

Abstract
Human ornithine aminotransferase (hOAT), a pyridoxal 5′-phosphate-dependent enzyme, plays a critical role in the progression of hepatocellular carcinoma (HCC). Pharmacological selective inhibition of hOAT has been shown to be a potential therapeutic approach for HCC. Inspired by the discovery of the nonselective aminotransferase inactivator (1R,3S,4S)-3-amino-4-fluoro cyclopentane-1-carboxylic acid (1), in this work, we rationally designed, synthesized, and evaluated a novel series of fluorine-substituted cyclohexene analogues, thereby identifying 8 and 9 as novel selective hOAT time-dependent inhibitors. Intact protein mass spectrometry and protein crystallography demonstrated 8 and 9 as covalent inhibitors of hOAT, which exhibit two distinct inactivation mechanisms resulting from the difference of a single fluorine atom. Interestingly, they share a similar turnover mechanism, according to the mass spectrometry-based analysis of metabolites and fluoride ion release experiments. Molecular dynamics (MD) simulations and electrostatic potential (ESP) charge calculations were conducted, which elucidated the significant influence of the one-fluorine difference on the corresponding intermediates, leading to two totally different inactivation pathways. The novel addition-aromatization inactivation mechanism for 9 contributes to its significantly enhanced potency, along with excellent selectivity over other aminotransferases.
Keywords: Mechanism-based inactivators, Inactivation mechanism, Human ornithine aminotransferase, Hepatocellular carcinoma, Fluorinated cyclohexenes
Graphical Abstract
INTRODUCTION
Hepatocellular carcinoma (HCC), which accounts for 90% of primary liver cancer, is the second most common cause of cancer death worldwide.1–4 To date, there is no effective treatment for HCC, as it is typically diagnosed at advanced disease stages and tumors are typically refractory to systemic treatment with the standard-of-care receptor tyrosine kinase inhibitor, sorafenib, and radiotherapy.5–8 Human ornithine aminotransferase (hOAT) is a pyridoxal 5′-phosphate (PLP)-dependent enzyme9 with roles in inborn errors of metabolism10 and hepatocellular carcinoma (HCC) progression.11 Mechanistically, two coupled half-reactions are involved in the transamination cycle of hOAT (Figure 1). In the first-half reaction, hOAT catalyzes the conversion of PLP and ornithine to pyridoxamine phosphate (PMP) and glutamyl-5-semialdehyde, which spontaneously cyclizes to Δ1-pyrroline-5-carboxylate (P5C).12 The P5C generated can be further converted to proline by pyrroline-5-carboxylate reductase (PYCR).13, 14 In the second-half reaction, PMP and α-ketoglutarate (α-KG) are converted to PLP and L-glutamate (L-Glu), which also can be converted to P5C by pyrroline-5-carboxylate synthase (P5CS). Mounting evidence indicates that proline metabolism plays an important role in metabolic reprogramming to sustain cancer proliferation along with the upregulated synthesis of P5C as a central intermediate.15–17 Metabolic reprogramming in HCC is characterized by the activation of the proline/hydroxyproline metabolic pathway, which supports HIF1a-dependent tumor progression and sorafenib resistance.18 Furthermore, the glutamate generated by hOAT can be converted to glutamine by glutamine synthetase (GS) (Figure 1),which supports anabolic and proliferative cell programs.19 hOAT and glutaminogenic enzymes are found to be strongly activated and commonly overexpressed in HCC because of aberrant, oncogenic Wnt/β-catenin signaling.20, 21 The selective inhibition of hOAT has been shown to effectively suppress HCC tumor growth in vivo.11 More recently, the specific knockdown of hOAT was also found to suppress the growth of non‐small cell lung cancer (NSCLC) in vitro and in vivo.22 Taken together, hOAT plays an important role in the metabolic reprograming of HCC via proline and glutamine metabolic pathways, and the selective inhibition of hOAT serves as a promising therapeutic strategy for the treatment of HCC and other related cancers.
Figure 1.
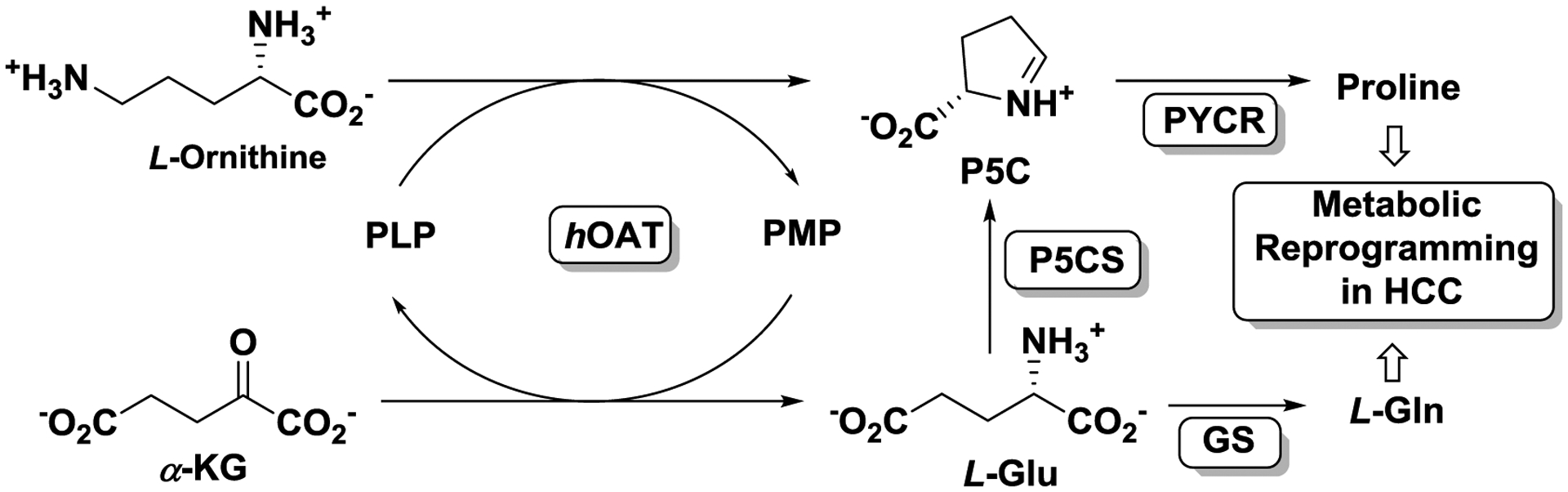
Metabolic pathway for ornithine
Among 14 known aminotransferases, hOAT belongs to the same subgroup as γ-aminobutyric acid aminotransferase (GABA-AT) because of their similarities in primary structures.23 hOAT and GABA-AT have very similar active sites,24 so it is not surprising that some potent inhibitors of GABA-AT also inhibit hOAT.11 Over many years, our laboratory has been focusing on the rational design of mechanism-based inactivators (MBIs) of GABA-AT.25 MBIs are unreactive alternate substrates for target enzymes, which are converted to active species in the catalytic site and then lead to inactivation via covalent bonding with the enzyme, tight-binding inhibition, or any functionally irreversible inhibition mechanism.26 For example, (1R,3S,4S)-3-amino-4-fluorocyclopentane-1-carboxylic acid (1) was reported to inactivate hOAT27 and GABA-AT28 via the same enamine mechanism, as shown in Scheme 1. The proposed inactivation mechanism is initiated by the formation of Schiff base 2, which is subjected to rate-determining deprotonation and in situ elimination of fluoride ion to afford intermediate 4. Active enamine 5 is formed via a Schiff base exchange reaction and the subsequent nucleophilic addition of 5 to the Lys-bound PLP complex yields covalent adduct 6.
Scheme 1.
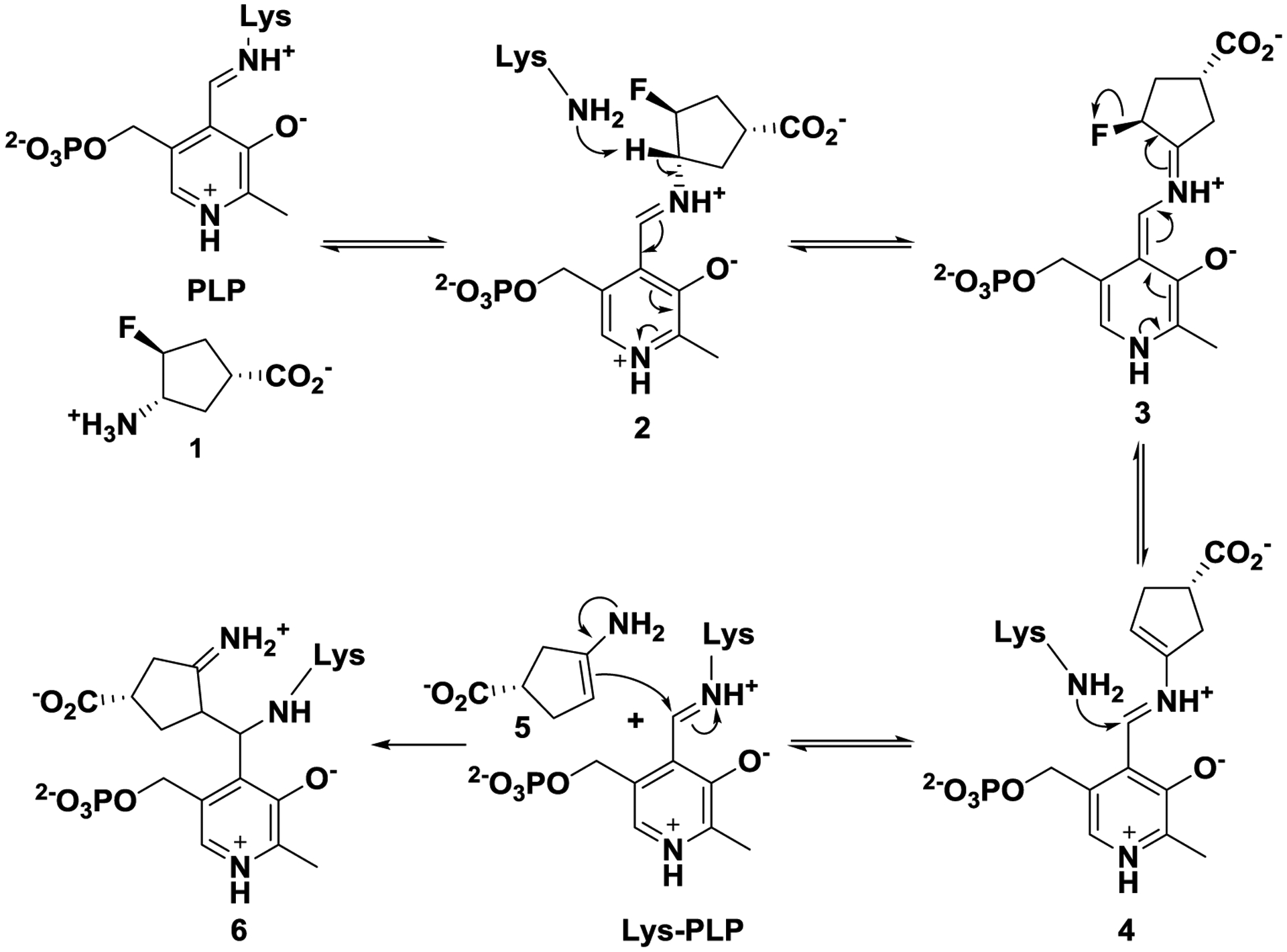
Mechanism of inactivation of hOAT and GABA-AT by 1
Because MBIs are inert until they are activated in the active site, undesirable off-target effects can be greatly reduced. More importantly, these inactivators can demonstrate higher potency and selectivity than traditional inhibitors even at a lower dosage.11, 29, 30 In 2015, GABA analogue 7 (Figure 2) was reported as a selective MBI of hOAT, which dramatically reduced alpha-fetoprotein levels (a biomarker for HCC) and suppressed in vivo HCC tumor growth at doses of 0.1 and 1.0 mg/kg.11 Very recently, the inactivation mechanism of 7 was also revealed.31
Figure 2.
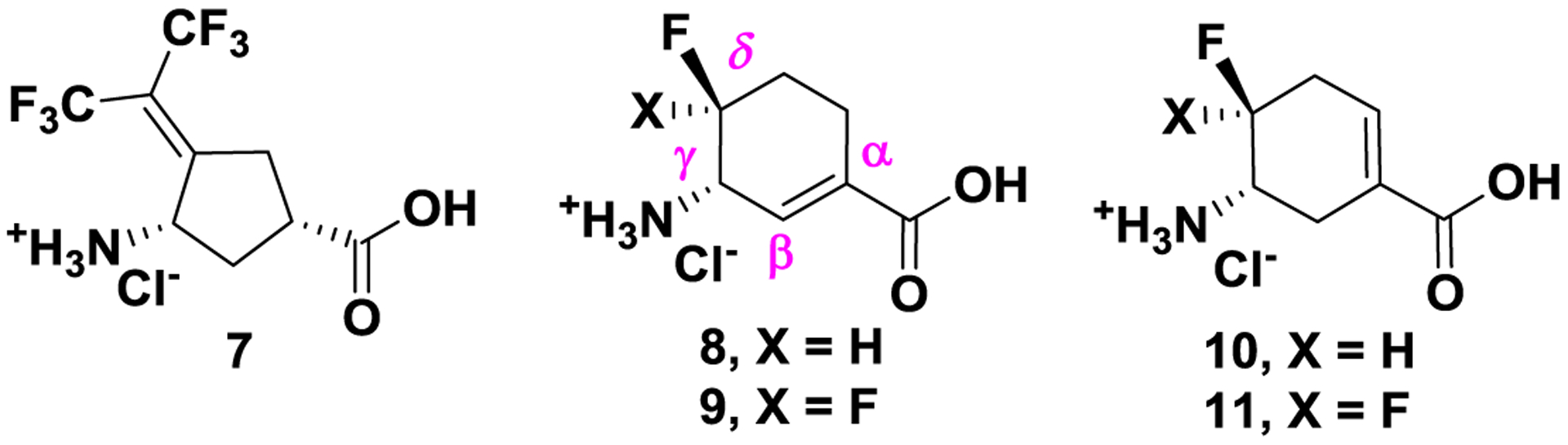
Structures of selective hOAT inactivators 7 and fluorine-substituted cyclohexene analogues 8–11
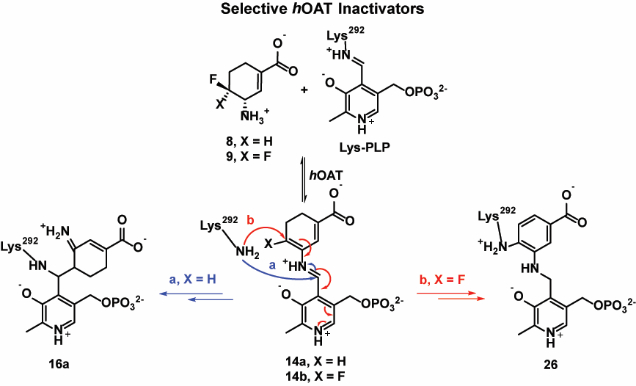
We have been interested in discovering potent and selective hOAT inactivators that have novel inactivation mechanisms. Herein, we rationally designed and synthesized fluorine-substituted cyclohexene-based GABA analogues 8–11 (Figure 2) based on the cyclopentane-based analogue 1. Among them, compound (9) is 23 times more efficient as an inactivator of hOAT than 7, along with excellent selectivity over other aminotransferases (e.g., GABA-AT). We also elucidated the inactivation and turnover mechanisms for 8 and 9 through mass spectrometry, crystallography, and various other biochemical methods, and can conclude that a difference of a single fluorine atom dramatically alters the inactivation mechanism.
RESULTS AND DISCUSSION
Design of Novel OAT Inactivators
Similar binding pockets are observed in hOAT and GABA-AT, which produce virtually identical distances between the anchor points of the ligands. Interestingly, Tyr55 in hOAT partially occupies a similar steric space as does Phe351 in GABA-AT. The more hydrophobic Phe351 residue intrudes into the active site of GABA-AT so that only the smaller GABA molecule can fit; the hOAT catalytic site is more flexible and larger to accommodate ornithine that is one carbon longer than GABA.32 Furthermore, the unique Tyr55 residue provides the potential of forming extra hydrogen bonds with the more hydrophilic substrate in the active site of hOAT.31 Previously, cyclopentane-based GABA analogue 1 was identified as a dual inhibitor against hOAT and GABA-AT. Because of the relatively small size of 1, it can fit into the catalytic pockets of hOAT and GABA-AT, consistent with its poor selectivity.27 Thus, we hypothesized that the relatively larger sizes of cyclohexene derivatives 8–11 could potentially improve their hOAT selectivity over GABA-AT (Figure 2). In addition, the incorporation of a double bond at the α/β-position (8/9) could possibly enhance the inhibitory activity33 compared with the double bond isomers 10/11, potentially by increasing the acidity of the proton adjacent to the amino group.
MBIs usually act as substrates initially, followed by the formation of active intermediates at the catalytic site, which then inactivate the enzymes through different mechanistic pathways. Therefore, a proposal of the inactivation mechanism plays an important role in the early stage of rational design of potential MBIs. Three possible inactivation mechanisms for 8/9 are proposed in Scheme 2. All of the mechanisms are initiated by Schiff base formation (12a/12b) with subsequent HF elimination to give reactive species 14a/14b, based on the known mechanism of 1 (Scheme 1). Mechanistic pathway a is similar to the inactivation mechanism of 1, in which active enamine 15a/15b could be released after transimination of 14a/14b with Lys292. The subsequent enamine addition could lead to inactivation with the formation of adducts 16a/16b. The second fluoride ion of adduct 16b could be further released to afford aromatic adduct 17. Mechanistic pathway b is a direct aromatization of 14a/14b to yield a tight binding adduct 20a/20b, which is similar to the inactivation mechanism of another fluorinated cyclohexene analogue.34 Mechanistic pathway c involves nucleophilic addition of Lys292 to conjugated olefin 14a/14b, yielding adducts 21a/21b.
Scheme 2.
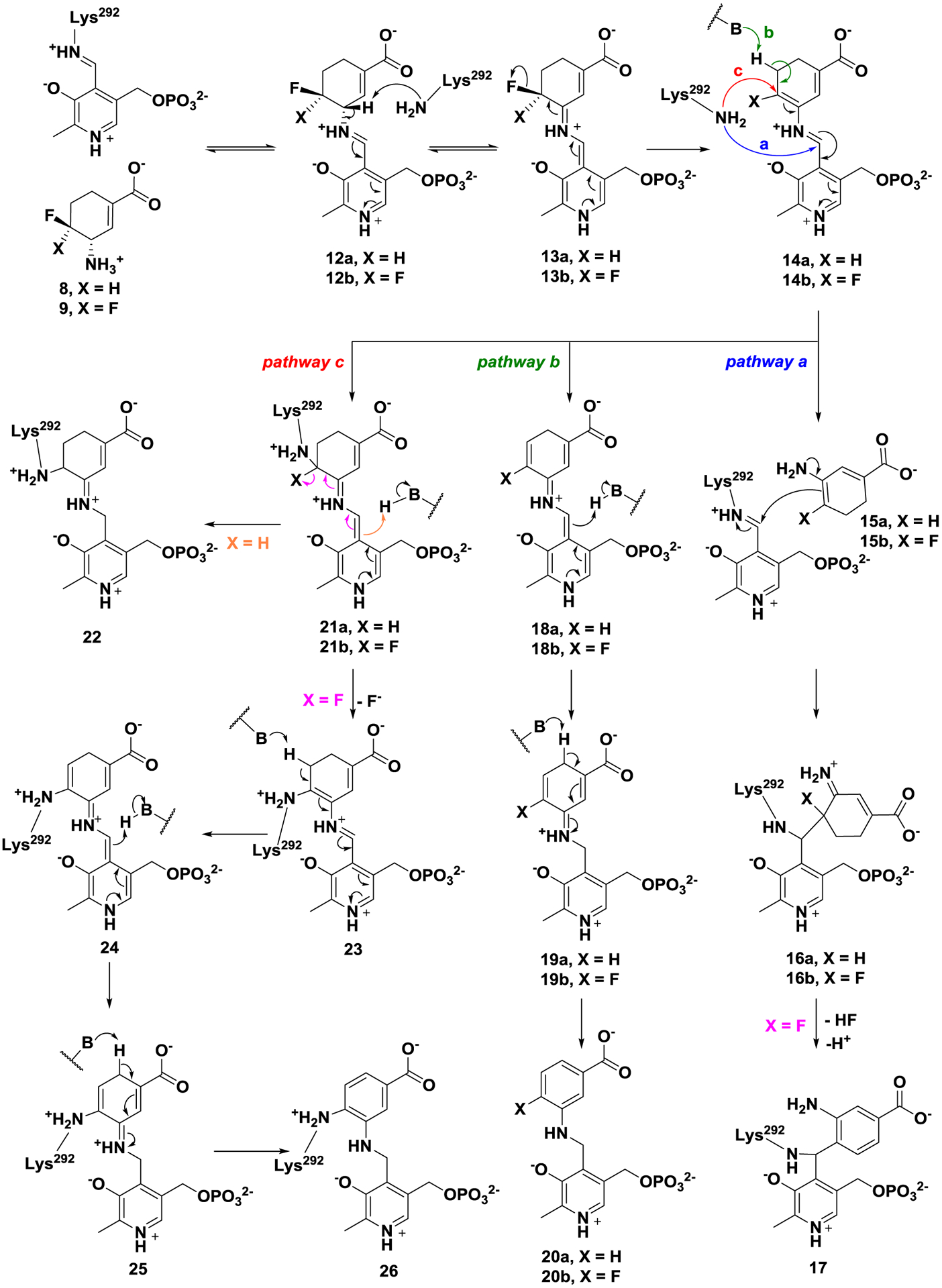
Possible inactivation mechanisms for 8 and 9
In the case of 21b, it is possible to form a more stable covalent aromatic adduct (26) after elimination of the second fluoride ion.
Molecular Docking
To better understand the difference in steric hindrance between ring sizes, molecular docking studies35 for 1 and 8 were employed to mimic their binding poses at the active site of GABA-AT and hOAT, compared with the native substrates. As shown in Figure S1A, GABA binds in the active site of GABA-AT and establishes hydrogen bonds with residues Arg192, Tyr69, and Glu270. This putative binding pose of GABA positions its γ-amino group close to the Lys329-PLP complex and facilitates the subsequent Schiff base exchange. The binding model of 1 demonstrates similar hydrogen bonds with these residues (Figure S1B). However, molecular docking shows 8 forms a more stable hydrogen bond with His206 to avoid the potential clash with residue Phe351 (Figure S1C), which potentially impedes its initial reaction with Lys329-PLP complex. Comparably, the binding model of ornithine with hOAT shows the δ-amino group goes deep into the active site and forms a distinct hydrogen bond with Thr322 (Figure S1D), which positions it close to the Lys292-PLP complex. The amino group of 8 forms a similar hydrogen bond with Thr322 in the docking model (Figure S1E), while the docking pose of 1 indicates it forms a hydrogen bond with Glu325 instead (Figure S1F). These docking results indicate that the larger cyclohexene analogues as substrates are favored in the hOAT active site but disfavored in the GABA-AT active site when compared with 1, which matches our design strategy described above.
The chirality of the γ-position of MBIs was found to be very important for the inactivation process and for retaining their inhibitory activity.25 Interestingly, the enantiomer of 8 shows a similar binding pose in the active site of hOAT (Figure S1G). Lys-assisted deprotonation at the γ-position proved to be the rate-determining step in earlier analogues.36 Thus, the molecule docking study was also conducted to predict the binding poses of active intermediates 12a (Figure S1H) and its enantiomer (Figure S1I) at the catalytic pocket of hOAT. Both carboxylate moieties of these intermediates establish hydrogen bonds with Tyr55 and Arg180, while the different chirality of the 12a-enantiomer forces its γ-proton to face the other side, which points away from the catalytic Lys292 (3.0Å vs 5.3Å). This docking simulation is also consistent with the observation of earlier inactivators and supports the synthesis of chirally-pure analogues.
Syntheses of Fluorine-substituted Cyclohexene Analogues 8–11
The synthetic route for 8–11 is shown in Scheme 3. The di-PMB intermediate (29) was afforded from chirally-pure starting material 27 by three sequential steps: intramolecular annulation, stereoselective epoxide ring-opening, and reductive amination with excess anisaldehyde. The obtained intermediate (29) was treated with XtalFluor-M and (HF)3Et3N to exclusively yield trans-isomer 30 in moderate yields, in which an aziridinium mechanism pathway might be involved.37 The direct conversion from di-PMB protected intermediate 30 to Boc-protected intermediate 31 was achieved by Pd(OH)2 catalyzed hydrogenation in the presence of Boc2O. Intermediate 31 was treated with PhSeCl and KHMDS, followed by oxidative elimination with m-CPBA to yield olefin isomers 32a and 32b, which were separated by chromatography and then deprotected to 8 and 10, respectively. On the basis of a previous report, alcohol 3338 can be prepared from chirally-pure 27; 33 was then oxidized by PCC to give ketone 34. After screening various fluorination reagents and reaction temperatures, key intermediate 35 was afforded in moderate yields, which avoided the production of monofluoroalkene impurities.39 Using the same method used to make 8 and 10, intermediate 35 was converted to difluoroolefin isomers 36a and 36b, which were further deprotected to 9 and 11, respectively.
Scheme 3.
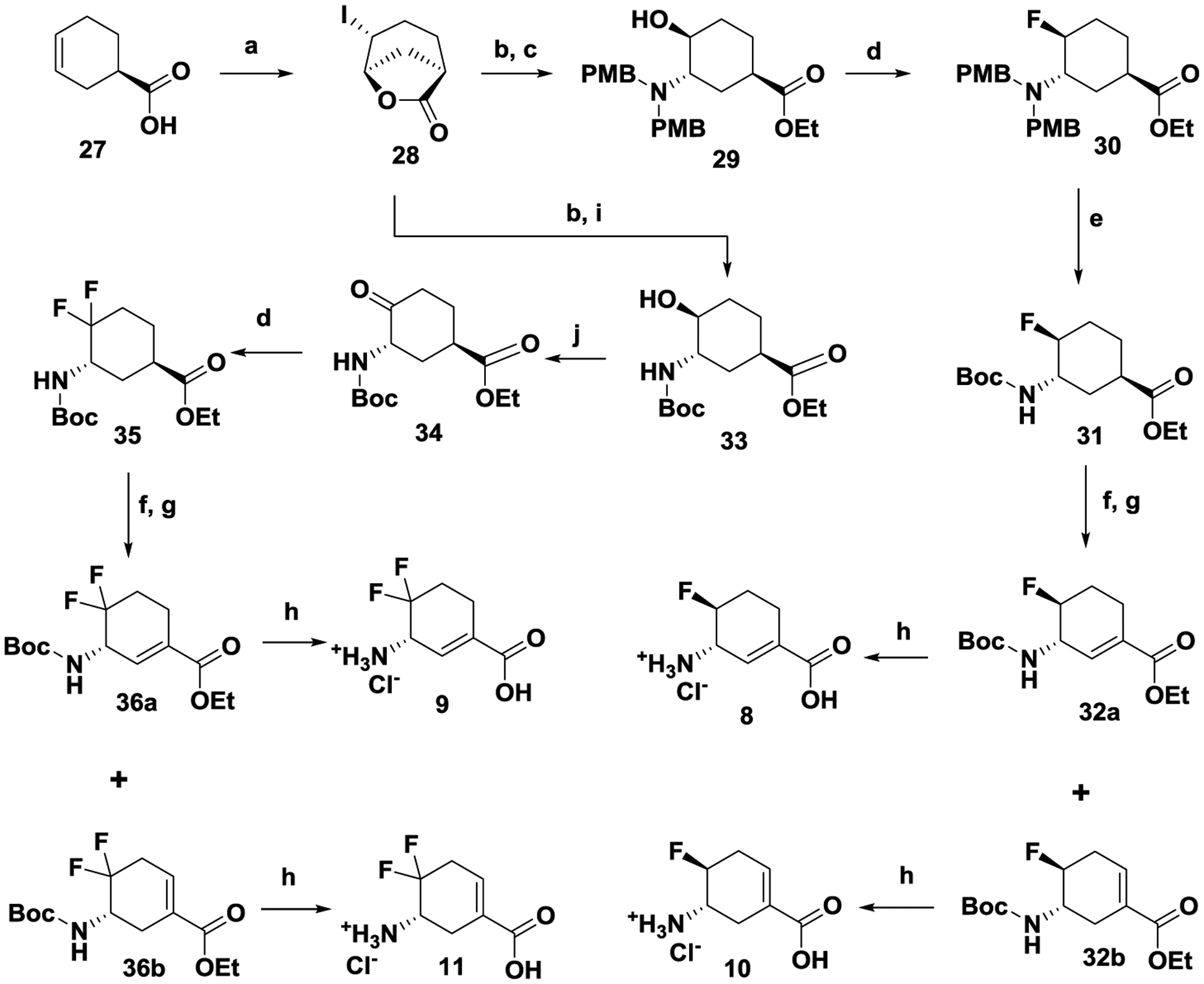
Syntheses of fluorine-substituted cyclohexene derivatives 8–11 Conditions: (a) NaHCO3, I2, KI, H2O, 0 ℃ - r.t., overnight; (b) NaOH, EtOH, 0 ℃ - r.t., 3 h; then ammonia solution (28–30%), EtOH, 45 ℃, overnight; (c) 4-anisaldehyde, AcOH, NaBH(OAc)3, DCE, 75 ℃, overnight; (d) XtalFluor-M, (HF)3Et3N, DCM, in a plastic container, r.t., overnight; (e) Pd(OH)2, Boc2O, MeOH, EtOAc, r.t., overnight; (f) PhSeCl, KHMDS (1M in THF), −78 ℃; (g) m-CPBA, DCM, r.t., 2h; ; (h) HCl (4M), AcOH, 80 ℃, overnight; (i) Boc2O, EtOH, 0 ℃ - r.t., 5 h; (j) PCC, DCM, r.t. overnight.
Kinetic Studies of Fluorine-substituted Cyclohexene Analogues
In vitro studies showed that fluorine-substituted cyclohexene analogues 8-11 inhibited hOAT with good selectivity over GABA-AT (Table 1). The results show that the six-membered ring analogues have a lower binding affinity to GABA-AT than 1 with much greater KI values, indicating that the larger ring size potentially interferes with binding interactions between the analogues and GABA-AT, which has a relatively small and rigid active site. The selectivity is consistent with our initial design strategy and docking results for the initial binding step (Figure S1 A–F).
Table 1.
Kinetic constants for the inactivation of hOAT and GABA-AT by 1, 7–11, and (S)-vigabatrina
| Compound | hOAT | GABA-AT | ||||
|---|---|---|---|---|---|---|
| KI (mM) | kinact (min−1) | kinact/KI (mM−1min−1) | KI (mM) | kinact (min−1) | kinact/KI (mM−1min−1) | |
| 8 | 0.031 ± 0.007 | 0.080 ± 0.007 | 2.56 | 2.53 ± 0.71 | 0.098 ± 0.011 | 0.039 |
| 9 | 0.0023 ± 0.0007 | 0.048 ± 0.004 | 20.33 | > 0.12b 1.1 ± 0.14c |
> 0.18b 0.20 ± 0.001c |
1.52 ± 0.09b 0.18c |
| 10 | 4.38 ± 0.86 | 0.075 ± 0.005 | 0.017 | 4.28 ± 2.05 | 0.028 ± 0.005 | 0.0065 |
| 11 | 0.54 ± 0.017 | 0.083 ± 0.013 | 0.15 | > 9.63b | > 0.29b | 0.030 ± 0.001b |
| 1 | 1.40 ± 0.041 | 0.086 ± 0.01 | 0.06 | 0.078 ± 0.04 | 0.017 ± 0.002 | 0.22 |
| 7 | 0.065 ± 0.010 | 0.057 ± 0.003 | 0.87 | - | - | - |
| (S)-vigabatrin | - | - | - | 0.29 ± 0.09 3.2cd |
0.21 ± 0.03 0.37cd |
0.72 0.11cd |
kinact and KI values were determined by the equation: kobs = kinact*[I]/(KI +[I]) and are presented as means and standard errors.
ratio of kinact/KI was determined by the slope of kobs = kinact*[I]/(KI +[I]). kinact is greater than maximum kobs, which was determined in a time-dependent assay; KI is greater than kobs(max)/ratio.
Assays tested at pH 6.5.
Ref40
The introduction of a double bond at the α/β-position or another fluorine at the δ-position significantly enhances the inactivation efficiency against both hOAT and GABA-AT, which could possibly result from the reduced acidity of the γ-position or the change in inactivation mechanisms. Among them, the best compound (9, kinact/KI = 20.33 min−1mM−1) is 23 times more efficient as an inactivator of hOAT than 7 (kinact/KI = 0.87 min−1mM−1), which exhibited potent in vivo antitumor efficacy.11 Moreover, 9 displayed good selectivity over GABA-AT with lower efficiency constants (pH 8.0, kinact/KI = 1.52 min−1mM−1; pH 6.5, kinact/KI = 0.18 min−1mM−1) and little or no inhibitory activities against aspartate aminotransferase (Asp-AT) and alanine aminotransferase (Ala-AT), even at high concentrations (5–20 mM, Figure S2).
Inactivation mechanisms of 8 and 9 were studied for a better understanding of the capabilities of the aminotransferase and for future rational design of new inactivators. To determine if any of the aforementioned mechanisms (Scheme 2) are responsible for the inactivation of hOAT by 8 and 9, we carried out a series of experiments, including dialysis, fluoride ion release studies, and obtained protein mass spectra and crystal structures of the inactivated enzymes.
Dialysis
Although 8 and 9 were designed to inactivate hOAT via a covalent or tight-binding mechanism, time-dependent reactivation of hOAT was carried out to determine if reversible components were also involved during the inactivation.30 After hOAT activity was partially or fully abolished by 4–25 equivalents of 8 or 0.6–2.0 equivalents of 9, they were dialyzed, and aliquots at different time intervals were collected and assayed for return of enzyme activity. No enzyme activity was recovered after 48 h of dialysis (Figure S3), indicating complete irreversible inhibition of hOAT.
Intact protein mass spectrometry of hOAT modified by 8 or 9
Intact protein mass spectrometry has served as an efficient tool to determine the covalent adducts or tight binding adducts of modified aminotransferases.31, 41 Notably, the hydrolysis of imine groups and loss of PLP or PMP were commonly observed.31 Taking into account the existing unstable groups in native hOAT or potential covalent adducts, the theoretical mass differences were calculated as shown in Table 2. When hOAT was inactivated by 8, one modified species (46506.08 ± 0.21 Da) was observed by mass spectrometry. A mass shift of +369.29 Da was observed, which corresponds to adduct 16a in pathway a (Scheme 2). In the case of 9, a mass shift of +366.34 Da was observed from the native enzyme (46138.32 ± 0.10 Da) and one modified species (46504.66 ± 0.13 Da) detected by mass spectrometry. This mass shift corresponds to either adduct 17 in pathway a or adduct 26 in pathway c (Scheme 2).
Table 2.
The mass differences of proposed adducts between experimental and theoretical mass values of native/modified hOAT
| Cmpd. | Mechanistic pathways | Adducts | Theoretical mass difference | Experimental mass of native hOATa | Experimental mass of modified hOATa | Experimental mass difference |
|---|---|---|---|---|---|---|
| 8 | a | 16a | 369.08 | 46136.79 ± 0.27 | 46506.08 ± 0.21 | 369.29 |
| b | 20a | 0 | ||||
| c | 22 | 368.06 or 138.03 | ||||
| 9 | a | 16b | 387.05 | 46138.32 ± 0.10 | 46504.66 ± 0.13 | 366.34 |
| 17 | 366.06 | |||||
| b | 20b | 0 | ||||
| c | 26 | 366.06 |
Values are presented as means and standard errors.
X-ray Crystallography of hOAT inactivated by 8 or 9
Although a highly accurate mass shift of +369.29 Da was observed in the intact protein mass spectrum of hOAT inactivated by 8, there is only a 1.02 Da difference between adducts 16a and 22. In addition, it is difficult to distinguish adducts 17 and 26 by intact protein mass spectrometry of inactivated hOAT by 9. To better interpret the inactivation mechanism(s) for 8/9, protein crystallography of hOAT inactivated by 8/9 was conducted to confirm the adducts formed in the active site, respectively.
The structure of hOAT inactivated by 8/9 was solved by molecular replacement using a monomer from a previously reported structure of hOAT (PDB code 1OAT), after all water molecules and ligand atoms were deleted. For hOAT-8, in space group C121, one asymmetric unit was found to contain 3 monomers. Two monomers in the asymmetric unit formed a biological assembly as a homodimer. The third monomer formed a homodimer with another monomer present in another asymmetric unit. For hOAT-9, in space group P3112, one asymmetric unit was also found to contain 3 monomers. The biological assemblies can be observed through crystallographic symmetry. Final models for hOAT-8 and hOAT-9 were refined to a resolution of 2.20Å and 1.90Å, with RFree/Rwork values of 15.70%/19.90% and 27.49%/23.98%, respectively. Final refinement statistics are presented in Table S1. A polder map42 for each structure was generated by omitting PLP, ligand, and Lys292 (Figure S4). The density for PLP was clearly represented in the polder map for both structures. Densities for 8 and 9 were also observed, clearly showing the presence of a ring and carboxylate moiety, as expected for the final adducts. The solved structures of hOAT-8/9 have been deposited in the PDB bank (PDB code: 6V8D & 6V8C).
Ternary adducts were observed within both active sites of hOAT-8 and hOAT-9. As seen in Figure S5, 8 and 9 reacted with and covalently linked to both PLP and Lys292, albeit at different positions, each forming ternary adducts. In the hOAT-8 crystal structure, PLP and Lys292 are linked with the inactivator at a central carbon atom, affording 16a. This result agrees with the MS data and rules out 22 as the final adduct, where PLP and Lys292 form covalent bonds at different positions with the inactivator. Therefore, pathway a is the most probable mechanism of inactivation by 8, rather than pathway c (Scheme 2). In the hOAT-9 crystal structure, Lys292 and PLP form covalent bonds with the inactivator to give 26. No density was observed to suggest a fluorine atom remained on the adduct. The complete loss of fluorine and the presence of covalent bonds on the adduct thereby excludes pathway b (Scheme 2) as a possible mechanism for inactivation, as was concluded by intact protein mass spectrometry. The ternary adduct in the active site of hOAT-9 is linked at two separate positions along the ring, rather than at a central atom as in 17. The electron density generated by the polder map (Fo-Fc) completely encloses the C-N bond formed between the lysine and PLP, suggesting 9 inactivates hOAT via pathway c to 26 (Scheme 2). Furthermore, Thr322 is positioned at the center of the six-membered ring and appears to form a lone-pair–aromatic interaction43 (2.9 Å) with the adduct (Figure S5), which supports the formation of 26.
As shown in Figure 3 (right), both oxygen atoms on the carboxylate of 26 establish hydrogen bonds with Tyr55 (2.7Å and 3.3Å), whereas only one oxygen atom on the carboxylate of 16a (Figure 3, left) was observed to engage in a hydrogen bond with Tyr55 (2.7Å). The interaction with Tyr55 has precedence; there is a similar interaction in the crystal structure of inactivated hOAT-7.31 The carboxylates of both 16a and 26 were observed to form an n-π* interaction with the peptide backbone of Gly320 (both 3.3Å) but no interaction with Arg180, which is essential for native substrate recognition.43 Tyr55 is a unique residue in the active site of hOAT, compared with that of GABA-AT, which is assumed to form a hydrogen bond with the α-amino group of ornithine.45 The interaction of GABA analogues 8 and 9 with Tyr55 contributes to their high potency, even though they are one carbon less than the native substrate ornithine.
Figure 3.
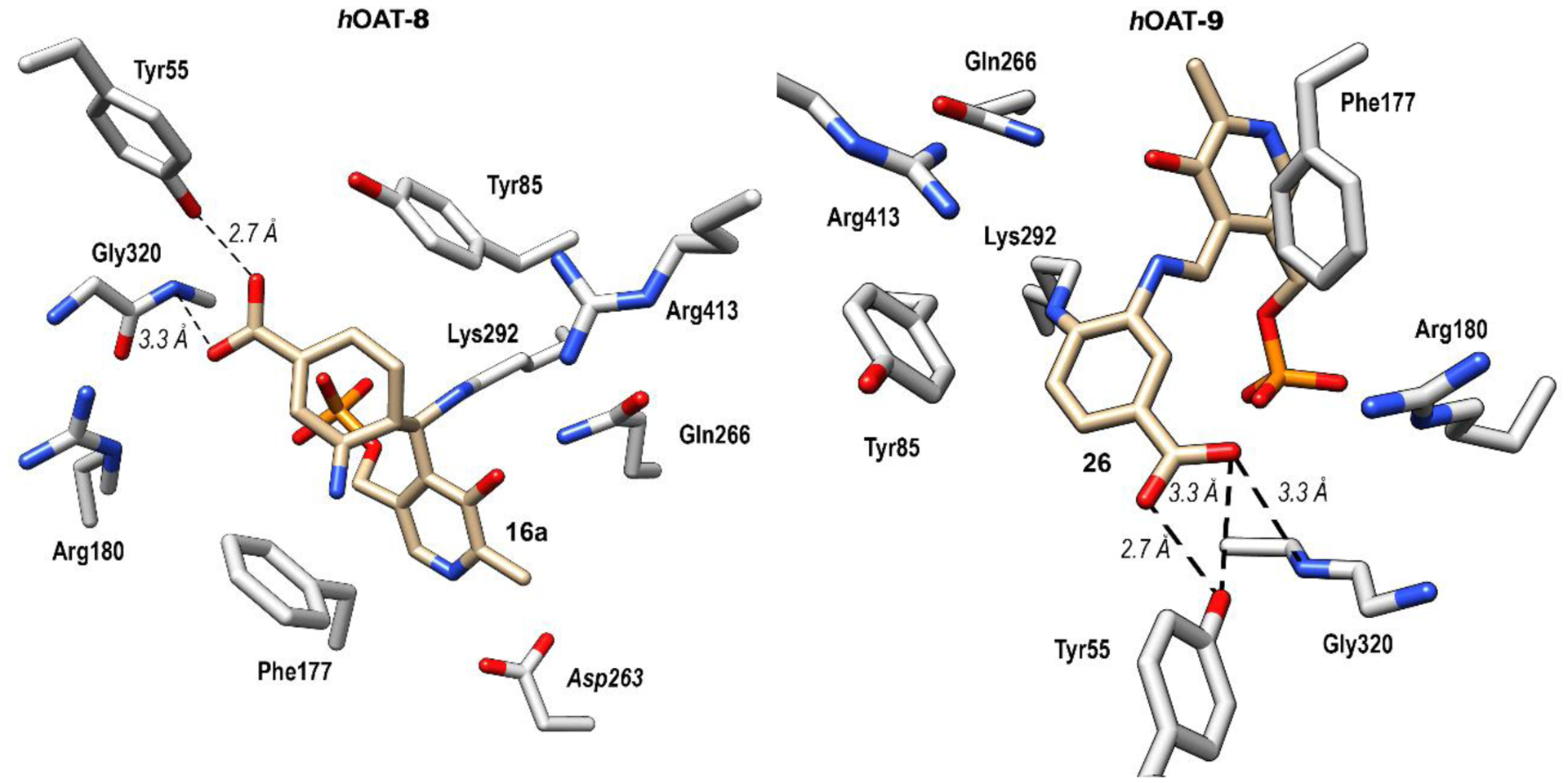
Co-crystal structures of hOAT inactivated by 8 (left) and 9 (right)
Turnover Mechanism
Previously, it was found that 1 can be converted to metabolites by aminotransferases, with the release of PLP or PMP.27 Considering the structural similarity between 1 and 8/9, two possible turnover pathways are proposed, as shown in Scheme 4. Schiff base formation of PLP with 8/9 and subsequent lysine-assisted deprotonation yields intermediates 13a/13b. Intermediates 13a/13b could be converted to intermediates 37a/37b via direct protonation of the coenzyme. The formed imines could be further hydrolyzed with the release of PMP and metabolites 38a/38b. This PMP turnover mechanism is similar to the degradation of ornithine by hOAT. Intermediates 13a/13b also could undergo elimination of fluoride ion and hydrolysis of the imine with release of enamines 15a/15b, which could be hydrolyzed to afford ketones 39a/39b. In this PLP turnover mechanism the regenerated PLP would further form Schiff bases with inactivators even in the absence of α-KG. To determine if any of the above turnover mechanisms are involved during the inactivation process of hOAT by 8 and 9, we carried out a partition ratio experiment, determination of fluoride ion release, and mass spectrometric analysis of metabolites for these two inactivators.
Scheme 4.
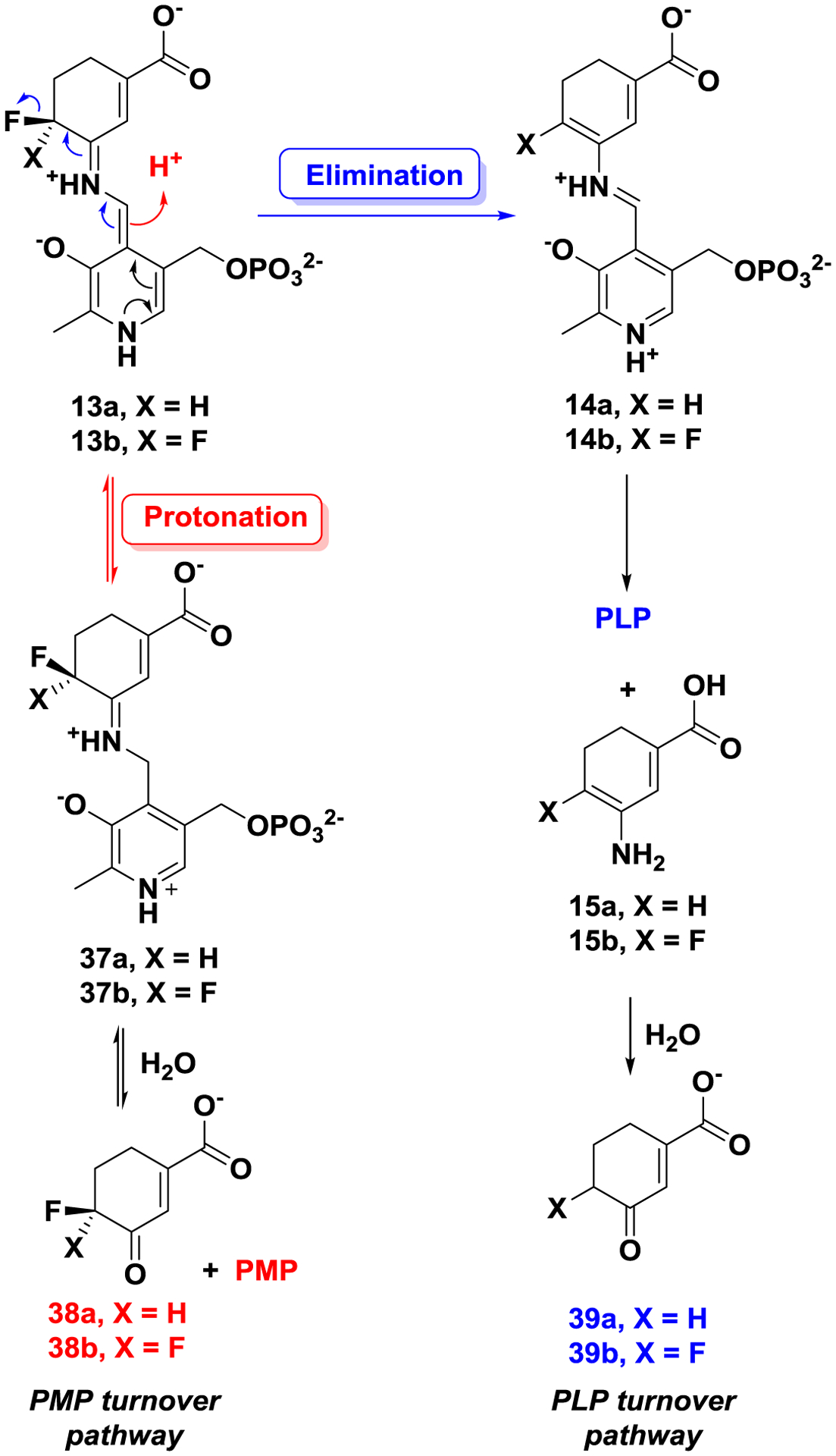
Proposed turnover mechanism for 8 or 9 by hOAT
Partition Ratio and Fluoride Release
The partition ratio is the ratio of the compound acting as a substrate relative to the compound inactivating the enzyme. Ideally, a plot of enzyme activity remaining vs. equivalents of compounds added will give a straight line from 100 to 0% enzyme activity remaining. The intercept with the x-axis gives the number of inactivator molecules required to inactivate each enzyme molecule (the turnover number). This number includes the one molecule of inactivator required to inactivate the enzyme; consequently, the partition ratio is the turnover number minus one (assuming there is a 1:1 stoichiometry of inactivator to enzyme). Therefore, the partition ratios of 8/9 were determined by titrating the enzyme with varying equivalents of inactivator with a known amount of hOAT (Figure S6). The linear relationship was extrapolated to yield the exact equivalents required to inactivate the enzyme completely. From this, we determined the partition ratio of 8 to be 22.0 and the partition ratio of 9 to be 0.8.
Different equivalents of fluoride ions can be released in PLP and PMP turnover pathways.29, 31 In the absence of α-KG, if PMP is formed in the turnover mechanism, it cannot be converted back to PLP, which results in less than one equivalent of fluoride ion released. If PLP is regenerated during the turnover mechanism after fluoride ion is released, multiple equivalents of fluoride ions will be released. The number of fluoride ions released per enzyme turnover can be detected using a fluoride ion selective electrode. When hOAT is inactivated with an excess of 8 in the absence of α-KG, 22.7 equivalents of fluoride ions were released per enzyme active site (Table S2). In the case of 9, 2.9 equivalents of fluoride ions were released per enzyme active site (Table S2). These results indicate that turnover mechanisms of 8 and 9 must regenerate PLP as part of the turnover mechanism with the release of additional equivalents of fluoride ion before inactivation, which is consistent with their partition ratios, respectively.
Mass Spectrometry-Based Analysis of Metabolites
After size exclusion filtration of inactivated hOAT samples 8/9, the filtrate was analyzed by untargeted metabolomics ((+/−ESI HRMS) to detect metabolites. As shown in Figure S7, metabolites 39a (m/z 139.0393, [M-H]−1) and 39b (m/z 157.0301, [M-H]−1) in the PLP turnover pathway were detected and confirmed by their fragmentation spectra. Standards of PLP and PMP were monitored by HRMS, and the HCD-based fragmentation of precursors was used to confirm metabolite detection and retention time (Figure S8A & S8B). In both samples of 8 and 9, only PLP was detected by mass spectrometry (m/z 248.0316, [M+H]+1); neither PMP nor adducts of PMP were observed (Figure S8C & S8D). Overall, metabolomic results indicate these two compounds undergo a similar PLP regeneration turnover mechanism (Scheme 4).
Plausible Mechanisms for 8 and 9 with hOAT
Based on the above inactivation and turnover mechanism studies, a modified mechanism pathway for inactivator 8 is shown in Scheme 5. Schiff base 12a is initially formed and subsequently deprotonated to afford intermediate 13a. Based on the fluoride ion release experiment and the metabolomics results, intermediate 13a is fully converted to intermediate 14a. Schiff base 14a is then attacked by Lys 292 at the PLP imine position instead of the conjugate olefin position. According to the partition ratio of 8, only 4% of active enamine 15a attacks the Lys-PLP complex for inactivation (pathway a), and 96% of that is hydrolyzed to yield 39a (pathway b).
Scheme 5.
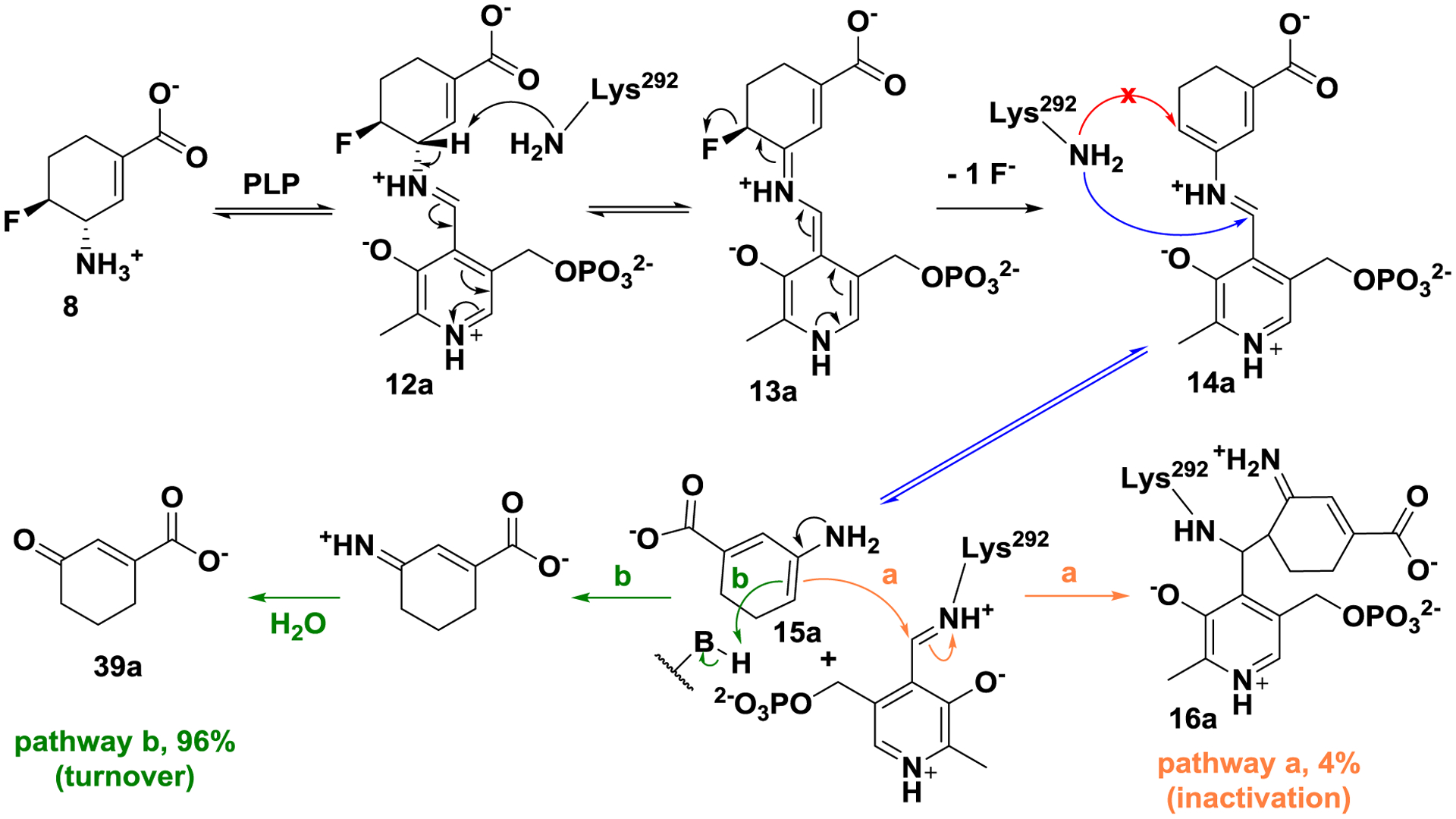
Plausible mechanism for 8 with hOAT
As shown in in Scheme 6, a modified mechanism pathway for inactivator 9 is proposed. Although active intermediate 14b is formed via a similar fluoride ion release process, inactivation occurs by lysine addition at the conjugated double bond. The formed adduct is further converted to the more stable aromatic adduct (26; 56%, pathway a), and 44% of intermediate 14b is converted to inert enamine 15b, which does not attack the Lys-PLP complex, and is hydrolyzed to yield metabolite 39b instead (pathway b).
Scheme 6.
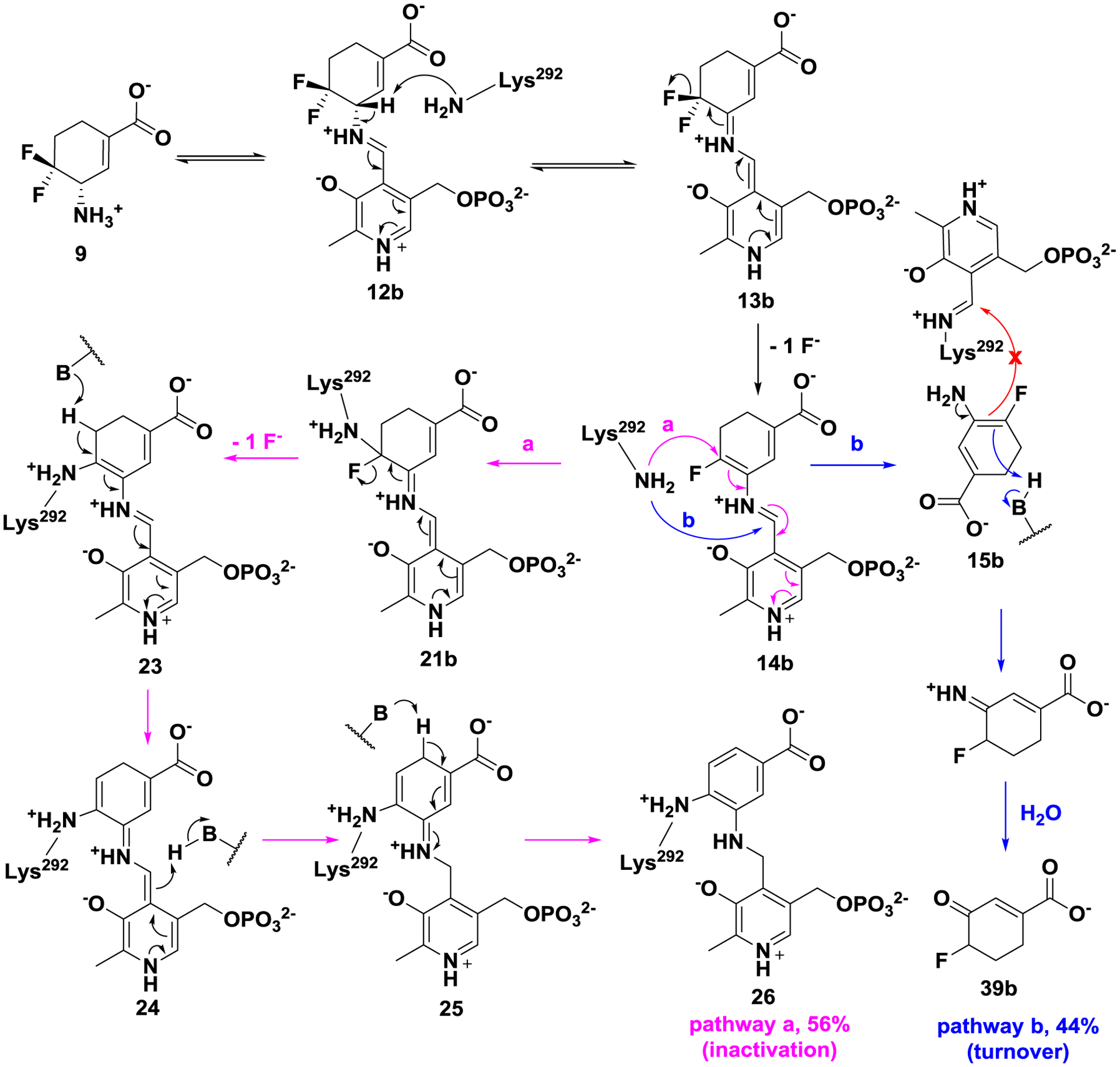
Plausible mechanism for 9 with hOAT
The structure of adduct 16a/26 was confirmed by intact protein MS and its crystal structure complex, and the corresponding metabolite 39a/39b was detected by mass spectrometry, along with multiple equivalents of fluorine ion released. Taken together, the difference of a single fluorine atom results in different inactivation mechanism pathways for 8 (enamine pathway) and 9 (addition-aromatization pathway) but by the same turnover mechanism (PLP pathway).
Molecular Dynamics (MD) Simulations and Electrostatic Potential (ESP) Charge Calculations
Although very similar intermediates (14a/14b and 15a/15b) are formed during inactivation by 8 and 9, the resulting covalent adducts (16a and 26, respectively) are quite different. Previously, different inactivation mechanisms for alanine racemase inhibitors were demonstrated as a result of a different number of fluorines (one-fluorine vs three-fluorines).46 For a better understanding of the impact the additional fluorine atom had on these intermediates, molecular docking/dynamics and ESP charge calculations were carried out to demonstrate their binding poses in the catalytic pocket of hOAT and reaction affinity with Lys292 or the Lys292-PLP complex.
As shown in Figure 4A, the docking poses of 14a and 14b are almost identical in the active site of hOAT, where Lys292 lies in the middle of two potential electrophilic sites: the carbon of the PLP imine moiety (C4`) and the terminal carbon of the conjugated olefin (Cδ). Molecular dynamics simulations were conducted to calculate the average distance between the nitrogen of the nucleophilic Lys292 (NLys) and the C4`/Cδ positions of 14a/14b, respectively (Figure 4B). The average distance of NLys-C4` (4.3 Å) and NLys-Cδ (4.1 Å) in 14a is very similar, whereas the C4` of 14b is closer to the NLys (5.0 Å) than the Cδ of 14b (6.6 Å), with an average difference of 1.6 Å. The average dihedral angles between the cyclohexadiene rings and the pyridine rings were also monitored during the molecular dynamics simulations (Figure S9). The average dihedral angle of 14b (−148.26°) is much closer to −180° (fully conjugated) than that of 14a (−91.64°; orthogonal and not in conjugation), indicating that the olefin in 14b has a greater propensity to conjugate with the pyridine ring. ESP charges (Figure S10) and electron density/electrostatic potential maps (Figure 5) for intermediates 14a/b and 15a/b were calculated to evaluate the impact of the additional fluorine atom. The significant differences are demonstrated between Cδ−14a (−0.28) and Cδ−14b (+0.49) with the introduction of an additional fluorine atom, as well as Cδ−15a (−0.70) and Cδ−15b (+0.07), while the charge at C4`−14a (+0.42) is close to that at C4`−14b (+0.45). Therefore, from the molecular dynamics simulation (Figure 4) the accessibility of Lys292 to Cδ−14a and C4`−14a is similar, but the dihedral angle (−91.64°) and carbon charges (−0.28 vs +0.42) significantly disfavor nucleophilic attack of Lys292 on the olefin of 14a; attack at C4’ gives enamine 15a and leads to final adduct 16a (Scheme 5). In the case of 14b, the distance NLys-C4` is favored for nucleophilic attack of Lys at C4` of 14b relative to Cδ of 14b (5.0 Å vs 6.6 Å), but the electrophilicity of the Cδ of 14b is greatly enhanced by the additional fluorine atom, resulting in the generation of final adduct 26 and inactive enamine 15b, which gets protonated and hydrolyzed to 39b (Scheme 6). The additional fluorine atom in 15b significantly decreases its nucleophilicity, which prevents the formation of adducts 16b and 17 (Scheme 2). Both molecular docking/dynamics and ESP charge calculations clearly rationalize our observations during the inactivation of hOAT by 8 and 9.
Figure 4.
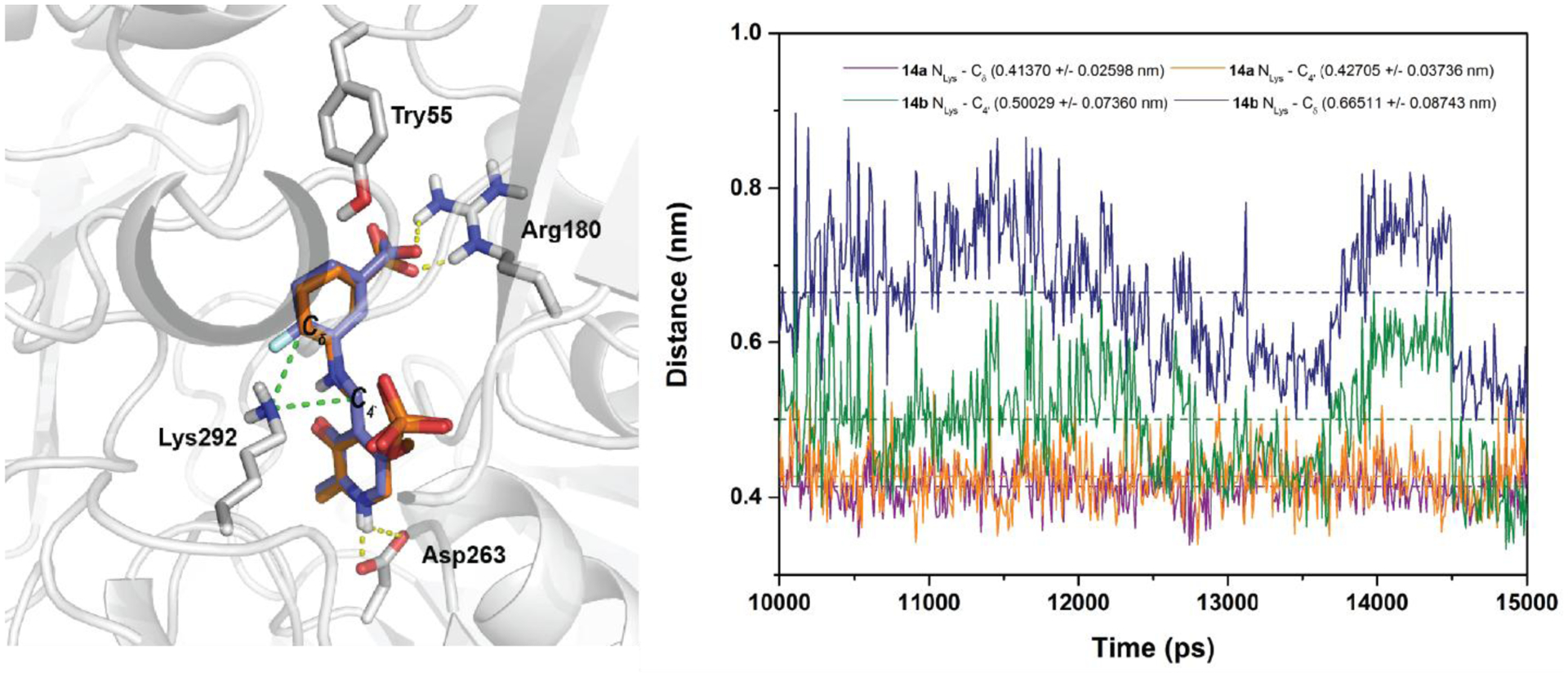
Molecular docking of 14a (orange) and 14b (blue) in the active site of hOAT (left); Distance changes between NLys and Cδ /C4` of 14a and 14b during molecular dynamics (right).
Figure 5.
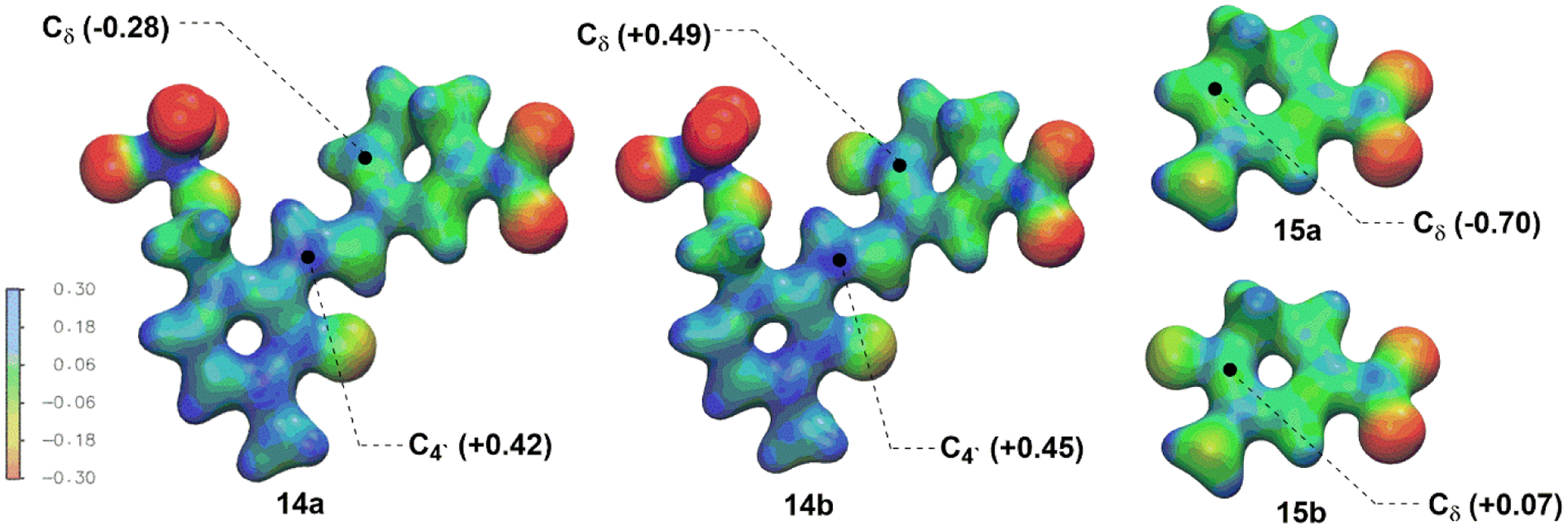
Electron density maps colored coded to the electrostatic potential of intermediates and ESP charges of Cδ and C4`.
CONCLUSIONS
Over the last few years, selective inhibition of human ornithine aminotransferase (hOAT) has been gradually recognized as a potential treatment for cancers, especially hepatocellular carcinoma (HCC).11 Based on the known inactivation mechanism for nonselective inactivator 1,31 a novel class of fluorine-substituted cyclohexene analogues (8–11) was rationally designed, with the aid of molecular dynamics simulations and docking, synthesized, and evaluated. Among them, analogues 8 and 9 showed significantly improved inhibitory activities against hOAT with excellent selectivity over GABA-AT, compared with 1 (Table 1). Inactivation pathways for 8 and 9 were elucidated by mass spectrometry and crystallography with the aid of a dialysis experiment, total turnover, and measurement of fluoride ion release. While monofluoro substituted analogue 8 inactivates hOAT via an enamine pathway (Scheme 2, pathway a), difluoro analogue 9 inactivates hOAT via a novel addition-aromatization mechanism (Scheme 2, pathway c), contributing to its significantly enhanced potency. Notably, strikingly accurate masses of intact protein were obtained with errors of less than 1 Da, thereby allowing the facile determination of adduct masses prior to their crystal structures (Table 2). Interestingly, despite the difference in inactivation mechanisms, the turnover mechanisms for 8 and 9 by hOAT are almost identical (Scheme 4, the PLP pathway), and the metabolites were identified with the aid of targeted mass spectrometry. The plausible inactivation and turnover mechanisms for 8 and 9, supported and rationalized using molecular dynamics (MD) simulations and electrostatic potential (ESP) charge calculations, indicate that a single fluorine atom difference in molecules can control enzyme inactivation mechanisms.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to the National Institutes of Health (grant R01 DA030604 to R.B.S. and grant P30 DA018310 to N.L.K.) and National Science Foundation (grant 2015210477 to P.F.D) for financial support. This work used the Extreme Science and Engineering Discovery Environment (XSEDE) Comet Bridges Stampede2 through allocation TG-CHE190070, which is supported by National Science Foundation grant number ACI-1548562. This work made use of the IMSERC at Northwestern University, which has received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF NNCI-1542205), the State of Illinois, and the International Institute for Nanotechnology (IIN). X-ray diffraction data collection used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under contract no. DE- AC02-06CH11357. The use of LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (grant 085P1000817). We thank Dr. Joseph Brunzelle at LS-CAT for help on data collection. We also thank Dr. Matthew J. Moschitto for helpful discussions and Dr. Cory T. Reidl for instruction in molecular docking studies using MOE.
ABBREVIATIONS
- PMB
p-methoxybenzyl
- XtalFluor-M
(diethylamino)difluorosulfonium tetrafluoroborate
- Boc
tert-butyloxycarbonyl
- KHMDS
potassium bis(trimethylsilyl)amide
- DCM
dichloromethane
- DCE
1,2-dichloroethane
- THF
tetrahydrofuran
- m-CPBA
meta-chloroperoxybenzoic acid
- PCC
pyridinium chlorochromate
- Boc2O
di-tert-butyl decarbonate
Footnotes
Supporting Information
Supporting Information is available free of charge on the ACS Publications website. Supplementary figures and tables, methods, syntheses, spectra, and crystallographic data.
The authors declare no competing financial interest.
REFERENCES
- 1.Sayiner M; Golabi P; Younossi ZM, Disease Burden of Hepatocellular Carcinoma: A Global Perspective. Digest. Dis. Sci 2019, 64, 910–917. [DOI] [PubMed] [Google Scholar]
- 2.Personeni N; Rimassa L, Hepatocellular Carcinoma: A Global Disease in Need of Individualized Treatment Strategies. J. Oncol. Pract 2017, 13, 368–370. [DOI] [PubMed] [Google Scholar]
- 3.Sherman M; Bruix J; Porayko M; Tran T; Comm APG, Screening for hepatocellular carcinoma: The Rationale for the American Association for the Study of Liver Diseases Recommendations. Hepatology 2012, 56, 793–796. [DOI] [PubMed] [Google Scholar]
- 4.Yang JD; Roberts LR, Hepatocellular carcinoma: a global view. Nat. Rev. Gastro. Hepat 2010, 7, 448–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leathers JS; Balderramo D; Prieto J; Diehl F; Gonzalez-Ballerga E; Ferreiro MR; Carrera E; Barreyro F; Diaz-Ferrer J; Singh D; Mattos AZ; Carrilho F; Debes JD, Sorafenib for Treatment of Hepatocellular Carcinoma A Survival Analysis From the South American Liver Research Network. J. Clin. Gastroenterol 2019, 53, 464–469. [DOI] [PubMed] [Google Scholar]
- 6.de Rosamel L; Blanc JF, Emerging Tyrosine Kinase Inhibitors for the Treatment of Hepatocellular Carcinoma. Expert Opin. Emerg. Dr 2017, 22, 175–190. [DOI] [PubMed] [Google Scholar]
- 7.Milgrom DP; Maluccio MA; Koniaris LG, Management of Hepatocellular Carcinoma (HCC). Curr. Surg. Rep 2016, 4, 1–8. [Google Scholar]
- 8.de Lope CR; Tremosini S; Forner A; Reig M; Bruix J, Management of HCC. J. Hepatol 2012, 56, S75–S87. [DOI] [PubMed] [Google Scholar]
- 9.Herzfeld A; Knox WE, Properties Developmental Formation and Estrogen Induction of Ornithine Aminotransferase in Rat Tissues. J. Biol. Chem 1968, 243, 3327–3332. [PubMed] [Google Scholar]
- 10.Ginguay A; Cynober L; Curis E; Nicolis I, Ornithine Aminotransferase, an Important Glutamate-Metabolizing Enzyme at the Crossroads of Multiple Metabolic Pathways. Biology (Basel) 2017, 6, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zigmond E; Ben Ya’acov A; Lee H; Lichtenstein Y; Shalev Z; Smith Y; Zolotarov L; Ziv E; Kalman R; Le HV; Lu HJ; Silverman RB; Ilant Y, Suppression of Hepatocellular Carcinoma by Inhibition of Overexpressed Ornithine Aminotransferase. ACS. Med. Chem. Lett 2015, 6, 840–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herzfeld A; Knox WE, The Properties, Developmental Formation, and Estrogen Induction of Ornithine Aminotransferase in Rat Tissues. J. Biol. Chem 1968, 243, 3327–3332. [PubMed] [Google Scholar]
- 13.De Ingeniis J; Ratnikov B; Richardson AD; Scott DA; Aza-Blanc P; De SK; Kazanov M; Pellecchia M; Ronai Z; Osterman AL; Smith JW, Functional Specialization in Proline Biosynthesis of Melanoma. Plos One 2012, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Phang JM; Donald SP; Pandhare J; Liu YM, The Metabolism of Proline, a Stress Substrate, Modulates Carcinogenic Pathways. Amino Acids 2008, 35, 681–690. [DOI] [PubMed] [Google Scholar]
- 15.Phang JM; Liu W; Hancock CN; Fischer JW, Proline Metabolism and Cancer: Emerging Links to Glutamine and Collagen. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 71–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Phang JM; Liu W; Zabirnyk O, Proline Metabolism and Microenvironmental Stress. Annu Rev. Nutr 2010, 30, 441–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Phang JM; Liu W; Hancock C; Christian KJ, The Proline Regulatory Axis and Cancer. Front Oncol. 2012, 2, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang L; Zeng J; Geng PY; Fang CN; Wang Y; Sun MJ; Wang CS; Wang J; Yin PY; Hu CX; Guo L; Yu JE; Gao P; Li EY; Zhuang ZP; Xu GW; Liu Y, Global Metabolic Profiling Identifies a Pivotal Role of Proline and Hydroxyproline Metabolism in Supporting Hypoxic Response in Hepatocellular Carcinoma. Clin. Cancer Res 2018, 24, 474–485. [DOI] [PubMed] [Google Scholar]
- 19.Heiden MGV; Cantley LC; Thompson CB, Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Colnot S; Decaens T; Niwa-Kawakita M; Godard C; Hamard G; Kahn A; Giovannini M; Perret C, Liver-targeted Disruption of Apc in Mice Activates beta-Catenin Signaling and Leads to Hepatocellular Carcinomas. P. Natl. Acad. Sci. USA 2004, 101, 17216–17221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cadoret A; Ovejero C; Terris B; Souil E; Levy L; Lamers WH; Kitajewski J; Kahn A; Perret C, New Targets of beta-Catenin Signaling in the Liver are Involved in the Glutamine Metabolism. Oncogene 2002, 21, 8293–8301. [DOI] [PubMed] [Google Scholar]
- 22.Liu YF; Wu L; Li K; Liu FR; Wang L; Zhang DL; Zhou J; Ma X; Wang SY; Yang SY, Ornithine Aminotransferase Promoted the Proliferation and Metastasis of Non-small cell Lung Cancer via Upregulation of miR-21. J. Cell Physiol 2019, 234, 12828–12838. [DOI] [PubMed] [Google Scholar]
- 23.Markova M; Peneff C; Hewlins MJE; Schirmer T; John RA, Determinants of Substrate Specificity in omega-Aminotransferases. J. Biol. Chem 2005, 280, 36409–36416. [DOI] [PubMed] [Google Scholar]
- 24.Mehta PK; Hale TI; Christen P, Aminotransferases - Demonstration of Homology and Division into Evolutionary Subgroups. Eur. J. Biochem 1993, 214, 549–561. [DOI] [PubMed] [Google Scholar]
- 25.Silverman RB, Design and Mechanism of GABA Aminotransferase Inactivators. Treatments for Epilepsies and Addictions. Chem. Rev 2018, 118, 4037–4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Silverman RB, Mechanism-Based Enzyme Inactivators. Methods Enzymol 1995, 249, 240–283. [DOI] [PubMed] [Google Scholar]
- 27.Mascarenhas R; Le HV; Clevenger KD; Lehrer HJ; Ringe D; Kelleher NL; Silverman RB; Liu D, Selective Targeting by a Mechanism-Based Inactivator against Pyridoxal 5’-Phosphate-Dependent Enzymes: Mechanisms of Inactivation and Alternative Turnover. Biochemistry 2017, 56, 4951–4961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Storici P; Qiu J; Schirmer T; Silverman RB, Mechanistic crystallography. Mechanism of inactivation of gamma-Aminobutyric Acid Aminotransferase by (1R,3S,4S)-3-Amino-4-fluorocyclopentane-1-carboxylic Acid as Elucidated by Crystallography. Biochemistry 2004, 43, 14057–14063. [DOI] [PubMed] [Google Scholar]
- 29.Juncosa JI; Takaya K; Le HV; Moschitto MJ; Weerawarna PM; Mascarenhas R; Liu DL; Dewey SL; Silverman RB, Design and Mechanism of (S)-3-Amino-4-(difluoromethylenyl)cyclopent-1-ene-1-carboxylic Acid, a Highly Potent gamma-Aminobutyric Acid Aminotransferase Inactivator for the Treatment of Addiction. J. Am. Chem. Soc 2018, 140, 2151–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pan Y; Gerasimov MR; Kvist T; Wellendorph P; Madsen KK; Pera E; Lee H; Schousboe A; Chebib M; Brauner-Osborne H; Craft CM; Brodie JD; Schiffer WK; Dewey SL; Miller SR; Silverman RB, (1S,3S)-3-Amino-4-difluoromethylenyl-1-cyclopentanoic Acid (CPP-115), a Potent gamma-Aminobutyric Acid Aminotransferase Inactivator for the Treatment of Cocaine Addiction. J. Med. Chem 2012, 55, 357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moschitto MJ; Doubleday PF; Catlin DS; Kelleher NL; Liu D; Silverman RB, Mechanism of Inactivation of Ornithine Aminotransferase by (1S,3S)-3-Amino-4-(hexafluoropropan-2-ylidenyl)cyclopentane-1-carboxylic Acid. J. Am. Chem. Soc 2019, 141, 10711–10721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee H; Juncosa JI; Silverman RB, Ornithine Aminotransferase versus GABA Aminotransferase: Implications for the Design of New Anticancer Drugs. Med. Res. Rev 2015, 35, 286–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bey P; Gerhart F; Jung M, Synthesis of (E)-4-Amino-2,5-Hexadienoic Acid and (E)-4-Amino-5-Fluoro-2-Pentenoic Acid - Irreversible Inhibitors of 4-Aminobutyrate-2-Oxoglutarate Aminotransferase. J. Org. Chem 1986, 51, 2835–2838. [Google Scholar]
- 34.Wang ZY; Yuan H; Nikolic D; Van Breemen RB; Silverman RB, (+/−)-(1S, 2R, 5S)-5-amino-2-fluorocyclohex-3-enecarboxylic acid. A Potent GABA Aminotransferase Inactivator that Irreversibly Inhibits via an Elimination-aromatization pathway. Biochemistry 2006, 45, 14513–14522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jones G; Willett P; Glen RC; Leach AR; Taylor R, Development and Validation of a Genetic Algorithm for Flexible Docking. J. Mol. Biol 1997, 267, 727–748. [DOI] [PubMed] [Google Scholar]
- 36.Qiu J; Silverman RB, A New Class of Conformationally Rigid Analogues of 4-amino-5-Halopentanoic Acids, Potent Inactivators of gamma-Aminobutyric Acid Aminotransferase. J. Med. Chem 2000, 43, 706–720. [DOI] [PubMed] [Google Scholar]
- 37.Metro TX; Duthion B; Pardo DG; Cossy J, Rearrangement of beta-Amino Alcohols via Aziridiniums: a review. Chem. Soc. Rev 2010, 39, 89–102. [DOI] [PubMed] [Google Scholar]
- 38.Wang X; Ma ML; Reddy AGK; Hu WH, An Efficient Stereoselective Synthesis of Six Stereoisomers of 3, 4-diaminocyclohexane carboxamide as Key Intermediates for the Synthesis of Factor Xa inhibitors. Tetrahedron 2017, 73, 1381–1388. [Google Scholar]
- 39.Beaulieu F; Beauregard LP; Courchesne G; Couturier M; LaFlamme F; L’Heureux A, Aminodifluorosulfinium Tetrafluoroborate Salts as Stable and Crystalline Deoxofluorinating Reagents. Org. Let 2009, 11, 5050–5053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pan Y; Qiu J; Silverman RB, Design, Synthesis, and Biological Activity of a Difluoro-substituted, Conformationally Rigid Vigabatrin Analogue as a Potent gamma-Aminobutyric Acid Aminotransferase Inhibitor. J. Med. Chem 2003, 46, 5292–5293. [DOI] [PubMed] [Google Scholar]
- 41.Lee H; Doud EH; Wu R; Sanishvili R; Juncosa JI; Liu DL; Kelleher NL; Silverman RB, Mechanism of Inactivation of gamma-Aminobutyric Acid Aminotransferase by (1S,3S)-3-Amino-4-difluoromethylene-1-cyclopentanoic Acid (CPP-115). J. Am. Chem. Soc 2015, 137, 2628–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liebschner D; Afonine PV; Moriarty NW; Poon BK; Sobolev OV; Terwilliger TC; Adams PD, Polder Maps: Improving OMIT Maps by Excluding Bulk Solvent. Acta. Crystallogr. D 2017, 73, 148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Egli M; Sarkhel S, Lone pair-aromatic Interactions: To Stabilize or not to Stabilize. Acc. Chem. Res 2007, 40, 197–205 [DOI] [PubMed] [Google Scholar]
- 44.Montioli R; Paiardini A; Giardina G; Zanzoni S; Cutruzzola F; Cellini B; Voltattorni CB, R180T Variant of delta-Ornithine Aminotransferase Associated with Gyrate Atrophy: Biochemical, Computational, X-ray and NMR Studies Provide Insight into its Catalytic Features. FEBS J. 2019, 286, 2787–2798. [DOI] [PubMed] [Google Scholar]
- 45.Storici P; Capitani G; Muller R; Schirmer T; Jansonius JN, Crystal Structure of Human Ornithine Aminotransferase Complexed with the Highly Specific and Potent Inhibitor 5-Fluoromethylornithine. J. Mol. Biol 1999, 285, 297–309. [DOI] [PubMed] [Google Scholar]
- 46.Faraci WS; Walsh CT, Mechanism of Inactivation of Alanine Racemase by β, β, β -Trifluoroalanine. Biochemistry 1989, 28, 431–437. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.