

Abstract
Objective:
Cardiac myosin (CM) is structurally similar to skeletal muscle myosin (SkM) which has procoagulant activity. Here we evaluated CM’s ex vivo, in vivo, and in vitro activities related to hemostasis and thrombosis.
Approach and Results:
Perfusion of fresh human blood over CM-coated surfaces caused thrombus formation and fibrin deposition. Addition of CM to blood passing over collagen-coated surfaces enhanced fibrin formation. In a murine ischemia/reperfusion injury model, exogenous CM, when administered intravenously, augmented myocardial infarction and troponin I release. In hemophilia A mice, intravenously administered CM reduced tail-cut-initiated bleeding. These data provide proof of concept for CM’s in vivo procoagulant properties. In vitro studies clarified some mechanisms for CM’s procoagulant properties. Thrombin generation assays showed that CM, like SkM, enhanced thrombin generation in human platelet-rich and platelet-poor plasmas and also in mixtures of purified factors Xa, Va and prothrombin. Binding studies showed that CM, like SkM, directly binds factor Xa, supporting the concept that the CM surface is a site for prothrombinase assembly. In tissue plasminogen activator-induced plasma clot lysis assays, CM was antifibrinolytic due to robust CM-dependent thrombin generation that enhanced activation of thrombin activatable fibrinolysis inhibitor.
Conclusion:
CM in vitro is procoagulant and prothrombotic. CM in vivo can augment myocardial damage and can be pro-hemostatic in the presence of bleeding. CM’s procoagulant and anti-fibrinolytic activities likely involve, at least in part, its ability to bind factor Xa and enhance thrombin generation. Future work is needed to clarify CM’s pathophysiology and its mechanistic influences on hemostasis or thrombosis.
Keywords: cardiac myosin, skeletal muscle myosin, thrombin, factor Xa, coagulation, TAFI, fibrinolysis, hemostasis, thrombosis, ischemia reperfusion
Subject Codes: Basic Science Research, Thrombosis
Graphic Abstract
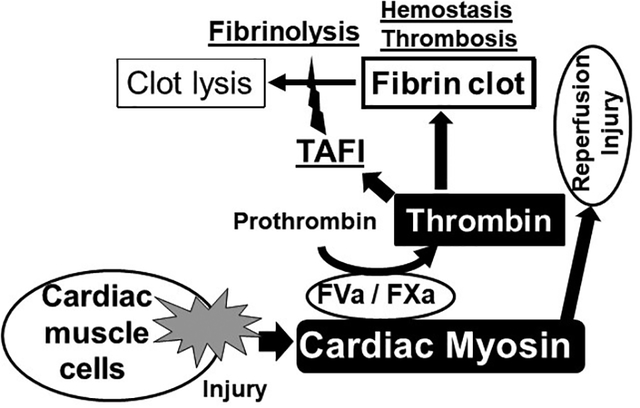
Introduction
Myocardial infarction is ultimately caused by occlusive thrombi.1–4 Based mainly on immunoassays5, extensive research by many groups has established that exposure and release of cardiac myosin (CM) are linked to myocardial infarction.6–9 In spite of extensive studies of CM as a prognostic or diagnostic biomarker for myocardial infarction as well as other coronary pathologies, knowledge about the direct contributions of CM to coronary physiology in terms of the regulation of hemostasis, thrombosis or inflammation is seriously lacking. During myocardial infarction, CM, one of the major cardiomyocyte components, is released from infarcted tissue as are troponins and myosin binding protein C.6, 10–12 Assuming that CM is less easily released from damaged coronary tissue than troponins or myosin binding protein C, it is very likely that blood is readily exposed to CM on the surface of damaged coronary tissue since myosin is much more abundant in coronary tissue. Following tissue damage, if CM were procoagulant, it might beneficially promote thrombin’s pro-hemostatic activity to prevent or limit bleeding into the tissue and reduce damage. Subsequently, or alternatively, extended exposure of blood to CM might promote thrombin-driven inflammation and/or thrombosis.13, 14
We recently discovered that SkM possesses procoagulant and prothrombotic activities by binding factor Xa.15 CM, which is structurally quite similar to skeletal muscle myosin,16–18 has procoagulant activity in vitro in whole blood in thromboelastograph studies.19 CM at high concentration was reported to have several effects on tPA-induced fibrinolysis.20, 21 However, these two reports have had no confirmation. Here we report extensive evaluation of CM’s ex vivo, in vivo, and in vitro activities related to hemostasis and thrombosis. Tail cut-induced bleeding assays using hemophilic mice provide in vivo proof of concept that CM can be pro-hemostatic. When given intravenously, exogenous CM augmented myocardial infarction and troponin I release in a murine ischemia/reperfusion injury model. Thrombin generation assays showed that CM, like SkM, enhanced thrombin generation in human platelet-rich and platelet-poor plasmas and also in mixtures of purified factors Xa, Va and prothrombin. Mechanistic studies support the concept that the CM surface is a site for prothrombinase assembly and that CM can inhibit clot lysis by indirectly promoting activation of thrombin activatable fibrinolysis inhibitor (TAFI). These provocative findings raise many questions about unexpected contributions of CM to myocardial pathophysiology.
MATERIALS AND METHODS
The authors declare that all supporting data are available for the article and its online supplementary files.
Materials
Bovine cardiac and rabbit skeletal myosins were purchased from Cytoskeleton (Denver, CO). Rabbit skeletal myosin motor proteins were also purchased from Sigma-Aldrich (St. Louis, MO). After reconstituting with water, myosins were dialyzed against buffer containing 600 mmol/L NaCl, 50 mmol/L Tris, pH 7.4. Myosins were aliquoted and stored at −80 °C. The dialyzed buffer was also stored at −80 °C as control vehicle. The concentration of myosins was determined using Coomassie protein assay reagent (Thermo Scientific, Rockford, IL). Rectangular glass cover slips # 1.5 were from Neuvitro (Vancouver, WA). Fibrillar collagen type I from equine tendons was from Nycomed (Munich, Germany). Quinacrine hydrochloride hydrate was from Cayman (Ann Arbor, MI). Anti-human fibrin β-chain monoclonal IgG1 antibody was purified from murine hybridoma HB-8545 (American Type Culture Collection) maintained at The Scripps Research Institute; the antibody was labelled with Alexa Fluor 546 (Molecular Probes, Eugene, OR) and is referred to as anti-fibrin Ab-AF546. Monoclonal antibody against myosin heavy chain was purified with immobilized-protein A from culture medium of the mouse hybridoma (MF20) (obtained from Developmental Studies Hybridoma Bank, Iowa City, IA). Polyclonal antibody against myosin heavy and light chains (PA1–28037) were from Fisher Scientific (Pittsburgh, PA). Trifluoperazine was from MP biochemical (Santa Ana, CA). Pooled normal, coagulation factor (F) X (FX)-, FIX-, FVIII-, FVII- and FV-deficient plasmas were from George King Bio-Medical (Overland Park, KS). TAFI-deficient plasma was from Affinity Biologicals (Lancaster, Ontario, Canada). Human prothrombin-immunodepleted plasma, corn trypsin inhibitor (CTI), human purified FVa, FX, FXa, Gla-domainless (DG)-factor Xa, plasmin, and thrombomodulin were from Haematologic Technologies (Essex Junction, VT). Prothrombin (FII) and chromogenic substrates Pefachrome® TH and Pefachrome® FXa were from 5-Diagnostics US Corp (Newtown, CT). Fluorogenic substrate I-1140 was from Bachem Bioscience Inc. (King of Prussia, PA). Innovin containing tissue factor (TF) was from DADE Behring (Marburg, Germany). Human TAFI was purified from normal pooled plasma as described previously.22 Human purified ɑ-thrombin was from Enzyme Research Laboratories (South Bend, IN). Human tPA was from Chromogenix (Mölndal, Sweden). Dialysis cassettes, Dulbecco Modified Eagle Medium (DMEM) and buffer ingredients were from Thermo Fisher Scientific (Waltham, MA). Chicken egg L-ɑ-phosphatidylcholine (PC), bovine brain L-ɑ-phosphatidylserine (PS), bovine brain L-ɑ-phosphatidylethanolamine (PE) were from Avanti (Alabaster, AL). Fatty acid-free bovine serum albumin (BSA) fraction V for in vitro studies was from Sigma (St. Louis, MO). Fraction V, low heavy metal, BSA for ex vivo blood flow assays and carboxypeptidase inhibitor from potato tubers (CPI) were from Calbiochem (La Jolla, CA). Rivaroxaban was provided by Bayer HealthCare AG, (Leverkusen, Germany).
Ex vivo blood perfusion studies
The ex vivo whole blood perfusion studies followed procedures, as described.15, 23 To produce control and immobilized protein test surfaces, glass cover slips were incubated with either control buffer, equine collagen type I in 100 mmol/L acetic acid (1.0 mg/ml), or bovine cardiac muscle myosin (150 nmol/L) in 20 mmol/L Hepes, 135 mmol/L NaCl, pH 7.4 (HBS) for 1 hr. All cover slips were subsequently washed with HBS and incubated for 1 hr with 1 mg/ml BSA/HBS to block activation of the coagulation contact phase by exposure of blood to glass. Whole human blood was collected from an antecubital vein into 1/50th final volume of citrate-phosphate-dextrose (CPD; 12.88 mmol/L citrate final concentration) and either incubated with the control buffer, myosin (50 nmol/L final) or carboxypeptidase inhibitor (CPI) from potato tubers (CPI; 10 μg/mL final) for 10 min at room temperature. Quinacrine hydrochloride hydrate at 40 μg/ml was added to blood to render platelets fluorescent. Fibrin was visualized by adding anti-fibrin Ab-AF546 to blood. Whole blood was recalcified to 1.15 mmol/L free Ca2+ immediately prior to perfusion over the coated glass cover slips assembled at the bottom of a rectangular flow chamber with a 125 μm height maintained by a silicon gasket. Blood was aspirated through the chamber with a syringe pump (Chemyx Fusion 200, Chemyx, Stafford, TX) at the wall shear rate of 300 s−1 and was followed by DMEM to remove red cells and improve platelet and fibrin visualization by confocal z-sectioning with a Zeiss Axiovert 135M/LSM 410 microscope (Carl Zeiss, Germany). Image processing and analysis was performed with ImageJ64 (downloaded at http://rsbweb.nih.gov/ij/).
Model for murine myocardial ischemia/reperfusion injury
Experimental protocols were approved by the Institutional Review Board (Institutional Animal Care and Use Committee [IACUC]) at the University of Colorado Denver, USA and followed NIH guidelines for the use of live animals. To eliminate gender- and age-related variations, we routinely used 12- to 16-week-old male C57BL/6J mice. Myocardial ischemia and reperfusion injury studies in C57BL/6J wildtype mice were performed as described previously.24–27 After 60 min of myocardial ischemia, reperfusion continued for 120 min. Mice (n=6 per group) received either vehicle or CM (5.4 mg/kg) via intraarterial infusion beginning at 15 min after initiation of reperfusion. Infarct sizes were determined by calculating the percentage of infarcted myocardium to the area at risk (AAR) using a double staining technique with Evan’s blue and triphenyltetrazolium chloride (TTC). Using planimetry via the NIH software Image 1.0 (National Institutes of Health, Bethesda, MA), the AAR and the infarct size were determined. All animals were under deep anesthesia while performing surgical procedures. After completion of reperfusion, blood was collected by central venous puncture for troponin I (cTnI) measurements using a quantitative rapid cTnI assay (Life Diagnostics, Inc., West Chester, PA, USA), which is highly specific for myocardial ischemia and has a well-documented correlation with the infarct size in mice24–27. After study completion, animals were euthanized with an overdose of pentobarbital and exsanguination.
Murine Tail Clip-induced bleeding studies
All animal protocols were approved by the Institutional Animal Care and Use Committee of The Scripps Research Institute. C57BL/6J mice acquired from the Rodent Breeding Colony at the Scripps Research Institute were anesthetized with isoflurane. Intravenous injection of anti-FVIII antibody (GMA-8015; 0.25 mg/kg) (25 female/19 male) or control vehicle (15 female/9 male) was given retro-orbitally to wild type 8–12 weeks old C57BL/6J mice at 2 hours prior to tail cutting.28 Bovine CM (Cytoskeletal Inc) (5.4 mg/kg, estimated to give ≤ 200 nmol/L final plasma concentration), was given i.v. at 15 min prior to tail cutting. Mice were anaesthetized with isoflurane and placed on a temperature-controlled flow heating pad (37 °C), and the distal portion of the tail was surgically removed via a scalpel blade at 1.5-mm tail diameter to induce a moderate bleeding effect28. Tails were immersed in 50 mL of saline, and blood was collected for 10 minutes. Body weight-corrected blood loss was determined as described.29 A final volume of 150 μL from the saline vials was mixed with equal volume of 2% acetic acid solution to induce erythrocyte lysis for measurement of hemoglobin concentration. Then 150 μL for each hemoglobin sample was placed in a PolySorp 96-well plate and the absorbance of 490 nm was measured. Data were obtained using a VERSAmax Microplate Reader (Molecular Devices Corporation, Menlo Park, CA). Total blood loss was expressed in microliter per gram of mouse and was obtained using a standard derived from defined blood volumes lysed and processed similarly as tail bleeding volumes above.
Phospholipid Vesicles
Small unilamellar vesicles of either 80% phosphatidyl choline (PC):20% phosphatidyl serine (PS) (w/v) or 60% PC:20% PS:20% phosphatidyl ethanolamine (PE) (w/v) in the buffer containing 150 mmol/L NaCl, 50 mmol/L Tris, pH 7.4 (TBS) were prepared by sonication under a flow of nitrogen using a microtip sonicator.
Collection of whole blood and preparation of platelet rich plasma (PRP) and platelet poor plasma (PPP)
Whole blood was obtained from healthy consenting adult donors by antecubital venipuncture in accord with The Scripps Research Institute IRB. Briefly, nine volumes of blood were collected into one volume containing 3.2% (w/v) sodium citrate, pH 7.4. The blood was centrifuged for 10 min at 265 g at room temperature and three-fourths of the upper phase was collected as PRP. The blood was also centrifuged for 20 min at 2,000 g at room temperature and three-fourths of the upper phase was collected as PPP.
Thrombin generation assay in PRP and PPP
Plasma thrombin generation assays were performed as described15, 30 with modifications. For some studies, PRP and PPP from the same individual donor were also prepared. PRP or PPP (30 μl) from individual donors was incubated with various concentrations of myosin for 10 min at 37°C without addition of phospholipid vesicles. For other studies, pooled plasma (George King Bio-Medical Inc., Overland Park, KS) (30 μl) was incubated with various concentrations of myosin for 10 min at 37°C in the presence of 4 μM phospholipid (80% PC:20% PS w/v) vesicles (PL). Human hemophilia A plasma (George King Bio-Medical) (50 μL) and C57BL/6J mouse plasma (BioIVT, Westbury, NY) were also used in some studies. Fluorogenic thrombin substrate solution (Z-Gly-Gly-Arg-AMC) (I-1140) with either tissue factor (TF) (Innovin, final 0.5 pM) containing Ca2+ or with only Ca2+ alone was added to the reaction mixture (total 110 μl) to initiate coagulation. Thrombin generation was followed continuously for 25 min using SPECTRAmax GEMINI XS fluorometer (Molecular Devices Corporation) or TECAN Spark® (Tecan Group Ltd., Männedorf, Switzerland) with excitation and emission wavelengths set at 360 and 460 nm, respectively. The first derivative of fluorescence versus time was used to produce thrombin generation curves with the correction for substrate consumption and inner filter effect.15, 30 Results also included determination of lag time in minutes defined as the interval from the addition of substrate and Ca2+ until the formation of 1 nmol/L of thrombin and determination of area under the curve (AUC) defined as the sum of thrombin generated from 1 to 25 min (nmol/L·min).
Activation of prothrombin by prothrombinase complex
CM was incubated with factor Va (5 nmol/L, final) and factor Xa (0.2 nmol/L, final) in TBS containing 0.5% BSA with 5 mmol/L Ca2+. Thrombin generation was initiated by the addition of prothrombin (0.75 μM, final, unless noted otherwise). The reaction was quenched by 10 mmol/L EDTA, and the rate of thrombin formation was quantified by measuring thrombin concentration as the rate of substrate (Pefachrome TH) hydrolysis. In separate experiments, des-Gla-domain (DG)-factor Xa was used in place of factor Xa following the same protocols.
Prothrombin (0.75 μM, final) activation in the absence of factor Va was determined using factor Xa alone. Prothrombin was mixed with various concentrations of CM at room temperature and then incubated with factor Xa (1.6 nM, final) for 60 min. Prothrombin (0.75 μM, final) activation in the presence of CM but without factor Xa was also measured when prothrombin was incubated with various concentrations of CM at room temperature for 60 min.
Immunodepletion of myosin using anti-myosin monoclonal antibody-coated beads
Anti-myosin monoclonal antibody (MF20) or mouse non-immune IgG were immobilized on magnetic beads using Dynabeads Antibody Coupling Kit (Life Technologies, Carlsbad, CA) according to the manufacture’s instruction. The rabbit SkM in TBS containing 0.5% bovine serum albumin was incubated at room temperature with either MF20-coated beads or non-immune mouse IgG coated beads. Then the beads were separated by DynaMag-Spin, and the supernatants were tested for prothrombin activation in purified systems and in plasma to determine the residual procoagulant activity.
Binding of factor Xa to immobilized CM in the presence and absence of factor Va
CM (100 μL at 10 μg/mL) in sodium bicarbonate, pH 9.3 was immobilized to the microtiter plate overnight, followed by blocking with BSA solution. Then various concentrations of factor Xa (0 to 50 nmol/L) in the presence or absence of factor Va (final 10 nmol/L) were added to microtiter plate-immobilized myosin for 1 hr at room temperature. Then, the factor Xa solution was removed followed by washing with TBS (200 μL ×3 times). The bound factor Xa was quantified by measuring factor Xa concentration as the rate of substrate (Pefachrome FXa) hydrolysis.
Plasma clot lysis
Clot lysis was studied in a plasma system in which tissue plasminogen activator (tPA)-mediated fibrinolysis of a thrombin-induced clot was measured using turbidity, as described.22, 31 An aliquot of 100 μL containing thrombin (5 nmol/L final, unless otherwise noted), Ca2+ (final free calcium concentration 2.3 mmol/L), PL vesicles (60% PC:20% PS:20% PE, final concentration 10 μM) and tPA (100 ng/mL final, unless otherwise noted) in a buffer of 25 mmol/L Hepes, pH 7.4, 137 mmol/L NaCl, 3 mmol/L KCl and 0.1% BSA was added to an aliquot of 100 μL containing 95 μL of citrated normal human pooled plasma (George King Bio Medical Inc.) plus 5 μL of myosin suspension in 50 nmol/L Tris, 600 mmol/L NaCl, pH 7.4 or matching control buffer that was preincubated at 37°C. After a brief rapid mixing, 100 μL of this reaction mixture was pipetted into a microtiter plate and then incubated at 37°C during which turbidity was measured over time at 405 nm on a Spectramax 340 kinetic microtiter plate reader (Molecular Devices Corporation). A rapid increase in turbidity represents clot formation followed eventually by a decrease in turbidity representing clot lysis. The clot lysis time (t1/2) was defined as the time between the maximum turbidity and the midpoint of the subsequent turbidity decrease that characterizes the lysis of fibrin determined using an in house Lysis Analysis software developed by van de Poel et al.32
Statistical analysis
Data have been analyzed for normality and equal variance as a justification for using parametric or non-parametric analyses using Prism software (Graph Pad Software Inc., San Diego, CA).
Two-way ANOVA with Tukey’s multiple comparison test, Mann-Whitney test and Km calculations were performed using Prism 7.01 software.
RESULTS
CM promotes ex vivo thrombus formation in flowing human blood
To assay the procoagulant effects of CM under shear, recalcified fresh whole human blood was perfused over either collagen-coated, BSA-coated, or myosin-coated surfaces in chambers at the initial wall shear rate of 300 s−1.15, 23 To assess if CM can support platelet deposition and fibrin formation directly, recalcified fresh whole human blood was perfused over a CM-coated surfaces in the absence of collagen. The results show that immobilized CM supported platelet deposition and fibrin formation in flow-oriented strands (Figure 1A–1B). In controls, recalcified fresh flowing human blood failed to form significant fibrin strands or to deposit platelets on BSA-coated surfaces. Typical patterns of platelet deposition and fibrin deposition on the CM-coated or BSA-coated surface were seen after 4 min perfusion as shown by images and by quantification (Figure 1A). The addition of CM at 50 nmol/L to blood before perfusion over a collagen-coated surface resulted in more extensive fibrin strand formation on the collagen surface with robust fibrin enrichment distal to the site of the blood entry to the chamber. Consistent with the published study for SkM15, CM primarily enhanced fibrin deposition, and no significant platelet deposition enrichment was seen in the presence of CM added to blood as compared to control blood (Figure 1C).
Figure 1. CM promotes ex vivo thrombus formation in flowing human blood.
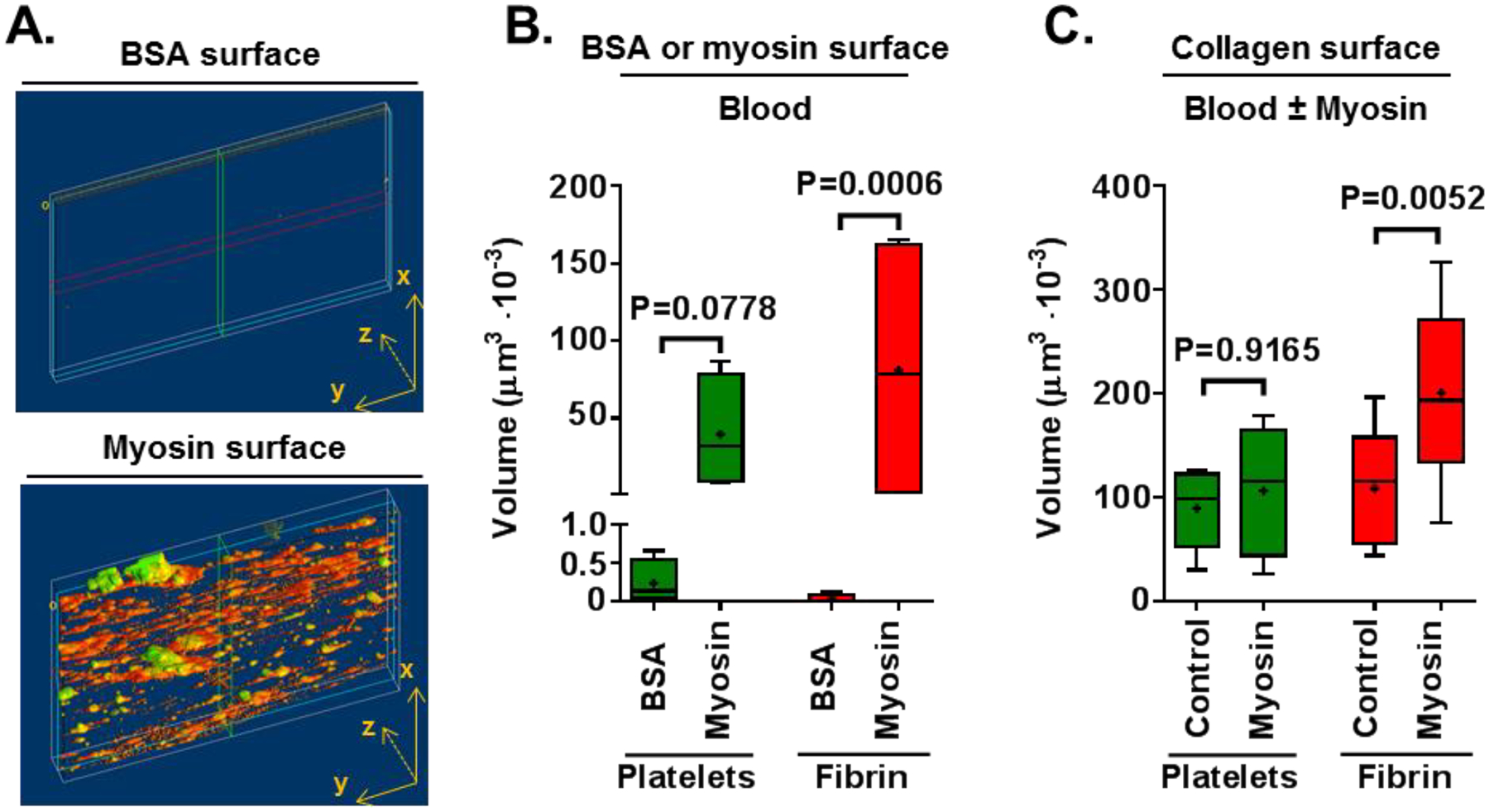
The procoagulant activity of CM was studied in flowing whole blood under initial wall shear of 300 s−1. (A) Whole blood was perfused over CM-coated surfaces or BSA-coated control surfaces and quantification of platelet adhesion and fibrin formation was made. Reperesentative images for CM-coated and BSA-coated surfaces are shown. (B) Amounts of platelet adhesion and fibrin formation were quantified from confocal microscopy images produced by an overlay of maximum fluorescence intensities at four separate positions within a channel. In separate experiments, (C) Whole blood to which either CM (50 nmol/L final) or control buffer was added was perfused past surfaces coated with collagen type I, followed by quantifying platelet adhesion and fibrin formation. P values were obtained by Two-way ANOVA with Tukey’s multiple comparison test, and Tukey method is used to create the whiskers with error bars ranging minimum and maximum.
CM promotes myocardial injury in vivo in a murine model for myocardial ischemia and reperfusion injury
The in vivo effects of CM on murine myocardial ischemia and reperfusion injury was studied. When compared to vehicle-treated control mice, mice which received CM injection shortly after the beginning of reperfusion exhibited significantly larger myocardial infarct size, as the median infarct size increased from 25.1% to 45.2% (Figure 2A). As further remarkable evidence of myocardial injury caused by CM infusion, the median serum cTnl level was increased from 54.2 ng/mL to 179 ng/mL (Figure 2B).
Figure 2. CM exacerbates myocardial injury in vivo in a murine ischemia / reperfusion injury model.
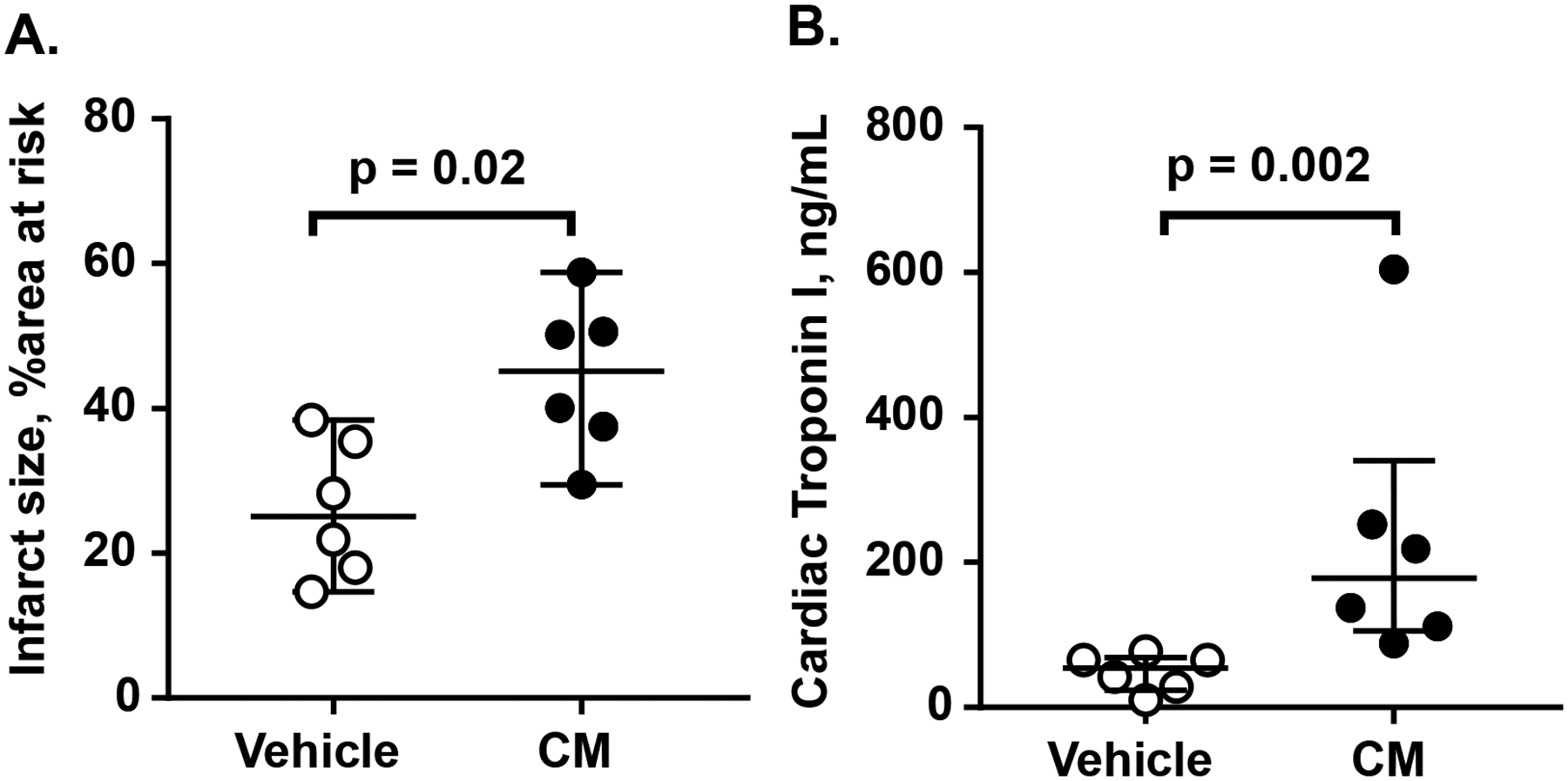
C57BL/6J wildtype mice (n=6 per group) were subjected to myocardial ischemia reperfusion injury and received either vehicle or CM (5.4 mg/kg) via intraarterial infusion 15 min after the initiation of reperfusion. Infarct size (A) and cardiac troponin I (B) were determined as noted in the Method section. Bars indicated median with interquartile range, and p values were calculated by Mann-Whitney test.
CM promotes hemostasis in vivo in a murine tail cut bleeding model
To study the in vivo pro-hemostatic activity of CM, an hemophilia A mouse model was employed.28 Intravenous injection of anti-FVIII antibody (GMA-8015; 0.25 mg/kg) or control vehicle was given retro-orbitally to wild type C57BL/6J mice at 2 hours prior to tail cutting. Mice given only anti-FVIII antibody (N = 22) had more blood loss (median = 6.7 μL/g) compared to control mice (N = 14) (median < 2 μL/g) (Figure 3). In this mouse model receiving anti-FVIII antibody (N = 22), CM (5.4 mg/kg) injected at 15 min prior to tail cutting significantly reduced the median blood loss from 6.7 to 3.2 μL/g (N = 20) (p < 0.001). There was no difference in sex for tail bleeding results in each group (data not shown). These in vivo data showing that intravenously administered CM enhanced hemostasis following tail cutting are consistent with in vitro thrombin generation assays where CM enhanced TF-induced thrombin generation in human hemophilia plasma and mouse hemophilia plasma (Supplemental Figure S1). The addition of only myosin (100–200 nmol/L) to human hemophilia A plasma greatly increased thrombin generation in a manner comparable to addition of only recombinant FVIII (Supplemental Figure S1A, S1B). In the wild-type C57BL/6J mouse plasma, anti-FVIII antibody (GMA-8015; 5 μg/mL final) reduced TF-induced thrombin generation as reported.33 When CM (12.5–200 nmol/L) was added to mouse plasma containing anti-FVIII antibodies, TF-induced thrombin generation was restored (Supplemental Figure 1C).
Figure 3. CM reduces blood loss following tail-cut-induced bleeding in hemophilia A mouse model.
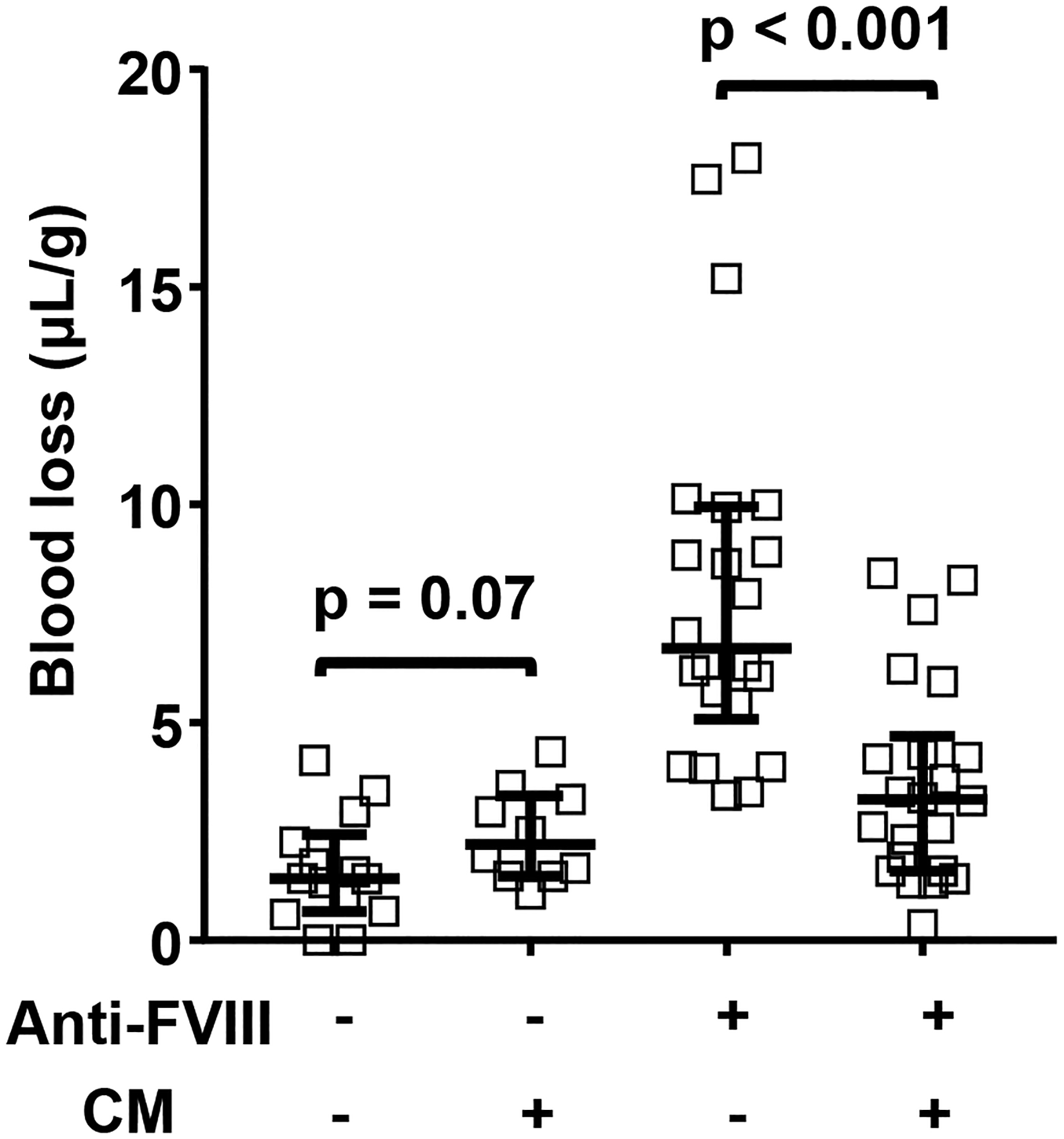
Wild type C57BL/6J mice were injected with 0.25 mg/kg of anti-FVIII (FVIII) antibody (acquired hemophilia A model) (36 female/28 male) or vehicle (saline) (20 female/14 male) at 2 hours prior to tail cut. Then, mice were injected with vehicle or 5.4 mg/kg CM. The distal portion of the tail was surgically removed via a scalpel blade at 1.5-mm tail diameter to induce a moderate bleeding effect. Tails were immersed in 50 mL of saline, pH 7.4 at 37°C. Total blood loss during 10 min was measured as the blood volume collected per mouse weight (μL/g). Bars indicated median with interquartile range, and p values were calculated by Mann-Whitney test.
CM promotes thrombin generation in human platelet rich and platelet poor plasmas
CM enhanced thrombin generation in freshly prepared platelet rich plasma (PRP) when either recalcification with low 0.5 pM TF (TF/Ca2+) (Figure 4A) or simple recalcification alone (Ca2+ alone) was used to induce thrombin generation (Figure 4B). Similarly, thrombin generation in freshly prepared platelet poor plasma (PPP) from the same donor was stimulated by myosin when TF/Ca2+ (Figure 4C) or Ca2+ alone (Figure 4D) was used to promote clotting. Thus, the ability of CM to enhance thrombin generation in freshly prepared plasma was primarily independent of platelets. When either 0.5 pM TF/Ca2+ ions or Ca2+ alone in the presence of added 4 μM PL was used to induce thrombin generation in commercially available, previously frozen pooled human plasma (George King Biomedical), CM enhanced thrombin generation (Supplemental Figure S2A, S2C). Under those conditions, SkM also increased thrombin generation (Supplemental Figure S2B, S2D) as previously described15. Thus, CM enhanced thrombin generation, even in the presence of added 4 μM PL, and the potency of CM to enhance thrombin generation in plasma was very similar to SkM’s procoagulant activity.
Figure 4. CM promotes thrombin generation in platelet rich and platelet poor plasma.
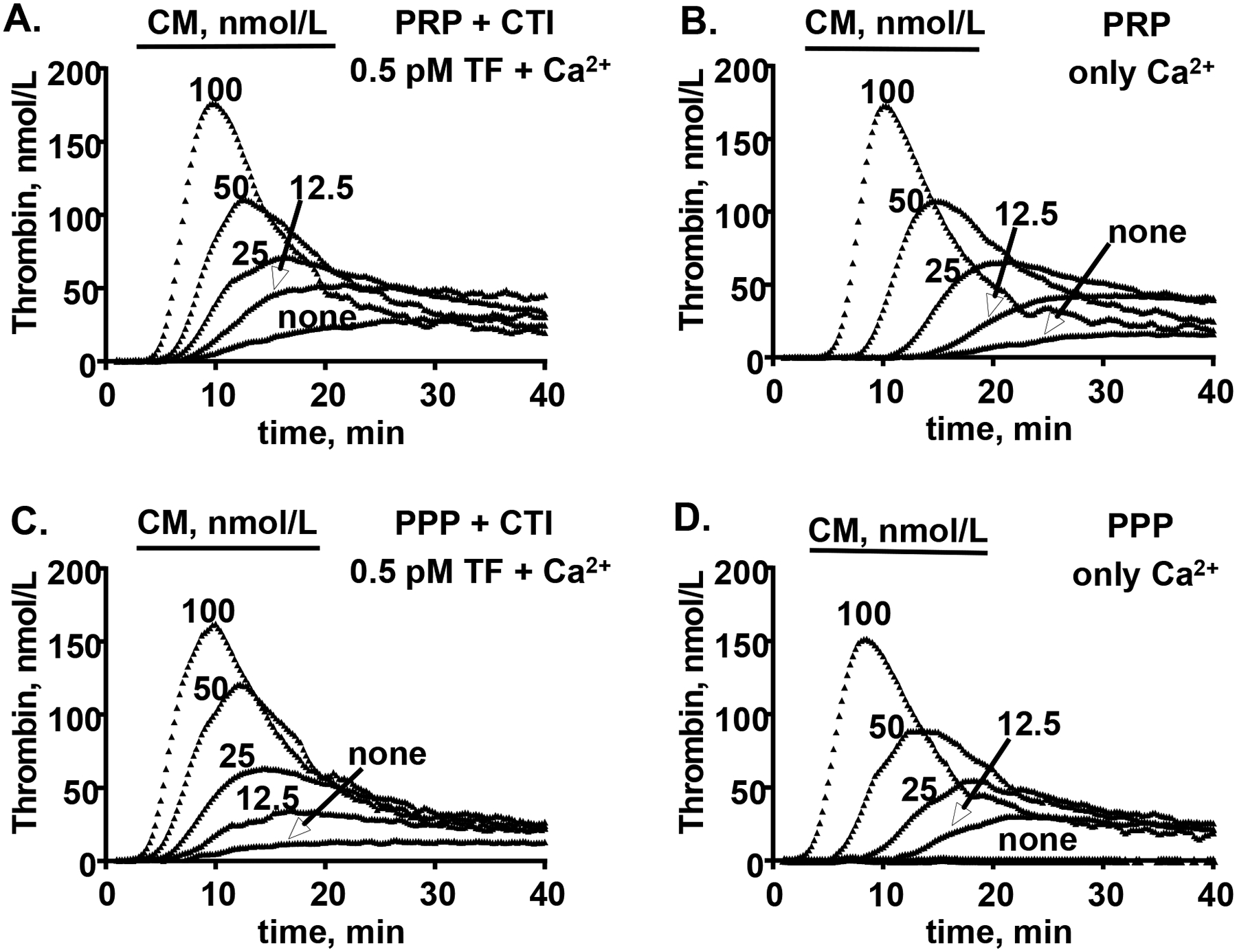
Whole human blood was collected by venipuncture from normal donors (1:10 sodium citrate) and serially centrifugated to prepare fresh platelet rich plasma (PRP) and platelet poor plasma (PPP). (A, C) After incubation with various indicated concentrations of myosin and CTI for 10 min at 37°C, thrombin generation was initiated by recalcification and addition of TF (0.5 pM final) to either (A) PRP or (C) PPP. (B, D) In parallel experiments, thrombin generation after incubation with various indicated concentrations of myosin alone was initiated with addition of calcium to (B) PRP or (D) PPP. Reactions using blood from a single representative male donor are shown here and are representative of data for three other different donors (one male and two female) studied.
To prove that the myosin molecule itself in the CM preparation promoted thrombin generation in plasma, CM was depleted from plasma plus CM reaction mixtures using monoclonal anti-myosin heavy chain MF20 antibody-beads (Supplemental Figure S2E). Removal of CM reduced peak thrombin generation from 170 nmol/L thrombin seen for incubation with non-immune control IgG beads to 120 nmol/L thrombin for anti-myosin MF20 beads. Thrombin generation in the presence of CM and no beads generated 170 nmol/L peak thrombin, while recalcification with no myosin gave 105 nmol/L peak thrombin. Thus, MF20 antibody-induced depletion of myosin caused a total relative loss of >75 % CM-induced excess thrombin generation, indicating that the myosin molecule itself in CM preparations was responsible for most of the observed enhancement of thrombin generation.
CM enhances prothrombin activation by factors Xa and Va in purified reaction mixtures
The potential for CM to enhance activation of purified prothrombin by purified factor Xa, factor Va, and Ca2+ (termed the prothrombinase complex) was tested in kinetic studies of prothrombin activation at varying concentrations of prothrombin and CM (Figures 5A, 5B). CM strongly stimulated prothrombin activation by factors Xa:Va:Ca2+. Prothrombin and CM variations enabled good fits for kinetic parameters for prothrombin activation by the enzymatic complex comprising factors Xa:Va:Ca2+. Analysis of the kinetic data showed that CM increased Vmax in a dose dependent fashion and yielded a value for Km of ~ 0.4 μmol/L prothrombin for CM-enhanced prothrombin activation (Supplemental Table S1). Based on curve fitting of data from Figure 5A summarized in Supplemental Table S1, Vmax was 74.1, yielding a value for kcat of 371 ± 51/min (Supplemental Figure S3). When prothrombinase assays were done with factor Xa, prothrombin, Ca++ and CM (2 – 40 nM), but without addition of factor Va, negligible activation of prothrombin (<2%) was observed (data not shown). CM itself (40 nM) in the absence of factor Xa, whether or not factor Va (5 nmol/L) was present, did not detectably activate prothrombin (<1%) during 60 min incubation (data not shown).
Figure 5. CM promotes thrombin generation in purified clotting factor reaction mixtures.
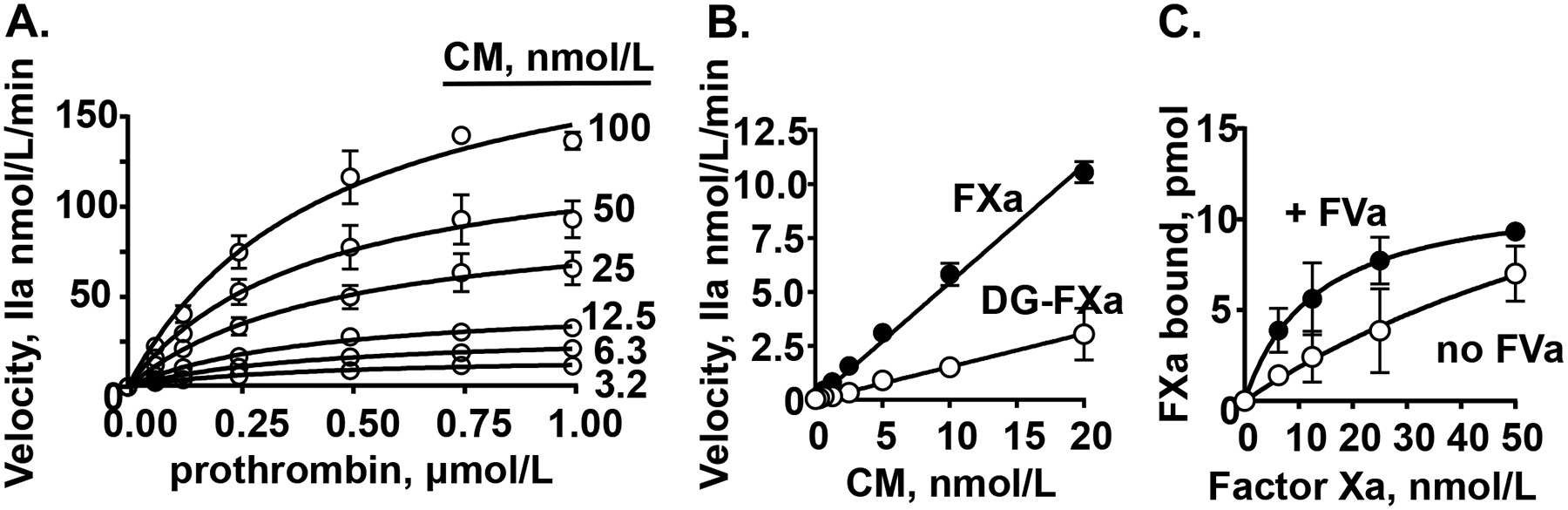
(A) The effect of varying concentrations of myosin and prothrombin on the initial rate of prothrombin activation by factor Xa/factor Va. (B) The effect of varying concentrations of myosin on thrombin generation from prothrombin (0.75 μM final) by factor Xa (FXa) or Gla domain-less factor Xa (DG-FXa) (0.2 nmol/L final) and factor Va (5 nmol/L final). (C) Binding studies of factor Xa to immobilized-myosin in the presence or absence of factor Va (FVa) (10 nmol/L final) where bound factor Xa was determined using factor Xa chromogenic activity assays. Each value represents the mean [SD] of at least triplicate determinations.
When Gla domainless (DG)-factor Xa was assayed and compared to factor Xa in purified prothrombinase assays in the presence of CM, it retained > 25 % prothrombinase activity (Figure 5B) which is similar to previous results for the ability of SkM to support DG-factor Xa activity.15 Thus, whereas deletion of the Gla domain of factor Xa reduces its prothrombinase activity in the presence of PL by > 99%, factor Xa’s Gla domain is not required for CM’s enhancement of prothrombinase activity, emphasizing that the CM surface is not simply acting like a PL surface.
To prove that the myosin molecule itself in the CM preparation is procoagulant in the purified prothrombinase assay mixtures, different myosin-targeting reagents were employed to test for ability to block CM’s activity. Two myosin inhibitors, namely the myosin allosteric inhibitor trifluoperazine which alters myosin’s conformation and functional activity activity34, 35 and anti-myosin polyclonal antibodies, each inhibited by > 90 % CM’s prothrombinase-enhancing action while neither inhibited prothrombin activation by factor Xa/factor Va in controls in the absence of CM (Supplemental Figure S4). In other studies, pulldown of CM from myosin solutions using monoclonal anti-myosin heavy chain antibody-beads but not control nonimmune monoclonal antibody-beads reduced by ~70 % CM’s enhancement of prothrombinase activity (Supplemental Figure S4). Thus, the myosin molecule itself in the CM preparations was required for the observed enhancement of prothrombin activation by factors Xa and Va in the purified prothrombinase assay mixtures.
Binding of factor Xa to immobilized myosin in the presence or absence of factor Va
To define direct interactions between CM and factor Xa, CM was immobilized on the surface of microtiter plates and factor Xa binding was quantified. Factor Xa bound to CM with a KDapp of 108 nmol/L (Figure 5C) and to SkM with a KDapp of 80.4 nmol/L (Supplemental Figure S5). Biolayer Interferometry binding studies for binding of factor Xa to SkM previously gave a similar value of 51 nmol/L.15 When binding of factor Xa to immobilized-CM in the presence of 10 nmol/L factor Va was analyzed, factor Va increased the affinity, and KDapp values for CM and SkM were 21 nmol/L and 13 nmol/L, respectively), indicating that factor Va stabilized the factor Xa:myosin complex, presumably due to ternary complexation.
CM attenuates tPA-induced plasma clot lysis
To determine whether CM plays a role in tPA-induced plasma clot lysis, pooled normal human plasma was incubated with either 100 nmol/L CM or control buffer. Clotting was initiated with recalcification plus 5 nmol/L thrombin, variable tPA, and PL vesicles (60% PC: 20% PS: 20% PE); then clot lysis was followed at 37°C in the presence of 0, 50, 100 or 200 ng/mL tPA or control buffer (Figure 6A). When tPA-mediated clot lysis times (t1/2) for different levels of tPA were quantified as previously described32 and compared for the presence of CM versus no myosin, the data showed that CM (100 nmol/L final) significantly attenuated tPA-mediated plasma clot lysis in the presence of 50–100 ng/mL tPA (Figure 6B). The efficacy for CM to attenuate 100 ng/mL tPA-mediated clot lysis was tested in the presence of varying concentrations of CM (Figure 6C). CM variation enabled good fits for biochemical parameters for CM’s effect on t1/2 compared to controls (Figure 6D). Analysis of the data showed that CM effects plateaued at maximum values of 185 percent difference in t1/2 fibrinolysis and analysis yielded EC50 values for 50% of maximum t1/2 difference of ~ 58 nmol/L CM (Figure 6D). SkM also showed a similar prolongation of tPA-induced clot lysis with a dose-response similar to that seen for CM (Supplemental Figure S6).
Figure 6. CM attenutes tPA-induced plasma clot lysis.
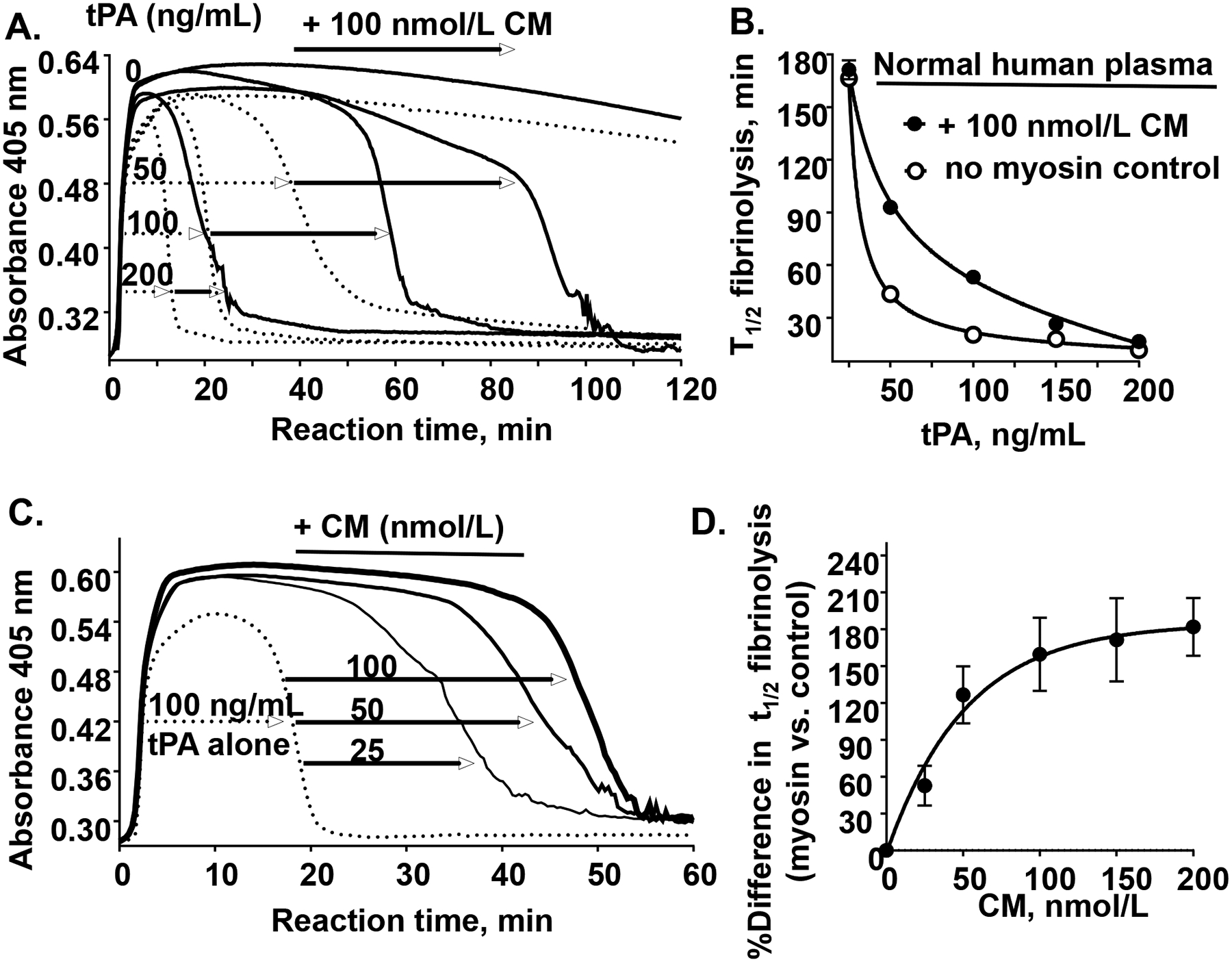
(A) As described in Materials and Methods, a 100 μL aliquot containing thrombin, Ca2+, phospholipid vesicles (60% PC: 20% PS: 20% PE), 0.1% BSA and different levels of tPA (as indicated, 0, 50, 100, or 200 ng/mL) was added to a 100 μL aliquot containing citrated normal human pooled plasma which had been pre-incubated for 30 min at 37 °C with CM (100 nmol/L) (solid lines) or control buffer (dotted lines). Following rapid mixing, the reaction mixture was monitored for turbidity at 405 nm at 37°C for 180 min during which a clot was rapidly formed (turbidity increase) and then that clot was subsequently lysed (turbidity decrease). (B) The plasma clot lysis time (t1/2, min for 50 % clot lysis) was quantified in the presence of CM (●) at different tPA levels or control buffer (○). Each value represents the mean [SD] of triplicate determinations. (C) Plasma was pre-incubated with different amounts of CM (as indicated, 0, 25, 50, or 100 nmol/L) followed by thrombin-initiated clot formation (as above), and the clot was lysed in the presence of 100 ng/mL tPA. (D) The percent difference in plasma clot lysis (t1/2) between values for the presence of CM at the indicated concentrations are shown where the plasma clot lysis time at no CM was defined as 100%. Each value represents the mean [SD] of at least triplicate determinations.
Prothrombin is required for CM’s inhibition of tPA-induced plasma clot lysis
To determine the importance of the initial levels of thrombin during the plasma clot formation phase on myosin’s effect on plasma clot lysis, normal human plasma was incubated with either control buffer or 100 nmol/L CM and clot was initiated with different levels of thrombin from 5 to 40 nmol/L combined with 100 ng/mL tPA (Supplemental Figure S7A). The data indicated that 5 nmol/L thrombin was useful for further assays. To assess a potential role for CM-induced thrombin generation in these clot lysis assays, CM’s effect was studied using prothrombin-deficient plasma supplemented with either control buffer or prothrombin at physiological plasma levels and compared to normal human plasma (Supplemental Figure S7B). Time to clot lysis in the presence of myosin was normalized to control buffer setting and expressed as percent difference in t1/2 fibrinolysis for both normal pooled human plasma and prothrombin-deficient plasmas (Supplemental Figure S7C). These data suggested the importance of myosin-induced prothrombin-dependent thrombin generation during the assay. To test the factor X importance in plasma, FX-deficient plasma in the presence of either FX replacement or control buffer or of normal plasma pretreated with a FX inhibitor (Rivaroxaban) were studied using clot lysis assays as were other deficient plasmas (FIX-, FVIII- and FV-deficient plasmas) (Supplemental Figure S7D, S7E). These various clot lysis studies all supported the conclusion that clotting factors X, IX, VIII, and V, as well as prothrombin, are vital for CM’s effect on clot lysis attenuation under the studied conditions.
Thrombin activatable fibrinolysis inhibitor activity is required for CM’s inhibition of tPA-induced plasma clot lysis
Thrombin can activate the endogenous plasma fibrinolysis inhibitor, thrombin activatable fibrinolysis inhibitor (TAFI). To determine whether CM’s clot lysis attenuation effect is related to TAFI activation, extensive studies were made. When normal pooled plasma was pretreated with the carboxypeptidase inhibitor (CPI) prior to tPA-induced clot lysis assays, fibrinolysis inhibition by CM was no longer significant (< 5%) (Figure 7A). When varying levels of CM (Figure 7B) or SkM (Supplemental Figure S8) were used for assays in the presence or absence of CPI pretreatment of plasma, CM’s dose-dependent effects on clot lysis prolongation were abolished by CPI, the TAFI inhibitor. When TAFI-deficient plasma was used for the clot lysis assay, CM (100 nmol/L final) had no significant effect on tPA-induced lysis (Figure 7C). But reconstitution of TAFI into the TAFI-deficient plasma fully enabled a robust inhibition of tPA-induced clot lysis similar to CM’s effect for normal plasma (Figure 7C). This effect of restoration of myosin’s inhibition of clot lysis by TAFI reconstitution of the deficient plasma was repeated and confirmed when different levels of CM and different levels of thrombin were studied (Figure 7D), emphasizing that TAFI is required for CM’s ability to inhibit tPA-induced fibrinolysis.
Figure 7. TAFI activity is required for CM’s inhibition of tPA-induced plasma clot lysis.
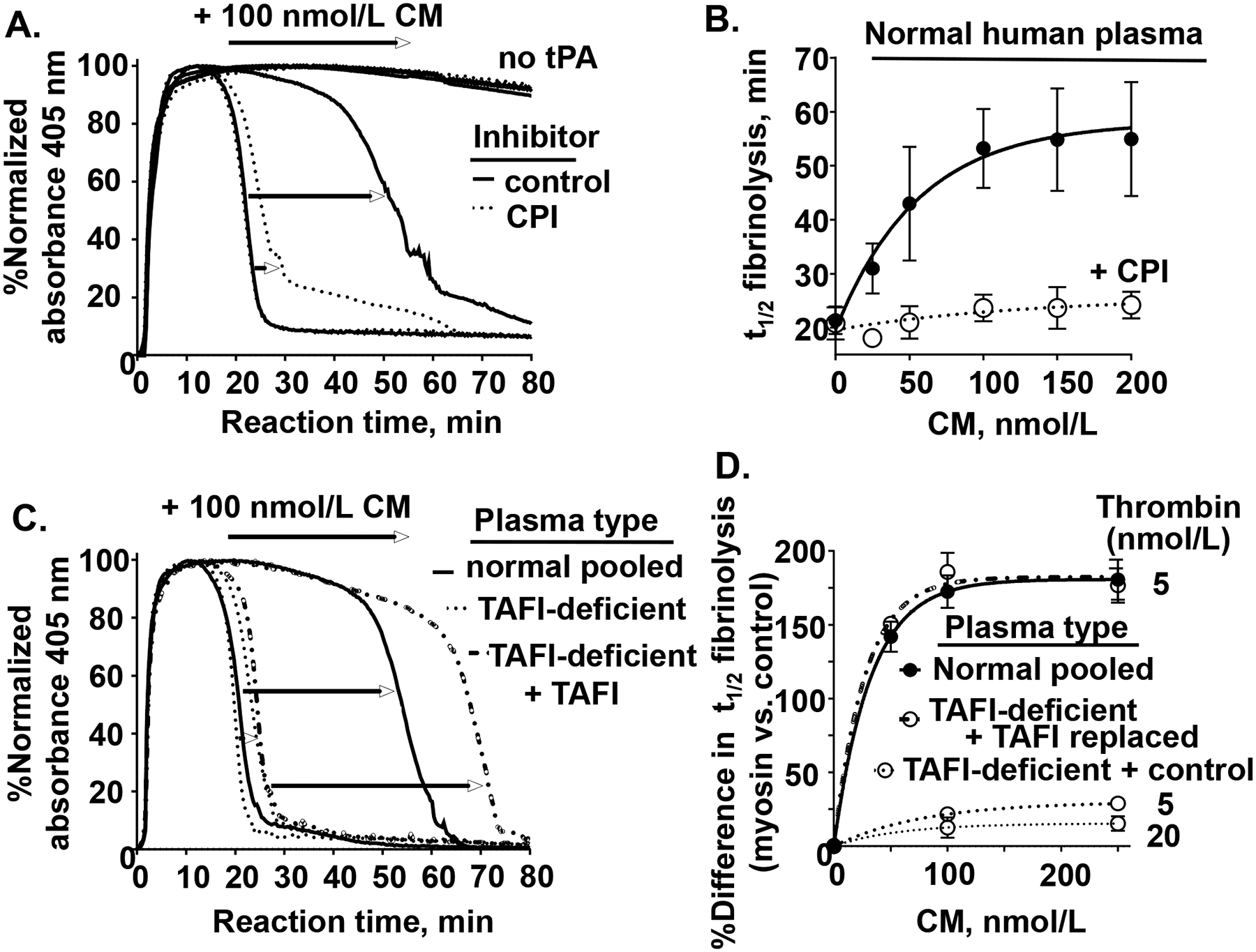
(A) Normal human pooled plasma was pre-incubated with either carboxypeptidase inhibitor (CPI) to inhibit TAFI (dotted lines) or control buffer (solid line) and clot lysis studies were done, as in Figure 6, with with 100 nmol/L CM and with 100 ng/mL tPA or without tPA (as indicated by “no tPA”). (B) Plasma clot lysis times (t1/2), determined as done in (A), were quantified in the presence of different levels of CM, 5 nmol/L thrombin, and 100 ng/mL tPA in the absence (●, solid line) or presence of the TAFIa inhibitor, CPI (○, dashed line). (C) TAFI-deficient plasma reconstituted with only control buffer or with reconsititued with normal levels of TAFI and normal plasma was used for plasma clot lysis studies (as above, using 5 nmol/L thrombin, and 100 ng/mL tPA) with either 0 or 100 nmol/L CM. (D) The percent difference in plasma clot lysis t1/2 between values for the presence of CM at the indicated concentrations (50, 100, 250 nmol/L) for the different indicated plasma types (normal (solid line), TAFI-deficient + control buffer (dotted lines for 5 nmol/L or 20 nmol/L thrombin), and TAFI-deficient + TAFI replacement (dash-dotted line)) are shown. Each value represents the mean [SD] of at least triplicate determinations.
To determine whether CM has a direct effect on TAFI activation by thrombin, purified TAFI was incubated with the combination of thrombin (20 nmol/L) and thrombomodulin (TM, 5 nmol/L), or with high levels of thrombin (300 nmol/L) or of plasmin (500 nmol/L), or with control buffer containing Ca2+, and TAFI activation was monitored as previously described22, 36 after different incubation times in the presence or absence of 100 nmol/L of CM (Supplemental Figure S9A). These studies showed that CM had no effect on the activation of TAFI by thrombin or plasmin. Once TAFI is activated, it spontaneously loses activity over 20 min. When the effect of CM on the relative stability of activated TAFI (TAFIa) was assessed after TAFI activation with either plasmin or thrombin:TM, it was apparent that CM had no significant direct effect on activated TAFIa’s transient stability or spontaneous decay (Supplemental Figure 9B). Thus, CM had no direct effects on either TAFI activation or TAFIa’s spontaneous loss of activity.
DISCUSSION
Studies here showed that CM exerts prothrombotic effects ex vivo when exposed to fresh flowing human blood, both when blood encountered immobilized myosin on a surface and, alternatively, when myosin was in the perfused blood that encountered a collagen-coated surface. Under each of these conditions, myosin notably enhanced fibrin formation greater than platelet deposition; this might imply that CM promotes activation of coagulation greater than activation of platelets, though this is an unproven implication. Consistent with this implication is the observation that in vitro studies showed that CM promotes thrombin generation in platelet poor plasma as well as it did in platelet rich plasma. CM also enhanced prothrombin activation in a simple mixture of purified clotting factors containing factor Xa, factor Va and prothrombin. Moreover, factor Xa binds directly to CM, implying that prothrombin activation is triggered by CM-bound factor Xa. CM’s procoagulant effects and its direct binding of factor Xa are very similar to those of SkM15 whose sequences are very similar to CM.37, 38
Tests for CM’s potential in vivo prothrombotic effects were provided by murine myocardial ischemia reperfusion studies showing that infused CM exacerbated myocardial injury. Tests for CM’s potential in vivo procoagulant effects were provided by murine acquired hemophilia A tail-cut initiated bleeding studies showing that when CM was infused shortly before tail cutting, it significantly reduced blood loss. Thus, in the in vivo setting of ongoing thrombosis, i.e., during ischemia reperfusion injury, and the in vivo setting of ongoing bleeding, CM appears to provide procoagulant activities. Studies here do not rule out that other biologic activities of CM might also contribute to the observed physiologic outcomes. The fact that CM has in vivo procoagulant activity raises many questions for future studies regarding its roles in coronary health and disease whose answers may provide novel insights into cardiovascular biology and may also point towards novel translational research.
Studies here for CM and previously for SkM15 imply that, beside traditional phospholipid membranes, these muscle myosins can also provide surfaces in support of prothrombin activation by factors Xa:Va. For CM’s enhancement of prothrombinase activity, the observed kcat value, 371 min−1, compares favorably to values reported for phospholipid or platelet enhancement of prothrombinase15, 39–42 (Supplemental Table S2). Biochemical studies recently identified the molecular surface site on SkM that binds factor Xa to enable prothrombin activation.43 The surface on SkM that binds factor Xa and promotes prothrombin activation is in the neck region of SkM, where the heavy chain binds both the essential and regulatory light chains. Future studies are needed to identify molecular surface sites for CM’s procoagulant protein-protein interactions and to determine the extent to which the same site43 on the neck region of CM supports prothrombinase activity.
Plasma clot lysis studies here confirm that CM inhibits tPA-induced clot lysis.20, 21 Extensive mechanistic studies for this effect of CM show that clotting factors which are needed for robust thrombin generation, i.e., prothrombin and factors X, V, IX, and VIII, are required for myosin’s attenuation of tPA-induced plasma clot lysis. Notably, myosin did not inhibit tPA-induced clot lysis in TAFI-deficient plasma; however, reconstitution of TAFI-deficient plasma with purified TAFI restored myosin’s inhibition of clot lysis. Addition of the carboxypeptidase inhibitor, CPI, a TAFI inhibitor, to the tPA-induced clot lysis assays blocked the effect of myosin on clot lysis. Thus, based on these extensive data, the primary mechanism for cardiac myosin’s inhibition of tPA-induced fibrinolysis, under the conditions studied, derives from the antifibrinolytic activity of TAFIa following robust thrombin generation which enables potent thrombin activation of TAFI.
To rescue myocardium from ischemic obstruction and infarction, reperfusion is essential. However, reperfusion itself can potentially cause injury, such as an additional myocardial infarction and/or other pathologic injuries that increase risk for morbidity and mortality.44, 45 Ischemia and reperfusion cause damage not only in cardiomyocytes but also in the coronary circulation. Debris and release of soluble factors from the infarcted region are key factors causing ischemia-reperfusion injury. Multiple reactions (e.g., microembolization, inflammation, platelet activation) can cause damage to the capillaries with microvascular obstruction and intramyocardial hemorrhage.44, 45 During ischemia-reperfusion injury, coagulation is activated with substantial thrombin generation.13, 46, 47 Thrombin is a central factor for the pathology of ischemic-reperfusion injury, as it can cause thrombus formation, inflammation and platelet activation.13, 14, 47 The observed increase of myocardial infarction and troponin I release caused by CM may reflect the likelihood that CM accelerates thrombin generation in the microvasculature, thereby contributing to accelerated development of ischemia-reperfusion injury with microvascular thrombosis and inflammation. However, there remain multiple other potential pathologic consequences of infused CM which need additional study.
In summary, CM is a reasonably potent procoagulant factor due to its ability to bind factor Xa and promote prothrombin activation. When blood encounters CM, thrombin generation is enhanced, and increased levels of thrombin may be either protectively pro-hemostatic and reduce bleeding or may be pathologically prothrombotic. Elucidation of the relevance of CM’s procoagulant actions for cardiovascular biology will benefit from both preclinical and clinical studies in the future. Hopefully, such studies may point towards novel translational insights.
Supplementary Material
Highlights:
Cardiac myosin is prothrombotic ex vivo for human blood and reduces bleeding in vivo in a murine hemophilia A bleeding model.
Exogenous cardiac myosin added to circulating blood exacerbates myocardial injury caused by myocardial ischemia/reperfusion in mice.
Cardiac myosin enhances thrombin generation in platelet-rich and platelet-poor plasma and in purified clotting factor mixtures.
Cardiac myosin is antifibrinolytic in vitro due to robust myosin-dependent thrombin generation in plasma that increases activation of thrombin activatable fibrinolysis inhibitor.
Sources of Funding:
National Institutes of Health (R01HL133728, R01HL142975, U01HL077863, HL104165, R01HL117722, R01HL101972, R01GM116184, R01HL122472 and F31HL136230) and American Heart Association (19POST34380105).
Non Standard Abbreviations:
- CM
cardiac myosin
- SkM
Skeletal muscle myosin
- TAFI
thrombin activatable fibrinolysis inhibitor
- HBS
20 mmol/L Hepes 135 mmol/L NaCl
- CPI
carboxypeptidase inhibitor from potato tubers
- TBS
Tris buffered saline
- PC
L-ɑ-phosphatidylcholine
- PS
bovine brain L-ɑ-phosphatidylserine
- PE
bovine brain L-ɑ-phosphatidylethanolamine
- PRP
platelet rich plasma
- PPP
platelet poor plasma
- TF
tissue factor
- DG
des-Gla-domain
- tPA
tissue plasminogen activator
- MI
myocardial infarction
Footnotes
Disclosures: None. All authors declare no competing financial interest.
References
- 1.Fuster V, Badimon L, Cohen M, Ambrose JA, Badimon JJ and Chesebro J. Insights into the pathogenesis of acute ischemic syndromes. Circulation. 1988;77:1213–1220. [DOI] [PubMed] [Google Scholar]
- 2.Herrick JB. Landmark article (JAMA 1912). Clinical features of sudden obstruction of the coronary arteries. By James B. Herrick. Jama. 1983;250:1757–1765. [PubMed] [Google Scholar]
- 3.Gorlin R, Fuster V and Ambrose JA. Anatomic-physiologic links between acute coronary syndromes. Circulation. 1986;74:6–9. [DOI] [PubMed] [Google Scholar]
- 4.Baumgartner HR. New aspects in thrombogenesis. VASA Zeitschrift fur Gefasskrankheiten. 1974;3:60–64. [PubMed] [Google Scholar]
- 5.Khaw BA, Gold HK, Fallon JT and Haber E. Detection of serum cardiac myosin light chains in acute experimental myocardial infarction: radioimmunoassay of cardiac myosin light chains. Circulation. 1978;58:1130–1136. [DOI] [PubMed] [Google Scholar]
- 6.Trahern CA, Gere JB, Krauth GH and Bigham DA. Clinical assessment of serum myosin light chains in the diagnosis of acute myocardial infarction. The American journal of cardiology. 1978;41:641–645. [DOI] [PubMed] [Google Scholar]
- 7.Ravkilde J, Nissen H, Horder M and Thygesen K. Independent prognostic value of serum creatine kinase isoenzyme MB mass, cardiac troponin T and myosin light chain levels in suspected acute myocardial infarction. Analysis of 28 months of follow-up in 196 patients. J Am Coll Cardiol. 1995;25:574–581. [DOI] [PubMed] [Google Scholar]
- 8.Hirayama A, Arita M, Takagaki Y, Tsuji A, Kodama K and Inoue M. Clinical assessment of specific enzyme immunoassay for the human cardiac myosin light chain II (MLC II) with use of monoclonal antibodies. Clinical biochemistry. 1990;23:515–522. [DOI] [PubMed] [Google Scholar]
- 9.Yamada T, Matsumori A, Tamaki S and Sasayama S. Myosin light chain I grade: a simple marker for the severity and prognosis of patients with acute myocardial infarction. American heart journal. 1998;135:329–334. [DOI] [PubMed] [Google Scholar]
- 10.Sonel A, Sasseen BM, Fineberg N, Bang N and Wilensky RL. Prospective study correlating fibrinopeptide A, troponin I, myoglobin, and myosin light chain levels with early and late ischemic events in consecutive patients presenting to the emergency department with chest pain. Circulation. 2000;102:1107–1113. [DOI] [PubMed] [Google Scholar]
- 11.Lavin F, Kane M, Forde A, Gannon F and Daly K. Comparison of five cardiac markers in the detection of reperfusion after thrombolysis in acute myocardial infarction. British heart journal. 1995;73:422–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jacquet S, Yin X, Sicard P, Clark J, Kanaganayagam GS, Mayr M and Marber MS. Identification of cardiac myosin-binding protein C as a candidate biomarker of myocardial infarction by proteomics analysis. Mol Cell Proteomics. 2009;8:2687–2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jackson SP, Darbousset R and Schoenwaelder SM. Thromboinflammation: challenges of therapeutically targeting coagulation and other host defense mechanisms. Blood. 2019;133:906–918. [DOI] [PubMed] [Google Scholar]
- 14.Petzold T and Massberg S. Thrombin: A Gas Pedal Driving Innate Immunity. Immunity. 2019;50:1024–1026. [DOI] [PubMed] [Google Scholar]
- 15.Deguchi H, Sinha RK, Marchese P, Ruggeri ZM, Zilberman-Rudenko J, McCarty OJ, Cohen MJ and Griffin JH. Prothrombotic skeletal muscle myosin directly enhances prothrombin activation by binding factors Xa and Va. Blood. 2016;128:1870–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hernandez OM, Jones M, Guzman G and Szczesna-Cordary D. Myosin essential light chain in health and disease. American journal of physiology Heart and circulatory physiology. 2007;292:H1643–H1654. [DOI] [PubMed] [Google Scholar]
- 17.Yamauchi-Takihara K, Sole MJ, Liew J, Ing D and Liew CC. Characterization of human cardiac myosin heavy chain genes. Proceedings of the National Academy of Sciences of the United States of America. 1989;86:3504–3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sellers JR. Myosins: a diverse superfamily. Biochimica et biophysica acta. 2000;1496:3–22. [DOI] [PubMed] [Google Scholar]
- 19.Coleman JR, Moore EE, Zilberman-Rudenko J, Samuels JM, Cohen MJ, Silliman CC, Banerjee A, Sauaia A, Griffin JH and Deguchi H. Cardiac and Skeletal Muscle Myosin Exert Procoagulant Effects. Shock. 2019;52:554–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kolev K, Tenekedjiev K, Ajtai K, Kovalszky I, Gombas J, Varadi B and Machovich R. Myosin: a noncovalent stabilizer of fibrin in the process of clot dissolution. Blood. 2003;101:4380–4386. [DOI] [PubMed] [Google Scholar]
- 21.Machovich R, Ajtai K, Kolev K and Owen WG. Myosin as cofactor and substrate in fibrinolysis. FEBS letters. 1997;407:93–96. [DOI] [PubMed] [Google Scholar]
- 22.Mosnier LO, von dem Borne PA, Meijers JC and Bouma BN. Plasma TAFI levels influence the clot lysis time in healthy individuals in the presence of an intact intrinsic pathway of coagulation. Thrombosis and haemostasis. 1998;80:829–835. [PubMed] [Google Scholar]
- 23.Rothmeier AS, Marchese P, Petrich BG, Furlan-Freguia C, Ginsberg MH, Ruggeri ZM and Ruf W. Caspase-1-mediated pathway promotes generation of thromboinflammatory microparticles. The Journal of clinical investigation. 2015;125:1471–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eckle T, Grenz A, Kohler D, Redel A, Falk M, Rolauffs B, Osswald H, Kehl F and Eltzschig HK. Systematic evaluation of a novel model for cardiac ischemic preconditioning in mice. American journal of physiology Heart and circulatory physiology. 2006;291:H2533–H2540. [DOI] [PubMed] [Google Scholar]
- 25.Eckle T, Kohler D, Lehmann R, El Kasmi K and Eltzschig HK. Hypoxia-inducible factor-1 is central to cardioprotection: a new paradigm for ischemic preconditioning. Circulation. 2008;118:166–175. [DOI] [PubMed] [Google Scholar]
- 26.Kohler D, Eckle T, Faigle M, Grenz A, Mittelbronn M, Laucher S, Hart ML, Robson SC, Muller CE and Eltzschig HK. CD39/ectonucleoside triphosphate diphosphohydrolase 1 provides myocardial protection during cardiac ischemia/reperfusion injury. Circulation. 2007;116:1784–1794. [DOI] [PubMed] [Google Scholar]
- 27.Oyama Y, Bartman CM, Bonney S, Lee JS, Walker LA, Han J, Borchers CH, Buttrick PM, Aherne CM, Clendenen N, Colgan SP and Eckle T. Intense Light-Mediated Circadian Cardioprotection via Transcriptional Reprogramming of the Endothelium. Cell reports. 2019;28:1471–1484.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wyseure T, Cooke EJ, Declerck PJ, Behrendt N, Meijers JCM, von Drygalski A and Mosnier LO. Defective TAFI activation in hemophilia A mice is a major contributor to joint bleeding. Blood. 2018;132:1593–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.von Drygalski A, Cramer TJ, Bhat V, Griffin JH, Gale AJ and Mosnier LO. Improved hemostasis in hemophilia mice by means of an engineered factor Va mutant. Journal of thrombosis and haemostasis : JTH. 2014;12:363–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hemker HC and Kremers R. Data management in thrombin generation. Thromb Res. 2013;131:3–11. [DOI] [PubMed] [Google Scholar]
- 31.von dem Borne PA, Meijers JC and Bouma BN. Feedback activation of factor XI by thrombin in plasma results in additional formation of thrombin that protects fibrin clots from fibrinolysis. Blood. 1995;86:3035–3042. [PubMed] [Google Scholar]
- 32.Mosnier LO, Meijers JC and Bouma BN. The role of protein S in the activation of thrombin activatable fibrinolysis inhibitor (TAFI) and regulation of fibrinolysis. Thrombosis and haemostasis. 2001;86:1040–1046. [PubMed] [Google Scholar]
- 33.Bhat V, von Drygalski A, Gale AJ, Griffin JH and Mosnier LO. Improved coagulation and haemostasis in haemophilia with inhibitors by combinations of superFactor Va and Factor VIIa. Thrombosis and haemostasis. 2016;115:551–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang W, Wilson GJ, Brown LJ, Lam H and Hambly BD. EPR and CD spectroscopy of fast myosin light chain conformation during binding of trifluoperazine. European journal of biochemistry. 1998;257:457–465. [DOI] [PubMed] [Google Scholar]
- 35.Patel H, Margossian SS and Chantler PD. Locking regulatory myosin in the off-state with trifluoperazine. The Journal of biological chemistry. 2000;275:4880–4888. [DOI] [PubMed] [Google Scholar]
- 36.Mosnier LO. Platelet factor 4 inhibits thrombomodulin-dependent activation of thrombin-activatable fibrinolysis inhibitor (TAFI) by thrombin. The Journal of biological chemistry. 2011;286:502–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weiss A and Leinwand LA. The mammalian myosin heavy chain gene family. Annu Rev Cell Dev Biol. 1996;12:417–439. [DOI] [PubMed] [Google Scholar]
- 38.Weiss A, Schiaffino S and Leinwand LA. Comparative sequence analysis of the complete human sarcomeric myosin heavy chain family: implications for functional diversity. Journal of molecular biology. 1999;290:61–75. [DOI] [PubMed] [Google Scholar]
- 39.Krishnaswamy S, Church WR, Nesheim ME and Mann KG. Activation of human prothrombin by human prothrombinase. Influence of factor Va on the reaction mechanism. The Journal of biological chemistry. 1987;262:3291–3299. [PubMed] [Google Scholar]
- 40.Moyer MP, Tracy RP, Tracy PB, van’t Veer C, Sparks CE and Mann KG. Plasma lipoproteins support prothrombinase and other procoagulant enzymatic complexes. Arteriosclerosis, thrombosis, and vascular biology. 1998;18:458–465. [DOI] [PubMed] [Google Scholar]
- 41.Tracy PB, Eide LL and Mann KG. Human prothrombinase complex assembly and function on isolated peripheral blood cell populations. The Journal of biological chemistry. 1985;260:2119–2124. [PubMed] [Google Scholar]
- 42.Bunce MW, Toso R and Camire RM. Zymogen-like factor Xa variants restore thrombin generation and effectively bypass the intrinsic pathway in vitro. Blood. 2011;117:290–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deguchi H, Guo Z, Hayat M, Pflimlin E, Lear S, Shen W and Griffin JH. Molecular interaction site on procoagulant myosin for factor Xa-dependent prothrombin activation. The Journal of biological chemistry. 2019;294:15176–15181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ibanez B, Heusch G, Ovize M and Van de Werf F. Evolving therapies for myocardial ischemia/reperfusion injury. J Am Coll Cardiol. 2015;65:1454–1471. [DOI] [PubMed] [Google Scholar]
- 45.Hausenloy DJ, Chilian W, Crea F, Davidson SM, Ferdinandy P, Garcia-Dorado D, van Royen N, Schulz R and Heusch G. The coronary circulation in acute myocardial ischaemia/reperfusion injury: a target for cardioprotection. Cardiovasc Res. 2019;115:1143–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ten Cate H, Hackeng TM and Garcia de Frutos P. Coagulation factor and protease pathways in thrombosis and cardiovascular disease. Thrombosis and haemostasis. 2017;117:1265–1271. [DOI] [PubMed] [Google Scholar]
- 47.Raivio P, Lassila R and Petaja J. Thrombin in myocardial ischemia-reperfusion during cardiac surgery. The Annals of thoracic surgery. 2009;88:318–325. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.