

Abstract
The immune system uses members of the toll-like receptor (TLR) family to recognize a variety of pathogen- and host-derived molecules in order to initiate immune responses. Although TLR-mediated, pro-inflammatory immune responses are essential for host defense, prolonged and exaggerated activation can result in inflammation pathology that manifests in a variety of diseases. Therefore, small molecule inhibitors of the TLR signaling pathway might have promise as anti-inflammatory drugs. We have previously identified a class of triaryl pyrazole compounds that inhibit TLR signaling by modulation of the protein-protein interactions essential to the pathway. We have now systematically examined the structural features essential for inhibition of this pathway, revealing characteristics of compounds that inhibited all TLRs tested (pan-TLR signaling inhibitors) as well as compounds that selectively inhibited certain TLRs. These findings reveal interesting classes of compounds that could be optimized for particular inflammatory diseases governed by different TLRs.
Keywords: toll-like receptor signaling, immune response, heterocycles, structure-activity-relationships, drug design
Graphical Abstract:

Dysregulation and sustained toll-like receptor (TLR) signalling is linked to a variety of inflammatory diseases. Herein, we report the structure-activity-relationships of triaryl pyrazole-core compounds with basic side chain appendages that inhibit TLR signalling. Interestingly, some compounds influence downstream activity of all TLRs whereas others are selective, making them promising candidates for further development as anti-inflammatory therapeutics.
Introduction
Toll-like receptors (TLRs) comprise a family of receptors that play key functions in the immune defense against pathogens. In humans, there are 10 functional TLRs that detect different pathogen- and host-derived molecules in order to mediate inflammation and initiate an immune response. For example, TLR9 detects viral and bacterial unmethylated CpG-DNA motifs, while TLR7 detects viral single stranded RNA; TLR2 and TLR4 are responsible for sensing bacterial lipoproteins and lipopolysaccharide (LPS), respectively.[1] TLRs also sense endogenous ligands, such as S100A8/S100A9 proteins (TLR4), in order to detect sterile tissue injury or to amplify pathogen-mediated immune responses.[2] TLRs are expressed by different cell types and control production of various effector mechanisms, including pro- and anti-inflammatory cytokines and interferons.[3]
While inflammation triggered by infections is essential for immune defense and health, prolonged TLR-mediated inflammation has been linked to a variety of diseases, including bacterial sepsis, systemic lupus erythematosus, cerebral malaria, and pancreatitis.[4] In the case of such pathologic inflammation, it is important to have effective pharmacologic means to moderate TLR activity. One potential therapeutic strategy is the development of TLR-signaling inhibitors (TSIs); however, at least in part due to the complexity of the signal transduction pathway, there are no clinically approved TSIs available to date.
Signal transduction through TLRs is initiated by dimerization of Toll/interleukin-1 receptor (TIR) domains contained in the cytoplasmic part of TLRs and cytoplasmic adaptor proteins, including MyD88 and TIRAP.[5] While some receptors, such as TLR7 and TLR9, bind directly to MyD88, TLR2 and TLR4 use the intermediate protein TIRAP to activate MyD88.[6] Dimerization of MyD88 leads to recruitment of members of the IRAK family, whose oligomerization in turn leads to recruitment and activation of the ubiquitin ligase TRAF6 to transduce signaling to downstream pathways.[6] As such, the up-stream mechanisms involved in TLR signaling rely heavily on protein-protein interactions (PPI), which are inherently challenging targets for drug development.
Recently, we have employed a drug screening method that relies on the inducible dimerization of the hierarchically acting proteins TIRAP, MyD88 and TRAF6 to identify TSIs.[7] Specific activation of these proteins allows assignment of compound activity to the level of signal transduction. Using this approach, we identified a class of compounds, i.e. triarylpyrazoles appended with a basic side chain, which interfere with TLR signaling at the level of TIR interaction. Specifically, we elucidated that these TSIs act mechanistically upstream of TRAF6 by interfering with TIR dimerization. Interestingly, initial structure-activity-relationships (SAR) showed that alterations to the structure could lead to compounds with selective activity against certain TIR-domain interactions, and thus selective TLR inhibition.[7] Herein, we describe the expansion of the SAR for the development of TSI with general TLR inhibitory activity (Pan-TLR), and TSI with more selective TLR inhibitory profile.
Results and Discussion
1,3,5-Triaryl pyrazoles: inhibition of Toll-Like Receptor signaling vs. binding to the estrogen receptor.
From our initial screen of 4364 unique compounds, we identified the lead compound, methyl-piperidino-pyrazole (MPP), a basic side chain-containing pyrazole (BSC-pyrazole) that had been previously optimized as a high affinity estrogen receptor-alpha (ERα) selective antagonist.[7–8] Therefore, our initial goal was to decouple ER binding affinity from activity against TLR signaling. In total, we investigated 90 pyrazoles. Initially, each compound was evaluated for ERα and ERβ binding affinities by a competitive radiometric ligand-binding assay (expressed as a relative binding affinity (RBA) value, with estradiol being 100) and for TLR9 inhibitory activity based on CpG-DNA-mediated TNFα production in RAW264.7 macrophage cells. Compounds that showed promising inhibition of TLR9-mediated TNFα release were then subjected to additional dose-response inhibition assays that used ligands for TLR7, TLR2 and TLR4, i.e., R848, Pam3Cys, and LPS, respectively; in parallel, the toxicity (Tox.) of each compound was measured using an Alamar blue assay. IC50 values were established in all of the cell assays. For each TLR signaling pathway, we were able to identify compounds with low ER affinity (RBA < 0.003) and moderate to good TLR signaling inhibitory potency (IC50 < 5 μM). Looking more deeply, it is clear that there is no significant correlation between binding to either ERα or ERβ, and TLR signaling inhibition (Figure 1), evidence being the abundance of inhibitors for all of the TLR signaling pathways with low IC50 values that also have low RBA values. These results established that ER binding and TLR signaling modulation can be decoupled, and selective TSIs can be optimized.
Figure 1.

Correlation of RBA for ERα (a) and ERβ (b) with TLR signaling inhibition. Binding values for ERα and ERβ are relative to that of estradiol (100). TSI inhibition values (IC50), assayed for the four TLRs studied, are color coded, as specified.
In our initial published study, we probed the requirement for and placement of the BSC around the pyrazole core, revealing the necessity of the BSC.[7] Although there was some activity when the BSC was appended to the N1 and C4 positions, we focused most of our early efforts on the derivatives with a BSC on the C5 ring, as found in our initial hit molecule MPP (compound 1), and eventually moving to derivatives with a BSC on the C3 ring, as they exhibited less affinity for the ERs (Figure 2).[7] All of the pyrazoles we studied contained three aromatic rings at positions N1, C3, and C5, because early in our studies we found that when the phenyl group appended to the N1 position was removed, potency decreased significantly (Supplemental Table S1). Therefore, the presence of three aryl rings appears important for TLR signaling inhibition.
Figure 2.

1,3,5-Triaryl pyrazoles studied, and structure of MPP (1), the initial hit.
Pyrazoles with BSC at C5.
We screened a variety of C5 close derivatives of MPP from a library previously developed as ER ligands to examine different alkyl lengths on C4 and side chains identities (Table 1).[8–9] As the alkyl group on C4 decreased in length, the TLR9 signaling inhibition potency increased (compounds 1-4), with similar activity against TLR2/4, where tested. The removal of the alkyl group on C4 (1) decreased affinity for ER somewhat. The identity of the C5 appended side chain did not influence the TLR activity significantly, as long as it was still basic. The length (2 vs. 15), ring linking atom (1 vs. 14), ring size (3 vs. 6), or the presence of a ring (vs. a dialkyl amine; compounds 7-9) on the BSC did not result in large differences in TLR inhibition. Increasing the length of the BSC by one methylene unit (1 vs. 15) reduced affinity towards ER without influencing TLR activity. We confirmed the necessity of a basic side chain by replacing with morpholino (10), lactam (11, 13), diol (12), and cyclohexyl (16) groups, resulting in reduced or even loss of activity against TLR9.
Table 1.
TLR signaling inhibition by MPP and derivatives with C5 basic side chains.
 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cmpd | R | BSC | IC50 (μM) | RBAa | ||||||
| TLR9 | TLR7 | TLR2 | TLR4 | Toxicity | ERα | ERβ | ||||
| 2 | H |  |
1.47 ± 0.25 | 3.01 ± 0.06 | 11.2 ± 0.3 | 8.67 ± 0.12 | 11.7 ± 3.7 | 5.72 ± 0.36 | 0.015 ± 0.001 | |
| 1 (MPP) | Me |  |
2.33 ± 0.64 | 3.67 ± 0.18 | 10.2 ± 0.2 | 8.8 ± 1.5 | 11.5 ± 2.4 | 12.0 ± 2.0 | 0.05 ± 0.01 | |
| 3 | Et |  |
2.85 ± 0.05 | 2.08 ± 0.10 | n.d. | n.d. | 3.89 ± 2.41 | 11.5 ± 5.8 | 0.49 ± 0.13 | |
| 4 | Pr |  |
4.72 ± 0.15 | 4.84 ± 0.13 | n.d. | n.d. | 29.3 ± 6.6 | 6.8 ± 1.1 | 0.54 ± 0.13 | |
| 5 | Me |  |
9.97 ± 0.38 | 16.1 ± 0.3 | n.d. | n.d. | 47.9 ± 3.0 | 1.15 ± 0.04 | 0.047 ± 0.010 | |
| 6 | Et |  |
3.32 ± 0.11 | 2.34 ± 0.45 | n.d. | n.d. | 12.4 ± 4.2 | 16.0 ± 2.6 | 0.17 ± 0.02 | |
| 7 | Me | 3.06 ± 0.33 | 3.70 ± 0.28 | n.d. | n.d. | 21.1 ± 6.4 | 7.77 ± 0.25 | 0.051 ± 0.008 | ||
| 8 | Et | 3.04 ± 0.30 | 3.64 ± 0.29 | n.d. | n.d. | 16.6 ± 4.0 | 16.6 ± 3.5 | 0.17 ± 0.04 | ||
| 9 | Et | 4.21 ± 0.27 | 30.9 ± 0.6 | n.d. | n.d. | 39.0 ± 2.2 | 9.12 ± 0.30 | 0.64 ± 0.14 | ||
| 10 | Et | 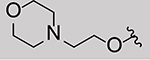 |
> 50 | n.d. | n.d. | n.d. | > 50 | 9.46 ± 0.77 | 0.083 ± 0.020 | |
| 11 | Et |  |
> 50 | n.d. | n.d. | n.d. | > 50 | 13.3 ± 3.6 | 0.43 ± 0.07 | |
| 12 | Et |  |
> 50 | n.d. | n.d. | n.d. | > 50 | 7.0 ± 1.9 | 0.246 ± 0.008 | |
| 13 | Me |  |
>50 | n.d. | n.d. | n.d. | > 50 | 1.61 ± 0.05 | 0.015 ± 0.005 | |
| 14 | Me |  |
1.07 ± 0.02 | 1.37 ± 0.16 | 9.70 ± 0.10 | 8.91 ± 0.13 | 8.76 ± 1.38 | 5.8 ± 1.7 | 0.026 ± 0.001 | |
| 15 | H |  |
1.44 ± 0.27 | 3.23 ± 0.33 | 7.12 ± 0.54 | 9.26 ± 0.36 | 12.4 ± 2.1 | 1.75 ± 0.43 | 0.011 ± 0.001 | |
| 16 | H |  |
12.7 ± 0.3 | 10.8 ± 0.4 | 15.8 ± 0.2 | n.d. | 20.9 ± 2.5 | 0.45 ± 0.12 | 0.037 ± 0.010 | |
RBA = relative binding affinity values, where estradiol = 100.
n.d. = not determined
In addition, we screened the methoxy precursors to the MPP derivatives above (Table 2). As expected, the relative binding affinity for ERs decreased significantly when the phenols on the MPP derivatives were masked as methoxy groups. However, it may not be prudent to rely on this functional group, because methoxy groups can readily be metabolized into phenols in vivo, restoring high affinity for ERα. Nevertheless, these compounds gave us additional insight into the structure-activity-relationships (SAR). First, as observed above, the smaller group at C4 provided more potent activity against TLR (17 vs.18). Extending the BSC by one methylene retained similar activity (19 vs. 17), with some increased activity towards TLR7. Removal of all oxygen-containing substituents decreased activity against TLR9/7 (22). The presence of the electron-withdrawing chloro group on the N1 ring also decreased TLR activity across the board (23, 24).
Table 2.
TLR signaling inhibition by other MPP derivatives with C5 basic side chains.
 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cmpd | R1 | R3 | R4 | n | IC50 (μM) | RBAa,b | |||||
| TLR9 | TLR7 | TLR2 | TLR4 | Tox. | ERα | ERβ | |||||
| 17 | OMe | OMe | H | 1 | 2.46 ± 0.97 | > 50 | 12.6 ± 0.4 | 9.85 ± 0.05 | 16.2 ± 3.4 | n.d. | n.d. |
| 18 | OMe | OMe | Me | 1 | 4.15 ± 0.56 | > 50 | n.d. | n.d. | 29.3 ± 0.6 | n.d. | n.d. |
| 19 | OMe | OMe | H | 2 | 3.58 ± 0.20 | 10.9 ± 0.5 | 10.1 ± 0.4 | n.d. | 10.0 ± 0.3 | 0.002 | 0.001 |
| 20 | H | OMe | H | 1 | 3.26 ± 0.18 | 14.4 ± 1.0 | 31.3 ± 1.5 | n.d. | 17.2 ± 3.0 | 0.004 | 0.001 |
| 21 | H | OH | H | 1 | 3.31 ± 0.26 | 27.7 ± 0.8 | 8.94 ± 0.18 | 10.2 ± 0.1 | 26.2 ± 2.0 | 0.002 | 0.001 |
| 22 | H | H | H | 1 | 8.92 ± 0.33 | 8.03 ± 0.32 | n.d. | n.d. | 23.9 ± 2.4 | n.d. | n.d. |
| 23 | Cl | OMe | H | 1 | 9.31 ± 0.36 | 31.4 ± 0.3 | 29.2 ± 0.1 | n.d. | 26.4 ± 0.6 | 0.002 | 0.001 |
| 24 | Cl | OH | H | 1 | 3.04 ± 0.30 | n.d. | > 50 | > 50 | > 50 | 0.007 | 0.002 |
RBA = relative binding affinity values, where estradiol = 100.
single determinations for RBA < 0.01
n.d. = not determined
Due to their low binding affinity for ER, we focused most of our synthetic efforts on diversifying the C3 BSC-containing pyrazole derivatives. From our initial screen for C5 BSC containing compounds, the size of the alkyl chain influenced activity, with smaller groups being preferred. Therefore, we focused on compounds without an alkyl chain at C4. These compounds were assembled by a four-step modular synthesis that allowed for easy derivatization on the C5 and N1 rings (Scheme 1). In the first step, an aldol condensation was performed between 4-hydroxyacetophenone (26) and the appropriate benzaldehyde (25). Then, the basic side chain was installed on the chalcone (27). Reaction of the BSC-chalcone (28) with the appropriate hydrazine resulted in the corresponding pyrazoline that was oxidized to the pyrazole (29–71) by treatment with manganese oxide. A total of 42 pyrazoles containing a 4-O(CH2)2-piperdinyl BSC at C3 (Tables 3–6, Supplemental Table S2) were synthesized and examined for their ability to inhibit TLR signaling. In addition, each compound had low ER binding affinity (RBA < 0.06, Supplemental Table S1).
Scheme 1.

General synthetic scheme for C3 4-O(CH2)2-piperdinyl pyrazole derivatives. Reagents and conditions: a) KOH, TBAB, MeOH, reflux, overnight. b) 1-(2-chloroethyl)-piperidine hydrochloride, K2CO3, Cs2CO3, DMF, overnight. c) phenylhydrazine hydrochloride, DMF, 85 °C, overnight. d) MnO2, benzene, reflux, 5–10 h.
Table 3.
TLR signaling inhibition by C3 BSC pyrazoles with free phenols.
 | |||||||
|---|---|---|---|---|---|---|---|
| Cmpd | R1 | R5 | IC50 (μM) | ||||
| TLR9 | TLR7 | TLR2 | TLR4 | Tox. | |||
| 29 | p-OH | p-OH | 2.65 ± 0.39 | 7.01 ± 1.41 | 9.13 ± 3.88 | 6.73 ± 0.66 | 35.0 ± 3.2 |
| 30 | H | p-OH | 2.61 ± 1.37 | 2.99 ± 0.62 | 6.73 ± 1.97 | 5.38 ± 0.64 | 33.3 ± 2.6 |
| 31 | p-OH | H | 6.80 ± 0.65 | 15.5 ± 1.0 | 7.94 ± 2.19 | 15.5 ± 3.1 | 19.9 ± 5.2 |
| 32 | p-OH | m-OH | 30.2 ± 0.1 | n.d. | > 50 | n.d. | > 50 |
| 33 | p-Me | p-OH | 3.97 ± 0.27 | 18.4 ± 0.6 | 23.5 ± 0.6 | 22.8 ± 0.7 | 26.3 ± 3.7 |
| 34 | p-Cl | p-OH | 3.79 ± 0.30 | 34.8 ± 0.1 | 11.3 ± 0.1 | 16.7 ± 0.5 | 23.6 ± 0.4 |
| 35 | H | 3,5-(OMe)2- 4-OH |
3.23 ± 0.08 | 10.7 ± 0.4 | 5.66 ± 0.75 | 2.92 ± 0.22 | 17.7 ± 4.4 |
| 36* | p-OH |
p-NH C(O)Me |
1.18 ± 0.18 | 7.67 ± 0.21 | 11.0 ± 0.5 | 6.17 ± 0.14 | 9.72 ± 0.15 |
compound 36 was a 1:1 mixture of pyrazole:pyrazoline
n.d. = not determined
Table 6.
TLR signaling inhibition by C3 BSC pyrazoles with electron-donating groups.
 | ||||||
|---|---|---|---|---|---|---|
| Cmpd | R | IC50 (μM) | ||||
| TLR9 | TLR7 | TLR2 | TLR4 | Tox. | ||
| 61 | 3,4-(OMe)2 | 8.26 ± 0.34 | 4.68 ± 1.16 | > 50 | > 50 | 40.9 ± 6.4 |
| 62 | 3,4,5-(OMe)3 | 5.45 ± 0.80 | 2.25 ± 0.27 | 2.49 ± 0.06 | 1.13 ± 0.39 | 16.90 ± 0.01 |
| 63 | 2,4,5-(OMe)3 | 7.62 ± 0.41 | 5.97 ± 1.33 | > 50 | 27.3 ± 2.8 | 22.8 ± 7.0 |
| 64 | 3,4,6-(OMe)3 | 8.40 ± 0.01 | > 50 | > 50 | > 50 | 26.5 ± 4.3 |
| 65 | 2,3,4-(OMe)3 | 10.8 ± 0.1 | > 50 | > 50 | > 50 | > 50 |
| 66 | 3,5-(OMe)2-4-OEt | 9.68 ± 0.82 | > 50 | > 50 | > 50 | > 50 |
| 67 | 3,4,5-(OEt)3 | 11.0 ± 1.1 | 1.48 ± 0.02 | > 50 | > 50 | 20.0 ± 0.2 |
| 68 | 3-OMe-4-OEt | 12.8 ± 0.3 | > 50 | > 50 | > 50 | > 50 |
| 69 | 5-OMe-3,4-dioxymethylene | 7.03 ± 0.29 | 0.79 ± 0.09 | > 50 | > 50 | > 50 |
| 70 | 3,5-(Me)2-4-OMe | 7.04 ± 0.24 | 2.25 ± 0.17 | 15.3 ± 0.3 | 17.3 ± 0.5 | 13.9 ± 0.9 |
| 71 | 3-Cl-4-OMe | 7.78 ± 0.34 | > 50 | > 50 | > 50 | 19.9 ± 1.4 |
The position and number of free phenols on the aromatic rings had some influence on activity (Table 3). Removal of the p-OH from the N1 ring was well tolerated (30 vs. 29); however, on the C5 ring, removal of the p-OH (31) or movement of it to the meta position (32) decreased activity. Replacement of the phenol on N1 with a methyl group (33 vs. 29) or electron-withdrawing chloro group (34 vs. 29) retained activity against TLR9 but reduced activity against TLR7/2/4. When additional electron-donating groups were added to the C5 ring (35 vs. 29), the activity against TLR4 increased but the general toxicity was also increased. The replacement of the phenol on C5 with an amide (29 vs. 36) improved activity against TLR9 but also resulted in increased toxicity.
We next expanded the SAR to include a variety of methoxy derivatives at N1 (R1) and C5 (R5) positions, retaining the same 4-O(CH2)2-piperidinyl BSC on C3 and a H at C4 (Table 4). The removal of the para-methoxy on the N1 phenyl group increased activity against TLR7 (38). Placement of the methoxy on the meta position slightly improved activity against TLR2 and TLR7 (39). All substitutions at the para position regardless of electronics (38, 40-45) retained comparable activity against TLR9; however, the activity against the other TLRs was variable, albeit not very potent. Of particular note was the N-Boc protected derivative (44) that was completely selective for TLR9 and exhibited no toxicity in the cells. Overall, changes to the C5 (R5) position were generally well tolerated, although they resulted in some increased toxicity (compounds 46-52). Of note, increased electron-donating methoxy groups tended to increase activity towards TLR7, TLR2, and TLR4 (47, 49, 50), with the tri-methoxy (49) being a pan-TLR inhibitor. An amide group on C5 also produced a pan-TLR inhibitor (48). Replacement of some methoxy groups with an electron-withdrawing chloro (51) or slightly donating methyl groups (52) seemed to decrease activity, with some selectivity for TLR9/7.
Table 4.
TLR signaling inhibition by C3 BSC pyrazoles with methoxy substituents.
 | |||||||
|---|---|---|---|---|---|---|---|
| Cmpd | R1 | R5 | IC50 (μM) | ||||
| TLR9 | TLR7 | TLR2 | TLR4 | Tox. | |||
| 37 | p-OMe | p-OMe | 4.26 ± 0.04 | > 50 | 28.4 ± 0.1 | n.d. | 28.2 ± 2.8 |
| 38 | H | p-OMe | 4.44 ± 0.36 | 4.96 ± 0.33 | 28.6 ± 0.1 | n.d. | 15.8 ± 2.9 |
| 39 | m-OMe | p-OMe | 5.75 ± 0.23 | 12.3 ± 0.1 | 11.6 ± 0.3 | n.d. | 14.0 ± 2.8 |
| 40 | p-Me | p-OMe | 3.95 ± 0.11 | 11.3 ± 0.1 | 24.3 ± 0.6 | 17.7 ± 0.7 | 13.3 ± 2.5 |
| 41 | p-OCF3 | p-OMe | 5.81 ± 0.55 | 12.1 ± 0.5 | 16.0 ± 0.1 | 19.1 ± 0.4 | 16.6 ± 2.3 |
| 42 | p-Cl | p-OMe | 4.49 ± 0.69 | 34.7 ± 2.4 | 16.0 ± 0.1 | n.d. | 20.0 ± 5.1 |
| 43 | p-NO2 | p-OMe | 3.02 ± 0.11 | 22.3 ± 0.9 | 26.5 ± 1.6 | 26.6 ± 0.3 | 31.4 ± 3.1 |
| 44 | 4-NHBoc | p-OMe | 3.00 ± 0.24 | > 50 | > 50 | > 50 | > 50 |
| 45 | 4-NH2 | p-OMe | 4.08 ± 0.38 | 19.6 ± 0.8 | 13.1 ± 1.0 | 19.3 ± 1.7 | 15.8 ± 1.4 |
| 46 | p-OMe | H | 4.61 ± 0.23 | > 50 | 27.4 ± 0.6 | n.d. | 22.6 ± 3.5 |
| 47 | p-OMe | 2,4-(OMe)2 | 3.73 ± 0.11 | 3.51 ± 0.07 | 15.5 ± 0.8 | 13.5 ± 0.7 | 8.61 ± 1.91 |
| 48 | p-OMe | 4-NH C(O)Me |
1.24 ± 0.07 | 3.19 ± 0.08 | 5.18 ± 0.48 | 5.60 ± 0.34 | 10.7 ± 0.8 |
| 49 | p-OMe | 3,4,5- (OMe)3 |
3.60 ± 0.49 | 3.14 ± 0.63 | 3.79 ± 1.10 | 1.26 ± 0.39 | 14.7 ± 0.1 |
| 50 | p-OMe | 3,4- (OMe)2 |
3.03 ± 0.38 | 17.0 ± 1.1 | 17.9 ± 0.7 | 13.5 ± 0.6 | 10.1 ± 2.1 |
| 51 | p-OMe | 3-Cl- 4-OMe |
6.00 ± 0.04 | 7.30 ± 0.33 | > 50 | > 50 | 18.1 ± 1.2 |
| 52 | p-OMe | 3,5-(CH3)2- 4-OMe |
7.53 ± 0.49 | 8.42 ± 0.55 | > 50 | > 50 | 17.1 ± 1. 4 |
n.d. = not determined
Next, we focused on carboxylic acid derivatives on the C5 position, keeping a phenyl ring at N1 (Table 5). The methyl ester (53) lost some potency, while the carboxylic acid (54) was completely inactive. The amide (55) and free amine (57) were selective for TLR9/7, while the nitrile (56) afforded selectivity for TLR9. Additional modifications of the amine as a carbamate (58) or amide (59, 60) retained selectivity for TLR9/7.
Table 5.
TLR signaling inhibition by C3 BSC pyrazoles with C5 carboxylic acid derivatives.
 | ||||||
|---|---|---|---|---|---|---|
| Cmpd | R5 | IC50 (μM) | ||||
| TLR9 | TLR7 | TLR2 | TLR4 | Tox. | ||
| 53 | CO2Me | 6.29 ± 0.19 | 16.9 ± 0.4 | 15.7 ± 0.5 | > 50 | 30.8 ± 1.3 |
| 54 | CO2H | > 50 | n.d. | n.d. | n.d. | > 50 |
| 55 | CONH2 | 3.43 ± 0.15 | 5.96 ± 0.74 | 27.8 ± 2.5 | > 50 | 17.7 ± 7.7 |
| 56 | CN | 7.89 ± 0.36 | > 50 | > 50 | > 50 | 33.5 ± 3.5 |
| 57 | NH2 | 3.81 ± 0.30 | 5.93 ± 0.04 | > 50 | > 50 | 33.1 ± 3.5 |
| 58 | NHCO2Me | 3.46 ± 0.31 | 3.18 ± 0.21 | > 50 | > 50 | 25.7 ± 5.9 |
| 59 | NHC(O)Me | 3.38 ± 0.14 | 7.25 ± 0.21 | > 50 | 17.4 ± 0.33 | 14.9 ± 2.4 |
| 60 | NHC(O)Et | 2.93 ± 0.84 | 7.92 ± 0.15 | > 50 | > 50 | 38.8 ± 4.5 |
n.d. = not determined
As the increased electron-donating capability on C5 seemed to have some effect on activity, we examined a wide range of derivatives with electron-donating groups on various positions on the C5 ring (Table 6). Depending on the position, number, and identity of the ethers, we could obtain TLR9-selective (compounds 64–66, 68, 71), TLR9/7-selective (compounds 61, 63, 67, 69, 70), or pan-TLR (62) inhibition. Many of the TLR9/7-selective inhibitors showed a preference for TLR7 inhibition (67, 69, 70). We also examined some different aromatic rings, 6-methoxyl-2-naphthyl and 3-thienyl, appended to C5, which showed moderate preference for TLR9 (Supplemental Table S2).
From our investigations of the C5 site above, we found that variation in the BSC, as long as it was basic, was well tolerated. Therefore, we appended different basic groups in the C3 position (Table 7). There were not significant changes to TLR signaling inhibition with modifications of the length or identity of the linker to the piperidinyl group, although there was a modest increase in toxicity (72-74). Surprisingly, replacement of the piperidinyl group with a pyrazole (75) resulted in a completely TLR7-specific compound without any toxicity issues. As reported elsewhere, we have examined this compound further in biological studies.[7]
Table 7.
TLR signaling inhibition by pyrazoles with different C3 basic side chains.
 | |||||||
|---|---|---|---|---|---|---|---|
| Cmpd | R | BSC | IC50 (μM) | ||||
| TLR9 | TLR7 | TLR2 | TLR4 | Tox. | |||
| 72 | OH |  |
3.36 ± 0.09 | 4.62 ± 0.12 | 5.88 ± 0.31 | 6.11 ± 0.48 | 26.0 ± 0.5 |
| 73 | OMe |  |
3.42 ± 0.34 | 3.52 ± 0.03 | 12.8 ± 0.3 | n.d. | 8.69 ± 2.41 |
| 74 | OMe |  |
3.46 ± 0.18 | 5.55 ± 0.22 | n.d. | n.d. | 4.99 ± 1.70 |
| 75 | OMe |  |
> 50 | 2.27 ± 0.95 | > 50 | > 50 | > 50 |
n.d. = not determined
In order to avoid any promiscuous chemotypes that might fail in future studies, we subjected all of our compounds to three publicly available in silico filters to identify Pan Assay INterference compoundS (PAINS).[10] All of the compounds passed two screens (http://www.cbligand.org/PAINS/ and http://zinc15.docking.org/patterns/home). In the third screen (http://fafdrugs3.mti.univ-paris-diderot.fr/), the two aniline-containing compounds (45, 57) were identified as high risk, and the phenol-containing compounds and thiophene compound (S4) were identified as low risk. As discussed above, future studies will not focus on phenolic compounds due to their off-target effects on ER. In addition, derivatives 45, 57, and S4 were not among the most promising TSIs. These results, coupled with our toxicity and ER RBA values, make us confident that the SAR concluded here is appropriate.
Conclusions
Previously, we identified triaryl-pyrazoles with an appended basic side chain (BSC) as TLR signaling inhibitors through the inhibition of MyD88 dimerization at the TIR domain.[7] Here, we expanded the SAR of these pyrazole-based TLR signaling inhibitors. We conclude that three aryl rings are necessary for activity, the BSC is optimal on the C3 or C5 ring, smaller substituents at the C4 position are best tolerated, and adjustments to the electronics around the aromatic groups result in variable TLR signaling inhibition (Figure 3). Binding to the estrogen receptor is essentially eliminated in the more extensively investigated series with the BSC on the C3 ring.
Figure 3.

Summary of Structure-TSI Activity-Relationships of triaryl-pyrazoles.
Interestingly, we identified some TSI with selectivity for one signaling pathway over the others. For example, we identified compounds with selectivity for nucleic acid-recognizing TLRs, i.e., TLR7 and TLR9. Most interesting are TLR7-selective inhibitors that result from additional electron-donating groups on the C5 position or replacement of the piperidinyl BSC with a pyrazolyl BSC (compound 76). Both TLR7 and TLR9, possibly acting in a complementary fashion, have been linked to the autoimmune disease systemic lupus erythematosus, and compounds targeting their signaling pathways are expected to have therapeutic potential.[11] On the other hand, other inflammatory diseases, such as polymicrobial sepsis or cerebral malaria, are likely driven by the combined activity of different TLRs, including TLR2, TLR4 and TLR9, making Pan-TLR inhibitors therapeutically attractive candidates.[12] While we speculate that TSI with selectivity for specific TLRs, e.g. TLR7, target specific TIR-interactions, more detailed molecular studies will be required to explore their mechanism of action.
Experimental Section
General Synthetic Methods.
All reagents were used as purchased. DMF used in reactions was dried using a solvent delivery system (neutral alumina column).[13] MeOH and benzene used in reactions were anhydrous, purchased from Aldrich. Solvents used for extraction and flash chromatography were reagent or ultima grade, purchased from either Aldrich or Fisher Scientific. All reactions were run under dry N2 atmosphere except where noted. Flash column chromatography was performed on Silica P Flash Silica Gel (40–64 μM, 60 Å) from SiliCycle® or using a Teledyne ISCO CombiFlash MPLC system equipped with Redisep Gold silica gel columns. 1H NMR and 13C NMR spectra were obtained on 400 or 500 MHz Varian® FT-NMR spectrometers. The chemical shifts are reported in ppm and are referenced to either tetramethylsilane or the solvent. Except where noted, both low and high resolution mass spectra were obtained using electrospray ionization on either a Micromass Q-Tof Ultima or Waters Quattro instrument.
The basic synthetic transformations outlined in Scheme 1 are given below. The detailed synthesis and spectroscopic characterization of specific compounds are given in the Supporting Information.
Chalcone Formation (Scheme 1 step i) Typical Procedure:
To a solution of p-hydroxyacetophenone (7.34 mmol) and the appropriate benzaldehyde (8.81 mmol) in methanol (70 mL), KOH (29.38 mmol) and catalytic tetrabutylammonium bromide (TBAB) was added. The reaction mixture was refluxed overnight. After the completion of reaction, the mixture was cooled and poured into iced cold water to which dil. HCl was added. The precipitate obtained was then filtered and washed with excess of water, methanol, dried in air and finally recrystallized from methanol to obtain pure chalcones in 65–90 % yield.
BSC Installment (Scheme 1 step ii) Typical Procedure:
To the solution of the respective chalcone (2.36 mmol) in DMF (12 ml), K2CO3 (3.54 mmol) and Cs2CO3 (2.36 mmol) were added, followed by 1-(2-chloroethyl)piperidine-hydrochloride (2.60 mmol). Reaction mixture was allowed to stir for overnight at room temperature. After completion of the reaction, extraction was done using ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate, concentrated under vacuum, and partially purified by flash column chromatography using methanol/DCM to remove the nonpolar impurities or any traces of unreacted chalcone. The synthesized chalcones with basic side chains (85–95 % yield) were then directly taken to the next step.
Pyrazole Formation (Scheme 1 steps iii-iv) Typical Procedure:
A mixture of substituted chalcone (0.55 mmol) and appropriate phenylhydrazine hydrochloride (0.82 mmol) in 5 mL of DMF was heated under a N2 atmosphere at 85 °C for 4–10 h. The reaction solution was concentrated under vacuum and partitioned between ethyl acetate and water twice. The organic layers were combined, washed with brine, dried (Na2SO4) and concentrated under vacuum. The residue was then subjected to oxidation using MnO2 (7.15 mmol) in benzene (15 ml) under reflux for 5–10 h. After completion, the reaction the mixture was allowed to cool to RT and then passed through a pad of Celite to remove particulates. The clear solution was concentrated under vacuum and purified by flash chromatography using DCM/methanol to obtain the pyrazoles in 75–90 % yield.
Cell Culture and Reagents.
RAW264.7 cells were cultured in RPMI 1640 (Life Technologies), supplemented with 10% (v/v) FCS (Hyclone), 50 μM 2-mercaptoethanol, penicillin G (100 IU/mL) and streptomycin sulfate (100 IU/mL). CpG-DNA refers to the phosphothioate backbone containing oligonucleotide 1668 (TCCATGACGTTCCTGATGCT) (TIB Molbiol). Other agonists used were LPS (Escherichia coli 0127:B8) (Sigma-Aldrich), R484 (GLSynthesis), and tripalmitoyl cysteinyl lipopeptide (Pam3Cys) (EMC Microcollections).
TNFα Measurement Assays.
RAW264.7 cells were seeded in complete RPMI 1640 (without phenol red) medium in 96 well plates at a cell density of 50,000 per well at least 12 hours before stimulation. Cell were treated with TSI in dose response settings (50 μM to 0.023 μM in three-fold dilutions) or DMSO for 30 min followed by stimulation with various TLR agonists at pre-determined concentrations: CpG-DNA (1 μM), R848 (300 nM), Pam3Cys (100 ng/mL), or LPS (10 ng/mL). TNFα concentrations were determined in cell culture supernatants by ELISA 6 hours after stimulation. Cell viability (Tox.) was analyzed by the Alamar blue assay system (Invitrogen). Data are presented as means ± SD from 3 independent experiments.
Estrogen Receptor Binding Assays.
Competitive radiometric ligand binding assays were performed on 96-well microtiter filter plates (Millipore), using full-length human estrogen receptor α and β, with titrated estradiol as tracer, as previously described.[14] After incubation on ice for 18–24 h, ER-bound tracer was absorbed onto hydroxyapatite (BioRad), washed with buffer, and measured by scintillation counting. Most RBA values are the average of 2–3 determinations ± SD. As indicated in Table footnotes, single determinations were done with compounds having <1/2000 or <1/10,000 the affinity of estradiol (RBA <0.05 or <0.01).
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health (PHS 5R01 DK015556 to JAK and T32ES007326 to JAP), the St. Jude Children’s Infection Defense Center (CIDC) and the American Lebanese Syrian Associated Charities (ALSAC).
Footnotes
Supporting information for this article is given via a link at the end of the document.
Contributor Information
Dr Hans Hӓcker, Email: Hans.Haecker@stjude.org.
Prof John A. Katzenellenbogen, Email: jkatzene@illinois.edu.
References:
- [1].a) Dunne A, O’Neill LA, Science’s STKE : signal transduction knowledge environment 2003, 2003(171), re3. [DOI] [PubMed] [Google Scholar]; b) Kawai T, Akira S, Nature Immunol. 2010, 11(5), 373–384. [DOI] [PubMed] [Google Scholar]
- [2].Vogl T, Tenbrock K, Ludwig S, Leukert N, Ehrhardt C, van Zoelen MA, Nacken W, Foell D, van der Poll T, Sorg C, Roth J, Nat. Med. 2007, 13(9), 1042–1049. [DOI] [PubMed] [Google Scholar]
- [3].Beutler B, Nature 2004, 430(6996), 257–263. [DOI] [PubMed] [Google Scholar]
- [4].a)Bamboat ZM, Balachandran VP, Ocuin LM, Obaid H, Plitas G, DeMatteo RP, Hepatology 2010, 51(2), 621–632 [DOI] [PMC free article] [PubMed] [Google Scholar]; b)Calcaterra C, Sfondrini L, Rossini A, Sommariva M, Rumio C, Menard S, Balsari A, J. Immunol 2008, 181(9), 6132–6139 [DOI] [PubMed] [Google Scholar]; c)Hoque R, Sohail M, Malik A, Sarwar S, Luo Y, Shah A, Barrat F, Flavell R, Gorelick F, Husain S, Mehal W, Gastroenterology 2011, 141(1), 358–369 [DOI] [PMC free article] [PubMed] [Google Scholar]; d)Knapp S, Wiener medizinische Wochenschrift 2010, 160(5–6), 107–111. [DOI] [PubMed] [Google Scholar]
- [5].Yamamoto M, Takeda K, Akira S, Mol. Immunol. 2004, 40(12), 861–868. [DOI] [PubMed] [Google Scholar]
- [6].Kumar H, Kawai T, Akira S, Biochem. Biophys. Res. Comm 2009, 388(4), 621–625. [DOI] [PubMed] [Google Scholar]
- [7].Ippagunta SK, Pollock JA, Sharma N, Lin W, Chen T, Tawaratsumida K, High AA, Min J, Chen Y, Guy RK, Redecke V, Katzenellenbogen JA, Hacker H, Sci. Signal 2018. 11(543), eaaq1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sun J, Huang YR, Harrington WR, Sheng S, Katzenellenbogen JA, Katzenellenbogen BS, Endocrinol. 2002, 143(3), 941–947. [DOI] [PubMed] [Google Scholar]
- [9].a) Stauffer SR, Huang YR, Aron ZD, Coletta CJ, Sun J, Katzenellenbogen BS, Katzenellenbogen JA, Bioorg. Med. Chem 2001, 9(1), 151–161 [DOI] [PubMed] [Google Scholar]; b) Zhou HB, Carlson KE, Stossi F, Katzenellenbogen BS, Katzenellenbogen JA, Bioorg. Med. Chem. Lett 2009, 19(1), 108–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Baell J, Walters MA, Nature 2014, 153, 481–483 [DOI] [PubMed] [Google Scholar]; b) McGovern SL, Caselli E, Grigorieff N, Shoichet BK J. Med Chem 2002, 45, 1712–1722 [DOI] [PubMed] [Google Scholar]; c) Basell JB, Holloway GA, J. Med. Chem 2010, 53(7), 2719–2740 [DOI] [PubMed] [Google Scholar]; d) Lagorce D, Sperandio O, Baell JB, Miteva MA, Villoutreix BO; Nucleic Acids Res 2015, 43, W200–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].a) Nickerson KM, Christensen SR, Shupe J, Kashgarian M, Kim D, Elkon K, Shlomchik MJ, J. Immunol 2010, 184(4), 1840–1848 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Pawar RD, Ramanjaneyulu A, Kulkarni OP, Lech M, Segerer S, Anders HJ, J. Am. Soc. Nephrol 2007, 18(6), 1721–1731 [DOI] [PubMed] [Google Scholar]; c) Santiago-Raber ML, Dunand-Sauthier I, Wu T, Li QZ, Uematsu S, Akira S, Reith W, Mohan C, Kotzin BL, Izui S, J. Autoimmun 2010, 34(4), 339–348. [DOI] [PubMed] [Google Scholar]
- [12].a) Gong Y, Zou L, Feng Y, Li D, Cai J, Chen D, Chao W, Anesthesiology 2014, 121(6), 1236–1247 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lohner R, Schwederski M, Narath C, Klein J, Duerr GD, Torno A, Knuefermann P, Hoeft A, Baumgarten G, Meyer R, Boehm O, Mediators Inflamm. 2013, 2013, 261049. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Sahu PK, Satpathi S, Behera PK, Mishra SK, Mohanty S, Wassmer SC, Front. Cell Infect. Microbiol 2015, 5, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ, Organometallics 1996, 15(5), 1518–1520. [Google Scholar]
- [14].Carlson KE, Choi I, Gee A, Katzenellenbogen BS, Katzenellenbogen JA, Biochemistry 1997, 36(48), 14897–14905. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.