

Abstract
The causative agent of severe acute respiratory syndrome (SARS) has been identified as a novel coronavirus, SARS-CoV. The main proteinase of SARS-CoV, 3CLpro, is an attractive target for therapeutics against SARS owing to its fundamental role in viral replication. We sought to identify novel inhibitors of 3CLpro to advance the development of appropriate therapies in the treatment of SARS. 3CLpro was cloned, expressed, and purified from the Tor2 isolate. A quenched fluorescence resonance energy transfer assay was developed for 3CLpro to screen the proteinase against 50,000 drug-like small molecules on a fully automated system. The primary screen identified 572 hits; through a series of virtual and experimental filters, this number was reduced to five novel small molecules that show potent inhibitory activity (IC50 = 0.5–7 μM) toward SARS-CoV 3CLpro.
Introduction
The first wave of Severe Acute Respiratory Syndrome (SARS) was successfully contained by the summer of 2003 [1], but not until close to 8500 people worldwide were infected, and over 900 had died [2]. Upon the emergence of this syndrome, international response was quick to identify and characterize its causative agent as a novel coronavirus, SARS-CoV 3, 4, 5, 6, 7. The reemergence of SARS in the Guangdong province of China in December 2003 [8] and the spring of 2004 [9], while not necessarily indicative of another global outbreak, illustrates the need to continue efforts to study this virus and develop appropriate therapeutics for its treatment.
SARS-CoV is an enveloped, positive-stranded RNA virus whose genome is predominated by two open reading frames that are connected by a ribosomal frameshift site and that encode the two replicase proteins, pp1a and pp1ab 10, 11. These polyproteins are cleaved by the main proteinase 3CLpro [11] (also called Mpro 12, 13) in the first step of the formation of the crucial replication-transcription complex. The activity of 3CLpro, so named for its similarity to 3C proteinases of Picornaviridae [14], is an attractive target for the development of therapeutics against SARS due to its fundamental role in viral replication. Homology modeling with other 3CLpro's 12, 15, X-ray crystallography [13], and a recent mutagenesis study [16] have identified 3CLpro as a cysteine protease with a Cys-His catalytic dyad in the active site. Structure-based sequence alignments of pp1a and pp1ab of SARS-CoV with the equivalent polyproteins from human coronavirus 229E (HCoV), porcine transmissible gastroenteritis virus (TGEV), bovine coronavirus (BCoV), and avian infectious bronchitis virus (IBV) have identified the consensus sequence for proteolysis as (S, T, V, P, A)-(L, I, V, F, M)-Q/(A, S, G, N, C) [12]. In vitro studies have confirmed the ability of 3CLpro to process sites of this type, and that the proteinase has the highest specificity for the cleavage sites flanking 3CLpro in the pp1a/pp1ab polyprotein 11, 17.
A structural model of 3CLpro based on sequence homology with TGEV [12] as well as the solved crystal structure [13] have both been used in a number of studies to dock substrate mimics 12, 13, 18 and for virtual screening of collections of synthetic compounds, natural products, and approved antiviral therapies to evaluate their ability to inhibit 3CLpro 15, 19, 20, 21, 22. Compounds identified as potential inhibitors of 3CLpro from these studies include the HIV-1 protease inhibitor L-700,417 [15], the reverse transcriptase inhibitors calanolide A and nevirapine [20], glycovir, an α-glucosidase inhibitor [20], sabadinine, a natural product [21], and the general antiviral ribavirin [20] . Ribavirin has been shown to exhibit anti-SARS-CoV activity in vitro, but at concentrations that are cytotoxic [23]. At the onset of the initial SARS outbreak, this compound was used as a first-line defense both as a monotherapy [24] and in combination with corticosteroids [25] or Kaletra (an approved HIV protease inhibitor) [24]; however, it is now generally believed that ribavirin is not an effective treatment for SARS (26, 27 and references therein). At the time of writing, it is not known whether the other compounds identified through virtual means have yet been tested with 3CLpro or SARS-CoV to evaluate their actual inhibitory capabilities.
Several in vitro screens have also been recently reported that have assessed the ability of existing proteinase inhibitors to inhibit replication of SARS-CoV and, in some cases, 3CLpro specifically. The HIV-1 protease inhibitors indinavir, saquinavir, ritonavir, lopinavir, TYA5, TYB5, and KNI-272 were shown to be ineffective at inhibiting replication of SARS-CoV in vitro 23, 28. Another HIV-1 protease inhibitor, nelfinavir, was shown to inhibit SARS-CoV replication with an EC50 of 48 nM [28], yet it has also been reported that complete inhibition of viral replication was not seen with compound concentrations up to 10 μM [23]. Another in vitro screen that similarly tested a library of 500 protease inhibitors resulted in only one compound that inhibited 3CLpro; this molecule had been developed as a transition-state analog inhibitor for HIV-1 protease and had a Ki of 0.6 μM with 3CLpro [29].
Although a number of existing drugs that could potentially act as inhibitors of 3CLpro were identified through virtual screening, other than ribavirin, it is not known how active these compounds actually are toward the protease. Additionally, in vitro screening campaigns illustrate that out of over 500 existing protease inhibitors, only two were identified as active toward 3CLpro. These findings illustrate the need to look toward the development of innovative inhibitors for this protease. One such study has recently identified unique keto-glutamine analogs as inhibitors of 3CLpro with IC50's in the low micromolar range (J.C. Vederas, submitted). In an effort to advance this initiative, we sought to identify novel small molecules that specifically target 3CLpro. In this paper, we describe the cloning, expression, and purification of 3CLpro from the SARS-CoV Tor2 isolate, and subsequent screening campaign against a library of 50,000 small molecules. We herein report the finding of five novel inhibitors of 3CLpro.
Results and Discussion
In order to screen SARS-CoV 3CLpro against a library of 50,000 compounds, it was necessary to use an activity assay for the proteinase that could be adapted to an automated system. Ideally, such an assay would (1) work over a time scale of several minutes at most to facilitate the rapid evaluation of thousands of potential inhibitors; (2) be homogeneous for ease of automation; and (3) be sensitive to minimize background signal from the presence of the compounds tested. Quenched fluorescence resonance energy transfer (FRET) assays have become a common tool to monitor proteinase activity [30] and fulfill the necessary criteria for use in an efficient and robust high-throughput screening (HTS) campaign. The basis for such an assay is the modification of a peptide substrate to include a fluorescent label and quencher on opposing sides of the proteinase cleavage site (Figure 1) . Using this substrate, the activity of a proteinase can be measured directly without the need for additional steps to purify and/or characterize the products. This type of assay is also beneficial as it permits enzymatic reactions to be monitored in real time to obtain accurate reaction rates. This aspect of the quenched-FRET assay is particularly attractive since, in evaluating the activity of a proteinase in terms of a rate, the potential for false negatives to occur in the screen due to the presence of inherent fluorescence from the compounds being tested is reduced.
Figure 1.

The Quenched-FRET Assay Used to Monitor 3CLpro Activity
(A) The substrate is a 9 amino acid peptide labeled with 2-aminobenzoyl (Abz) and a nitro-tyrosine on either side of the scissile peptide bond (dashed) to act as donor and acceptor, respectively, for FRET. In the intact substrate, excited state energy of Abz is transferred to the nitro-tyrosine via FRET and fluorescence is quenched.
(B) Once the substrate is cleaved by 3CLpro, the donor and acceptor are spatially separated; subsequent fluorescence from the Abz functionality is not quenched and can be detected.
Two fluorogenic peptide substrates were synthesized to develop a continuous assay of 3CLpro proteolytic activity based on FRET. The peptides were designed principally on the sequences flanking 3CLpro in the polyprotein and solubility considerations. A peptide incorporating the anthranilate-nitrotyrosine donor-acceptor pair (Abz-SVTLQSG-Tyr(NO2)R) (Figure 1) was over an order of magnitude more sensitive in FRET-based assays than the equivalent Edans-Dabcyl peptide (data not shown) and was the preferred substrate for HTS. In the analysis of kinetic data using the Abz-Tyr(NO2) substrate, the primary data were corrected for inner filter effect [31]. This adjustment was essential as it was observed that at even relatively low concentrations of substrate (2 μM), the fluorescence of the cleaved product was significantly quenched by the nitrotyrosine moiety of the intact substrate.
The Km for the Abz-Tyr(NO2) substrate with 3CLpro was 820 ± 130 μM (Figure 2A) . This value is comparable to those seen recently for nonlabeled 11-mer peptides that also mimic the cleavage sites of the natural substrates of 3CLpro (0.286–1.94 mM) [17], implying that the labels incorporated for FRET were not detrimental to the association between enzyme and substrate. The calculated values of the apparent kcat and kcat/Km for the labeled substrate were 1.01 ± 0.09 min−1 and 1.2 ± 0.2 mM−1 min−1 respectively, and were also similar to those reported for the 11-mer peptides (0.847–12.2 min−1 and 0.202–10.6 mM−1min−1) [17]. These kinetic parameters are reported as apparent values since it has been recently shown 17, 32 that at the concentration of 3CLpro used in this study (1 μM) much of the enzyme is thought to be monomeric, whereas it is the dimeric form of the enzyme that is believed to be catalytically competent.
Figure 2.

Activity of 3CLpro with the Fluorogenic Peptide Substrate
(A) Michaelis-Menton plot to determine Km and the observed kcat.
(B) A plot of observed assay rates containing 100 μM substrate and varying concentrations of 3CLpro demonstrates assay linearity with respect to proteinase concentration.
For the purposes of screening, it is desirable to use the lowest concentration of substrate that yields a reliable, reproducible signal. It was found that assays using 100 μM of the fluorogenic peptide gave a strong signal that was linear with varying 3CLpro concentration (Figure 2B). This enabled us to screen at substrate concentrations well below Km, which is advantageous for identifying inhibitors that compete with substrate for the enzyme active site [33] and are therefore more likely to be specific toward 3CLpro.
The compound library used to screen 3CLpro contained 50,000 small molecules with an average molecular mass of 325 g/mol. This particular collection was screened because of its high quality, diversity, drug-likeness [34], resupply availability, and past success with other targets [35]. The campaign was run in duplicate in 384-well microplate format on a fully automated system.
The statistical parameter Z′, which is described as a measure of the quality of an HTS campaign [36], is defined as
| 1 − Z′ = 3σc+ + 3σc−|μc+ − μc−| | 1 |
where σc+, σc−, μc+, and μc− are the standard deviations (σ) and averages (μ) of the high (c+) and low (c−) controls. This value reflects both the error associated with the controls of a screen, as well as the size of the “hit window” (Figure 3) . A Z′ of 0.5 or greater is indicative of a quality screen with a well-defined hit window [36]. The Z′ for the primary screen of SARS-CoV 3CLpro was 0.60 for each of the two replicates indicating that the assay used to detect activity of 3CLpro was robust and amenable to HTS. The replicate plot of residual enzyme activity with each of the tested library compounds (Figure 4) also illustrates the quality of the screen in terms of the reproducibility between the duplicate sets of data. This figure also indicates that 3CLpro appears to have been inhibited to some degree by a large number of compounds. The cutoff to determine the hit threshold was set to 50% residual enzyme activity in order to focus on the most potent inhibitors.
Figure 3.
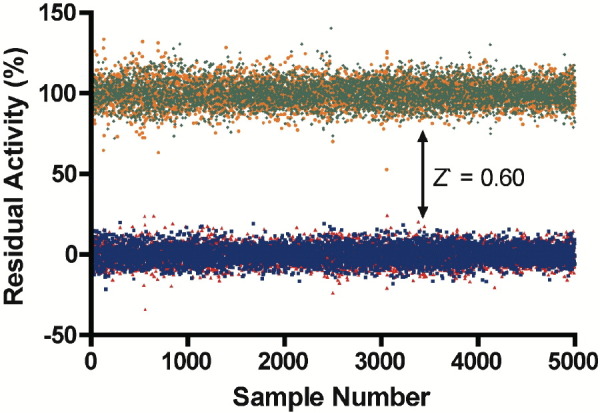
Plot of the Residual Activity of the Controls from the Screen
High (Replicate 1, orange circles; Replicate 2, green diamonds) and low (Replicate 1, red triangles; Replicate 2, blue squares) controls are clustered around 100% and 0% residual activity, respectively. The Z′ of 0.60 is reflected in the well-defined hit window.
Figure 4.
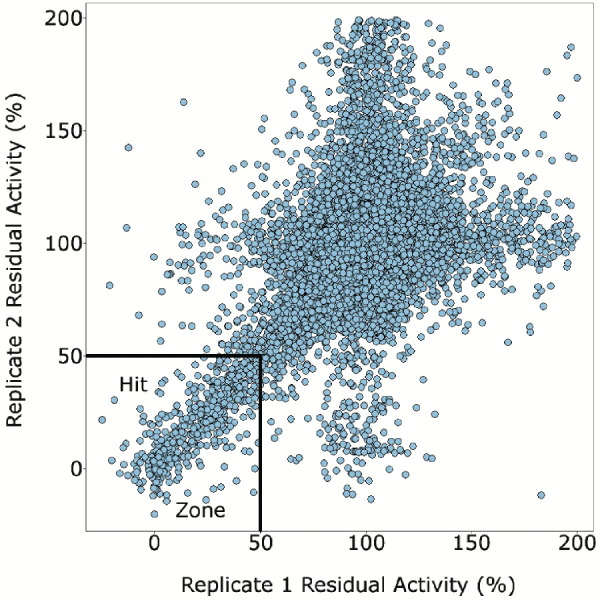
Replicate Plot for the Primary Screen of SARS-CoV 3CLpro
The outlined hit zone was defined as 50% residual enzyme activity.
Five hundred and seventy-two hits (1.1% hit rate) were identified from the primary screen of 3CLpro. For the preliminary analyses of these hits, we opted to restrict the number of compounds entering secondary screening by a series of virtual and experimental filters. The hits were first classified by recursive partitioning into 126 groups based on similarities between molecular structure and activity in the primary screen 37, 38. Within each of these groups, the compound that strongly inhibited 3CLpro was chosen as a representative candidate for further study in secondary analyses. The dose-response curves of the 126 representative compounds were determined using standard assay conditions (100 μM substrate, 1 μM 3CLpro) and 90 nM–100 μM of tested compound. The dose-response relationship of 72 compounds resulted in the typical sigmoidal semilogarithmic curve associated with desirable inhibitors; the 54 compounds that did not result in this typical curve were filtered out.
To eliminate nonspecific inhibitors, the effect of the remaining 72 compounds on 3CLpro activity in the presence of bovine serum albumin (BSA) was determined. If the degree of inhibition of 3CLpro by a compound is decreased in the presence of BSA, inhibition is not likely to be targeted exclusively toward 3CLpro. These so-called “promiscuous” inhibitors likely form aggregates in solution that nonspecifically absorb or adsorb the target enzyme [39]. Only three compounds showed any marked reduction in enzyme inhibition in the presence of BSA, with the greatest effect being a decrease of only 20%; these compounds were removed from the prospective inhibitor pool.
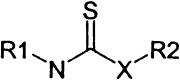
The remaining 69 potential candidates were predominated by compounds with at least one potentially reactive center (Table 1) . The thiol side chain of the active site cysteine of 3CLpro is necessarily a good nucleophile and therefore has the capacity to combine with a variety of electrophilic functionalities to form covalent adducts. This reaction is the basis for several classes of existing peptidic inhibitors of cysteine proteases which include α-β-unsaturated ketones, esters, and amides 40, 41 (Table 1, 1–4) and nitriles [42] (Table 1, 15). In addition, N-substituted maleimides (Table 1, 14) have long been recognized as being reactive toward thiols in general, and cysteine proteases in particular [43]. The two identified sulfur containing compounds (Table 1, 13) are also capable of forming disulfide linkages with the catalytic thiol of 3CLpro to inactivate the enzyme [44] The remaining carbonyl functionalities and corresponding nitrogen- and sulfur-containing analogs listed in Table 1, 5–12, may also have enough electrophilic character to promote the nucleophilic attack of the active site thiol of 3CLpro. Due to the potential inherent reactivity of the 69 candidate inhibitors, we sought to identify and eliminate those compounds whose high reactivity rendered them nonspecific toward the active site of 3CLpro by including 1,4-dithio-D,L-threitol (DTT) in the inhibition reaction. A reduction in the inhibition of 3CLpro by any candidate compound in the presence of DTT suggests that such a compound would probably react nonspecifically with any biologically available thiol functionality 40, 41. Although this renders such compounds as unlikely drug candidates, they may be useful in studying the active site architecture of 3CLpro. Of the remaining 69 candidate inhibitors, the inhibitory action of 5 of these was not significantly affected by the inclusion of 1 mM DTT; these were selected for further characterization.
Table 1.
Some of the Functional Groups of the 69 Filtered Hits from the Screening Campaign that Are Potentially Reactive with the Catalytic Thiol in the Active Site of 3CLpro
| General Structurea | Group | X | Y | Z | No. of Hits in Groupb |
|---|---|---|---|---|---|
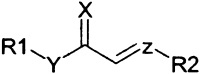 |
1 | O | |||
| 2 | O | CH2 | CH | 10 | |
| 3 | O | O | CH | 5 | |
| 4 | O | N | CH | 9 | |
| 5 | S | N | CH | 1 | |
| 6 | S | N | N | 1 | |
 |
7 | CBr2 | — | — | 1 |
| 8 | O | — | — | 5 | |
| 9 | N | — | — | 12 | |
| 10 | S | — | — | 5 | |
 |
11 | S | — | — | 4 |
| 12 | N | — | — | 1 | |
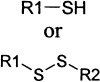 |
13 | — | — | — | 2 |
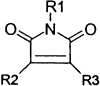 |
14 | — | — | — | 6 |
| 15 | — | — | — | 16 | |
R1, R2, and R3 are a variety of aromatic and aliphatic substituents.
The sum of the number of compounds in each group is greater than 69 as several compounds contained more than one of the listed functionalities.
The dose-response curves for these five compounds with 3CLpro yielded IC50 values of 0.5–7 μM (Figure 5) . To evaluate the selectivity of each compound, we investigated their ability to inhibit four other proteinases with varying structural and mechanistic relatedness to SARS-CoV 3CLpro. The Hepatitis A virus (HAV) 3Cpro, like all picornaviral 3Cpros, is similar in structure, mechanism, and substrate specificity to 3CLpro 11, 14, 45. The Hepatitis C Nonstructural 3 proteinase (NS3pro) and chymotrypsin are serine proteases with the same two β-barrel fold as 3CLpro 13, 46. Finally, papain is a paradigm cysteine proteinase with an active site Cys/His/Asp catalytic triad whose structural fold does not resemble that shared by chymotrypsin, NS3pro, 3CLpro, and 3Cpro. It was difficult in some cases to obtain a full dose-response curve for every inhibitor with each of the proteinases due to interfering compound fluorescence and/or compound insolubility under each of the assay conditions. In such cases, if the lack of data at higher compound concentrations precluded interpolation of a reliable IC50 value, the IC50 is reported as a lower limit (Table 2) . Alternatively, those compounds for which enzyme inhibition was not seen at the highest concentration tested, are indicated accordingly. It is important to note that in the absence of full mechanistic analyses, the IC50 values are intended to reveal profound differences in potency to provide insight into the selectivity of the inhibitors, and to aid in their prioritization as leads for continuing studies.
Figure 5.

Dose-Response Curves for the Five Secondary Hits from the Screening Campaign
IC50 values were extracted from the curves using nonlinear regression analysis (see Experimental Procedures). IC50 values for the five secondary hits were 0.5 ± 0.3 μM (MAC-5576), 4.3 ± 0.5 μM (MAC-8120), 7 ± 2 μM (MAC-13985), 2.6 ± 0.4 μM (MAC-22272), and 7 ± 3 μM (MAC-30731). The corresponding structure of each compound is shown on each dose-response curve.
Table 2.
Summary of IC50 Values of the Five Secondary Hits with SARS-CoV 3CLpro and Four Other Proteases
|
IC50 (μM)a |
||||||
|---|---|---|---|---|---|---|
| Compound | Structure | 3CLpro | HAV 3Cpro | NS3pro | Chymb | Papain |
| MAC-5576 | 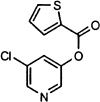 |
0.5 ± 0.3 | 0.54 ± 0.09† | N.I. (500)c | 13 ± 5 | N.I. (500) |
| MAC-8120 | 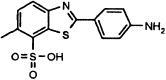 |
4.3 ± 0.5 | N.I. (5) | N.I. (5) | N.I. (500) | N.I. (500) |
| MAC-13985 | 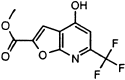 |
7 ± 2 | N.I. (5) | N.I. (5) | N.I. (500) | N.I. (500) |
| MAC-22272 | 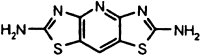 |
2.6 ± 0.4 | 0.9 ± 0.1 | >500d | 29 ± 12 | 18 ± 9 |
| MAC-30731 | 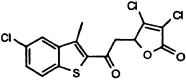 |
7 ± 3 | 54 ± 16 | 71 ± 14 | >800 | N.I. (500) |
To calculate IC50 values, dose-response data were fit to a 3-parameter equation (Equation 2), excluding †, in which a 4-parameter equation (Equation 3) was used (see Experimental Procedures).
Bovine pancreas chymotrypsin.
No inhibition seen at the concentrations of compound tested; highest concentration tested in brackets (in μM).
Protease inhibition was seen, but insufficient data were obtained to calculate a reliable IC50 value.
Compound MAC-22272 showed the least selectivity with respect to 3CLpro (IC50 = 2.6 μM), as it was the only inhibitor capable of inactivating all of the proteinases studied, although the response with NS3pro (IC50 > 500 μM) was significantly lower than with the other enzymes. MAC-5576 showed some selectivity toward those proteinases with a chymotrypsin-like fold, as it was seen to inhibit 3CLpro (IC50 = 0.5 μM), HAV 3Cpro (IC50 = 0.5 μM), and chymotrypsin (IC50 = 13 μM), although no effect was seen with NS3pro, which also shares this structural motif. Papain, which lacks this fold, was unaffected by MAC-5576. An important difference between 3CLpro and 3Cpro is that the coronaviral enzyme is only functionally active as a dimer whereas 3Cpro is active as a monomer in vitro. The interaction between the 3CLpro subunits is sufficiently weak that disrupting the dimerization interface has been proposed as a promising target for novel antiviral agents [32]. In view of the relatedness between 3CLpro and HAV 3Cpro, it is therefore likely that of the five secondary hits, those that are also active toward HAV 3Cpro do not interact with 3CLpro at its dimer interface, but at a site that is common to both proteinases.
Considering the mechanistic and structural relationships between 3CLpro and HAV 3Cpro, and to a lesser extent NS3pro, we were interested to learn that some of the compounds showed highest activity against these related enzymes. In addition to inhibiting 3CLpro with an IC50 of 7 μM, compound MAC-30731 was observed to have relatively potent activity against HAV 3Cpro (IC50 = 54 μM) and NS3pro (IC50 = 71 μM), with only weak impact on chymotrypsin (IC50 > 800 μM).
MAC-8120 and MAC-13985 also demonstrated interesting selectivity profiles with good activity against SARS-CoV 3CLpro (IC50 values were 4.3 and 7 μM, respectively) and no detectable inhibition of any of the other proteinases tested. These particular compounds, however, proved to be difficult in the assays of HAV 3Cpro and NS3pro due to background fluorescence, and could not be tested at concentrations greater than 5 μM. Nevertheless, no detectable inhibition was seen at that concentration, indicating that an IC50 for these molecules would necessarily be much higher.
The protease selectivity data, therefore, point to two molecules with apparent selectivity for 3CLpro, MAC-8120 and MAC-13985. These may well be the most promising leads for further mechanistic characterization and optimization efforts toward a protease-based antiviral for SARS-CoV. One molecule, MAC-30371, has an interesting selectivity for the picornaviral-like proteinases and could prove to be a viable lead for this family of proteolytic enzymes.
A search of the literature for any studies involving the five secondary hits showed that at present, none have been identified as proteinase inhibitors. Of particular significance, however, is the finding that MAC-8120 has been identified as an inhibitor of triosephosphate isomerase from Trypanosoma cruzi, T. brucei, human, and yeast (IC50 = 56 μM to 4 mM) [47]. This study examined the inhibition of the isomerase by a series of substituted benzothiazoles, and indicated that MAC-8120 interacted with cysteine residues on the enzyme that were previously shown to be central in dimerization. This interaction, although as yet uncharacterized, was reported to perturb the contact interface of the subunits of the homodimer to inactivate the enzyme. It is therefore quite possible that MAC-8120 associates with the active site cysteine of 3CLpro in a similar manner to disrupt its catalytic activity. The exact mechanism of inhibition of 3CLpro by MAC-8120 and the remaining secondary hits requires further study and characterization in order to evaluate their potential to act as leads for the development of novel therapeutics against SARS-CoV, and potentially, other picornaviral-like proteinases. Additionally, the core structure of these compounds can be chemically modified to improve key physical properties such as potency and solubility.
It is clearly evident that the 3CLpro screening campaign has generated a wealth of data that can be mined for a number of studies. While it was our intention to identify novel small molecules that would lead to the development of therapeutics effective against SARS-CoV, the results generated by the screening campaign can be applied to other types of studies of 3CLpro and related proteinases including the picornaviral 3Cpro. To this end, we have made the complete primary screening data set and corresponding compound structures available for download from the McMaster HTS Lab (http://hts.mcmaster.ca/sars). Interested parties are invited to review the data and mine it for their own studies.
Significance
In response to the initial SARS outbreak of 2003, a concerted effort by the international scientific community identified the causative agent of this infection as a novel coronavirus, SARS-CoV. Characterization of this virus and the development of appropriate therapies soon followed and remains a continuing global effort. The main proteinase of SARS-CoV, 3CLpro, plays a crucial role in viral replication and is therefore an excellent target for therapeutics effective in treating SARS. Our contribution to the anti-SARS initiative was to employ a high-throughput screening strategy to identify small molecules that specifically target 3CLpro in order to generate new therapeutic leads. HTS is an efficient and rapid means of assessing the effect of a large number of compounds on a target of interest; we feel that such campaigns should play an integral part of any initial investigations undertaken to combat newly emerging infectious agents.
A quenched-FRET assay was developed and used to evaluate the activity of 3CLpro in the presence of 50,000 small drug-like molecules; of these compounds, 572 were shown to inhibit 3CLpro. Utilizing a series of filters, this number of compounds was reduced to five potent hits (IC50 = 0.5–7 μM) that represent novel leads. Secondary studies of these leads will investigate their mode of inhibition of 3CLpro and their potential to provide a basis for innovative antiviral therapies.
By virtue of the number of molecules evaluated, this campaign has resulted in an extensive data set. As well as identifying inhibitors of 3CLpro, the primary data from the screen can be utilized in numerous studies, such as identifying structure-activity relationships, by others investigating 3CLpro in particular and other main proteinases in general. For this reason, the authors have made the primary screening data set publicly available in the hope that researchers will find this information useful in future studies.
Experimental Procedures
Chemicals and Reagents
All chemicals were of analytical grade and used without further purification. Restriction enzymes were purchased from New England Biolabs (Pickering, ON, Canada). All peptide substrates and activating peptides were synthesized by the Peptide Synthesis Laboratory, NAPS Unit, University of British Columbia (Vancouver, BC, Canada).
Strains, Media, and Growth
DNA was propagated using Escherichia coli DH5α. The protease was expressed using E. coli GJ1158 [48] transformed with pT7-7 derivatives [49]. E. coli strains were cultured at 37°C and 250 rpm in Luria-Bertani (LB) broth supplemented with the appropriate antibiotics. For expression, E. coli GJ1158 was cultured in LB broth with 100 μg/mL ampicillin.
Construction of Plasmids and Overexpression
DNA was manipulated using standard protocols [50]. A vector was designed to express 3CLpro as an N-terminal, cleavable his-tagged protein. The expression vector was derived from pT7HP20, a derivative of pT7-7 [49] containing an approximately 300 bp XbaI/HindIII fragment from pLEHP20 [51]. A plasmid containing the coding sequence of the SARS-CoV 3CLpro was a generous gift from Genome Sciences Centre (Vancouver, BC, Canada). The gene encoding 3CLpro was amplified by PCR using the following oligonucleotides: SPH (5′-CCCAGCTAGCGGTTTTAGGAAAATGGCATTC-3′) and 3SP (5′-TACGAAGCTTGTCGACTTATTGGAAGGTAACACCAGAGCA-3′). SPH introduces an NheI site (underlined) at the beginning of the gene and 3SP introduces a HindIII site (underlined) and a stop codon (boldface) at the end of the coding sequence. The PCR reaction was performed using the Expand High Fidelity DNA polymerase system (Roche Applied Sciences, Laval, PQ, Canada) according to the manufacturer's instructions using an annealing temperature of 50°C. The resulting amplicon was digested with NheI and HindIII and cloned in pT7HP20 yielding the expression plasmid pT7HSP1. The cloned gene was sequenced using an ABI 373 Strech (Applied Biosystems, Foster City, CA) using Big-Dye terminators to verify its authenticity. The amplified gene and the original clone had a deletion (corresponding to guanine at position 32 of the coding sequence). The mutation was repaired in pT7HSP1 using a convenient BsmI site. Accordingly, the oligonucleotide S3CDM (5′-GAAAATGGCATTCCCGTCAGGCAAAGTTGAAGG-3′) contains the BsmI restriction site (underlined) and the missing nucleotide (boldface). A second PCR reaction was performed using S3CDM and 3SP and Pwo DNA polymerase (Roche Applied Sciences) according to the manufacturer's conditions, using an annealing temperature of 50°C. The resulting amplicon was digested using BsmI and HindIII, and was used to replace the corresponding fragment in pT7HSP1. This yielded pT7HSP2, whose nucleotide sequence was confirmed as described above.
The his-tagged 3CLpro (ht-3CLpro) was expressed in E. coli GJ1158 freshly transformed with pT7HSP2. The cultures were grown to an OD600 of ∼0.5 in LB with low salt before inducing the expression of the protein by addition of 0.3 M NaCl. The cultures were incubated for an additional 12 hr before harvesting.
Protein Purification
All buffers were prepared using water purified on a Barnstead NANOpure UV apparatus to a resistivity of greater than 17 MΩcm. Unless otherwise specified, all manipulations involving the protein were performed at 4°C. Cells from 4 liter of culture were harvested by centrifugation. The cell pellet (approximately 10 g) was washed twice with 500 ml of 20 mM sodium phosphate (pH 8.0), 10% glycerol and then was resuspended in 10 ml of the same buffer, containing 1 mM MgCl2, 1 mM CaCl2, and 0.1 mg/ml DNase I. The cells were disrupted by three successive passages through a French Press (Spectronic Instruments Inc., Rochester, NY) using an operating pressure of 20,000 p.s.i. The cell debris was removed by ultracentrifugation at 37,000 rpm for 90 min in a T1250 rotor (DuPont Instruments, Wilmington, DE). The clear supernatant fluid (raw extract) was decanted.
The raw extract (approximately 20 ml) was loaded onto a glass Econo-Column column (Bio-Rad, Hercules, CA) containing 5 ml of Ni-NTA agarose resin (Qiagen Inc., Mississauga, ON, Canada) preequilibrated with 20 mM sodium phosphate (pH 8.0) and 10% glycerol. The column was then washed by the successive passage of 4 column volumes (CV) each of 20 mM sodium phosphate (pH 8.0); 20 mM sodium phosphate (pH 6.3) and 300 mM NaCl; and 20 mM sodium phosphate (pH 8.0), 20 mM imidazole, and 300 mM NaCl. The column was then washed with 2 CV of 20 mM sodium phosphate (pH 8.0) and 20 mM imidazole. The proteinase was eluted using 2 CV of 20 mM sodium phosphate (pH 8.0) and 150 mM imidazole. The eluted protein was exchanged into 50 mM Tris-HCl (pH 8.0) by performing three rounds of concentration-dilution using a stirred cell concentrator equipped with a YM10 membrane (Amicon, Etobicoke, ON, Canada). The protein solution was concentrated to 16.8 mg/mL and frozen as beads in liquid nitrogen. A yield of 185 mg of ht-3CLpro was obtained from 4 liter of cell culture. Removal of the his-tag did not significantly affect 3CLpro cleavage activity.
SARS-CoV 3CLpro Kinetic Assay
SARS-CoV 3CLpro activity was measured by a quenched fluorescence resonance energy transfer (FRET) assay with the peptide substrate 2-aminobenzoyl-SVTLQSG-Tyr(NO2)-R. The rate of enzyme activity was determined by the increase in fluorescence upon continuous monitoring of reactions in 384-well black microplates (Nalge Nunc International, Rochester, NY) (excitation wavelength = 330 nm, emission wavelength = 420 nm). Total assay volume was 50 μL, and contained 1 μM 3CLpro, 100 μM peptide substrate, 4.2% dimethyl sulfoxide (DMSO) (v/v) (Sigma, Oakville, ON, Canada), and 100 mM potassium phosphate (pH 8.0) (Sigma), unless noted otherwise. Reactions were done at room temperature, and were initiated by the addition of substrate. Steady-state kinetic parameters were evaluated using 16 μM–1 mM substrate and 1 μM 3CLpro; these data were corrected for inner filter effect by the method of Liu et al. [31] with the use of Abz-SVRLQ to calculate correction factors.
Primary Screening
The screen of 3CLpro against 50,000 small molecules was fully automated with the use of a SAGIAN Core System (Beckman Coulter, Inc., Fullerton, CA) equipped with an ORCA arm for labware transportation, a Biomek FX with a 96-channel head for liquid handling, and an Analyst HT (Molecular Devices Corp., Sunnyvale, CA) for fluorescence detection; the entire system was integrated through SAMI (v. 3.5, Beckman Coulter). Reactions were done in duplicate, and contained 10 μM library compounds sourced from Maybridge plc (Cornwall, UK), and peptide and 3CLpro as above. Wells containing high and low controls, where neat DMSO was added instead of library compounds, were included on each microplate; buffer was added to the reaction instead of 3CLpro for low controls. Primary hits were defined as those compounds that reduced the activity of 3CLpro to half of the average residual activity of the high controls. Activity Base (v. 5.0.5, ID Business Solutions Limited, Emeryville, CA), SARgen (v. 1.0, ID Business Solutions Limited), and Spotfire DecisionSite (v. 7.1.1, Spotfire Inc., Somerville, MA) were used for data analysis. Compounds identified as primary hits were grouped by functionality using ChemTree (v. 3.2.1, Golden Helix, Bozeman, MT); a representative compound from each cluster was selected for secondary screening.
Secondary Screening
The dose-response curves of the representative hits from primary screening were determined by 3CLpro activity assays (as described above), in the presence of seven concentrations of inhibitor (0.1–100 μM) and 100 μM substrate. Data was fit to
| v = a1 + IIC50s | 2 |
using GraFit (v. 4.0.10, Erithacus Software Ltd., Surrey, UK) (where v is the background corrected reaction rate, a is the reaction rate in the absence of inhibitor, [I] is the concentration of inhibitor, and s is the slope factor) to calculate IC50 values. Compounds exhibiting sigmoidal semilogarithmic dose-response curves were tested by the described 3CLpro activity assay (with 31.2 μM of tested compound) in the presence and absence of 0.01% (w/v) bovine serum albumin (BSA) (Sigma) and 1 mM 1,4-dithio-D,L-threitol (DTT) (BioShop Canada Inc., Burlington, ON, Canada) in two separate studies. Those compounds whose inhibition of 3CLpro was not affected by the presence of BSA or DTT were classified as secondary hits.
Evaluation of Hit Specificity
The dose-response of the secondary hits was determined with papain (Calbiochem, La Jolla, CA), bovine pancreas chymotrypsin (Calbiochem), Hepatitis A 3C proteinase (HAV 3Cpro) (purified according to [52]) and the Hepatitis C Nonstructural 3 proteinase (NS3pro) (181 amino-terminal portion of the enzyme purified according to [46]) in the presence of five to eleven concentrations of each inactivator (5 nM–500 μM). Activity assays for papain (5 nM) and chymotrypsin (200 pM) were done at room temperature in a total volume of 50 μl in black 384-well microplates (Corning, Corning, NY). Activity assays for HAV 3Cpro (0.5 μM) and NS3pro (0.23 μM) were done in black 96-well microplates in a total volume of 200 μl at 37°C and 30°C, respectively. The substrate for papain and chymotrypsin was a casein derivative extensively labeled with the green-fluorescent BODIPY FL dye (Molecular Probes, Eugene, OR) (50 and 10 nM substrate for papain and chymotrypsin assays, respectively). Substrates for HAV 3Cpro and NS3pro were 10 μM Dabcyl-GLRTQSND(Edans)G and 100 μM Abz-DDIVPCSMSY(NO2)T, respectively. Enzyme activity for all proteases was determined by the increase in fluorescence upon continuous monitoring of reactions using either the Analyst HT (Molecular Devices Corp.) or Victor2 (PerkinElmer, Woodbridge, ON, Canada) fluorescence plate readers. Excitation and emission wavelengths, respectively, for each proteinase assay were 485 and 530 nm (papain and chymotrypsin), 355 and 460 nm (HAV 3Cpro), and 340 and 430 nm (NS3pro). Buffer systems for each protease were as follows: papain, 10 mM MES (pH 6.2), 200 mM NaCl, 2 mM EDTA, and 1 mM DTT; chymotrypsin, 50 mM Tris/HCl (pH 7.8); HAV 3Cpro, 100 mM potassium phosphate (pH 7.5), and 2 mM EDTA; NS3pro, 50 mM HEPES (pH 7.3), 150 mM NaCl, 0.1% Triton X-100, and 1 mM DTT. For the NS3pro activity assay, the enzyme was incubated with 15 μM 4A activating peptide (acetyl-KKKGSVVIVGRIILSGR-NH2) at 30°C for 15 min in reaction buffer, then incubated with inactivator for an additional 15 min before initiation of the reaction with substrate. Dose-response data for these proteases with the secondary hits were fit to Equation 2 above, excluding HAV 3Cpro with MAC-5576, which was fit to
| v = a1 + IIC50s + c | 3 |
(where c is the calculated background) since enzyme activity at the highest concentration of inhibitor did not fall to zero.
Mass Spectrometry
The mass of each secondary hit was confirmed by the McMaster Regional Centre for Mass Spectrometry (Hamilton, ON, Canada) using a Quattro Ultima triple quadrupole mass spectrometer (Waters/Micromass, Manchester, UK) using an electrospray ionization source.
Acknowledgements
This work was supported by Strategic grants from the Natural Sciences and Engineering Research Council of Canada (NSERC) and the Protein Engineering Network of Centres of Excellence of Canada (PENCE). C.H. and P.D.F. are recipients of NSERC postgraduate scholarships. E.D.B. holds a Canada Research Chair in Microbial Biochemistry. Dr. John Hobbs, Debbie Adam, and Dr. Krystyna Piotrowska of the NAPS Unit at the University of British Columbia are thanked for their diligent help in oligonucleotide synthesis, nucleotide sequencing, and peptide synthesis, respectively. Thanks also to Carla Martinez for her assistance in the McMaster HTS Lab and Martin Richer at UBC for purifying NS3pro. Finally, we thank Gerry Wright for thoughtful discussions on the manuscript.
Published: October 15, 2004
References
- 1.World Health Organization (August 14, 2003). Alert, verification and public health management of SARS in the post-outbreak period (http://www.who.int.csr/sars/postoutbreak/en/).
- 2.World Heath Organization (August 15, 2003). Summary table of SARS cases by country, 1 November 2002 to 31 July 2003 (http://www.who.int/csr/sars/country/2003_08_15/en/).
- 3.Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S., Urbani C., Comer J.A., Lim W., Rollin P.E. A novel coronavirus associated with Severe Acute Respiratory Syndrome. N. Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 4.Drosten C., Günther S., Preiser W., van der Werf S., Brodt H.-R., Becker S., Rabenau H., Panning M., Kolesnikova L., Fouchier R.A.M. Identification of a novel coronavirus in patients with Severe Acute Respiratory Syndrome. N. Engl. J. Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 5.Fouchier R.A.M., Kuiken T., Schutten M., van Amerongen G., van Doornum G.J.J., van den Hoogen B.G., Peiris M., Lim W., Stöhr K., Osterhaus A.D.M.E. Koch's postulates fulfilled for SARS virus. Nature. 2003;423:240. doi: 10.1038/423240a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marra M.A., Jones S.J.M., Astell C.R., Holt R.A., Brooks-Wilson A., Butterfield Y.S.N., Khattra J., Asano J.K., Barber S.A., Chan S.Y. The genome sequence of the SARS-associated coronavirus. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 7.Rota P.A., Oberste M.S., Monroe S.S., Nix W.A., Campagnoli R., Icenogle J.P., Peñaranda S., Bankamp B., Maher K., Chen M.-H. Characterization of a novel coronavirus associated with Severe Acute Respiratory Syndrome. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 8.World Health Organization (January 31, 2004). New case of laboratory-confirmed SARS in Guangdong, China, update 5 (http://www.who.int/csr/don/2004_01_31/en/).
- 9.World Health Organization (April 30, 2004). China confirms SARS infection in another previously reported case; summary of cases to date, update 5 (http://www.who.int/csr/don/2004_04_30/en/).
- 10.Thiel V., Herold J., Schelle B., Siddell S.G. Viral replicase gene products suffice for coronavirus discontinuous transcription. J. Virol. 2001;75:6676–6681. doi: 10.1128/JVI.75.14.6676-6681.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thiel V., Ivanov K.A., Putics Á., Hertzig T., Schelle B., Bayer S., Weißbrich B., Snijder E.J., Rabenau H., Doerr H.W. Mechanisms and enzymes involved in SARS coronavirus genome expression. J. Gen. Virol. 2003;84:2305–2315. doi: 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- 12.Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main proteinase (3Clpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 13.Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z., Sun L., Mo L., Ye S., Pang H. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. USA. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ziebuhr J., Snijder E.J., Gorbalenya A.E. Virus-encoded proteinases and proteolytic processing in the Nidovirales. J. Gen. Virol. 2000;81:853–879. doi: 10.1099/0022-1317-81-4-853. [DOI] [PubMed] [Google Scholar]
- 15.Jenwitheesuk E., Samudrala R. Identifying inhibitors of the SARS coronavirus proteinase. Bioorg. Med. Chem. Lett. 2003;13:3989–3992. doi: 10.1016/j.bmcl.2003.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang C., Wei P., Fan K., Liu Y., Lai L. 3C-like proteinase from SARS coronavirus catalyzes substrate hydrolysis by a general base mechanism. Biochemistry. 2004;43:4568–4574. doi: 10.1021/bi036022q. [DOI] [PubMed] [Google Scholar]
- 17.Fan K., Wei P., Feng Q., Chen S., Huang C., Ma L., Lai B., Pei J., Liu Y., Chen J. Biosynthesis, purification, and substrate specificity of severe acute respiratory syndrome coronavirus 3C-like proteinase. J. Biol. Chem. 2004;279:1637–1642. doi: 10.1074/jbc.M310875200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chou K.-C., Wei D.-Q., Zhong W.-Z. Binding mechanism of coronavirus main proteinase with ligands and its implication to drug design against SARS. Biochem. Biophys. Res. Commun. 2003;308:148–151. doi: 10.1016/S0006-291X(03)01342-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xiong B., Gui C.-S., Xu X.-Y., Luo C., Chen J., Luo H.-B., Chen L.-L., Li G.-W., Sun T., Yu C.-Y. A 3D model of SARS-CoV 3CL proteinase and its inhibitors design by virtual screening. Acta Pharmacol. Sin. 2003;24:497–504. [PubMed] [Google Scholar]
- 20.Lee V.S., Wittayanarakul K., Remsungnen T., Parasuk V., Sompornpisut P., Chantratita W., Sangma C., Vannarat S., Srichaikul P., Hannongbua S. Structure and dynamics of SARS coronavirus proteinase: the primary key to the designing and screening for anti-SARS drugs. ScienceAsia. 2003;29:181–188. [Google Scholar]
- 21.Toney J.H., Navas-Martin S., Weiss S.R., Koeller A. Sabadinine: a potential non-peptide anti-Severe Acute-Respiratory-Syndrome agent identified using structure-aided design. J. Med. Chem. 2004;47:1079–1080. doi: 10.1021/jm034137m. [DOI] [PubMed] [Google Scholar]
- 22.Sirois S., Wei D.-Q., Du Q., Chou K.-C. Virtual screening for SARS-CoV protease based on KZ7088 pharmacophore points. J. Chem. Inf. Comput. Sci. 2004;44:1111–1122. doi: 10.1021/ci034270n. [DOI] [PubMed] [Google Scholar]
- 23.Tan E.L.C., Ooi E.E., Lin C.-Y., Tan H.C., Ling A.E., Lim B., Stanton L.W. Inhibition of SARS coronavirus infection in vitro with clinically approved antiviral drugs. Emerg. Infect. Dis. 2004;10:581–586. doi: 10.3201/eid1004.030458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vastag B. Old drugs for a new bug; influenza, HIV drugs enlisted to fight SARS. JAMA. 2003;290:1695–1696. doi: 10.1001/jama.290.13.1695. [DOI] [PubMed] [Google Scholar]
- 25.Lee N., Hui D., Wu A., Chan P., Cameron P., Joynt G.M., Ahuja A., Yung M.Y., Leung C.B., To K.F., Lui S.F., Szeto C.C., Chung S., Sung J.J.Y. A major outbreak of Severe Acute Respiratory Syndrome in Hong Kong. N. Engl. J. Med. 2003;348:1986–1994. doi: 10.1056/NEJMoa030685. [DOI] [PubMed] [Google Scholar]
- 26.Knowles S.R., Phillips E.J., Dresser L., Matukas L. Common adverse events associated with the use of ribavirin for Severe Acute Respiratory Syndrome in Canada. Clin. Infect. Dis. 2003;37:1139–1142. doi: 10.1086/378304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lau A.C.-W., So L.K.-Y., Miu F.P.-L., Yung R.W.-H., Poon E., Cheung T.M.-T., Yam L.Y.-C. Outcome of coronavirus-associated severe acute respiratory syndrome using a standard treatment protocol. Respirology. 2004;9:173–183. doi: 10.1111/j.1440-1843.2004.00588.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamamoto N., Yang R., Yoshinaka Y., Amari S., Nakano T., Cinatl J., Rabenau H., Doerr H.W., Hunsmann G., Otaka A. HIV protease inhibitor nelfinavir inhibits replication of SARS-associated coronavirus. Biochem. Biophys. Res. Commun. 2004;318:719–725. doi: 10.1016/j.bbrc.2004.04.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu C.-Y., Jan J.-T., Ma S.-H., Kuo C.-J., Juan H.-F., Cheng Y.-S.E., Hsu H.-H., Huang H.-C., Wu D., Brik A. Small molecules targeting severe acute respiratory syndrome human coronavirus. Proc. Natl. Acad. Sci. USA. 2004;101:10012–10017. doi: 10.1073/pnas.0403596101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gershkovich A.A., Kholodovych V.V. Fluorogenic substrates for proteases based on intramolecular fluorescence energy transfer (IFETS) J. Biochem. Biophys. Methods. 1996;33:135–162. doi: 10.1016/s0165-022x(96)00023-1. [DOI] [PubMed] [Google Scholar]
- 31.Liu Y., Kati W., Chen C.-M., Tripathi R., Molla A., Kohlbrenner W. Use of a fluorescence plate reader for measuring kinetic parameters with inner filter effect correction. Anal. Biochem. 1999;267:331–335. doi: 10.1006/abio.1998.3014. [DOI] [PubMed] [Google Scholar]
- 32.Shi J., Wei Z., Song J. Dissection study on the Severe Acute Respiratory Syndrome 3C-like protease reveals the critical role of the extra domain in dimerization of the enzyme. J. Biol. Chem. 2004;279:24765–24773. doi: 10.1074/jbc.M311744200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Copeland R.A. Mechanistic considerations in high-throughput screening. Anal. Biochem. 2003;320:1–12. doi: 10.1016/s0003-2697(03)00346-4. [DOI] [PubMed] [Google Scholar]
- 34.Lipinski C.A., Lombardo F., Dominy B.W., Feeny P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1996;23:3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 35.Zolli-Juran M., Cechetto J.D., Hartlen R., Daigle D.M., Brown E.D. High-throughput screening identifies novel inhibitors of Escherichia coli dihydrofolate reductase that are competitive with dihydrofolate. Bioorg. Med. Chem. Lett. 2003;13:2493–2496. doi: 10.1016/s0960-894x(03)00480-3. [DOI] [PubMed] [Google Scholar]
- 36.Zhang J.-H., Chung T.D.Y., Oldenburg K.R. A simple statistical parameter for use in evaluation and validation of high-throughput screening assays. J. Biomol. Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 37.Hawkins D.M., Kass G.V. In. In: Hawkins D.M., editor. Topics in Applied Multivariate Analysis. Cambridge University Press; New York: 1982. pp. 269–302. [Google Scholar]
- 38.Breiman L., Friedman J.H., Olshen R.A., Stone C.J. Classification and Regression Trees. Chapman & Hall; New York: 1984. [Google Scholar]
- 39.McGovern S.L., Caselli E., Grigorieff N., Shoichet B.K. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J. Med. Chem. 2002;45:1712–1722. doi: 10.1021/jm010533y. [DOI] [PubMed] [Google Scholar]
- 40.Dragovich P.S., Webber S.E., Babine R.E., Fuhrman S.A., Patick A.K., Matthews D.A., Lee C.A., Reich S.H., Prins T.J., Marakovits J.T. Structure-based design, synthesis, and biological evaluation of irreversible human rhinovirus 3C protease inhibitors. 1. Michael acceptor structure-activity studies. J. Med. Chem. 1998;41:2806–2818. doi: 10.1021/jm980068d. [DOI] [PubMed] [Google Scholar]
- 41.Matthews D.A., Dragovich P.S., Webber S.E., Fuhrman S.A., Patick A.K., Zalman L.S., Hendrickson T.F., Love R.A., Prins T.J., Marakovits J.T. Structure-assisted design of mechanism-based irreversible inhibitors of human rhinovirus 3C protease with potent antiviral activity against multiple rhinovirus serotypes. Proc. Natl. Acad. Sci. USA. 1999;96:11000–11007. doi: 10.1073/pnas.96.20.11000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moon J.B., Coleman R.S., Hanzlik R.P. Reversible covalent inhibition of papain by a peptide nitrile. 13C NMR evidence for a thioimidate ester adduct. J. Am. Chem. Soc. 1986;108:1350–1351. [Google Scholar]
- 43.Webb J.L. Enzyme and Metabolic Inhibitors. Volume. 1966;3 (New(York: Academic Press) Inc. [Google Scholar]
- 44.Otto H.-H., Schirmeister T. Cysteine proteases and their inhibitors. Chem. Rev. 1997;97:133–171. doi: 10.1021/cr950025u. [DOI] [PubMed] [Google Scholar]
- 45.Allaire M., Chernaia M.M., Malcolm B.A., James M.N.G. Picornaviral 3C cysteine proteinases have a fold similar to chymotrypsin-like serine proteinases. Nature. 1994;369:72–76. doi: 10.1038/369072a0. [DOI] [PubMed] [Google Scholar]
- 46.Richer M.J., Juliano L., Hashimoto C., Jean F. Serpin mechansim of Hepatitis C virus Nonstructural 3 (NS3) protease inhibition. J. Biol. Chem. 2004;279:10222–10227. doi: 10.1074/jbc.M313852200. [DOI] [PubMed] [Google Scholar]
- 47.Téllez-Valencia A., Ávila-Ríos S., Pérez-Montfort R., Rodríguez-Romero A., de Gómez-Puyou M.T., López-Calahorra F., Gómez-Puyou A. Highly specific inactivation of triosphosphate isomerase from Trypanosoma cruzi. Biochem. Biophys. Res. Commun. 2002;295:958–963. doi: 10.1016/s0006-291x(02)00796-9. [DOI] [PubMed] [Google Scholar]
- 48.Bhandari P., Gowrishankar J. An Escherichia coli host strain useful for efficient overproduction of cloned gene products with NaCl as the inducer. J. Bacteriol. 1997;179:4403–4406. doi: 10.1128/jb.179.13.4403-4406.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tabor S., Richardson C. A bacteriophage T7 RNA polymerase/promotor system for controlled exclusive expression of specific genes. Proc. Natl. Acad. Sci. USA. 1985;82:1074–1078. doi: 10.1073/pnas.82.4.1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ausubel F.M., Brent R., Kingston R.E., Moore D.D., Seidman J.G., Smith J.A., Struhl K. Current Protocols in Molecular Biology. Greene Publishing Associates; New York: 1991. [Google Scholar]
- 51.Eltis L.D., Iwagami S.G., Smith M. Hyperexpression of a synthetic gene encoding a high potential iron sulfur protein. Protein Eng. 1994;7:1145–1150. doi: 10.1093/protein/7.9.1145. [DOI] [PubMed] [Google Scholar]
- 52.Malcolm B.A., Chin S.M., Jewell D.A., Stratton-Thomas J.R., Thudium K.B., Ralston R., Rosenberg S. Expression and characterization of recombinant Hepatitis A virus 3C proteinase. Biochemistry. 1992;31:3358–3363. doi: 10.1021/bi00128a008. [DOI] [PubMed] [Google Scholar]