

Abstract
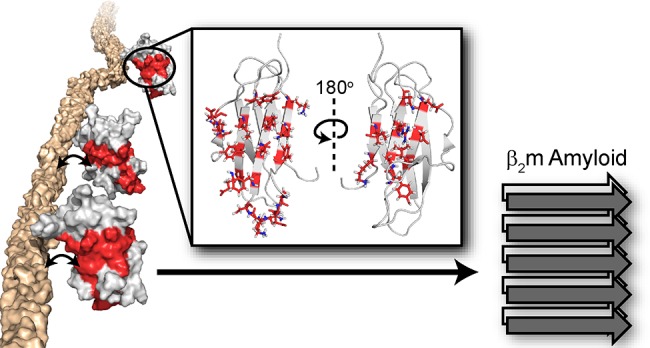
Amyloidogenesis is significant in both protein function and pathology. Amyloid formation of folded, globular proteins is commonly initiated by partial or complete unfolding. However, how this unfolding event is triggered for proteins that are otherwise stable in their native environments is not well understood. The accumulation of the immunoglobulin protein β2-microglobulin (β2m) into amyloid plaques in the joints of long-term hemodialysis patients is the hallmark of dialysis-related amyloidosis (DRA). While β2m does not form amyloid unassisted near neutral pH in vitro, the localization of β2m deposits to joint spaces suggests a role for the local extracellular matrix (ECM) proteins, specifically collagens, in promoting amyloid formation. Indeed, collagen and other ECM components have been observed to facilitate β2m amyloid formation, but the large size and anisotropy of the complex, combined with the low affinity of these interactions, have limited atomic-level elucidation of the amyloid-promoting mechanism(s) by these molecules. Using solution NMR approaches that uniquely probe weak interactions in large molecular weight complexes, we are able to map the binding interfaces on β2m for collagen I and detect collagen I-induced μs–ms time-scale dynamics in the β2m backbone. By combining solution NMR relaxation methods and 15N-dark-state exchange saturation transfer experiments, we propose a model in which weak, multimodal collagen I−β2m interactions promote exchange with a minor population of amyloid-competent species to induce fibrillogenesis. The results portray the intimate role of the environment in switching an innocuous protein into an amyloid-competent state, rationalizing the localization of amyloid deposits in DRA.
Introduction
Several proteins self-associate into amyloid fibrils, which in some cases have functional roles,1−3 but for others are associated with debilitating human diseases4−6 including Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, type II diabetes, cataracts, and dialysis-related amyloidosis (DRA). The protein precursors of amyloid diseases have unrelated primary sequences and structures,7 spanning natively unfolded (intrinsically disordered) states, such as α-synuclein, amyloid-β peptide, and tau,8−10 to stable, globular proteins, such as β2-microglobulin (β2m), transthyretin, and immunoglobulin light chains.11−13 Initiation of amyloid formation of the latter class of proteins requires complete or partial unfolding of monomeric precursors, which can transiently assume amyloid-competent state(s). This kinetic barrier may be lower for intrinsically disordered proteins. However, what triggers the initial unfolding and subsequent amyloidogenesis of natively folded globular proteins remains poorly understood.
Accumulation of β2m amyloid plaques in the joints of long-term hemodialysis patients leads to DRA and arthritic symptoms.14−17 In healthy individuals, β2m dissociates from the major histocompatibility complex-I (MHC-I), is released into the plasma, and is carried to the kidneys for degradation.18,19 However, when hemodialysis or peritoneal dialysis are required due to kidney failure, β2m is not efficiently removed from the plasma, leading to increased concentrations of the protein by up to 60-fold.16,20,21 Remarkably, despite being transported throughout the body, β2m accumulates into amyloid plaques specifically in skeletal tissues of dialysis patients.16,21−24 The mechanism(s) by which β2m fibrillizes in vivo is not well understood, since in isolation, the wild-type protein (the major culprit of DRA) resists amyloid formation in vitro under physiological conditions, even at high (100 μM) concentrations.25,26 It has been proposed that β2m amyloid localized in the joints could result, at least in part, from interactions with the major components of the extracellular matrix (ECM) in bone and cartilage: collagens I and II20−23 and glycosaminoglycans (GAGs).27−29 The binding affinities of β2m to these collagens have been shown to be in the μM–mM range,30 with preference for collagen I.27 Although the interaction is weak, it is nonetheless pathologically significant, as images of ex vivo DRA plaques reveal β2m amyloid covering the surface of collagen I fibrils.21 Indeed, recent kinetic studies have revealed that ECM components, such as collagens21,28,29 and GAGs28,29,31,32 as well as preformed fibril seeds and other cofactors,25,26,28,31−49 induce and modulate β2m amyloid formation. However, atomic details of how these components interact with, and induce, amyloid formation of β2m have remained an open question.
The weak nature of the interaction and large, anisotropic shape of the β2m–collagen I complex creates a challenge for deriving atomic-level information on how collagen I−β2m interactions initiate β2m amyloidogenesis. The immunoglobulin fold of monomeric β2m has dimensions of ∼4 nm × 2 nm × 2 nm, whereas the simplest triple helical unit of collagen I has strikingly larger dimensions of 300 nm × 1.5 nm × 1.5 nm. Collagen I triple helices assemble into even larger, structured fibrils that have diameters ranging from 10–500 nm and lengths on the μm-scale. Collagen I therefore presents as a large surface with numerous reactive groups for β2m interactions. These challenges are not insurmountable, however, as powerful solution nuclear magnetic resonance (NMR) spectroscopy methods can indirectly probe large, lowly populated complexes in site-specific detail that are invisible by other biophysical techniques.
In this study, by utilizing NMR spectroscopy experiments designed to probe large complexes, we are able to pinpoint the binding interfaces of wild-type β2m for collagen I at physiological pH and have shown the interfaces to involve both β-sheets of the native protein, suggestive of different binding modes between these two proteins. Residues identified at the binding interface include both hydrophobic and hydrophilic side chains. Through 15N relaxation experiments, we have also found that collagen I increases the number of residues in β2m involved in conformational exchange on the μs–ms time scale. These regions include residues 6–11 (β-strand A), 36–39 (β-strand C), 51 (β-strand D), and 91–94 (β-strand G) in the edge β-strands and loop residues 15–20 (loop AB), 35 (loop BC), 52–53 (loop DE), 63 (loop DE), and 78 (loop EF), the dynamics and conformations of which are known to be important for β2m amyloid formation.31,38,50,51 We propose that the weak interactions of collagen I with the β2m β-sheets and loops promote exchange of the native protein with minor populations of more amyloid-competent species that induce fibrillogenesis. This study illuminates how a protein component, collagen I, local to the environment in which β2m plaques are found, can interact with a stable, globular protein to initiate debilitating amyloid formation.
Results
Collagen I Induces β2m Amyloid Formation in a Concentration-Dependent Manner
Since the direct interaction of β2m with collagen in the joint space has been proposed to induce β2m amyloid formation,21,27 we probed the β2m–collagen I interaction under physiological pH conditions (pH 7.4) using solid-phase enzyme-linked immunosorbent assays (ELISA) (Figures 1A and S1). This is a colorimetric assay that detects an HRP-conjugated anti-β2m primary antibody and indicates the presence of β2m bound to collagen I immobilized in a 96-well plate. Importantly, the results suggest a dose-dependent interaction of the two proteins, consistent with previously published results,27 under the conditions employed here. We observe that the β2m–collagen I binding does not easily saturate with increasing concentrations of β2m (up to 100 μM; Figure S1), consistent with the low affinity of the interaction at pH 7.4 (Kd ≈ 410 μM)30 measured previously by surface plasmon resonance. The adhesion of β2m to casein was monitored as a negative control, for which no significant binding was observed (Figure 1A).
Figure 1.

Detection of collagen I-driven β2m amyloid formation. (A) ELISA probing dose-dependent adhesion of β2m (10–80 μg/mL) to collagen I (10 μg/mL) or casein (10 μg/mL, used as a negative control), at pH 7.4. The average absorbance at 450 nm from triplicates within the same plate are reported with the standard deviation given as error bars. (B) ThT fluorescence curves of 85 μM β2m (black), 85 μM β2m + 3.4 mg/mL (8.5 μM) collagen I (blue), or 3.4 mg/mL collagen I alone (orange) over 650 h in 10 mM sodium phosphate buffer, pH 7.4, shaking at 600 rpm at 37 °C. Three representative curves are given for each condition. The inset shows a zoom-in of the baseline of the ThT fluorescence curves to highlight the lack of fluorescence enhancement for both β2m (black) and collagen I (orange) alone. (C–E) Representative amplitude-modulated AFM images of (C) β2m coincubated with collagen I fibrils, (D) collagen I fibrils alone, and (E) β2m alone after incubation for 96 h at 37 °C with shaking.
Having verified the β2m–collagen I interaction under physiological pH conditions, we next monitored amyloid growth of β2m in the presence or absence of collagen I by thioflavin T (ThT) fluorescence (Figure 1B). In the presence of 3.4 mg/mL collagen I (1:0.1 molar ratio β2m:collagen I), β2m amyloid is formed within 400–600 h at pH 7.4 (Figure 1B, blue), as evident by enhanced ThT fluorescence. This is not observed in the absence of collagen I in the same conditions, and collagen I alone does not show ThT fluorescence enhancement (Figure 1B). Notably, at lower concentrations of collagen I, lower β2m concentrations, or shorter time scales, fibrils are not observed.29,31 Collagen I-induced β2m amyloid formation is accelerated at pH 6.2 relative to at pH 7.4 (Figure S2A,B), enabling measurement of the dependence of the rate of fibril formation on the concentration of collagen I. The results showed that at this pH, β2m amyloid formation is dependent on collagen I concentration, as addition of 3.4 mg/mL collagen I significantly reduces the lag time and half time of β2m aggregation relative to 0.34 or 0.17 mg/mL collagen I (Figure S2C,D). This trend is consistent with our previously published data,29 in which increasing concentrations of collagen I (0–0.47 mg/mL) accelerated β2m amyloid formation at pH 6.2 in the presence of a constant concentration of the glycosaminoglycan, heparin. These results reinforce earlier proposals that collagen I−β2m interactions play a significant role in triggering β2m fibril formation.21 Atomic force microscopy (AFM) images also showed that β2m interacts with collagen I fibrils, consistent with previous results,21 showing that β2m coats the collagen I fibril surface before detectable fibril formation occurs (monitored by ThT fluorescence), obscuring the characteristic collagen I fibril D-banding that is clearly observed in the absence of β2m (Figure 1C,D). Importantly, control experiments showed that β2m alone (Figure 1E) does not aggregate in the conditions employed, with no fibrils or high molecular weight assemblies observed by AFM. Together, these data confirm that adhesion of collagen I and β2m induces amyloid formation under physiological conditions in vitro, while β2m is not able to form amyloid within these time scales in vitro in the absence of collagen I.
Weak, but Specific β2m–Collagen I Interactions Observed through 15N-R2 Perturbations
In order to understand the mechanistic details by which collagen I interacts with β2m and initiates amyloid formation, we used solution NMR methods, which provide an excellent toolbox of approaches able to characterize residue-specific features of weak protein–protein interactions on multiple time scales.52−55 A titration of collagen I into a β2m monomer solution showed no significant chemical shift perturbations in 1H–15N heteronuclear single quantum correlation (HSQC) spectra (Figure S3). However, a residue-specific attenuation of the peak intensities was observed with increasing collagen I concentrations (Figure 2A), suggesting specific, although weak, β2m–collagen I interactions. To minimize collagen I aggregation during the NMR experiments and to capture the most specific interactions, we proceeded with low collagen I concentrations (0.6–1.2 mg/mL) that displayed consistent residue-specific perturbations and kept samples at 10 °C, allowing NMR spectra to be acquired for over 1 week without visible alterations in spectral quality. Under these conditions, addition of 1.2 mg/mL collagen I to 300 μM β2m resulted in a reduction in resonance intensity of all peaks, consistent with transient formation of a high molecular weight complex (Figure 2A). However, the greatest reduction in peak intensities occurred for residues in the eight β-strands of the wild-type protein (Figure 2A). These peak intensity losses arise, in part, from increased 15N-transverse relaxation rates (R2), which are sensitive to changes in internal motions on the ps–ns time scale and conformational exchange on the μs–ms time scale. Indeed, at these concentrations, we observe an overall increase in 15N-R2 values of β2m relative to their magnitude in the absence of collagen I, but importantly, the increase is not uniform across all residues, but is residue specific, involving predominantly residues 2–3 (N-terminus), 7–11 (β-strand A), 16–19 (loop AB), 23–26 (β-strand B), 35–39 (β-strand C), 50–52 (β-strand D), 64, 66–69 (β-strand E), 79–82 (β-strand F), 85, 87 (loop FG), and 91–94 (C-terminal β-strand G) (Figure 2B,C). The increased 15N-R2 at these specific sites could have multiple origins, arising from reduced backbone mobility upon direct interaction with collagen I and/or to line broadening due to exchange between species with different chemical shifts, especially since the observed 15N-ΔR2 is dependent on magnetic field (700 MHz vs 900 MHz, Figure S4). In order to disentangle these contributions to the increase in 15N-R2, we proceeded with two sets of NMR experiments: 15N-dark-state exchange saturation transfer (DEST) experiments, which can identify residues in β2m interacting with the large collagen I fibrils and in-phase Hahn-echo experiments, which detect conformational exchange on the μs–ms time scale.
Figure 2.

Characterizing residue-specific β2m–collagen I binding through 15N-R2 measurements. (A) Amide backbone signal intensity ratios from 1H–15N HSQC spectra of 300 μM β2m in the presence of 1.2 mg/mL collagen I compared with values in the absence of collagen I (gray bars). The dashed line is drawn at the average signal intensity ratio over the entire protein. Dips in the signal intensity reflect regions maximally perturbed by the presence of collagen I. Error bars are propagated from the noise level of the spectra. The secondary structural elements of β2m are indicated above the plot. (B) 15N-R2 measurements of 300 μM β2m in the presence (red) or absence (black) of 1.2 mg/mL collagen I. The errors are propagated from the fitting errors. The dashed lines indicate the mean 15N-R2 values of β2m in the presence or absence of 1.2 mg/mL collagen I over the entire protein. All experiments were conducted in TBS, pH 7.4 containing 0.5 mg/mL casein as a nonspecific binding blocking agent at 10 °C. Note that in these conditions, several residues in the DE loop do not have observable peak intensities in the 1H–15N HSQC spectrum due to inherent conformational exchange, consistent with previous results.49 (C) Solution NMR structure of the WT-β2m monomer (PDB: 2XKS)49 highlighting residues that show an increase in 15N-R2 higher than 13.4 s–1 (the mean Δ15N-R2) upon addition of 1.2 mg/mL collagen I.
Pinpointing the Collagen I Interaction Interface on β2m through 15N-DEST
In order to determine
which residues of β2m interact most intimately with
collagen I, we used 15N-DEST experiments.56,57 This experiment is optimal when there is a measurable increase in R2 due to formation of a transient, large molecular
weight complex that is NMR-invisible because of its high R2 and detects the exchange between an observable “light”
state (free monomeric β2m) and the NMR-invisible
“dark” state (the high molecular weight collagen I−β2m complex). In the DEST experiment, resonances of high molecular
weight species with high R2 values, such
as the collagen I−β2m complex, can be partially
saturated by weak radiofrequency (RF) fields at frequency offsets
far from monomeric β2m resonances. Saturation transfer
to the observable monomeric species by chemical exchange is detected
as a loss in monomeric β2m signal intensity. The
broadening of these DEST saturation profiles (reduced signal intensities
at further frequency offsets) in the presence of collagen I, relative
to in its absence, is therefore indicative of residues at the interaction
interface (Figure 3A,B). The “broadness” of the profiles was measured
by calculating the DEST difference (Θ) for each residue, which
is a measure of the relative effects of on-resonance and off-resonance 15N saturation. Using a saturation frequency of 350 Hz, we
measured Θ as 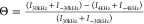 , where ±30
kHz were the most off-resonance 15N offsets, and 15N offsets of ±4 kHz provide
enough saturation transfer from bound to unbound β2m to show significant intensity loss without eliminating the signal
in most cases. Notably, we observe that the broadening of the DEST
saturation profiles is residue-specific and not uniform across all
β2m residues, with some residues showing no change
in the DEST difference in the presence of collagen I (Figure 3A,B). Examples of DEST profiles
in the presence or absence of collagen I for a residue that shows
DEST due to collagen I binding (V82 in β-strand F) and one that
does not (K41 in the C–C′ loop) are given in Figure 3B. In Figure 3A, those residues with ΔΘ
larger than the mean, and likely have the most direct contacts with
the collagen in the β2m–collagen I complex
(shaded red), include residues 6–11 (β-strand A), 15–20
(loop AB), 21–26 (β-strand B), 35 (loop BC), 36–39
(β-strand C), 51 (β-strand D), 52–53 (loop DE),
63 (loop DE), 64–69 (β-strand E), 78 (loop EF), 79–83
(β-strand F), and 91–94 (β-strand G).
, where ±30
kHz were the most off-resonance 15N offsets, and 15N offsets of ±4 kHz provide
enough saturation transfer from bound to unbound β2m to show significant intensity loss without eliminating the signal
in most cases. Notably, we observe that the broadening of the DEST
saturation profiles is residue-specific and not uniform across all
β2m residues, with some residues showing no change
in the DEST difference in the presence of collagen I (Figure 3A,B). Examples of DEST profiles
in the presence or absence of collagen I for a residue that shows
DEST due to collagen I binding (V82 in β-strand F) and one that
does not (K41 in the C–C′ loop) are given in Figure 3B. In Figure 3A, those residues with ΔΘ
larger than the mean, and likely have the most direct contacts with
the collagen in the β2m–collagen I complex
(shaded red), include residues 6–11 (β-strand A), 15–20
(loop AB), 21–26 (β-strand B), 35 (loop BC), 36–39
(β-strand C), 51 (β-strand D), 52–53 (loop DE),
63 (loop DE), 64–69 (β-strand E), 78 (loop EF), 79–83
(β-strand F), and 91–94 (β-strand G).
Figure 3.

15N-DEST to identify collagen-binding interface on 15N-β2m. (A) ΔΘ calculated from 15N-β2m DEST intensities at ±30 kHz and ±4 kHz 15N offsets with a 350 Hz saturation frequency in the presence or absence of collagen I (gray bars). The secondary structure is shown above the plot. (B) Examples of 15N-DEST profiles of 300 μM 15N-β2m in the presence (solid line, solid circles) or absence (dashed line, open circles) of 0.6 mg/mL collagen I. V82 in β-strand F shows enhancement of the DEST effect upon addition of collagen, whereas K41 in the C–C′ loop does not. (C) 15N-R2bound for each residue determined from fitting 15N-DEST profiles to the McConnell equations. (D) Examples of fit DEST profiles using the same residues as in (B). Data points are shown as circles and the fits as solid lines (gray, 150 Hz saturation; black, 350 Hz saturation). All experiments were carried out in TBS, pH 7.4, 10 °C at 700 MHz 1H Larmor frequency.
In addition, the full DEST profiles can be used to quantify residue-specific transverse relaxation values of β2m in the collagen I-bound state (R2bound) and exchange kinetics between the bound and unbound β2m. Since the ΔR2 may be due to more complex processes than collagen I binding alone, such as an overall increased viscosity due to the presence of the large collagen I molecules, we fit only the 15N DEST profiles of each residue with 150 and 350 Hz RF saturation to the McConnell equations.56,57 The use of two RF fields provides an additional constraint to distinguish between models with equal fits to data acquired at only one RF field and provides more accurate determination of kinetic parameters.57 Fitting to a simple two-state model, the population of the unbound, monomeric β2m was determined to be 94 ± 2% with an apparent first-order rate constant for the conversion of β2m from unbound to collagen I-bound conformation (konapp) of 6.4 ± 0.8 s–1. We interpret the direct binding interface to be the residues with the highest R2bound. The 15N-R2bound profile shows a similar trend to the ΔΘ profile (Figure 3A,C) and suggests that binding interfaces for collagen I on β2m occur on both β-sheets. Examples of fitting to the experimental values of residues V82 (in a binding region) and K41 (away from interface) are shown in Figure 3D.
Collagen I-Induced Conformational Exchange in β2m Revealed by 15N Relaxation
The enhanced observed 15N-R2 of β2m may not only be due to the increased intrinsic 15N-R2 caused by slowed molecular motions upon binding with a high molecular weight species (such as in a large complex) but also to an increased contribution of chemical exchange dynamics on the μs–ms time scale, since the 15N-ΔR2 is dependent on the magnetic field (Figure S4). Chemical exchange describes the interconversion of a residue between multiple states with distinct chemical shifts, such as in the case of conformational conversion or complex formation. In order to qualitatively determine which residues in β2m are in conformational exchange in the presence of collagen I, we use 15N in-phase Hahn echo experiments (R2HE) to estimate the chemical exchange contribution to the observed 15N-R2, denoted Rex. An increase in Rex in the presence of collagen I, relative to in its absence, suggests collagen I-induced conformational exchange on a per residue basis. At pH 7.4 and 10 °C, few residues in β2m have Rex values >10 s–1 in the absence of collagen I as measured by the in-phase Hahn echo experiments (Figure 4A). In the absence of collagen I, the N-terminal seven amino acids and residues in the BC and DE loops (for which several signals are unobservable) are inherently in conformational exchange (Figure 4A). Upon addition of 0.6 mg/mL collagen I, the regions with high Rex are expanded to include the N-terminus and full β-strand A, part of β-strand B to part of β-strand C, including the connecting BC loop, β-strand D, the DE loop, the C-terminal residue of β-strand F into the FG loop, and the C-terminal β-strand G (Figure 4B). Conformational dynamics in specific regions of β2m, including the N-terminal region, edge β-strands C and D, and the BC loop that contains cis Pro32, have been shown previously to be crucial in controlling the amyloidogenicity of the protein.49,58−60 Thus, the enhanced conformational exchange induced by the presence of collagen I may facilitate minor populations of amyloid-component states of β2m.
Figure 4.
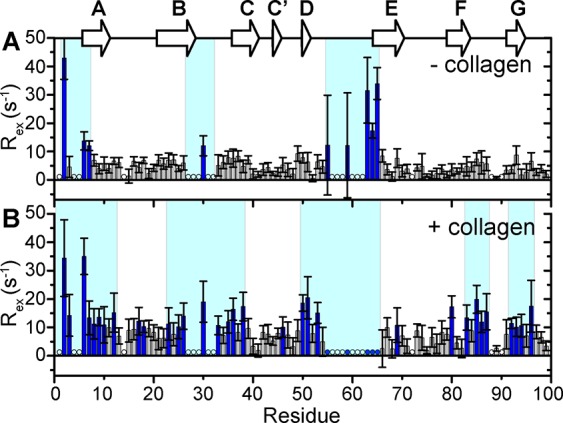
Conformational exchange in β2m induced by collagen I. Relaxation exchange rates (Rex) obtained by 15N-R2 Hahn echo experiments for each residue in 300 μM 15N-β2m in the absence (A) or presence (B) of 0.6 mg/mL collagen I at pH 7.4, 10 °C, 700 MHz 1H Larmor frequency. Rex values over 10 s–1 are indicated in blue bars. Regions shaded in cyan in both panels contain several residues with Rex > 10 s–1 in the respective conditions. Residues with unobservable cross-peaks in the absence of collagen I are indicated by open circles. Residues that are observable in the absence of collagen I, but are reduced to the level of the noise in the presence of collagen I are indicated by filled blue circles. Error bars are propagated from fitting errors.
Discussion
A Novel Collagen I Binding Surface on β2m
Amyloid formation of β2m at physiological pH in vitro requires assistance by cofactors.21,25,26,28,29,31−49 In particular, ECM molecules, such as collagens and GAGs, have been targeted as amyloid-inducing cofactors, since β2m amyloid formation is localized to musculoskeletal tissues.16,22−24 While previous experiments have focused on the kinetics of amyloid formation in the presence of these molecules,21,28,29,31,61 a detailed atomistic description of the interactions involved and how these may enhance β2m conformational dynamics and amyloid formation had not been elucidated. Here, we have used complementary NMR relaxation-based experiments to pinpoint residues of β2m involved in the collagen I binding interface and collagen I-induced dynamics that lead to enhanced β2m amyloid formation at neutral pH in vitro. The 15N-DEST experiments indicate that residues in β-strands A, B, C, D, E, F, and G form interaction surfaces with collagen I. These provide two surfaces of mixed hydrophilic and hydrophobic composition (Figure S5). Both contain hydrophobic patches, with the ABED β-sheet displaying several aromatic residues on the interaction surface (Figure S5). Since both β-sheets on opposite sides of β2m were shown to interact with the collagen I surface, binding must be multimodal involving interaction surfaces formed by K6, Q8, Y10, F22, N24, Y26, S52, Y63, L65, Y67, and E69 on the ABED β-sheet and E36, D38, L40, A79, R81, N83, I92, and K94 on the GFC β-sheet (Figures 3 and S5). Comparison of the molecular dimensions of the interacting molecules (4 nm × 2 nm × 2 nm for β2m, 300 nm × 1.5 nm for a collagen I triple helix, and microns in length and up to 500 nm in diameter for mature collagen I fibrils) highlights the potential for a myriad of binding modes, enabling independent binding of several β2m molecules to the same collagen molecule (Figure 5A,B). Importantly, the collagen I triple helix surface is interspersed with numerous hydrophilic and hydrophobic residues along its length (Figure 5A). The collagen I fibril surface maintains this repeating pattern of surface chemistries (Figure 5B), enhancing the potential for multiple binding modes to complementary surfaces on β2m (Figure 5C).
Figure 5.

Potential surface contacts for β2m–collagen I interaction. (A) Surface model of the collagen I monomer (PDB: 3HR2)62 color-coded by amino acid type (top, hydrophilic; bottom, hydrophobic). A surface representation of β2m is shown for size comparison (PDB: 2XKS).49 As an example, electrostatic surfaces of β2m are shown weakly interacting with electrostatic surfaces of collagen I. (B) Surface models of the collagen I fibril repeating unit (built from PDB: 3HR2),62 color-coded by amino acid type (top, hydrophilic; bottom, hydrophobic). The repeating unit is ∼67 nm in length, however mature fibrils can be microns long and ∼500 nm in diameter. Distinct bands of electrostatic residues are observed within the repeating unit. (C) Surface representation of β2m monomer (PDB: 2XKS)49 color-coded by amino acid type (left, hydrophilic; right, hydrophobic). All models are color-coded as red, acidic; blue, basic; purple, uncharged-polar; and green, hydrophobic.
Collagen I is known to interact with multiple immunoglobulin-like protein folds through binding interfaces that include both hydrophobic and hydrophilic residues. Interactions of collagen I with osteoclast-associated receptor (OSCAR), leukocyte-associated immunoglobulin-like receptor-1 (LAIR-1), and glycoprotein VI (GPVI) play functional roles in immune system regulation63−66 and platelet activation.67−69 Similar to the β-sheet binding interface on β2m for collagen I identified here, the collagen I binding sites on OSCAR and LAIR-1 are also found in β-sheet regions.70,71 In the case of the OSCAR–collagen I interactions, Tyr and Arg residues that line the interacting β-sheet of OSCAR have been suggested to play a primary role.70 LAIR-1 binds primarily to collagen fragments rich in Gly, Pro, and hydroxyproline (GPO) content, but also has been shown to interact with multiple binding motifs in collagen II and III toolkit peptides, some of which are not GPO rich.71 NMR and mutagenesis studies on LAIR-1 have shown that depletion of Arg or Glu at the putative β-sheet interface showed decreased collagen binding, suggesting a role for electrostatic interactions.72 GPVI also recognizes GPO-rich collagen motifs, however through a unique hydrophobic groove formed by a β-strand connecting loop that is flanked by hydrophilic residues.73−75 Thus, although these proteins all share a similar immunoglobulin fold, each shows a unique binding interface to collagen, interacting in grooves formed by β-sheets or loops and having both hydrophobic and hydrophilic residues that each plays fundamental roles in binding.
Collagen-Induced Conformational Dynamics in β2m Reflect Amyloid Prone Dynamics
Beyond the structured collagen I-binding interface of β2m, using 15N relaxation experiments, we observe enhanced dynamics in the N- and C- terminal regions, the BC and FG loops, and the β-strand D of β2m upon complex formation. Enhanced dynamics in each of these regions has been proposed to play key roles in the aggregation mechanism of wild-type β2m.38,49−51,59,60,76−86 Amyloid formation of β2m is nucleation dependent and proceeds through a near native folding intermediate, IT, that is in part defined by a non-native trans-His31-Pro32 peptide bond in the BC loop.38,50,51,87,88 NMR studies of a P32G-β2m variant, which inherently has a trans-His31-Gly32 peptide bond, showed significant line broadening in β-strands A and D and the BC and FG loops relative to WT-β2m.38 This was interpreted to result from conformational conversion between the native and IT conformations.38 The observation of increased Rex of these same regions upon addition of collagen I to WT-β2m in this study is consistent with the same regions undergoing conformational exchange from the native state to enhance the formation of non-native species with enhanced amyloid potential.
The cis–trans isomerization of Pro32 is aided by displacement of the N-terminal six residues, as in the naturally occurring amyloidogenic variant, ΔN6,49,76,89,90 which destabilizes the protein, allowing β2m to sample multiple amyloidogenic conformations that enhance the rate of aggregation.38,50,51,76,81−83,87,88 In addition, NMR relaxation experiments show enhanced dynamics in β-strand D and the BC and DE loops of amyloidogenic ΔN6, which have been proposed to contribute to its higher aggregation propensity.49 Consistent with this, collagen I-induced enhancement of conformational exchange in the BC and DE loops of WT-β2m in this work may rationalize the increased β2m amyloidogenicity in the presence of collagen I.
Another proposed mechanism of the initiation of aggregation of WT-β2m involves destabilization of the edge β-strands A, D, C, and G to expose the β-strands with the highest aggregation propensity, namely β-strands B and E, unleashing their amyloid potential. Protection of the aggregation-prone β-strands was suggested to play a role in the amyloidogenicity of pathological variant D76N-β2m, which rapidly self-assembles into amyloid fibrils at neutral pH in vitro in the absence of cofactors,59 and an aggregation-resistant variant, W60G-β2m.60 Indeed, a recent study on D76N-β2m proposed that the enhanced aggregation propensity of this protein arises, in part, due to destabilization of the edge strands and conformational exchange in β-strand A, the BC loop, and the EF loop.59 In line with this hypothesis, reduced aggregation propensity of W60G-β2m, relative to WT-β2m, has been suggested to result from increased protection of the solvent exposed residues in the aggregation-prone β-strands, along with reduced dynamics of the protein, including in edge β-strands and the aggregation-prone β-strand B.60 Analogously, we find that collagen I enhances conformational exchange in the edge β-strands, A, D, C, and G, which may allow increased sampling of alternative conformations in these strands, deprotecting the aggregation-prone β-strands B and E, to enhance amyloidogenicity.
A Proposed Mechanism of Collagen I-Driven β2m Amyloidogenesis
With the new insights into the binding interface of collagen I on β2m and its impact on β2m dynamics described here, we propose a mechanistic view by which collagen I might drive amyloidogenesis of β2m (Figure 6). In the presence of collagen I, the β-sheets of β2m are available for binding to the collagen I surface, with both β-sheets providing potential binding interfaces, indicative of multiple binding modes, rather than a unique and specific binding interface. Through modification of the dynamics of β2m in the BC loop and the edge strand regions, concomitant sampling of amyloid-competent species, including the IT state,87,88 may be increased by a heightened probability of cis–trans isomerization of Pro3238,50,51 and/or exposure of highly amyloidogenic B and E β-strands, enabling them to realize their amyloid potential. Binding to collagen I may also concentrate bound β2m molecules, enhancing the probability of self-association, or may increase the potential for secondary nucleation on the collagen I surface, enhancing fibril formation. All, or several, of these mechanisms may be at play to catalyze β2m aggregation both in vitro and provide a molecular rationale for the deposition of β2m in collagenous-rich joints in dialysis patients.16,21−24 More generally, they also serve as an exemplar of the key role of the physiological environment in amyloid formation, by rationalizing the often remarkably specific deposition of amyloid to different tissues,1 and in some cases, of different variants of the same protein in different tissues.91,92 The methods used here to interrogate the weak-transient interaction of the large, β2m–collagen I complex can be extended to future studies to gain atomic-level insight into how other physiologically relevant cofactors promote amyloid formation of globular proteins involved in other amyloid diseases.
Figure 6.
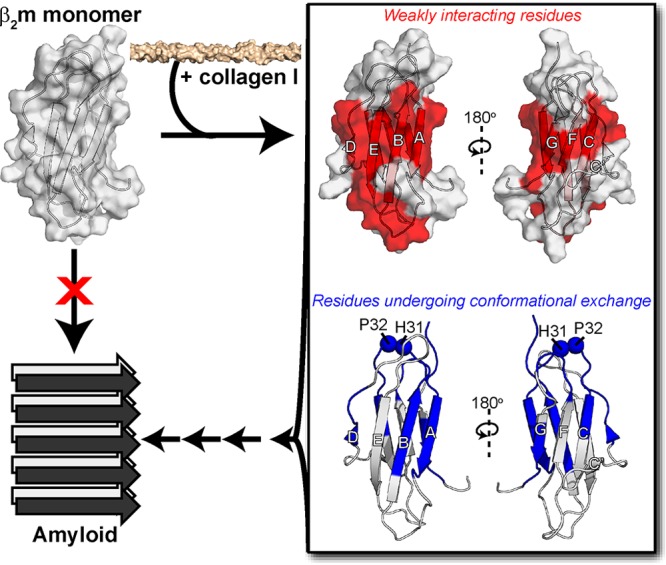
Proposed mechanism for collagen-driven β2m amyloidogenesis. Alone, the β2m monomer (PDB: 2XKS)49 does not readily aggregate into amyloid fibrils. Upon addition of collagen I, we have observed interaction interfaces to include both β-sheets of β2m through the 15N-DEST experiment (red). Collagen also induces conformational exchange in regions colored in blue, as assessed by 15N relaxation experiments. The interaction of collagen I with the structured regions of β2m enhances conformational exchange, promoting formation of amyloid-competent species and inducing aggregation.
Materials and Methods
Expression and Purification of β2m
Wild-type β2m was expressed recombinantly in Escherichia coli BL21(DE3) pLysS cells by induction with 1 mM IPTG overnight at 37 °C, following methods described previously.39 Cells were lysed in 25 mM Tris-HCl buffer, pH 8.0 and with an Avestin Emulsiflex-C5 homogenizer. β2m is accumulated in inclusion bodies. To extract the β2m from inclusion bodies, the cell pellet was washed five times with 25 mM Tris-HCl buffer, pH 8.0 and solubilized in 25 mM Tris-HCl, pH 8.0 buffer containing 8 M urea, rocking overnight at room temperature. The protein was verified to be in the soluble fraction by SDS-PAGE. β2m was refolded by dialyzing against 25 mM Tris-HCl buffer, pH 8.0 at 4 °C and purified by anion exchange (HiTrap Q HP, GE Healthcare). The protein was further purified by size exclusion chromatography with a HiLoad 26/600 Superdex 75 gel filtration column (GE Life Sciences). Protein purity was verified by SDS-PAGE, and concentrations for experiments were determined by measuring the absorbance at 280 nm using a molar extinction coefficient of 19,060 M–1 cm–1. [U–15N]-enriched β2m was expressed recombinantly for NMR using the same protocol in HCDM1 minimal media supplemented with 15N-ammonium chloride.
ELISA
Relative adhesion of variable concentrations of β2m to collagen I was determined by ELISA experiments. Nunc Maxisorp 96-well plates (Thermo Scientific) were coated with 100 μL of collagen I from rat tail tendon (BD Biosciences; 10 μg/mL in 10 mM acetic acid) overnight at 4 °C. Uncoated areas on the plates were blocked with 200 μL of 0.5% w/v casein in binding buffer at room temperature for 1 h. The binding and washing buffer consisted of PBS at pH 7.4 with 0.05% v/v Tween 20 (PBS-T) and 0.05% w/v casein as a nonspecific blocking agent. After washing the wells three times with 200 μL washing buffer, 100 μL β2m (10 μg/mL, 40 μg/mL, or 80 μg/mL) in PBS-T and 0.05% w/v casein was added to the wells and incubated for 1 h at room temperature. For the dose-dependent β2m–collagen I binding curve in Figure S1, β2m concentrations of 1, 10.5, 21.6, 33.2, 45.6, 58.6, 72.4, and 91.7 μM were used. After three washes with 200 μL washing buffer, 100 μL mouse anti-β2m monoclonal antibody (1:2000 v/v in PBS-T and 0.05% w/v casein, Millipore Sigma) was bound to β2m in each well by incubating at room temperature for 1 h. Subsequently, following three washes with 200 μL washing buffer, 100 μL of goat HRP-conjugated antimouse secondary antibody (1:5000 v/v dilution in PBS-T and 0.05% w/v casein, Genscript) was incubated in the wells at room temperature for 30 min. After washing for a final four times with 200 μL washing buffer, the binding of β2m to collagen I was detected through a colorimetric assay using a 3,3′,5,5′-tetramethylbenzidine substrate kit (Pierce) according to the manufacturer’s protocol and measuring the absorbance at 450 nm using a Tecan Infinite F50 plate reader with Magellan software.
ThT Fluorescence
Amyloid fibril formation was monitored by ThT fluorescence assays of β2m in the presence or absence of collagen I fibrils. Purified recombinant β2m lyophilized powder was dissolved in 100 μL of 10 mM sodium phosphate buffer, pH 7.4 or pH 6.2 to 1 mg/mL (85 μM). Varying concentrations of collagen I fibrils were separately prepared by incubating collagen I (3.4 mg/mL, 0.34 mg/mL, or 0.17 mg/mL; BD Biosciences) in 100 μL PBS, pH 7.4 at 37 °C for 1 h. Each 100 μL fibril suspension was sonicated in a bath sonicator for 10 min and centrifuged at 16,500 rpm for 10 min to isolate fibrils. Each collagen I fibril pellet was resuspended in 100 μL of 10 mM sodium phosphate buffer, pH 7.4 or pH 6.2 in the presence or absence of 1 mg/mL β2m. Three to six samples were prepared for each condition and were transferred to a 96-well plate. ThT was added to each sample to a final concentration of 10 μM. ThT fluorescence was monitored over 650 h at 37 °C with shaking at 600 rpm in a POLARstar Omega fluorimeter (BMG Labtech).
NMR
For all NMR experiments, purified recombinant [U–15N]-labeled β2m was diluted to 300 μM in TBS, pH 7.4 with 0.5 mg/mL casein and 10% v/v D2O. Before mixing, collagen I from rat tail tendon was dialyzed against TBS, pH 7.4. The concentration of collagen I after dialysis was determined by bicinchoninic acid assay (Pierce). All experiments were performed at 10 °C. All data were collected on 700 MHz Bruker AVIII or 900 MHz AVI NMR spectrometers equipped with TCI-cryo-probes. Data were processed in NMRPipe93 and analyzed in Sparky.94
1H–15N HSQC Spectra
1H–15N HSQC spectra95,96 of [U–15N]-labeled β2m were acquired with different concentrations of collagen I (0, 0.12 mg/mL, and 1.2 mg/mL) in TBS, pH 7.4 with 0.5 mg/mL casein and 10% D2O at 10 °C. The intensity ratio is taken as the intensity of a given cross-peak in the 1H–15N HSQC spectrum of β2m in the presence of collagen I relative to the intensity of the same cross-peak in the absence of collagen I, determined in Sparky.94 The errors were propagated from the signal-to-noise ratios.
15N-R2 and 15N-R2HE
[U–15N]-labeled β2m 15N transverse relaxation rates (R2) were measured from a series of HSQC-based 2D 1H–15N spectra using the Carr–Purcell–Meiboom–Gill pulse sequence97 with varying relaxation delays: in the absence of collagen I at 700 MHz- 0, 16, 16, 32, 48, 64, 64, 80, 96, and 112 ms and 900 MHz- 0, 16, 32, 32, 32, 48, 64, 80, 96, 112, and 128 ms and in the presence of 1.2 mg/mL collagen I at 700 mHz- 0, 16, 16, 32, 48, 48, 64, and 80 ms and at 900 MHz- 0, 16, 32, 32, 32, 48, 64, 80, and 96 ms. Relaxation delays used to quantify 15N-R2 rates of β2m in the presence of 0.6 mg/mL collagen I at 700 MHz were: 0, 8, 8, 16, 24, 32, and 56 ms and in the absence of collagen I: 0, 8, 8, 16, 24, 40, and 56 ms. 15N-R2HE informs on the chemical exchange contribution to R2 by using an in-phase Hahn echo experiment.98 Relaxation delays used in the R2HE experiment both in the presence and absence of 0.6 mg/mL collagen I were: 0.768, 7.68, 7.68, 15.4, 23, 38.5, and 61.4 ms. In each case, the R2 rates were determined by fitting peak intensities to a single exponential decay function. The chemical exchange contribution (Rex) for each β2m residue in the absence and presence of 0.6 mg/mL collagen I was determined as Rex = R2HE – R2.
DEST Experiments
The 15N-DEST experiment56,57 was applied to [U–15N]-labeled β2m in the presence or absence
of 0.6 mg/mL collagen I. In this experiment,
an 15N saturation pulse of 150 or 350 Hz was applied for
0.9 ms at different 15N frequency offsets: 0, ±1,
±2, ±4, ±8, ±14, ±21, and ±30 kHz. An
experiment in which the 15N saturation pulse was set to
0 Hz with an offset of 30 kHz was also included as a reference. The 15N-DEST profiles were extracted for each residue as the peak
intensity at each 15N saturation offset and were fitted
to a two-state model using the destfit program56,57 by Clore and co-workers to obtain R2bound, pbound, and konapp. The ΔΘ profile
was obtained by measuring Θ for each β2m residue
in the presence and absence of 0.6 mg/mL collagen I as 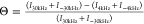 and taking ΔΘ = Θ+col – Θ–col.
and taking ΔΘ = Θ+col – Θ–col.
Acknowledgments
We acknowledge Arthur Palmer for helpful discussions. We also acknowledge with thanks the many discussions with our group members. We thank Ana Monica Nunes for contributions in the beginning of this project and Nuria Benseny-Cases, who provided critical insights in the early stages of the work.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.9b10421.
Figures that show weak, dose-dependent binding of β2m to collagen I by ELISA (Figure S1); ThT fluorescence experiments to assess the pH and collagen I concentration dependence of β2m amyloid formation (Figure S2); an overlay of 1H–15N HSQC spectra of [U–15N]-β2m in the presence of 0 mg/mL, 0.12 mg/mL, or 1.2 mg/mL collagen I (Figure S3); 15N-R2 measurements of [U–15N]-β2m in the absence or presence of collagen I at 700 or 900 MHz (Figure S4); and a model showing the amino acid composition of the interacting β2m β-sheets (Figure S5) (PDF)
This work was supported by American Heart Association Postdoctoral Fellowship 17POST33410326 to C.L.H. and NIH grant GM045302 to J.B. Additional support was provided by the Wellcome Trust (204963 and 092896) and the European Research Council (ERC) under European Union’s Seventh Framework Programme (FP7/2007–2013) ERC grant agreement no. 322408 to S.E.R. Some of the work presented here was conducted at the Center on Macromolecular Dynamics by NMR Spectroscopy located at the New York Structural Biology Center, supported by a grant from the NIH NIGMS (P41 GM118302) and ORIP/NIH facility improvement grant CO6RR015495. The 900 MHz NMR spectrometers were purchased with funds from NIH grant P41 GM066354, the Keck Foundation, New York State Assembly, and U.S. Department of Defense.
The authors declare no competing financial interest.
Supplementary Material
References
- Chiti F.; Dobson C. M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006, 75, 333–66. 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- Pham C. L.; Kwan A. H.; Sunde M. Functional amyloid: widespread in Nature, diverse in purpose. Essays Biochem. 2014, 56, 207–19. 10.1042/bse0560207. [DOI] [PubMed] [Google Scholar]
- Fowler D. M.; Koulov A. V.; Balch W. E.; Kelly J. W. Functional amyloid - from bacteria to humans. Trends Biochem. Sci. 2007, 32 (5), 217–224. 10.1016/j.tibs.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Iadanza M. G.; Jackson M. P.; Hewitt E. W.; Ranson N. A.; Radford S. E. A new era for understanding amyloid structures and disease. Nat. Rev. Mol. Cell Biol. 2018, 19 (12), 755–773. 10.1038/s41580-018-0060-8. [DOI] [PubMed] [Google Scholar]
- Knowles T. P.; Vendruscolo M.; Dobson C. M. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 2014, 15 (6), 384–96. 10.1038/nrm3810. [DOI] [PubMed] [Google Scholar]
- Chiti F.; Dobson C. M. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu. Rev. Biochem. 2017, 86, 27–68. 10.1146/annurev-biochem-061516-045115. [DOI] [PubMed] [Google Scholar]
- Eisenberg D. S.; Sawaya M. R. Structural Studies of Amyloid Proteins at the Molecular Level. Annu. Rev. Biochem. 2017, 86, 69–95. 10.1146/annurev-biochem-061516-045104. [DOI] [PubMed] [Google Scholar]
- Nizynski B.; Dzwolak W.; Nieznanski K. Amyloidogenesis of Tau protein. Protein Sci. 2017, 26 (11), 2126–2150. 10.1002/pro.3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsak M.; Kozyreva T. Beta Amyloid Hallmarks: From Intrinsically Disordered Proteins to Alzheimer’s Disease. Adv. Exp. Med. Biol. 2015, 870, 401–21. 10.1007/978-3-319-20164-1_14. [DOI] [PubMed] [Google Scholar]
- Darling A. L.; Uversky V. N. Intrinsic Disorder in Proteins with Pathogenic Repeat Expansions. Molecules 2017, 22 (12), 2027. 10.3390/molecules22122027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iannuzzi C.; Maritato R.; Irace G.; Sirangelo I. Misfolding and amyloid aggregation of apomyoglobin. Int. J. Mol. Sci. 2013, 14 (7), 14287–300. 10.3390/ijms140714287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiti F.; Dobson C. M. Amyloid formation by globular proteins under native conditions. Nat. Chem. Biol. 2009, 5 (1), 15–22. 10.1038/nchembio.131. [DOI] [PubMed] [Google Scholar]
- Iadanza M. G.; Silvers R.; Boardman J.; Smith H. I.; Karamanos T. K.; Debelouchina G. T.; Su Y.; Griffin R. G.; Ranson N. A.; Radford S. E. The structure of a beta2-microglobulin fibril suggests a molecular basis for its amyloid polymorphism. Nat. Commun. 2018, 9 (1), 4517. 10.1038/s41467-018-06761-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dember L. M.; Jaber B. L. Dialysis-related amyloidosis: late finding or hidden epidemic?. Semin Dial 2006, 19 (2), 105–9. 10.1111/j.1525-139X.2006.00134.x. [DOI] [PubMed] [Google Scholar]
- Dzido G.; Sprague S. M. Dialysis-related amyloidosis. Minerva Urol. Nefrol. 2003, 55 (2), 121–129. [PubMed] [Google Scholar]
- Gejyo F.; Yamada T.; Odani S.; Nakagawa Y.; Arakawa M.; Kunitomo T.; Kataoka H.; Suzuki M.; Hirasawa Y.; Shirahama T.; et al. A new form of amyloid protein associated with chronic hemodialysis was identified as beta 2-microglobulin. Biochem. Biophys. Res. Commun. 1985, 129 (3), 701–6. 10.1016/0006-291X(85)91948-5. [DOI] [PubMed] [Google Scholar]
- Muñoz-Gómez J.; Bergadá-Barado E.; Gómez-Pérez R.; Llopart-Buisán E.; Subías-Sobrevía E.; Rotés-Querol J.; Solé-Arqués M. Amyloid arthropathy in patients undergoing periodical haemodialysis for chronic renal failure: a new complication. Ann. Rheum. Dis. 1985, 44 (11), 729–733. 10.1136/ard.44.11.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker J. W.; Reeke G. N. Jr. Three-dimensional structure of beta 2-microglobulin. Proc. Natl. Acad. Sci. U. S. A. 1985, 82 (12), 4225–9. 10.1073/pnas.82.12.4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floege J.; Bartsch A.; Schulze M.; Shaldon S.; Koch K. M.; Smeby L. C. Clearance and synthesis rates of beta 2-microglobulin in patients undergoing hemodialysis and in normal subjects. J. Lab Clin Med. 1991, 118 (2), 153–165. [PubMed] [Google Scholar]
- Homma N.; Gejyo F.; Isemura M.; Arakawa M. Collagen-binding affinity of beta-2-microglobulin, a preprotein of hemodialysis-associated amyloidosis. Nephron 2004, 53 (1), 37–40. 10.1159/000185699. [DOI] [PubMed] [Google Scholar]
- Relini A.; Canale C.; De Stefano S.; Rolandi R.; Giorgetti S.; Stoppini M.; Rossi A.; Fogolari F.; Corazza A.; Esposito G.; Gliozzi A.; Bellotti V. Collagen plays an active role in the aggregation of beta2-microglobulin under physiopathological conditions of dialysis-related amyloidosis. J. Biol. Chem. 2006, 281 (24), 16521–9. 10.1074/jbc.M513827200. [DOI] [PubMed] [Google Scholar]
- Hadjipavlou A.; Lander P.; Begin L.; Bercovitch D.; Davidman M.; Jakab E. Skeletal amyloidosis due to beta microglobulinemia in a patient on hemodialysis. A case report. J. Bone Joint Surg Am. 1988, 70 (1), 119–21. 10.2106/00004623-198870010-00019. [DOI] [PubMed] [Google Scholar]
- Bardin T.; Kuntz D.; Zingraff J.; Voisin M.-C.; Zelmar A.; Lansaman J. Synovial amyloidosis in patients undergoing long-term hemodialysis. Arthritis Rheum. 1985, 28 (9), 1052–1058. 10.1002/art.1780280913. [DOI] [PubMed] [Google Scholar]
- Gejyo F.; Odani S.; Yamada T.; Honma N.; Saito H.; Suzuki Y.; Nakagawa Y.; Kobayashi H.; Maruyama Y.; Hirasawa Y.; et al. Beta 2-microglobulin: a new form of amyloid protein associated with chronic hemodialysis. Kidney Int. 1986, 30 (3), 385–90. 10.1038/ki.1986.196. [DOI] [PubMed] [Google Scholar]
- Eakin C. M.; Miranker A. D. From chance to frequent encounters: origins of beta2-microglobulin fibrillogenesis. Biochim. Biophys. Acta, Proteins Proteomics 2005, 1753 (1), 92–9. 10.1016/j.bbapap.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Platt G. W.; Radford S. E. Glimpses of the molecular mechanisms of beta2-microglobulin fibril formation in vitro: aggregation on a complex energy landscape. FEBS Lett. 2009, 583 (16), 2623–9. 10.1016/j.febslet.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moe S. M.; Chen N. X. The role of the synovium and cartilage in the pathogenesis of beta(2)-microglobulin amyloidosis. Semin Dial 2001, 14 (2), 127–30. 10.1046/j.1525-139x.2001.00032.x. [DOI] [PubMed] [Google Scholar]
- Relini A.; De Stefano S.; Torrassa S.; Cavalleri O.; Rolandi R.; Gliozzi A.; Giorgetti S.; Raimondi S.; Marchese L.; Verga L.; Rossi A.; Stoppini M.; Bellotti V. Heparin strongly enhances the formation of beta2-microglobulin amyloid fibrils in the presence of type I collagen. J. Biol. Chem. 2008, 283 (8), 4912–20. 10.1074/jbc.M702712200. [DOI] [PubMed] [Google Scholar]
- Benseny-Cases N.; Karamanos T. K.; Hoop C. L.; Baum J.; Radford S. E. Extracellular matrix components modulate different stages in beta2-microglobulin amyloid formation. J. Biol. Chem. 2019, 294 (24), 9392–9401. 10.1074/jbc.RA119.008300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgetti S.; Rossi A.; Mangione P.; Raimondi S.; Marini S.; Stoppini M.; Corazza A.; Viglino P.; Esposito G.; Cetta G.; Merlini G.; Bellotti V. Beta2-microglobulin isoforms display an heterogeneous affinity for type I collagen. Protein Sci. 2005, 14 (3), 696–702. 10.1110/ps.041194005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers S. L.; Jones S.; Jahn T. R.; Morten I. J.; Tennent G. A.; Hewitt E. W.; Radford S. E. A systematic study of the effect of physiological factors on beta2-microglobulin amyloid formation at neutral pH. Biochemistry 2006, 45 (7), 2311–21. 10.1021/bi052434i. [DOI] [PubMed] [Google Scholar]
- Yamamoto S.; Yamaguchi I.; Hasegawa K.; Tsutsumi S.; Goto Y.; Gejyo F.; Naiki H. Glycosaminoglycans enhance the trifluoroethanol-induced extension of beta 2-microglobulin-related amyloid fibrils at a neutral pH. J. Am. Soc. Nephrol. 2004, 15 (1), 126–33. 10.1097/01.ASN.0000103228.81623.C7. [DOI] [PubMed] [Google Scholar]
- Srikanth R.; Mendoza V. L.; Bridgewater J. D.; Zhang G.; Vachet R. W. Copper binding to beta-2-microglobulin and its pre-amyloid oligomers. Biochemistry 2009, 48 (41), 9871–81. 10.1021/bi901172y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antwi K.; Mahar M.; Srikanth R.; Olbris M. R.; Tyson J. F.; Vachet R. W. Cu(II) organizes beta-2-microglobulin oligomers but is released upon amyloid formation. Protein Sci. 2008, 17 (4), 748–59. 10.1110/ps.073249008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese M. F.; Miranker A. D. Metal binding sheds light on mechanisms of amyloid assembly. Prion 2009, 3 (1), 1–4. 10.4161/pri.3.1.8601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese M. F.; Eakin C. M.; Wang J. M.; Miranker A. D. A regulatable switch mediates self-association in an immunoglobulin fold. Nat. Struct. Mol. Biol. 2008, 15 (9), 965–71. 10.1038/nsmb.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese M. F.; Miranker A. D. Formation of a stable oligomer of beta-2 microglobulin requires only transient encounter with Cu(II). J. Mol. Biol. 2007, 367 (1), 1–7. 10.1016/j.jmb.2006.12.034. [DOI] [PubMed] [Google Scholar]
- Jahn T. R.; Parker M. J.; Homans S. W.; Radford S. E. Amyloid formation under physiological conditions proceeds via a native-like folding intermediate. Nat. Struct. Mol. Biol. 2006, 13 (3), 195–201. 10.1038/nsmb1058. [DOI] [PubMed] [Google Scholar]
- McParland V. J.; Kad N. M.; Kalverda A. P.; Brown A.; Kirwin-Jones P.; Hunter M. G.; Sunde M.; Radford S. E. Partially unfolded states of beta(2)-microglobulin and amyloid formation in vitro. Biochemistry 2000, 39 (30), 8735–46. 10.1021/bi000276j. [DOI] [PubMed] [Google Scholar]
- Morgan C. J.; Gelfand M.; Atreya C.; Miranker A. D. Kidney dialysis-associated amyloidosis: a molecular role for copper in fiber formation. J. Mol. Biol. 2001, 309 (2), 339–45. 10.1006/jmbi.2001.4661. [DOI] [PubMed] [Google Scholar]
- Yamamoto S.; Hasegawa K.; Yamaguchi I.; Tsutsumi S.; Kardos J.; Goto Y.; Gejyo F.; Naiki H. Low concentrations of sodium dodecyl sulfate induce the extension of beta 2-microglobulin-related amyloid fibrils at a neutral pH. Biochemistry 2004, 43 (34), 11075–82. 10.1021/bi049262u. [DOI] [PubMed] [Google Scholar]
- Borysik A. J.; Morten I. J.; Radford S. E.; Hewitt E. W. Specific glycosaminoglycans promote unseeded amyloid formation from beta2-microglobulin under physiological conditions. Kidney Int. 2007, 72 (2), 174–81. 10.1038/sj.ki.5002270. [DOI] [PubMed] [Google Scholar]
- Gosal W. S.; Morten I. J.; Hewitt E. W.; Smith D. A.; Thomson N. H.; Radford S. E. Competing pathways determine fibril morphology in the self-assembly of beta2-microglobulin into amyloid. J. Mol. Biol. 2005, 351 (4), 850–64. 10.1016/j.jmb.2005.06.040. [DOI] [PubMed] [Google Scholar]
- Ricagno S.; Colombo M.; de Rosa M.; Sangiovanni E.; Giorgetti S.; Raimondi S.; Bellotti V.; Bolognesi M. DE loop mutations affect beta2-microglobulin stability and amyloid aggregation. Biochem. Biophys. Res. Commun. 2008, 377 (1), 146–150. 10.1016/j.bbrc.2008.09.108. [DOI] [PubMed] [Google Scholar]
- Santambrogio C.; Ricagno S.; Colombo M.; Barbiroli A.; Bonomi F.; Bellotti V.; Bolognesi M.; Grandori R. DE-loop mutations affect beta2 microglobulin stability, oligomerization, and the low-pH unfolded form. Protein Sci. 2010, 19 (7), 1386–94. 10.1002/pro.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halabelian L.; Relini A.; Barbiroli A.; Penco A.; Bolognesi M.; Ricagno S. A covalent homodimer probing early oligomers along amyloid aggregation. Sci. Rep. 2015, 5, 14651. 10.1038/srep14651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leney A. C.; Pashley C. L.; Scarff C. A.; Radford S. E.; Ashcroft A. E. Insights into the role of the beta-2 microglobulin D-strand in amyloid propensity revealed by mass spectrometry. Mol. BioSyst. 2014, 10 (3), 412–20. 10.1039/C3MB70420C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narang D.; Singh A.; Swasthi H. M.; Mukhopadhyay S. Characterization of Salt-Induced Oligomerization of Human beta2-Microglobulin at Low pH. J. Phys. Chem. B 2016, 120 (32), 7815–23. 10.1021/acs.jpcb.6b05619. [DOI] [PubMed] [Google Scholar]
- Eichner T.; Kalverda A. P.; Thompson G. S.; Homans S. W.; Radford S. E. Conformational conversion during amyloid formation at atomic resolution. Mol. Cell 2011, 41 (2), 161–72. 10.1016/j.molcel.2010.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kameda A.; Hoshino M.; Higurashi T.; Takahashi S.; Naiki H.; Goto Y. Nuclear magnetic resonance characterization of the refolding intermediate of beta2-microglobulin trapped by non-native prolyl peptide bond. J. Mol. Biol. 2005, 348 (2), 383–97. 10.1016/j.jmb.2005.02.050. [DOI] [PubMed] [Google Scholar]
- Sakata M.; Chatani E.; Kameda A.; Sakurai K.; Naiki H.; Goto Y. Kinetic coupling of folding and prolyl isomerization of beta2-microglobulin studied by mutational analysis. J. Mol. Biol. 2008, 382 (5), 1242–55. 10.1016/j.jmb.2008.08.003. [DOI] [PubMed] [Google Scholar]
- Janowska M. K.; Wu K. P.; Baum J. Unveiling transient protein-protein interactions that modulate inhibition of alpha-synuclein aggregation by beta-synuclein, a pre-synaptic protein that co-localizes with alpha-synuclein. Sci. Rep. 2015, 5, 15164. 10.1038/srep15164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu K. P.; Baum J. Detection of transient interchain interactions in the intrinsically disordered protein alpha-synuclein by NMR paramagnetic relaxation enhancement. J. Am. Chem. Soc. 2010, 132 (16), 5546–7. 10.1021/ja9105495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian L. Y. NMR studies of weak protein-protein interactions. Prog. Nucl. Magn. Reson. Spectrosc. 2013, 71, 59–72. 10.1016/j.pnmrs.2012.11.002. [DOI] [PubMed] [Google Scholar]
- Vinogradova O.; Qin J. NMR as a unique tool in assessment and complex determination of weak protein-protein interactions. Top. Curr. Chem. 2011, 326, 35–45. 10.1007/128_2011_216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawzi N. L.; Ying J.; Ghirlando R.; Torchia D. A.; Clore G. M. Atomic-resolution dynamics on the surface of amyloid-beta protofibrils probed by solution NMR. Nature 2011, 480 (7376), 268–72. 10.1038/nature10577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawzi N. L.; Ying J.; Torchia D. A.; Clore G. M. Probing exchange kinetics and atomic resolution dynamics in high-molecular-weight complexes using dark-state exchange saturation transfer NMR spectroscopy. Nat. Protoc. 2012, 7 (8), 1523–33. 10.1038/nprot.2012.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karamanos T. K.; Kalverda A. P.; Thompson G. S.; Radford S. E. Visualization of transient protein-protein interactions that promote or inhibit amyloid assembly. Mol. Cell 2014, 55 (2), 214–26. 10.1016/j.molcel.2014.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Marchand T.; de Rosa M.; Salvi N.; Sala B. M.; Andreas L. B.; Barbet-Massin E.; Sormanni P.; Barbiroli A.; Porcari R.; Sousa Mota C.; de Sanctis D.; Bolognesi M.; Emsley L.; Bellotti V.; Blackledge M.; Camilloni C.; Pintacuda G.; Ricagno S. Conformational dynamics in crystals reveal the molecular bases for D76N beta-2 microglobulin aggregation propensity. Nat. Commun. 2018, 9 (1), 1658. 10.1038/s41467-018-04078-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camilloni C.; Sala B. M.; Sormanni P.; Porcari R.; Corazza A.; De Rosa M.; Zanini S.; Barbiroli A.; Esposito G.; Bolognesi M.; Bellotti V.; Vendruscolo M.; Ricagno S. Rational design of mutations that change the aggregation rate of a protein while maintaining its native structure and stability. Sci. Rep. 2016, 6, 25559. 10.1038/srep25559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn T. R.; Tennent G. A.; Radford S. E. A common beta-sheet architecture underlies in vitro and in vivo beta2-microglobulin amyloid fibrils. J. Biol. Chem. 2008, 283 (25), 17279–86. 10.1074/jbc.M710351200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orgel J. P.; Irving T. C.; Miller A.; Wess T. J. Microfibrillar structure of type I collagen in situ. Proc. Natl. Acad. Sci. U. S. A. 2006, 103 (24), 9001–5. 10.1073/pnas.0502718103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merck E.; Gaillard C.; Gorman D. M.; Montero-Julian F.; Durand I.; Zurawski S. M.; Menetrier-Caux C.; Carra G.; Lebecque S.; Trinchieri G.; Bates E. E. OSCAR is an FcRgamma-associated receptor that is expressed by myeloid cells and is involved in antigen presentation and activation of human dendritic cells. Blood 2004, 104 (5), 1386–95. 10.1182/blood-2004-03-0850. [DOI] [PubMed] [Google Scholar]
- Kim N.; Takami M.; Rho J.; Josien R.; Choi Y. A novel member of the leukocyte receptor complex regulates osteoclast differentiation. J. Exp. Med. 2002, 195 (2), 201–9. 10.1084/jem.20011681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyaard L.; Hurenkamp J.; Clevers H.; Lanier L. L.; Phillips J. H. Leukocyte-associated Ig-like receptor-1 functions as an inhibitory receptor on cytotoxic T cells. J. Immunol 1999, 162 (10), 5800–5804. [PubMed] [Google Scholar]
- Poggi A.; Tomasello E.; Ferrero E.; Zocchi M. R.; Moretta L. p40/LAIR-1 regulates the differentiation of peripheral blood precursors to dendritic cells induced by granulocyte-monocyte colony-stimulating factor. Eur. J. Immunol. 1998, 28 (7), 2086–91. . [DOI] [PubMed] [Google Scholar]
- Ruggeri Z. M. Platelets in atherothrombosis. Nat. Med. 2002, 8 (11), 1227–34. 10.1038/nm1102-1227. [DOI] [PubMed] [Google Scholar]
- Moroi M.; Jung S. M. Platelet glycoprotein VI: its structure and function. Thromb. Res. 2004, 114 (4), 221–33. 10.1016/j.thromres.2004.06.046. [DOI] [PubMed] [Google Scholar]
- Kahn M. L. Platelet-collagen responses: molecular basis and therapeutic promise. Semin. Thromb. Hemostasis 2004, 30 (4), 419–25. 10.1055/s-2004-833477. [DOI] [PubMed] [Google Scholar]
- Zhou L.; Hinerman J. M.; Blaszczyk M.; Miller J. L.; Conrady D. G.; Barrow A. D.; Chirgadze D. Y.; Bihan D.; Farndale R. W.; Herr A. B. Structural basis for collagen recognition by the immune receptor OSCAR. Blood 2016, 127 (5), 529–37. 10.1182/blood-2015-08-667055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebbink R. J.; Raynal N.; de Ruiter T.; Bihan D. G.; Farndale R. W.; Meyaard L. Identification of multiple potent binding sites for human leukocyte associated Ig-like receptor LAIR on collagens II and III. Matrix Biol. 2009, 28 (4), 202–10. 10.1016/j.matbio.2009.03.005. [DOI] [PubMed] [Google Scholar]
- Brondijk T. H.; de Ruiter T.; Ballering J.; Wienk H.; Lebbink R. J.; van Ingen H.; Boelens R.; Farndale R. W.; Meyaard L.; Huizinga E. G. Crystal structure and collagen-binding site of immune inhibitory receptor LAIR-1: unexpected implications for collagen binding by platelet receptor GPVI. Blood 2010, 115 (7), 1364–73. 10.1182/blood-2009-10-246322. [DOI] [PubMed] [Google Scholar]
- Smethurst P. A.; Joutsi-Korhonen L.; O’Connor M. N.; Wilson E.; Jennings N. S.; Garner S. F.; Zhang Y.; Knight C. G.; Dafforn T. R.; Buckle A.; MJ I. J.; De Groot P. G.; Watkins N. A.; Farndale R. W.; Ouwehand W. H. Identification of the primary collagen-binding surface on human glycoprotein VI by site-directed mutagenesis and by a blocking phage antibody. Blood 2004, 103 (3), 903–911. 10.1182/blood-2003-01-0308. [DOI] [PubMed] [Google Scholar]
- Horii K.; Kahn M. L.; Herr A. B. Structural basis for platelet collagen responses by the immune-type receptor glycoprotein VI. Blood 2006, 108 (3), 936–42. 10.1182/blood-2006-01-010215. [DOI] [PubMed] [Google Scholar]
- O’Connor M. N.; Smethurst P. A.; Farndale R. W.; Ouwehand W. H. Gain- and loss-of-function mutants confirm the importance of apical residues to the primary interaction of human glycoprotein VI with collagen. J. Thromb. Haemostasis 2006, 4 (4), 869–73. 10.1111/j.1538-7836.2005.01764.x. [DOI] [PubMed] [Google Scholar]
- Eichner T.; Radford S. E. Understanding the complex mechanisms of beta2-microglobulin amyloid assembly. FEBS J. 2011, 278 (20), 3868–83. 10.1111/j.1742-4658.2011.08186.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdone G.; Corazza A.; Viglino P.; Pettirossi F.; Giorgetti S.; Mangione P.; Andreola A.; Stoppini M.; Bellotti V.; Esposito G. The solution structure of human beta2-microglobulin reveals the prodromes of its amyloid transition. Protein Sci. 2002, 11 (3), 487–99. 10.1110/ps.29002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricagno S.; Raimondi S.; Giorgetti S.; Bellotti V.; Bolognesi M. Human beta-2 microglobulin W60V mutant structure: Implications for stability and amyloid aggregation. Biochem. Biophys. Res. Commun. 2009, 380 (3), 543–7. 10.1016/j.bbrc.2009.01.116. [DOI] [PubMed] [Google Scholar]
- Esposito G.; Corazza A.; Viglino P.; Verdone G.; Pettirossi F.; Fogolari F.; Makek A.; Giorgetti S.; Mangione P.; Stoppini M.; Bellotti V. Solution structure of beta(2)-microglobulin and insights into fibrillogenesis. Biochim. Biophys. Acta, Proteins Proteomics 2005, 1753 (1), 76–84. 10.1016/j.bbapap.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Trinh C. H.; Smith D. P.; Kalverda A. P.; Phillips S. E.; Radford S. E. Crystal structure of monomeric human beta-2-microglobulin reveals clues to its amyloidogenic properties. Proc. Natl. Acad. Sci. U. S. A. 2002, 99 (15), 9771–6. 10.1073/pnas.152337399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodkinson J. P.; Jahn T. R.; Radford S. E.; Ashcroft A. E. HDX-ESI-MS reveals enhanced conformational dynamics of the amyloidogenic protein beta(2)-microglobulin upon release from the MHC-1. J. Am. Soc. Mass Spectrom. 2009, 20 (2), 278–86. 10.1016/j.jasms.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rennella E.; Corazza A.; Fogolari F.; Viglino P.; Giorgetti S.; Stoppini M.; Bellotti V.; Esposito G. Equilibrium unfolding thermodynamics of beta2-microglobulin analyzed through native-state H/D exchange. Biophys. J. 2009, 96 (1), 169–79. 10.1529/biophysj.108.142448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rennella E.; Corazza A.; Giorgetti S.; Fogolari F.; Viglino P.; Porcari R.; Verga L.; Stoppini M.; Bellotti V.; Esposito G. Folding and fibrillogenesis: clues from beta2-microglobulin. J. Mol. Biol. 2010, 401 (2), 286–97. 10.1016/j.jmb.2010.06.016. [DOI] [PubMed] [Google Scholar]
- Armen R. S.; Daggett V. Characterization of two distinct beta2-microglobulin unfolding intermediates that may lead to amyloid fibrils of different morphology. Biochemistry 2005, 44 (49), 16098–107. 10.1021/bi050731h. [DOI] [PubMed] [Google Scholar]
- Fogolari F.; Corazza A.; Viglino P.; Zuccato P.; Pieri L.; Faccioli P.; Bellotti V.; Esposito G. Molecular dynamics simulation suggests possible interaction patterns at early steps of beta2-microglobulin aggregation. Biophys. J. 2007, 92 (5), 1673–81. 10.1529/biophysj.106.098483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corazza A.; Rennella E.; Schanda P.; Mimmi M. C.; Cutuil T.; Raimondi S.; Giorgetti S.; Fogolari F.; Viglino P.; Frydman L.; Gal M.; Bellotti V.; Brutscher B.; Esposito G. Native-unlike long-lived intermediates along the folding pathway of the amyloidogenic protein beta2-microglobulin revealed by real-time two-dimensional NMR. J. Biol. Chem. 2010, 285 (8), 5827–35. 10.1074/jbc.M109.061168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiti F.; De Lorenzi E.; Grossi S.; Mangione P.; Giorgetti S.; Caccialanza G.; Dobson C. M.; Merlini G.; Ramponi G.; Bellotti V. A partially structured species of beta 2-microglobulin is significantly populated under physiological conditions and involved in fibrillogenesis. J. Biol. Chem. 2001, 276 (50), 46714–21. 10.1074/jbc.M107040200. [DOI] [PubMed] [Google Scholar]
- Chiti F.; Mangione P.; Andreola A.; Giorgetti S.; Stefani M.; Dobson C. M.; Bellotti V.; Taddei N. Detection of two partially structured species in the folding process of the amyloidogenic protein beta 2-microglobulin. J. Mol. Biol. 2001, 307 (1), 379–91. 10.1006/jmbi.2000.4478. [DOI] [PubMed] [Google Scholar]
- Esposito G.; Michelutti R.; Verdone G.; Viglino P.; Hernandez H.; Robinson C. V.; Amoresano A.; Dal Piaz F.; Monti M.; Pucci P.; Mangione P.; Stoppini M.; Merlini G.; Ferri G.; Bellotti V. Removal of the N-terminal hexapeptide from human beta2-microglobulin facilitates protein aggregation and fibril formation. Protein Sci. 2000, 9 (5), 831–845. 10.1110/ps.9.5.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monti M.; Amoresano A.; Giorgetti S.; Bellotti V.; Pucci P. Limited proteolysis in the investigation of beta2-microglobulin amyloidogenic and fibrillar states. Biochim. Biophys. Acta, Proteins Proteomics 2005, 1753 (1), 44–50. 10.1016/j.bbapap.2005.09.004. [DOI] [PubMed] [Google Scholar]
- Mangione P. P.; Esposito G.; Relini A.; Raimondi S.; Porcari R.; Giorgetti S.; Corazza A.; Fogolari F.; Penco A.; Goto Y.; Lee Y. H.; Yagi H.; Cecconi C.; Naqvi M. M.; Gillmore J. D.; Hawkins P. N.; Chiti F.; Rolandi R.; Taylor G. W.; Pepys M. B.; Stoppini M.; Bellotti V. Structure, folding dynamics, and amyloidogenesis of D76N beta2-microglobulin: roles of shear flow, hydrophobic surfaces, and alpha-Crystallin. J. Biol. Chem. 2013, 288 (43), 30917–30. 10.1074/jbc.M113.498857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson S. M.; Connelly S.; Fearns C.; Powers E. T.; Kelly J. W. The transthyretin amyloidoses: from delineating the molecular mechanism of aggregation linked to pathology to a regulatory-agency-approved drug. J. Mol. Biol. 2012, 421 (2–3), 185–203. 10.1016/j.jmb.2011.12.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio F.; Grzesiek S.; Vuister G. W.; Zhu G.; Pfeifer J.; Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6 (3), 277–293. 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Goddard T. D.; Kneller D. G.. SPARKY 3; University of California: San Fransisco, CA, 2008. [Google Scholar]
- Palmer A. G.; Cavanagh J.; Wright P. E.; Rance M. Sensitivity improvement in proton-detected two-dimensional heteronuclear correlation NMR spectroscopy. J. Magn. Reson. (1969-1992) 1991, 93 (1), 151–170. 10.1016/0022-2364(91)90036-S. [DOI] [Google Scholar]
- Kay L.; Keifer P.; Saarinen T. Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. J. Am. Chem. Soc. 1992, 114 (26), 10663–10665. 10.1021/ja00052a088. [DOI] [Google Scholar]
- Hansen D. F.; Vallurupalli P.; Kay L. E. An improved 15N relaxation dispersion experiment for the measurement of millisecond time-scale dynamics in proteins. J. Phys. Chem. B 2008, 112 (19), 5898–904. 10.1021/jp074793o. [DOI] [PubMed] [Google Scholar]
- Millet O.; Loria J. P.; Kroenke C. D.; Pons M.; Palmer A. G. The static magnetic field dependence of chemical exchange linebroadening defines the NMR chemical shift time scale. J. Am. Chem. Soc. 2000, 122 (12), 2867–2877. 10.1021/ja993511y. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.