)
Accession Codes
The atomic coordinates and structure factors have been deposited in the Protein Data Bank: PDB entry 6O6M for EgtBCth and 6O6L for EgtBCth in complex with hercynine.
REFERENCES
- (1).Melville DB; Horner WH; Lubschez R Tissue Ergothioneine. J. Biol. Chem 1954, 206, 221–228. [PubMed] [Google Scholar]
- (2).Shires TK; Brummel MC; Pulido JS; Stegink LD Ergothioneine Distribution in Bovine and Porcine Ocular Tissues. Comp. Biochem. Physiol., Part C: Pharmacol., Toxicol. Endocrinol 1997, 117, 117–120. [DOI] [PubMed] [Google Scholar]
- (3).Leone E; Mann T Ergothioneine in the Seminal Vesicle Secretion. Nature 1951, 168, 205–206. [DOI] [PubMed] [Google Scholar]
- (4).Grundemann D; Harlfinger S; Golz S; Geerts A; Lazar A; Berkels R; Jung N; Rubbert A; Schomig E Discovery of the Ergothioneine Transporter. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 5256–5261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Hartman PE Ergothioneine as Antioxidant. Methods Enzymol. 1990, 186, 310–318. [DOI] [PubMed] [Google Scholar]
- (6).Hand CE; Honek JF Biological Chemistry of Naturally Occurring Thiols of Microbial and Marine Origin. J. Nat. Prod 2005, 68, 293–308. [DOI] [PubMed] [Google Scholar]
- (7).Fahey RC Novel Thiols of Prokaryotes. Annu. Rev. Microbiol 2001, 55, 333–356. [DOI] [PubMed] [Google Scholar]
- (8).Scott EM; Duncan IW; Ekstrand V Purification and Properties of Glutathione Reductase of Human Erythrocytes. J. Biol. Chem 1963, 238, 3928–3933. [PubMed] [Google Scholar]
- (9).Cheah IK; Halliwell B Ergothioneine; Antioxidant Potential, Physiological Function and Role in Disease. Biochim. Biophys. Acta, Mol. Basis Dis 2012, 1822, 784–793. [DOI] [PubMed] [Google Scholar]
- (10).Libby P; Ridker PM; Hansson GK Progress and Challenges in Translating the Biology of Atherosclerosis. Nature 2011, 473, 317–325. [DOI] [PubMed] [Google Scholar]
- (11).Taubert D; Lazar A; Grimberg G; Jung N; Rubbert A; Delank K-S; Perniok A; Erdmann E; Schömig E Association of Rheumatoid Arthritis with Ergothioneine Levels in Red Blood Cells: A Case Control Study. J. Rheumatol 2006, 33, 2139–2145. [PubMed] [Google Scholar]
- (12).Tokuhiro S; Yamada R; Chang X; Suzuki A; Kochi Y; Sawada T; Suzuki M; Nagasaki M; Ohtsuki M; Ono M; Furukawa H; Nagashima M; Yoshino S; Mabuchi A; Sekine A; Saito S; Takahashi A; Tsunoda T; Nakamura Y; Yamamoto K An Intronic SNP in a RUNX1 Binding Site of SLC22A4, Encoding an Organic Cation Transporter, Is Associated with Rheumatoid Arthritis. Nat. Genet 2003, 35, 341–348. [DOI] [PubMed] [Google Scholar]
- (13).Peltekova VD; Wintle RF; Rubin LA; Amos CI; Huang Q; Gu X; Newman B; Van Oene M; Cescon D; Greenberg G; Griffiths AM; St George-Hyslop PH; Siminovitch KA Functional Variants of Octn Cation Transporter Genes Are Associated with Crohn Disease. Nat. Genet 2004, 36, 471–475. [DOI] [PubMed] [Google Scholar]
- (14).Leung E; Hong J; Fraser AG; Merriman TR; Vishnu P; Krissansen GW Polymorphisms in the Organic Cation Transporter Genes Slc22a4 and Slc22a5 and Crohn’s Disease in a New Zealand Caucasian Cohort. Immunol. Cell Biol 2006, 84, 233–236. [DOI] [PubMed] [Google Scholar]
- (15).Kaneko I; Takeuchi Y; Yamaoka Y; Tanaka Y; Fukuda T; Fukumori Y; Mayumi T; Hama T Quantitative Determination of Ergothioneine in Plasma and Tissues by TLC-densitometry. Chem. Pharm. Bull 1980, 28, 3093–3097. [DOI] [PubMed] [Google Scholar]
- (16).Briggs I Ergothioneine in the Central Nervous System. J. Neurochem 1972, 19, 27–35. [DOI] [PubMed] [Google Scholar]
- (17).Crossland J; Mitchell J; Woodruff GN The Presence of Ergothioneine in the Central Nervous System and Its Probable Identity with the Cerebellar Factor. J. Physiol 1966, 182, 427–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Moncaster JA; Walsh DT; Gentleman SM; Jen L-S; Aruoma OI Ergothioneine Treatment Protects Neurons Against N-methyl-d-aspartate Excitotoxicity in an in Vivo Rat Retinal Model. Neurosci. Lett 2002, 328, 55–59. [DOI] [PubMed] [Google Scholar]
- (19).Bastard J-P; Maachi M; Van Nhieu JT; Jardel C; Bruckert E; Grimaldi A; Robert J-J; Capeau J; Hainque B Adipose Tissue IL-6 Content Correlates with Resistance to Insulin Activation of Glucose Uptake Both in Vivo and in Vitro. J. Clin. Endocrinol. Metab 2002, 87, 2084–2089. [DOI] [PubMed] [Google Scholar]
- (20).Seebeck FP In Vitro Reconstitution of Mycobacterial Ergothioneine Biosynthesis. J. Am. Chem. Soc 2010, 132, 6632–6633. [DOI] [PubMed] [Google Scholar]
- (21).Vit A; Misson L; Blankenfeldt W; Seebeck FP Crystallization and Preliminary X-ray Analysis of the Ergothioneine-biosynthetic Methyltransferase Egtd. Acta Crystallogr., Sect. F: Struct. Biol. Commun 2014, 70, 676–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Goncharenko KV; Vit A; Blankenfeldt W; Seebeck FP Structure of the Sulfoxide Synthase Egtb from the Ergothioneine Biosynthetic Pathway. Angew. Chem., Int. Ed 2015, 54, 2821–2824. [DOI] [PubMed] [Google Scholar]
- (23).Vit A; Mashabela GT; Blankenfeldt W; Seebeck FP Structure of the Ergothioneine-Biosynthesis Amidohydrolase EgtC. ChemBioChem 2015, 16, 1490–1496. [DOI] [PubMed] [Google Scholar]
- (24).Vit A; Misson L; Blankenfeldt W; Seebeck FP Ergothioneine Biosynthetic Methyltransferase Egtd Reveals the Structural Basis of Aromatic Amino Acid Betaine Biosynthesis. ChemBioChem 2015, 16, 119–125. [DOI] [PubMed] [Google Scholar]
- (25).Goncharenko KV; Seebeck FP Conversion of a Non-heme Iron-dependent Sulfoxide Synthase Into a Thiol Dioxygenase by a Single Point Mutation. Chem. Commun 2016, 52, 1945–1948. [DOI] [PubMed] [Google Scholar]
- (26).Faponle AS; Seebeck FP; de Visser SP Sulfoxide Synthase versus Cysteine Dioxygenase Reactivity in a Non-heme Iron Enzyme. J. Am. Chem. Soc 2017, 139, 9259–9270. [DOI] [PubMed] [Google Scholar]
- (27).Liao C; Seebeck FP Convergent Evolution of Ergothioneine Biosynthesis in Cyanobacteria. ChemBioChem 2017, 18, 2115–2118. [DOI] [PubMed] [Google Scholar]
- (28).Hu W; Song H; Her AS; Bak DW; Naowarojna N; Elliott SJ; Qin L; Chen X; Liu P Bioinformatic and Biochemical Characterizations of C-S Bond Formation and Cleavage Enzymes in the Fungus Neurospora Crassa Ergothioneine Biosynthetic Pathway. Org. Lett 2014, 16, 5382–5385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Song H; Hu W; Naowarojna N; Her AS; Wang S; Desai R; Qin L; Chen X; Liu P Mechanistic Studies of a Novel C-s Lyase in Ergothioneine Biosynthesis: the Involvement of a Sulfenic Acid Intermediate. Sci. Rep 2015, 5, 11870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Irani S; Naowarojna N; Tang Y; Kathuria KR; Wang S; Dhembi A; Lee N; Yan W; Lyu H; Costello CE; et al. Snapshots of C-S Cleavage in Egt2 Reveals Substrate Specificity and Reaction Mechanism. Cell Chem. Biol 2018, 25, 519–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Naowarojna N; Cheng R; Chen L; Quill M; Xu M; Zhao C; Liu P Mini-review: Ergothioneine and Ovothiol Biosyntheses, an Unprecedented Trans-sulfur Strategy in Natural Product Biosynthesis. Biochemistry 2018, 57, 3309–3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Kessler D Enzymatic Activation of Sulfur for Incorporation Into Biomolecules in Prokaryotes. FEMS Microbiol. Rev 2006, 30, 825–840. [DOI] [PubMed] [Google Scholar]
- (33).Castellano I; Seebeck FP On ovothiol biosynthesis and biological roles: from life in the ocean to therapeutic potential. Nat. Prod. Rep 2018, 35, 1241–1250. [DOI] [PubMed] [Google Scholar]
- (34).Dunbar KL; Scharf DH; Litomska A; Hertweck C Enzymatic Carbon–Sulfur Bond Formation in Natural Product Biosynthesis. Chem. Rev 2017, 117, 5521–5577. [DOI] [PubMed] [Google Scholar]
- (35).Braunshausen A; Seebeck FP Identification and Characterization of the First Ovothiol Biosynthetic Enzyme. J. Am. Chem. Soc 2011, 133, 1757–1759. [DOI] [PubMed] [Google Scholar]
- (36).Naowarojna N; Huang P; Cai Y; Song H; Wu L; Cheng R; Li Y; Wang S; Lyu H; Zhang L; et al. In Vitro Reconstitution of the Remaining Steps in Ovothiol A Biosynthesis: C–S Lyase and Methyltransferase Reactions. Org. Lett 2018, 20, 5427–5430. [DOI] [PubMed] [Google Scholar]
- (37).Song H; Her AS; Raso F; Zhen Z; Huo Y; Liu P Cysteine Oxidation Reactions Catalyzed by a Mononuclear Non-heme Iron Enzyme (Ovoa) in Ovothiol Biosynthesis. Org. Lett 2014, 16, 2122–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Li R; Wijma HJ; Song L; Cui Y; Otzen M; Tian Y. e.; Du J; Li T; Niu D; Chen Y; et al. Computational Redesign of Enzymes for Regio-and Enantioselective Hydroamination. Nat. Chem. Biol 2018, 14, 664–670. [DOI] [PubMed] [Google Scholar]
- (39).Richter F; Leaver-Fay A; Khare SD; Bjelic S; Baker D De Novo Enzyme Design Using Rosetta3. PLoS One 2011, 6, No. e19230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Song H; Leninger M; Lee N; Liu P Regioselectivity of the Oxidative C–S Bond Formation in Ergothioneine and Ovothiol Biosyntheses. Org. Lett 2013, 15, 4854–4857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Finn RD; Coggill P; Eberhardt RY; Eddy SR; Mistry J; Mitchell AL; Potter SC; Punta M; Qureshi M; Sangrador-Vegas A; et al. The Pfam Protein Families Database: Towards a More Sustainable Future. Nucleic Acids Res. 2016, 44, D279–D285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Chen L; Naowarojna N; Chen B; Xu M; Quill M; Wang J; Deng Z; Zhao C; Liu P Mechanistic Studies of a Non-heme Iron Enzyme OvoA in Ovothiol Biosynthesis Using a Tyrosine Analogue, 2-Amino-3-(4-hydroxy-3-(methoxyl) phenyl) Propanoic Acid (MeOTyr). ACS Catal. 2019, 9, 253–258. [Google Scholar]
- (43).Chen L; Naowarojna N; Song H; Wang S; Wang J; Deng Z; Zhao C; Liu P Use of a Tyrosine Analogue To Modulate the Two Activities of a Nonheme Iron Enzyme OvoA in Ovothiol Biosynthesis, Cysteine Oxidation versus Oxidative C–S Bond Formation. J. Am. Chem. Soc 2018, 140, 4604–4612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Holm L; Rosenström, P. Dali Server: Conservation Mapping in 3D. Nucleic Acids Res. 2010, 38, W545–W549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Francis JW; Royer CJ; Cook PD Structure and Function of the Bacillithiol-S-transferase BstA from Staphylococcus aureus. Protein Sci. 2018, 27, 898–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Meury M; Knop M; Seebeck FP Structural Basis for Copper–Oxygen Mediated C– H Bond Activation by the Formylglycine-Generating Enzyme. Angew. Chem 2017, 129, 8227–8231. [DOI] [PubMed] [Google Scholar]
- (47).Zhang Y I-TASSER Server for Protein 3D Structure Prediction. BMC Bioinformatics 2008, 9, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Xu J; Yadan JC Synthesis of L-(+)-Ergothioneine. J. Org. Chem 1995, 60, 6296–6301. [Google Scholar]
- (49).Erdelmeier I; Daunay S; Lebel R; Farescour L; Yadan JC Cysteine as a Sustainable Sulfur Reagent for the Protecting-group-free Synthesis of Sulfur-containing Amino Acids: Biomimetic Synthesis of L-ergothioneine in Water. Green Chem. 2012, 14, 2256–2265. [Google Scholar]
- (50).Stampfli AR; Goncharenko KV; Meury M; Dubey BN; Schirmer T; Seebeck FP An Alternative Active Site Architecture for O2 Activation in the Ergothioneine Biosynthetic EgtB from Chloracidobacterium thermophilum. J. Am. Chem. Soc 2019, 141, 5275–5285. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.