

Abstract
Elexacaftor-tezacaftor-ivacaftor is a newly approved triple-combination cystic fibrosis transmembrane conductance regulator (CFTR) modulating therapy that contains 2 correctors and a potentiator of the CFTR channel. Its labeled indication for use is for persons 12 years of age and older with at least 1 F508del mutation for the CFTR gene. This drug combination provides potential therapy to many patients who had previously been excluded from CFTR modulation therapy due to the nature of their genetic mutations. The efficacy demonstrated in clinical trials surpasses the currently available therapies related to lung function, quality of life, sweat chloride reduction, and reducing exacerbations. The most common adverse events seen in clinical trials included rash and headache, and laboratory monitoring is recommended to evaluate liver function. Continued evaluation of patient data is needed to confirm its long-term safety and efficacy. Elexacaftor-tezacaftor-ivacaftor is a monumental and encouraging therapy for cystic fibrosis; however, approximately 10% of the CF population are not candidates for this or any other CFTR modulation therapy.
Keywords: CFTR modulator, cystic fibrosis, elexacaftor, ivacaftor, tezacaftor
Introduction
Cystic fibrosis (CF) is an autosomal recessive disorder characterized by defects in chloride ion transport affecting pancreatic, respiratory, gastrointestinal, reproductive, and skeletal function. This is caused by mutations in genes responsible for encoding of the cystic fibrosis transmembrane conductance regulator (CFTR) protein. To combat this disease, many life-prolonging therapies are required. These therapies range from non-pharmacologic airway clearance techniques to newer CFTR modulating small molecule therapies.1
There are more than 2000 gene variants of the disease that have been identified with the most predominant being the F508del mutation. The various mutations affect how the CFTR protein is created and how well it carries out its function on the cellular surface.2 Different mutations are currently divided into 6 classes and are represented in the Figure. Class I mutations (e.g., G542X) consist of primarily nonsense mutations that cause a disruption in synthesis of the CFTR protein.3 Class II mutations (e.g., F508del) allow for the CFTR protein to be synthesized; however, it is misfolded, which hinders its presentation to the cell surface.3 Class III mutations (e.g., G551D) allow for the formation and presentation of the CFTR protein to the cell surface, but the gating regulation is dysfunctional resulting in loss of functionality of ion transfer. Class IV mutations (e.g., R117H) provide a functional CFTR protein, but the pore conduction is diminished, leading to decreased ion transfer. Class V mutations (e.g., A455E) are often a result of splicing defects that lead to a decreased production of the CFTR protein. Class IV and Class V mutations are also known as residual function mutations. Class VI mutations (e.g., 120del23) cause reduced CFTR membrane stability, which increases turnover of active CFTR proteins.3
Figure.
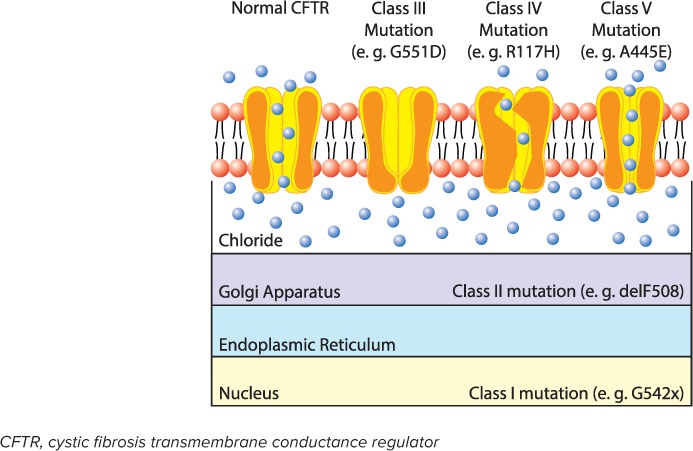
Classes of CFTR mutations. Class I and II mutations result in CFTR protein that does not reach the cell surface due to impaired protein translation in the cell nucleus (Class I) or misfolded protein in the golgi apparatus (Class II). Class III and IV mutations result in CFTR that reaches the cell surface but exhibits impaired function due to a gating defect (Class III) or decrease conductivity (Class IV). Class V mutations lead to a reduced production of normal CFTR.
Great advancements in CFTR modulating therapy have been made over the previous decade. Since development of the CFTR “potentiator” ivacaftor and the “correctors” lumacaftor and tezacaftor, roughly 50% of patients with CF were eligible for CFTR modulating therapy.4 A new option containing elexacaftor, tezacaftor, and ivacaftor (Trikafta, Vertex Pharmaceuticals, Boston, MA) has recently been approved and is indicated for patients with CF 12 years of age and older who have at least 1 mutation in the F508del gene,5 regardless of their second mutation type. This increases the eligibility for CFTR modulating therapy to around 90% of patients who have CF.1,4 Indications for CFTR modulators are detailed in Table 1. Additionally, the clinical benefit seen from elexacaftor-tezacaftor-ivacaftor combination therapy has exceeded the other combination therapy options currently available to patients and truly paves the way for changing the trajectory of CF.
Table 1.
FDA Indicated Populations for CFTR Modulators
| Drug | Age | Mutation(s) | Clinical Trial Outcomes |
|---|---|---|---|
| Ivacaftor | ≥6 mo | G511D, R117H, S1251N, and Class IV or Class V mutations | ppFEV1: ↑ 10% Exacerbations: ↓ 55% Weight: ↑ 2.7 kg Sweat chloride: ↓ 48 mmol/L |
| Lumacaftor-ivacaftor | ≥2 yr | Homozygous F508del6 | ppFEV1: ↑ 2.6% Exacerbations: ↓ 30% BMI: ↑ 0.1 kg/m2 Sweat chloride: ↓ 20.4 mmol/L |
| Tezacaftor-ivacaftor | ≥6 yr | Homozygous F508del OR F508del heterozygous with a Class IV or Class V mutation | ppFEV1: ↑ 6.8% Exacerbations: ↓ 35% BMI: ↑ 0.06 kg/m2 Sweat chloride: ↓ 10.1 mmol/L |
| Elexacaftor-tezacaftor-ivacaftor | ≥12 yr | At least 1 F508del allele | ppFEV1: ↑ 10%–13.8% Exacerbations: ↓ 63% BMI: ↑ 1.04 kg/m2 Sweat chloride: ↓ 41–45 mmol/L |
BMI, body mass index; ppFEV1, percent predicted forced expiratory volume in 1 second
Mechanism of Action
Ivacaftor functions as a potentiator of the CFTR protein for common gating mutations, allowing for an increase in chloride ion flow.7 Tezacaftor functions as a corrector to facilitate the folding and presentation of the mature CFTR protein to the cell surface, improving CFTR function for the F508del mutation.8 Elexacaftor is also a CFTR corrector that works at an alternate binding site than tezacaftor on the CFTR protein to further facilitate the functionality of the CFTR protein at the cell surface. When used in combination, elexacaftor, tezacaftor, and ivacaftor demonstrated an increase in function of the F508del mutated CFTR protein at the cell surface resulting in increased chloride ion transport.5
Pharmacokinetics
Elexacaftor, tezacaftor, and ivacaftor are absorbed and reach median Tmax concentrations in 6, 3, and 4 hours, respectively.5 Administration with a moderate-fat meal resulted in a 1.9- to 2.5-fold increase in elexacaftor concentration and 2.5- to 4-fold increase in ivacaftor concentration. No clinically significant change in concentration was observed with coadministration of food with tezacaftor.5 The area under the curve exposure of patients 12 to 18 years of age was similar to the pharmacokinetic profile of patients over the age of 18.5
Elexacaftor, tezacaftor, and ivacaftor are all approximately 99% protein bound. Elexacaftor and tezacaftor are primarily bound to albumin while ivacaftor is mostly bound to albumin, 1-acid glycoprotein, and human γ-globulin.5 Mean apparent volumes of distribution for elexacaftor, tezacaftor, and ivacaftor are 53.7 L, 82 L, and 293 L, respectively.
Metabolism is carried out primarily through the CYP3A4/5 system for all 3 drug components. Active metabolites are produced from each of the parent drugs with at least similar potency seen in the elexa-caftor and tezacaftor metabolites while the ivacaftor metabolite maintains approximately 1/6th the potency of ivacaftor.5 Primary metabolism through the CYP enzyme system creates the potential for many drug-drug interactions. Exposure to elexacaftor in patients with hepatic impairment has not been studied. Therefore, it is recommended to avoid use of Trikafta in patients with severe hepatic impairment (Child-Pugh Class C), and if used in moderate hepatic impairment (Child-Pugh Class B), Trikafta should be dose adjusted and used with caution.5 There is lack of data surrounding specific dosage recommendations for patients with moderate hepatic impairment and patients who have liver disease but do not meet Child-Pugh criteria.
The primary route of excretion is through the feces for elexacaftor (87.3%), tezacaftor (72%), and ivacaftor (87.8%) with renal elimination being the secondary path of excretion. Elexacaftor alone or in combination has not been studied in end stage renal disease or severe renal impairment. Population pharmacokinetic analysis has shown that clearance of elexacaftor and tezacaftor was similar in patients with mild to moderate renal dysfunction when compared with patients with normal renal function.5 Although elexacaftor and tezacaftor are administered once a day, ivacaftor requires twice daily dosing.
Clinical Experience in Children
Two randomized controlled trials have evaluated the safety and efficacy of elexacaftor-tezacaftor-ivacaftor in 2 distinct subsets of patients. The first trial included patients 12 years of age and older who were homozygous for the F508del mutation. The trial included 30 total patients aged 12 to 17 years old, accounting for 28% of the total participants. At baseline, mean percent predicted forced expiratory volume in 1 second (ppFEV1) was 60.9%, mean body mass index (BMI) was 21.82 kg/m2, and mean sweat chloride concentration was 90.7 mmol/L. Triple therapy (elexacaftor 200 mg once daily, tezacaftor 100 mg once daily, and ivacaftor 150 mg every 12 hours) was studied against the current recommended treatment (dual therapy tezacaftor 100 mg once daily and ivacaftor 150 mg every 12 hours) for patients with CF homozygous for F508del mutations.9 The trial consisted of a 4-week run-in phase where all patients received tezacaftor-ivacaftor so that study comparisons were between triple versus dual therapy. Following the 4-week run-in phase, the primary outcome of change in baseline ppFEV1 was assessed. It was concluded that treatment with elexacaftor-tezacaftor-ivacaftor resulted in an absolute improvement in ppFEV1 of 10% compared with dual therapy with tezacaftor-ivacaftor. Quality of life was evaluated using the Cystic Fibrosis Questionnaire-Revised and resulted in an increase of 17.4 points with elexacaftor-tezacaftor-ivacaftor versus tezacaftor-ivacaftor. This met the minimum 4-point improvement threshold to meet clinical significance. Additionally, there was a 45-mmol/L decrease in sweat chloride concentration from baseline, which brought the mean sweat chloride concentration (48 mmol/L) under the diagnostic level for CF (60 mmol/L).9
The second study assessed the safety and efficacy of elexacaftor 200 mg once daily, tezacaftor 100 mg once daily, and ivacaftor 150 mg every 12 hours against placebo in patients heterozygous for the F508del mutation and a minimal function (Class I or Class II) genotype. One hundred sixteen total patients aged 12 to 17 years old met inclusion criteria, which consisted of 28.8% of the total trial participants. At baseline, mean ppFEV1 was 60.9%, mean BMI was 21.4 kg/m2, and mean sweat chloride concentration was 102.6 mmol/L. The trial duration was 24 weeks, and the primary endpoint of baseline change in ppFEV1 was assessed at 4 weeks. It resulted in a mean treatment difference in absolute ppFEV1 of 13.8%, which was sustained through week 24 with a treatment difference of 14.3% compared with placebo.10 The reduction of sweat chloride from baseline was 41.8 mmol/L (compared with 0.4 mmol/L change from the placebo), resulting in an average sweat chloride concentration of 57.9 mmol/L in the treatment arm.10 The annualized rate of pulmonary exacerbations was 63% lower in the treatment group compared with placebo. Additionally, Cystic Fibrosis Questionnaire-Revised scores increased by 20.1 points, which substantially exceeds the clinically significant threshold of 4 points. BMI also improved by 1.04 kg/m2 over the 24-week period. These secondary outcomes demonstrate clinically significant improvements outside of lung function (ppFEV1) alone and show great promise to increase quality of life relative to placebo.10
There is an ongoing phase 3 study evaluating the safety, efficacy, and tolerability of elexacaftor-tezacaftor-ivacaftor in patients 6 to 11 years old. The study is expected to be completed in early 2020 (NCT03691779). Although elexacaftor-tezacaftor-ivacaftor has not been studied in patients heterozygous for the F508del mutation who are already eligible for tezacaftor-ivacaftor, the FDA approval includes this patient population. An ongoing clinical trial (NCT04058353) will evaluate potential benefit of triple therapy for this population. A phase 2 study (NCT03911713) is evaluating the efficacy of deuterated ivacaftor. Deuteration could provide greater ivacaftor stability in metabolism, allowing for once a day dosing.
Dosing and Administration
Elexacaftor-tezacaftor-ivacaftor is supplied as a fixed-dose combination tablet of elexacaftor 100 mg, tezacaftor 50 mg, and ivacaftor 75 mg copackaged with ivacaftor 150-mg tablets. Adults and children over the age of 12 should administer 2 fixed-dose combination tablets each morning with a fat-containing meal. The evening dose should be separated by approximately 12 hours from morning administration and consists of 1 ivacaftor 150-mg tablet taken with a fat-containing meal or snack. This medication is recommended to be taken with fat-containing foods, and pancreatic enzymes for those who are pancreatic insufficient, to maximize efficacy; the specific quantity of fat needed per dose is unclear. Fat-containing foods include nuts, whole milk, cheeses, meats, and preparations with oils or butter.5 Ivacaftor and tezacaftor can be crushed for administration, but at the time of this manuscript, data were unavailable on stability of elexacaftor when crushed.
If 6 hours or less have gone by since the scheduled dose, it is recommended to take the missed morning dose as soon as possible and continue the normal dosing schedule. If more than 6 hours have passed since the morning dose, it is recommended that the patient take the missed morning dose as soon as possible and skip the evening dose, resuming the regular dosing schedule the next morning. If more than 6 hours have passed since the evening dose, the patient should not take the missed dose and resume the normal dosing schedule beginning the next morning. All patient and caretakers should be reminded not to double up on doses in the event of a missed dose.5
Clinical Considerations
Adverse Effects. The most common side effects observed in a placebo-controlled phase 3 trial of elexacaftor-tezacaftor-ivacaftor were headache, diarrhea, upper respiratory tract infection, creatine phosphoki-nase elevation, and rash. Adverse effect profiles in this study were similar between treatment and placebo trial groups with a slightly lower incidence in the elexacaftor-tezacaftor-ivacaftor group.5,8
Rash was more common in the treatment group and occurred in 10% of study participants. The rash may be more common in women on hormonal contraception. Clinically, the rash may present as localized or diffuse and can appear as papular or may resemble hives. It can be accompanied by pruritis that can be managed with diphenhydramine. In postmarketing clinical practice, many people who have developed a rash have not discontinued the drug and have seen resolution of the rash within 1 to 2 weeks.
Creatine kinase elevations were often associated with exercise, but no one discontinued therapy due to this adverse effect. Two patients in the placebo group withdrew due to adverse effects. One was due to a rash, and the other was due to developing portal hypertension in a patient with underlying cirrhosis.10
When compared with tezacaftor-ivacaftor in patients homozygous for F508del mutations, elexacaftor-tezacaftor-ivacaftor showed no severe adverse events and no effects that led to discontinuation. Adverse effects were similar between both trial groups and most resolved by the end of the 4-week study.9
Elevated liver enzymes were observed in the safety and efficacy studies of elexacaftor-tezacaftor-iva-caftor.9,10 For patients initiating therapy with elexacaftor-tezacaftor-ivacaftor, it is recommended that alanine ami-notransferase (ALT), aspartate aminotransferase (AST), and bilirubin be checked at baseline, every 3 months for the first year of treatment, then yearly thereafter. It is recommended that treatment should be paused if AST or ALT reach >5 times the upper limit of normal or if AST or ALT reach >3 times the upper limit of normal with bilirubin >2 times upper limit of normal. Once liver function test abnormalities resolve, risk versus benefit must be assessed before treatment resumes.5
Because of lens opacities seen in juvenile rats treated with ivacaftor, children starting any ivacaftor-based regimen for the first time should be screened with an ophthalmology examination before starting therapy and in the first year of therapy. Those who have already been on ivacaftor-based therapy who have already completed these screening requirements do not need to be re-screened when starting elexacaftor-tezacaftor-ivacaftor.
Drug Interactions. As mentioned previously, elexacaftor, tezacaftor, and ivacaftor are extensively metabolized by the cytochrome P450 system with ivacaftor being the most sensitive substrate of CYP3A.5 Concomitant use of known weak to moderate inducers or inhibitors with elexacaftor-tezacaftor-ivacaftor warrants caution. Coadministration with strong CYP3A inducers such as rifampin, phenytoin, and carbamaze-pine are not recommended. When coadministered with the strong CYP3A inhibitor itraconazole, concentrations of elexacaftor, tezacaftor, and ivacaftor increased 2.8-fold, 4.5-fold, and 15.6-fold, respectively; therefore, it is recommended to administer a reduced dose of elexacaftor-tezacaftor-ivacaftor if used in conjunction with moderate or strong CYP3A inhibitors; the interaction is described in Table 2.5 Additional drug-drug interactions may include P-glycoprotein substrates such as digoxin, CYP2C9 substrates such as warfarin, and OATP1B1/3 substrates such as statins and insulin secretagogues.5 Due to the unique pre-packaging of elexacaftor-tezacaftor-ivacaftor, it is important to clearly educate the patient and/or caregiver on administration and monitoring parameters in such cases where dose adjustments are warranted. A summary of significant interactions is available in Table 2.
Table 2.
Elexacaftor-Tezacaftor-Ivacaftor Drug Interactions
| Concomitant Drugs | Mechanism | Expected Effect; Recommendations |
|---|---|---|
| Rifampin, rifabutin, phenytoin, St. John's wort, carbamazepine, phenobarbital | Strong CYP3A inducers | Decreased concentration of elexacaftor-tezacaftor-ivacaftor; concomitant use not recommended |
| Ketoconazole, posaconazole, itraconazole, voriconazole, clarithromycin, telithromycin | Strong CYP3A inhibitors | Increased concentration of elexacaftor-tezacaftor-ivacaftor; dose of elexacaftor-tezacaftor-ivacaftor should be reduced to 2 elexacaftor-tezacaftor-ivacaftor tablets twice weekly (dosed 3 to 4 days apart) and ivacaftor evening dose should not be taken |
| Fluconazole, erythromycin | Moderate CYP3A inhibitors | Increased concentration of elexacaftor-tezacaftor-ivacaftor; dose of elexacaftor-tezacaftor-ivacaftor should be reduced to 2 elexacaftor/tezacaftor/ivacaftor tablets every other day and 1 ivacaftor tablet every other day on alternating days |
| Warfarin | CYP2C9 substrate | Warfarin concentration affected; concomitant use not recommended |
| Digoxin, cyclosporin, sirolimus, tacrolimus, everolimus | P-glycoprotein substrates | Use with caution and monitor serum concentrations of substrates |
| Statins, glyburide, nateglinide, repaglinide | OATP1B1, OATP1B3 substrates | Use with caution and monitor substrates |
CYP, cytochrome P450
Other Considerations. It is unknown what impact elexacaftor-tezacaftor-ivacaftor will have on the need for other maintenance medications. Patients should be advised to continue all previous therapies, even if they begin to feel dramatically better. A study is planned to determine if certain inhaled therapies can be decreased for those who respond favorably to therapy.
At approximately $24,000 per month, elexacaftor-tezacaftor-ivacaftor is a costly therapy, similar to the other CFTR modulators. Patient assistance is available in the United States for patients who are uninsured or underinsured. There are also copay assistance programs and grant programs (e.g., Healthwell Foundation) to assist in paying prescription copayments.
Summary
The approval of the first triple-combination CFTR modulator for approximately 90% of patients with CF is a monumental advancement in the treatment of CF. With significant improvements in markers of lung function (ppFEV1) and quality of life, elexacaftor-tezacaftor-ivacaftor is a crucial therapy for all eligible patients. There remains a need for expanded indications for younger children and alternative dosage forms for those who cannot swallow tablets. Data analysis from ongoing extensions of both safety and efficacy trials concerning treatment of homozygous and heterozygous for F508del mutations are critical to evaluate long-term outcomes.9,10 The CF Foundation remains committed to discovering more inclusive and effective treatment options ultimately leading to a cure for all patients affected by CF.
ABBREVIATIONS
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- BMI
body mass index
- CF
cystic fibrosis
- CFTR
cystic fibrosis transmembrane conductance regulator
- CYP
cytochrome P450 isoenzyme system
- ppFEV1
percent predicted forced expiratory volume in 1 second
Footnotes
Disclosure The authors declare no conflicts or financial interest in any product or service mentioned in the manuscript, including grants, equipment, medications, employment, gifts, and honoraria.
REFERENCES
- 1.Ren CL, Morgan RL, Oermann C et al. Cystic fibrosis pulmonary guidelines: use of CFTR modulator therapy in patients with cystic fibrosis. Ann Am Thorac Soc. 2018;15(3):271–280. doi: 10.1513/AnnalsATS.201707-539OT. [DOI] [PubMed] [Google Scholar]
- 2.Elborn JS. Cystic fibrosis. Lancet. 2016;388(10059):2519–2531. doi: 10.1016/S0140-6736(16)00576-6. [DOI] [PubMed] [Google Scholar]
- 3.Burgener EB, Moss RB. Cystic fibrosis conductance regulator modulators: precision medicine in cystic fibrosis. Curr Opin Pulm Med. 2018;30(3):372–377. doi: 10.1097/MOP.0000000000000627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cystic Fibrosis Foundation Bethesda, MD: 2019. Research milestones. https://www.cff.org/Research/About-Our-Research/Research-Milestones/ Accessed January 19, 2020. [Google Scholar]
- 5.Trikafta [package insert] Boston, MA: Vertex Pharmaceuticals Incorporated; 2019. [Google Scholar]
- 6.Orkambi [package insert] Boston, MA: Vertex Pharmaceuticals Incorporated; 2015. [Google Scholar]
- 7.Kalydeco [package insert] Cambridge, MA: Vertex Pharmaceuticals Incorporated; 2012. [Google Scholar]
- 8.Symdeko [package insert] Boston, MA: Vertex Pharmaceuticals Incorporated; 2018. [Google Scholar]
- 9.Heijerman HG, Mckone EF, Downey DG et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial. Lancet. 2019;394(10212):1940–1948. doi: 10.1016/S0140-6736(19)32597-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Middleton PG, Mall MA, Drevinek P et al. Elexacaftor-tezacaftor-ivacaftor for cystic fibrosis with a single phe-508del allele. N Engl J Med. 2019;381(19):1809–1819. doi: 10.1056/NEJMoa1908639. [DOI] [PMC free article] [PubMed] [Google Scholar]