

Abstract
Background:
Sphingolipids have recently emerged as a biomarker of recurrence and mortality after myocardial infarction (MI). The increased ceramide levels in mammalian heart tissues during acute MI, as demonstrated by several groups, is associated with higher cell death rates in the left ventricle (LV) and deteriorated cardiac function. Ceramidase, the only enzyme known to hydrolyze pro-apoptotic ceramide, generates sphingosine, which is then phosphorylated by sphingosine kinase (Sphk) to produce the pro-survival molecule sphingosine-1-phosphate (S1P). We hypothesized that Acid Ceramidase (AC) overexpression would counteract the negative effects of elevated ceramide and promote cell survival, thereby providing cardioprotection after MI.
Methods:
We performed transcriptomic, sphingolipid and protein analyses to evaluate sphingolipid metabolism and signaling post MI. We investigated the effect of altering ceramide metabolism through a loss (chemical inhibitors) or gain (modified mRNA (modRNA)) of AC function post hypoxia or MI.
Results:
We found that several genes involved in de novo ceramide synthesis were upregulated and that ceramide (C16, C20, C20:1 and C24) levels had significantly increased 24 hours after MI. AC inhibition post hypoxia or MI resulted in reduced AC activity and increased cell death; by contrast, enhancing AC activity via AC modRNA treatment increased cell survival post hypoxia or MI. AC modRNA-treated mice had significantly better heart function, longer survival and smaller scar size than control mice 28 days post MI. We attributed the improvement in heart function post MI following AC modRNA delivery to decreased ceramide levels, lower cell death rates and changes in the composition of the immune cell population in the LV manifested by lowered abundance of pro-inflammatory detrimental neutrophils.
Conclusions:
Our findings suggest that transiently altering sphingolipid metabolism through AC overexpression is sufficient and necessary to induce cardioprotection post MI, thereby highlighting the therapeutic potential of AC modRNA in ischemic heart disease.
Keywords: sphingolipid metabolism, Acid Ceramidase, Modified mRNA, myocardial infarction, Cardioprotection
Introduction
Myocardial infarction is an acute, life-threatening medical condition caused by the blockage of a coronary artery, leading to ischemia and, eventually, to cell death in the affected heart area. Cardiac tissue has a low regenerative capacity. MI thus leads to cardiac scarring and cardiac remodeling that increase the risk of heart failure (HF). The extent of MI and the risk of developing HF after acute MI are positively correlated in patients.1 Several therapeutic strategies based on inhibiting controlled cell death, attenuating inflammation and inducing regeneration have been proposed for protecting MI survivors against HF.2, 3
Lipid levels and composition in patient blood during acute MI have been shown to predict the risk of complications.4 In particular, high plasma ceramide concentrations are associated with a higher probability of MI recurrence and death.4, 5 Ceramides are simple membrane sphingolipids that form the backbone of all complex sphingolipids.6 High cellular ceramide levels can trigger programmed cell death.7 In recent years, a few research groups have shown that ceramide levels are high in the heart tissues of rodents and humans during acute MI8–10 and that blocking de novo ceramide synthesis in rodents can improve heart function post MI.8, 9
Ceramidases hydrolyze ceramide to generate free fatty acids and sphingosine, which is then phosphorylated by sphingosine kinase (Sphk) to produce sphingosine 1-phosphate (S1P), a pro-survival lipid mediator with both intra- and extracellular functions.11, 12 More specifically, acid ceramidase (AC) is encoded by the Asah1 gene and catalyzes ceramide hydrolysis to free fatty acids and sphingosine, which is then phosphorylated by Sphk (1 and 2) to generate S1P.13 Asah1 gene mutations lead to ceramidase deficiency and cause Farber lipogranulomatosis, a lysosomal storage disease.13 AC is essential for embryogenesis, and Asah1−/− embryos undergo apoptotic death at the two-cell stage.12 AC is a lysosomal and secreted enzyme, with optimal in vitro activity in acidic conditions,14 and belongs to the N-terminal nucleophile hydrolase family. The autoproteolytic cleavage of AC generates two active subunits: the α subunit with a molecular weight of ~14 kDa and the β subunit with a molecular weight of ~43 kDa.15 The autoproteolytic cleavage of the precursor triggers a conformational change that reveals the active site and activates the enzyme toward sphingolipid metabolism.16 It has been suggested that interfering with the signal transduction pathways mediated by sphingolipids could prevent cell death post MI. Recent studies have suggested that S1P could be used as a therapeutic target in patients with heart failure17 and MI,18 to prolong cardiac cell survival and consequently improve heart function. While S1P lyase inhibition causes increased cardiac S1P levels and bradycardia in rats,19 S1P receptor agonist, FTY720, boosts myocardial salvage and enhances heart function in a porcine model of ischemia/reperfusion injury.20 Inhibiting de novo ceramide synthesis has also been suggested as a strategy for reducing the pro-apoptotic effect of ceramide post MI.9 Indeed, inhibiting acid sphingomyelinase, which hydrolyzes spingomyelin to generate ceramide, limits ceramide accumulation in post-ischemic hearts.10 Moreover, adiponectin seems to exert its anti-apoptotic effect on CMs through adiponectin receptor-mediated ceramidase activity.21
We investigated using AC and/or Sphk enzymes to inhibit cell death and initiate cell survival through ceramide hydrolysis and S1P production. Delivering AC or Sphk proteins is safe and controlled, but their effects are limited by these proteins’ half-lives. Conversely, using DNA or viruses (DNA or RNA viruses) is not controlled and may elicit an immune response that could compromise genome integrity. We therefore delivered AC and Sphk via synthetic modified mRNA (modRNA), a nucleic acid delivery tool, to transiently alter sphingolipid metabolism. Our group and others have shown that modRNA is a highly efficient system for delivery to the heart, rapidly yielding transient expression with no signs of innate immunoresponse.22–28 More specifically, we have shown that synthesized modRNAs, in which all uridine residues are replaced with pseudouridine-5’-triphosphate or N1-methylpseudouridine-5’-triphosphate (1-mψU), result in more efficient translation, with lower immunogenicity and greater resistance to RNase cleavage than unmodified mRNA in cardiac cells and tissue.27 ModRNA, which has a unique, transient, pulse-like pharmacokinetic profile, is translated within minutes and the resulting protein remains detectable for ~5–7 days in vitro and ~10 days in vivo.27 This distinctive kinetic profile makes modRNA approaches ideal for immediately and transiently altering sphingolipid metabolism in the heart post MI. While insulin-like growth factor 1 (IGF-1) modRNA delivered immediately after MI has been shown to decrease CM apoptosis, this treatment’s long-term effects on heart function are remain unclear.22 We recently showed that delivering IGF-1 modRNA to the heart post MI induces detrimental epicardial adipose tissue formation by differentiate epicardial derived cells into fat cells 28 days post MI.25
In this study, we investigated sphingolipid level dynamics and the expression of genes involved in sphingolipid metabolism and signaling pathways post MI. Loss (chemical inhibitors) or gain (modRNA) of AC function showed that inhibiting AC activity increased cell death and, by contrast, enhancing AC activity reduced cell death under hypoxia conditions in vitro or during MI in vivo. Moreover, we show that enhancing AC activity attenuated inflammation post MI by decreasing detrimental neutrophil levels in the LV, thereby improving heart function and long-term survival after ischemic injury.
Methods
All methods and materials used in this study are described in detail in the Supplementary Materials and Methods section. Below please see a brief summary of the most relevant ones.
Mice
All animal procedures were performed according to protocols approved by the Icahn School of Medicine at Mount Sinai Institutional Care and Use Committee. CFW male and female mice were used. For protein expression, mice were injected intramyocardially with 100 μg of Luc, Sphk1 or modRNA in citrate buffer. MI was induced by permanent LAD ligation. When required, 100 μg of modRNA was injected into the border zone immediately after LAD ligation. For the inhibitor assay, 10mg/kg of ARN14974 was injected IP at the time and 7 hours post LAD ligation. Heart function was determined using echocardiography and MRI.
ModRNA Synthesis
Clean PCR products generated with plasmid templates (GeneArt, Thermo Fisher Scientific) were used as the template for mRNA (for a complete list of the open reading frames used in this study, see Supplementary Table 1). ModRNAs were produced by transcription in vitro with a customized ribonucleoside blend and DNA PCR products with plasmid templates, followed by Antarctic Phosphatase treatment and purification. Please see supplemental materials methods section for more detailes.
nrCM isolation and hPSC differentiation
Ventrical nrCMs were isolated from 3- to 4-day-old Sprague-Dawley rats by multiple digestion series using 0.1% collagenase II. We obtained hPSCs-derived CMs by using the embryoid bodies (EBs) formatting method.
In vitro modRNA transfection
Neonatal rat hPSC-derived CMs were transfected with 2.5 μg of modRNA per well (on 24 well plate) encoding nGFP, AC, Sphk1, SphK2 or S1pr2 using RNAiMAX (Life Technologies).
Sphingolipid composition and AC activity
To determine the sphingolipid composition of mouse hearts, samples were analyzed at the Stony Brook Lipidomics facility. AC activity in heart tissue lysate was analyzed by HPLC-MS/MS.
Immunofluorescence
Hearts were perfused and fixed with 4% PFA, followed by incubation with 30% sucrose and mounting in OCT. Transversal 10 μm-thick sections were stained overnight with primary antibodies followed by appropriate secondary antibodies, as listed in Supplementary Table 2, depending on the experiment. To detect apoptotic cells in the heart tissue, TUNEL assay was performed according to manufacturer’s protocol. To stain cells, coverslips with previously seeded cells were fixed with 4%PFA and stained with appropriate antibodies. Images were quantified using ImageJ software.
Flow cytometry
Infarct zones of AC modRNA- or Luc modRNA-treated hearts were collected at 2, 7 and 14 days post MI and processed according to a previously described protocol29 with a few modifications. Isolated cells were first stained with hematopoietic lineage markers followed by a second staining with specific cell markers. Neutrophils were defined as CD45+ CD11b+ Ly6G+, Macrophages as CD45+ CD11b+ Lin- F4/80+ Ly6Clow/int and Monocytes as CD45+ CD11b+ Lin- F4/80- Ly6Clow / Ly6Chi. Data were acquired using Aurora (Cytek) Spectral Flow Cytometer.
Statistical analysis
All statistical analysis was performed with GraphPad-Prism software. Values are reported as means ± SD. Two-tailed Student’s t-tests (*p < 0.05 considered significant) or one-way ANOVA with Bonferroni correction (*p < 0.05 considered significant) were used for comparisons among groups. Log-rank (Mantel-Cox) tests were used to analyze survival.
Results
Cell death and sphingolipid signaling pathway expression dynamics post MI
We characterized the dynamics of cell death and gene expression during and after acute MI by ligating the left anterior descending artery (LAD) of mouse hearts to cause infarction and then harvesting hearts at various time points (Fig. 1A).
Figure 1. Characterizing cell death dynamics and sphingolipid metabolism in mouse hearts after MI.
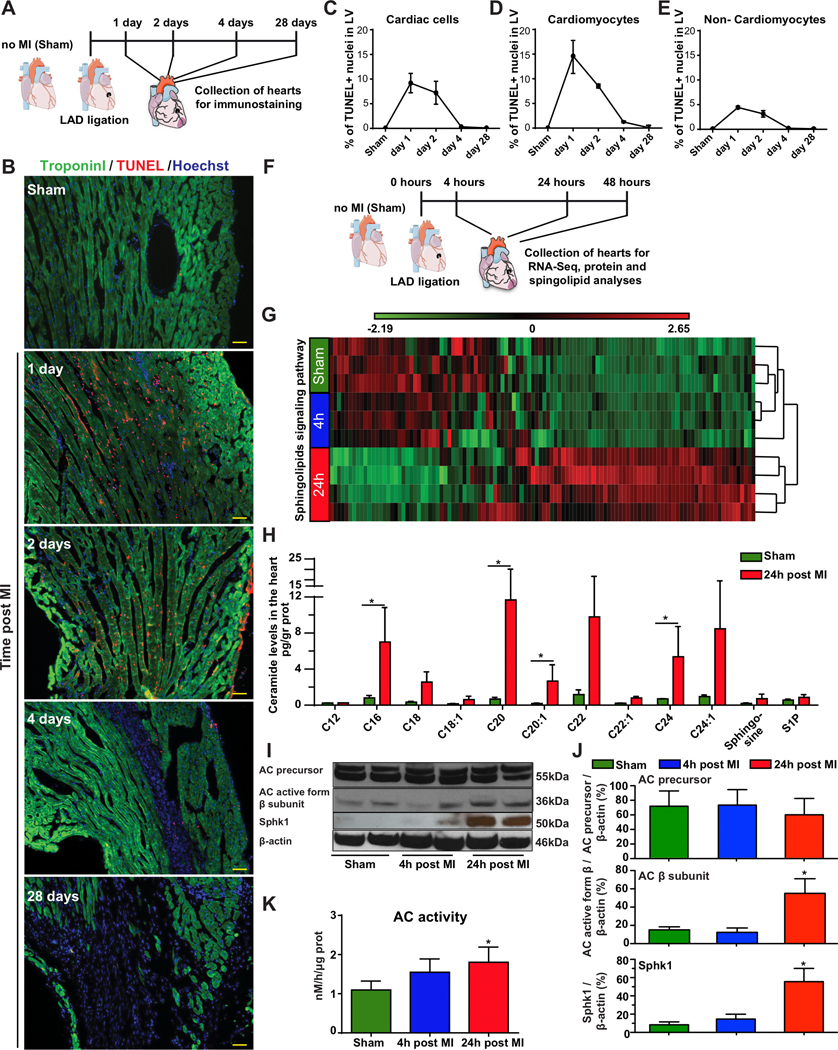
A, Hearts were harvested from sham-operated mice or 1, 2, 4 and 28 days post MI. B, TUNEL assays were performed to assess DNA fragmentation in hearts harvested from either sham-operated mice or 1, 2, 4 or 28 days post MI. Troponin-I immunostaining was used to distinguish between cardiomyocytes and non-cardiomyocytes. Percentage of dead cells in left ventricle at day 1, 2, 4 and 28 post MI was quantified within all cardiac cells (DAPI+ cells) n=3 (C); CMs only (DAPI+, Troponin I+) n=3 (D) and non-CM cells only (DAPI+, Troponin I-) n=3 (E). F, For RNASeq, protein analysis and mass spectrometry, hearts were harvested from sham-operated mice or 4 or 24 hours post MI. RNA, proteins and lipids were extracted for sphingolipid determination. G, Hierarchical clustering dendrogram for the sphingolipid signaling pathway transcriptome in sham-operated hearts and hearts harvested 4 or 24 hours post MI, n=3, 3, 4, respectively. H, Sphingolipid levels measured in the LV of sham-operated mice or 24 hours post MI, n=3. I, Western blot of the AC precursor, AC active subunit β and Sphk1 in the LV of sham-operated hearts and hearts harvested 4 or 24 hours post MI. J, Quantified protein levels of AC precursor, AC β subunit and Sphk1, n=4. K, HPLC-MS/MS determination of AC activity in the LV of sham-operated hearts and hearts harvested 4 or 24 hours post MI, n=3. *, P<0.05, One-way ANOVA, Tukey’s Multiple Comparison Test for (J&K) and Holm-Sidak correction for multiple comparisons (H). Scale bar = 50μm. The results include two independent experiments for H-K.
To assess cell death, we harvested hearts from both sham-operated and ligated mice 1, 2, 4 and 28 days post MI. We used TUNEL staining to assess DNA fragmentation in cardiac cells and troponin-I (cardiomyocytes marker) immunostaining to distinguish between cardiomyocytes (CMs) and non-CMs (Fig. 1B). DNA fragmentation levels were highest 24 hours post MI, with fragmented DNA detected in 9±2% of total cells in the LV (Fig. 1C), 15±3% of CMs (Fig. 1D) and 4±0.2% of non-CM cells (Fig. 1E). DNA fragmentation levels were lower 48 hours after MI, in both CMs and non-CM cells, and fell to basal levels by 28 days post MI (Fig. 1C–E).
An unbiased comparison of the LV transcriptome (Fig. 1F) 24 hours post MI identified a significant change (P=0.02) in the sphingolipid signaling pathway KEGG PATHWAY map04071.30 This pathway includes 112 genes, 38 of which had significantly changed expression after MI (Fig. 1G, Supplementary Table 3 and Supplementary Fig. 1F). In addition, 24 hours post MI, 12 of the 39 (30%) sphingolipid metabolism genes were significantly upregulated (Supplementary Fig. 1E, F). None of the genes involved in the sphingomyelin hydrolysis pathway were upregulated at 4 or 24 hours post MI, but four genes involved in the de novo ceramide synthesis pathway (Sptlc2, CerS2, 3 and 6) displayed 2.2-, 2.3-, 5.2- and 2.2-fold upregulation, respectively (Supplementary Table 3).
We next determined ceramide levels in the myocardium 24 hours post MI. The levels of C16-ceramide (8.75-fold increase), C20-ceramide (16.57-fold increase), C20:1-ceramide (8.38-fold increase) and C24-ceramide (8.95 increase) were significantly higher 24 hours post MI than in the absence of MI (Fig. 1H).
In this study, we focused on sphingolipid metabolism and the role of ceramide degradation by AC and sphingosine phosphorylation to S1P by sphingosine kinase 1 (Sphk1). We therefore used a separate mouse cohort to measure the mRNA and protein levels corresponding to these key ceramide metabolism genes. Consistent with our transcriptome analysis, relative levels of AC mRNA (Supplementary Fig. 1G and Supplementary Table 3) and AC precursor levels were unaffected after MI (Fig. 1I&J). However, the active AC-enzyme, as shown by β subunit, was significantly increased along with activity (Fig. 1K). AC-enzyme α subunit was also upregulated 24 hours post MI (Supplementary Fig. 1I). Moreover, the Sphk1, mRNA (Supplementary Fig. 1H) and protein (Fig. 1I&J) levels were notably upregulated 24 hours post MI. We also determined relative mRNA levels for AC and Sphk1 as well as Sphk1 protein levels in nrCMs under normoxia or hypoxia (Supplementary Fig. 1A–D). Though we observed no significant effect on AC and Sphk1 mRNA levels (Supplementary Fig. 1A–B), Sphk1 protein levels were moderately but significantly higher in cells subjected to hypoxia (Supplementary Fig. 1C–D).
Modulating sphingolipid metabolism in the heart during acute MI affects cell death
Ceramide metabolites’ effect on cardiomyocyte (CM) death and heart function were investigated post MI by altering ceramide metabolism via inhibiting or enhancing ceramide degradation and S1P generation. We used two previously described approaches to limit ceramide degradation in neonatal rat cardiomyocytes (nrCM): the pan-ceramidase inhibitor B1331 and the acid ceramidase-specific inhibitor ARN1497432. We also blocked S1P formation with a pan-sphingosine kinase inhibitor, SK1-II.33 Then, we assessed these inhibitors’ effects under normoxia (21% oxygen) and hypoxia (<2% oxygen) (Fig. 2A). In normoxia, inhibiting ceramidase activity with 50 μM B13 or 20 μM ARN14974 and inhibiting sphingosine kinase activity with 30 μM SK1-II for 48 hours had no significant effect on nrCM death rates, as shown by comparison with control cells treated with the solvent used for the inhibitors, DMSO (Fig. 2B). However, increasing B13 concentration to 100 μM or SK1-II concentration to 60 μM induced nrCM cell death 48 hours after treatment (Fig. 2B). Next, we assessed the combined inhibition of ceramidase and sphingosine kinase activities. When used together at low doses (50 μM for B13 and 30μM for SK1-II), these two inhibitors induced significantly higher levels of nrCM cell death than any other treatment except 100 μM B13, for which the same trend was observed but to a lesser extent (Fig. 2B). In hypoxic conditions, all inhibitor types, at all concentrations tested, significantly increased CM cell death levels 24 hours after treatment (Fig. 2C). By contrast, 48 hours after treatment, most cells were apoptotic regardless of the presence or absence of inhibitors, which no longer significantly affected CM death rates (Fig. 2D).
Figure 2. Inhibiting sphingolipid metabolism increases cell death in cardiac cells in vitro and after MI.

A, Primary cardiomyocytes were isolated from the hearts of two- to three-day-old rats. Two days after isolation, the cells were treated with ceramidase inhibitors (pan-ceramidase inhibitor B13 and AC-specific ARN 14974) or sphingosine kinase inhibitor (SK1-II) and transferred either to normoxia (21% oxygen) for 48 hours or hypoxia (<2% oxygen) for 24 or 48 hours. At the end of the incubation period, the cells were stained with an apoptosis marker (Annexin 5) and a cell viability marker (DAPI) to assess the effects of inhibitors and oxygen levels on cell death. Cell death was quantified in cells treated with chemical inhibitors under normoxia for 48 hours, n=4 (B), or hypoxia for 24 hours, n=4 (C) and 48 hours, n=4 (D). E, AC inhibitor was injected intraperitoneally at the time of MI and 7 hours post MI. Hearts were harvested 24 hours post MI. Proteins were extracted for AC activity assays, or tissue was fixed and stained with TUNEL to assess cell death in the LV. F, HPLC-MS/MS determined AC activity in LV lysates 24 hours post MI and treatment with various AC inhibitor concentrations, n=3. G, Representative images of heart sections from mice treated with AC inhibitor or DMSO (control) obtained 24 hours post MI. H, Quantified cell death levels in the LV after treatment with AC inhibitor or DMSO control, 24 hours post MI, n=4. I, Short-term post MI survival curve for mice injected with AC inhibitor or DMSO control (n=8). ****, P<0.0001,***, P<0.001,**, P<0.01, *, P<0.05, NS, not significant. One-way ANOVA, Tukey’s Multiple Comparison Test (B-D&F), two-tailed Student’s t-test (H), Mantel-Cox log-rank test (I). Scale bar 1mm.
We assessed loss of function in vivo using an AC-specific inhibitor (ARN14974) that has been shown to reduce enzyme activity in mouse hearts for more than seven hours after intraperitoneal injection (Fig. 2E).31 First, we first confirmed that the AC-specific inhibitor could affect AC in our assay (Fig. 2F). We then administered two IP injections (immediately and seven hours post MI) of the AC-specific inhibitor; this treatment resulted in twice as many cells containing fragmented DNA 24 hours post MI (Fig. 2G&H). Importantly, lower doses of AC-specific inhibitor lead to insignificant changes in cell death 24 hours post MI (Supplementary Fig. 2A–B). To evaluative AC’s role in mouse survival post MI, we compared mouse survival following treatment with either AC-specific inhibitor or DMSO control post MI. We show significantly reduced survival 72 hours post MI and treatment with AC-specific inhibitor (Fig 2I).
Additionally, to investigate the effect of overexpressing AC or Sphk1, alone or together, in nrCMs under hypoxic conditions (Fig. 3A), we used modRNAs encoding human AC and Sphk1. First, we confirmed the modRNAs were translated in nrCMs (Fig. 3B). We then cultured the transfected nrCMs in a hypoxia chamber to assess the modRNAs’ ability to prevent nrCM cell death. After 48 hours in hypoxia, 60% of the nrCMs transfected with nGFP (the control) were apoptotic, whereas cell death rates were 22% and 27% lower, respectively, in cells transfected with the AC modRNA and the Sphk1 modRNA. One of modRNA’s advantages as an upregulation tool is that several modRNAs can be transfected together into the same cell.25 The overexpression of both AC and Sphk1 decreased the number of apoptotic cells by 48% (Fig. 3C). Similarly, we investigated the effects of AC and Sphk1 overexpression on cell death in the LV post MI (Fig. 3D). We first used immunofluorescence staining to confirm that the various modRNAs were translated in the LV 24 hours post MI (Fig. 3E). We then confirmed that AC activity was significantly higher following AC modRNA delivery (Fig. 3F). Finally, we assessed DNA fragmentation by performing TUNEL assays 48 hours post MI and after the various treatments (Fig. 3G&H). AC overexpression in the left ventricle immediately after LAD ligation produced 54% fewer cells with fragmented DNA than hearts treated with Luc modRNA, 48 hours post MI (Fig. 3G&H). Neither Sphk1 overexpression nor lower doses of AC modRNA significantly changed DNA fragmentation (Fig. 3H and Supplementary Fig. 3A&B), while the combined approach, upregulating both AC and Sphk1, had an effect similar to that of AC alone (59% decrease) (Fig. 3G&H). In addition, we evaluated AC modRNA’s effect in human CMs by transfecting human-induced pluripotent stem cell-derived CMs (hiPS-CMs) with AC modRNA (Supplementary Fig. 4A). The AC modRNA was translated to generate AC protein (Supplementary Fig. 4B), resulting in lower cell death levels under hypoxia (Supplementary Fig. 4C).
Figure 3. Acid ceramidase overexpression using modRNA decreases cell death in cardiac cells in vitro and after MI.
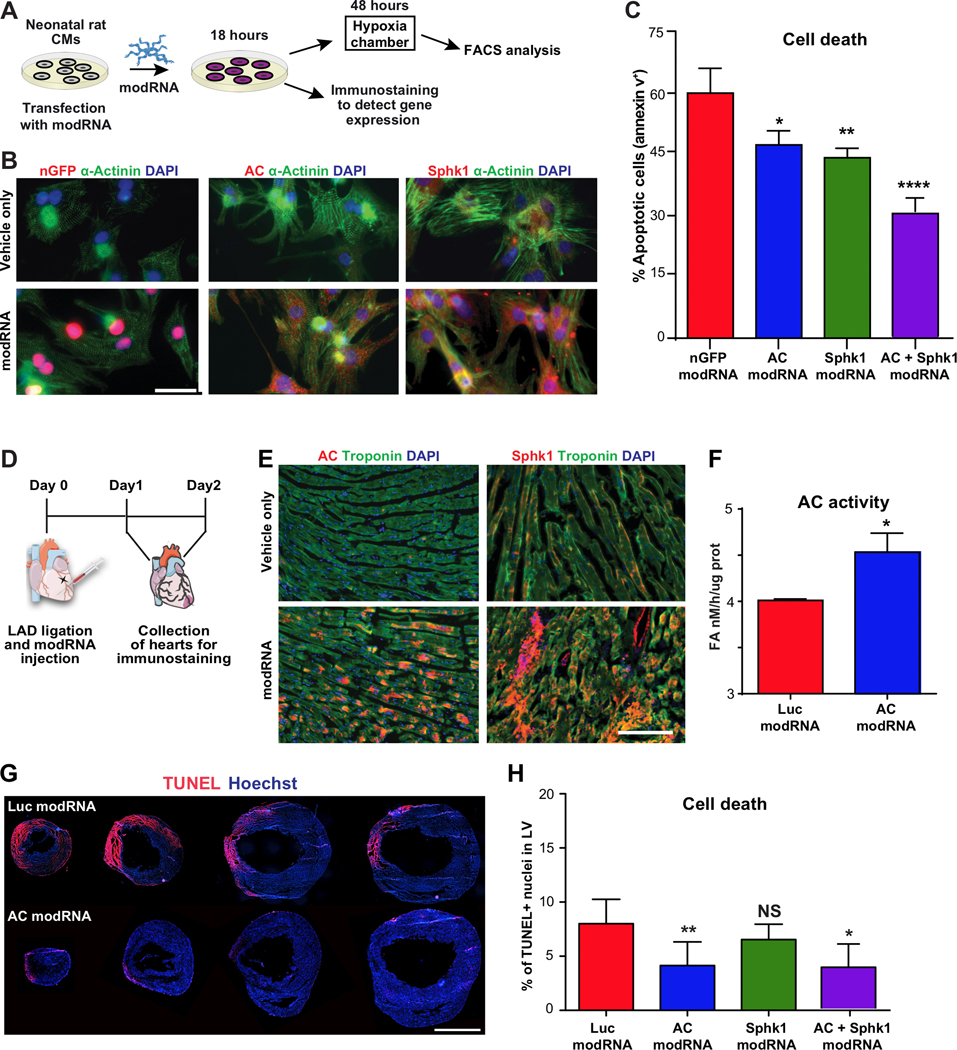
A, Primary cardiomyocytes were isolated from the hearts of two- to three-day-old rats. Two days after isolation, the cells were transfected with modRNAs for AC and SphK1. B, 18 hours post transfection in normoxia, CMs were fixed and immunostained to confirm modRNA translation. C, Cell death levels of CMs transfected with the nGFP modRNA (control), AC modRNA and SphK1 modRNA after 48 hours under hypoxia, n=4. D, modRNAs for Luc, AC and Sphk1 were injected into mouse hearts at the time of MI. To evaluate injected modRNA’s effects on the left ventricle, immunostaining (24 hours post MI), AC activity (24 hours post MI) and TUNEL assays were performed. E, Immunostaining for AC and SphK1 24 hours post modRNA injection. F, HPLC-MS/MS measurement of AC activity in the LV 24 hours post modRNA injection and MI, n=3 . G-H, Quantification and representative images of cell death levels in heart sections from mice treated with Luc modRNA (control), AC modRNA, Sphk1 modRNA or combination of AC + Sphk1 modRNAs 48 hours post injection, n=7. ****, P<0.0001, **, P<0.01, *, P<0.05, NS, not significant. One-way ANOVA, Tukey’s Multiple Comparison Test (C & H), Two-tailed Student’s t-test (F). Scale bars; 10μm (B), 50μm (E) and 1mm (G).
Transient AC overexpression improves heart function and survival rate in mice post MI
Based on the beneficial effects of AC both alone and with Sphk1, we investigated how these modRNAs affect cardiac function post MI by injecting AC, Sphk1, AC+Sphk1 or Luc modRNAs directly into the LV and evaluating cardiac function post MI (Fig. 4A). Two days post MI, no significant difference in either area at risk from the left ventricle (AAR/LV) or %FS (fractional shortening, expressed as a percentage) was observed between groups (Supplementary Fig. 5A&B). However, 28 days post MI, the %FS of the LV in mice treated with AC, Sphk1 or AC+ Sphk1 modRNAs was significantly higher than in the control (Supplementary Fig. 5C). Left ventricular internal diameter end diastole (LVIDd) did not differ significantly between treated and control mice 28 days post MI (Fig. 4B). Yet left ventricular internal diameter end systole (LVIDs) was significantly lower in mice treated with AC or AC+Sphk1 modRNAs than in control mice (Fig. 4C). Comparing %FS values between 2 and 28 days post MI revealed significantly improved heart function in mice treated with the Sphk1 modRNA and highly improved heart function in mice treated with AC or AC + Sphk1 modRNAs (Fig. 4D). To evaluate scar size after MI and the various treatments, we performed Masson trichrome staining (Fig. 4E) and found that scars were significantly smaller after treatment with AC, Sphk1 or AC+Sphk1 modRNAs (Fig. 4F). Further, we found no sign of CM hypertrophy in the LV of treated mice relative to the control (Supplementary Fig. 5D&E). AC modRNA alone yielded the most significant beneficial effect on the LV post MI. We therefore used magnetic resonance imaging (MRI) to compare the percent ejection fraction (%EF) of mice treated with AC modRNA with that of mice treated with the Luc control modRNA (Fig. 4G). We observed significantly higher EF (Fig. 4H), with better-preserved wall thickness, specifically in the areas into which the AC modRNA was injected (posterior and lateral) relative to the injection-free areas (anterior and septal) (Fig. 4I & Supplementary Movies 1&2). Finally, we determined that survival rates three months post MI were significantly higher for mice treated with the AC modRNA than for mice receiving other treatments or for control mice (Fig. 4J).
Figure 4. AC modRNA improves heart function and mouse survival post MI.

A, modRNAs for Luc, AC, Sphk1 or a combination of modRNAs for AC and Sphk1 were injected into the LV immediately after MI. Heart function was assessed by echocardiography and MRI. At day 29 post MI, hearts were collected for histological analysis. B-C, Echochardiography to measure left ventricular internal dimension end diastole (LVIDd) and left ventricular internal dimension end systole (LVIDs) 28 days post MI, n=6. D, Echocardiography to determine changes in left ventricle fractional shortening (FS) between day 2 and day 28 post MI, n=6 . E, Representative images of Masson’s trichrome staining and F, percentage of left ventricle area occupied by scar tissue 28 days post MI and modRNA injection, n=6. G-H, Representative images of MRI scans and quantification of left ventricle ejection fraction (LVEF) 29 days post MI and injection with AC or Luc modRNA. I, MRI-based measurement of fractional wall thickening percentage 29 days post MI and injection with AC or Luc modRNA, n=6. J, Long-term survival of mice after MI and transfection with different modRNAs, n=10. ****, P<0.0001, **, P<0.01, *, P<0.05, NS, not significant. One-way ANOVA, Bonferroni post-hoc tests (B, C, D, F) and two-tailed Student’s t-tests (H). The results include two independent experiments. Scale bar = 1mm.
Transient AC overexpression reduces ceramide levels and attenuates inflammation post MI
To investigate the molecular basis of improved heart function, we collected hearts from mice treated with AC or Luc control modRNAs and analyzed the effect of AC overexpression on protein levels and sphingolipid composition 24 hours post MI; we also analyzed whole-transcriptome dynamics 4, 24 and 48 hours post MI (Fig. 5A). Strikingly, AC overexpression decreased the levels of all ceramides investigated – significantly for C20 and C22 – in the LV post MI (Fig. 5B). We investigated this effect’s possible correlation with apoptosis markers, such as caspase 3 dimerization,34 and cleavage. We found that caspase 3 dimerization levels were higher in control mice than in sham-operated mice 24 hours post MI and that AC-treated mice had lower caspase 3 dimer levels than did control mice (Fig. 5C and Supplementary Fig. 6A). Unbiased transcriptome analysis revealed that only 30 genes displayed at least 1.5-fold expression changes (p<0.05) in either direction (Supplementary Fig. 6B and Supplementary Table 4) four hours after MI. By contrast, the number of genes displaying at least 1.5-fold expression (p<0.05) in either direction rose to 299 by 24 hours post MI (Supplementary Fig. 6B and Supplementary Table 5) and to 1675 by 48 hours post MI (Supplementary Fig. 6B and supplementary Table 6). GO enrichment analysis revealed that AC overexpression altered many immune-related pathways 24 hours post MI (Supplementary Table 7). Two days post MI, most of the genes displaying expression changes were involved in the cell cycle, cell apoptosis and immune response (Supplementary Fig. 6C). Out of all the genes that were upregulated two days post MI in mice that received ligation rather than sham, 144 genes had lower expression levels in AC modRNA-treated hearts than in Luc control modRNA-treated hearts. GO enrichment analysis with Enrichr35, 36 revealed that among the 144 genes with lower expression levels are genes involved in lipid metabolism-related genes and cell death. Interestingly, neutrophil chemotaxis and inflammatory response also show lower expression in the AC modRNA-treated hearts (Fig. 5D and Supplementary Fig. 6D). The partial transcriptome for sphingolipid metabolism-related genes was unchanged 4 and 24 hours post MI in AC modRNA-treated hearts (data not shown). However, 48 hours post MI, we observed significant expression changes for many of the sphingolipid metabolism-related genes included in RNA sequencing analysis (Supplementary Fig. 6D). To verify RNAseq data, we selected the Ngp gene encoding the neutrophilic granule protein, this gene’s expression was downregulated at 4, 24 and 48 hours post MI in AC modRNA-treated hearts relative to Luc modRNA-treated hearts. We confirmed changes in this gene’s expression patterns by performing RT-qPCR (please see primers in Supplemental Table 8) on RNA isolated from the hearts two days post MI; the results obtained were consistent with our transcriptomic results (Fig. 5E).
Figure 5. AC modRNA reduces ceramide levels and alters the immune cell composition in the left ventricle post MI.

A, modRNAs for Luc or AC were injected into mouse hearts immediately after MI. Lipids and proteins were extracted from the hearts 24 hours post MI to analyze sphingolipids by mass spectrometry and proteins by western blot. For RNA sequencing, hearts were harvested 4, 24 or 48 hours post MI. B, Sphingolipid levels after Luc or AC modRNA treatment, assessed 24 hours post MI, n=3. C, Western blot and quantification of AC α subunits and caspase 3 dimers in sham-operated hearts and in hearts treated with Luc or AC modRNA, 24 hours post MI, n=3. D, Scatterplot and GO enrichment analysis of all genes downregulated in AC-treated vs. Luc-treated out of all upregulated genes 2 days post MI, n=4. E, Relative Ngp gene expression levels 2 days post MI in the LV of mice treated with Luc or AC, quantified using qPCR, n=4. F, To analyze modRNA treatment’s effect on immune cell content after MI, hearts were collected at 2, 7 and 14 days post MI and Luc or AC modRNA injection and analyzed using flow cytometry. G, Gating strategy for immune cell populations in the infarct zone of modRNA-treated hearts. Neutrophils are defined as CD45+ CD11b+ Ly6G+, macrophages as CD45+ CD11b+ Lin- F4/80+ Ly6Clow/int and monocytes as CD45+ CD11b+ Lin- F4/80- Ly6Clow / Ly6Chi. H, Representative plots of neutrophil populations in Luc modRNA or AC modRNA-treated hearts 2 days post MI. I-N, Flow cytometric quantification of immune cell subsets in the infarct zone of Luc modRNA- and AC modRNA-treated hearts 2 days post MI included following populations: neutrophils, n=16 (I); Ly6Clow monocytes, n=16 (J); Ly6Chi monocytes, n=16 (K); total macrophages, n=16 (L), MHCII-, n=16 (M) and MHCII+, n=16 (N) macrophage subsets, n=16. O, Summary of proposed AC modRNA action mechanism during and after acute MI. ***, P<0.001, *, P<0.05, n=3 (B, C), n=4 (E), n=16–18 (I-N). Holm-Sidak correction for multiple comparisons (B), one-way ANOVA, Tukey’s Multiple Comparison Test (C) and two-tailed Student’s t-tests (E&I-N). Scale bar 20μm. The results include two and three (FACS) independent experiments.
The significantly decreased expression of inflammation-related genes in AC modRNA-treated hearts led us to investigate the composition of the immune cell populations in the infarct zone of the heart at days 2, 7 and 14 post MI (Fig. 5F). At day 2 post MI, neutrophil infiltration was reduced significantly in the infarct zone of AC modRNA-treated hearts compared to those from controls (Fig. 5H–I). No significant differences were found in Ly6Chi and Ly6Clow monocyte populations (Fig. 5J–K), nor macrophages (Fig. 5L–N). Seven days post MI, macrophage numbers increased dramatically whereas neutrophils decreased; both cells remained near these levels by day 14 post MI. (Supplementary Fig. 6E–F).
Discussion
CM cell death and cardiac inflammation are key to the severity of cardiac dysfunction post MI. Decreasing CM cell death and inflammation in patients with acute MI or other cardiovascular diseases is highly desirable.2, 37 There is growing evidence that blood ceramide levels can predict the extent of cardiovascular disease, complications and mortality.4, 5, 38, 39 Total ceramide levels, particularly of ceramides with fatty acid chains of 16 or more carbon atoms, are high in the myocardium in rodent models of MI8, 10 and in human patients with HF.8, 10
We characterized the significant changes to programmed cell death, the expression of genes involved in sphingolipid metabolism or signaling, sphingolipid levels and AC activity, 24 hours post MI (Fig. 1). We observed significant increases in the levels of C16, C20, C20:1 and C24 ceramides (Fig. 1H). Further, we found that the heart accommodated the increased ceramide levels through two different molecular mechanisms. The first involved sharply upregulated Sphk1 mRNA and protein levels (Fig. 1I&J and Supplementary Fig. 1H), and the second involved moderately upregulated AC enzymatic activity despite the absence of change in AC mRNA or protein levels (Fig. 1I–K and Supplementary Fig. 1G&I). These findings suggest that sphingolipid metabolism, especially Sphk1 and AC enzymatic activity, are important factors in cardiac tissue response to ischemic conditions.
Moderate concentrations of either ceramidase or sphingosine kinase inhibitors were not toxic to nrCMs under normal culture conditions. However, incubating nrCM cells with a higher concentration of either inhibitor separately, or lower concentrations of the two inhibitors together, induced cell death. Curtailing these enzymes may have caused ceramide and/or sphingosine to rise to toxic levels. The significant increase in nrCM cell death observed in conditions of moderate ceramidase or sphingosine kinase inhibition in hypoxic conditions suggests that a combination of hypoxia and enzyme inhibition can increase the rate of pro-apoptotic sphingolipid accumulation and cell death (Fig. 2C). Furthermore, in our in vivo loss-of-function study, limiting AC activity in mouse hearts led to elevated cardiac cell death 24 hours post MI. These results suggest that AC activity is a crucial cardioprotective factor in the ischemic heart (Fig. 2E–H). That inhibiting AC post MI significantly decreases survival indicates AC’s active role is vital to mouse survival post MI (Fig. 2I).
Ceramide has been shown to induce cell death in various cell types,7 including murine and human CMs.40, 41 Conversely, the phosphorylation of sphingosine, a ceramide degradation product, generates a major cell survival and cardioprotective agent, S1P.20,42 Klevsting et al. suggested that Ceramide accumulation in the myocardium may be partially reliant due to sphingomyelinase activity, and a heterozygous knockout of Smpd1, which encodes acid sphingomyelinase, reduced ceramide accumulation in mouse hearts, albeit insufficiently to improve either heart function or survival 30 days post MI.10 Recent studies in mouse models have demonstrated constraining de novo ceramide synthesis improves heart function post MI8 and moderately decreases the infarct area and inflammation levels following myocardial I/R injury.9 Our transcriptomic analysis revealed upregulated genes involved in de novo ceramide synthesis, including the Sptlc2 and CerS2 3 and 6 genes, with no change in mRNA levels for the natural sphingomyelinase (Smpd3) and lower transcript levels for other sphingomyelinases (including Smpd1) 24 hours post MI (Supplementary Table 3). We here use AC for ceramide hydrolysis43 to prevent ceramide toxicity and promote survival.
Consistent with its anti-apoptotic effects, sphingosine may disassemble mitochondrial ceramide channels,44, 45 but it has also been implicated in apoptotic or necrotic cell death.46 Benaim et al. suggested that sphingosine might disturb cellular calcium homeostasis by blocking the activity of the sarcoendoplasmic reticulum Ca2+-ATPase (SERC2a),47 which plays a crucial role in correct cardiac function.48, 49 Sphingosine kinase catalyzes the phosphorylation of sphingosine to generate S1P and has cardioprotective properties.50 Accordingly, adenovirus-mediated Sphk1 overexpression in rat hearts has been shown to protect treated hearts from I/R injury.51 In this study, we decided to decrease ceramide levels in the ischemic heart by increasing ceramide hydrolysis via modRNA used as a gene delivery tool to induce transient overexpression of AC. This strategy not only decreased ceramide levels but also expanded the sphingosine reservoir, the principal source of raw materials for producing the pro-survival molecule S1P. We found that AC modRNA increased AC expression in vitro and in vivo (Fig. 3B, E). The resulting high levels of AC protein were associated with more AC activity, leading to less cardiac cell death post MI (Fig. 3C and Fig. 3F–H). We examined AC and/or Sphk1 overexpression’s in vitro and in vivo effects on reduced cell death. Sphk1 overexpression generated via modRNA was found to prevent ischemic induced cell death in vitro but not in vivo. (Fig. 3C and Fig. 3H). This may be because Sphk1 mRNA and protein levels are naturally higher post MI in vivo than in in vitro hypoxia settings. (Supplementary Fig. 1B–D, H).
Interestingly, S1P receptor 2 (S1pr2) or Sphk2 overexpression did not significantly reduce cardiac cell death either in vitro or in vivo (Supplementary Fig. 7&8). Sphk1 is, thus, effective for preventing apoptotic death but not redundant with Sphk2 for this function. Consistent with this conclusion, Xia et al. showed that Sphk1 interacted with TNF receptor-associated factor 2 (TRAF2) and that this interaction was required for TRAF2’s anti-apoptotic activity.52 Our pathway analysis for sphingolipid signal transduction revealed upregulated TRAF2 post MI (Supplementary Table 3). Guo et al. recently reported a cardioprotective role for TRAF253. Future studies are required to investigate the interaction between Sphk1 and TRAF2 in the context of MI. Cell death levels did not change following S1pr2 upregulation, possibly due to this receptor’s high physiological expression levels in the heart.
We observed a clear therapeutic effect 28 days after AC modRNA delivery in mice with MI (Fig. 4), including preserved posterior and lateral wall thicknesses, which were most marked in the area into which the AC modRNA was injected. AC modRNA had no significant effect on %LVFS two days post MI (Supplementary Fig. 5A), whereas its effect was significant 28 days post MI (Supplementary Fig. 5B). This suggests that AC modRNA may not only prevent cell death immediately post MI but also influence other cardiac functions in this context. We also show a non-significant reduction of LVIDd 30 days post-MI (Fig. 4b) and reduce scar size (Fig. 4e) that implies but not prove a reduced remodeling. Further investigation on the long-term effects of AC treatment will clarify the effect on heart remodeling. In further sphingolipid analyses, we found that AC modRNA affected cell death by decreasing ceramide levels post MI, particularly for ceramides 20 and 22, (Fig. 5B), and by strongly curtailing caspase 3 dimer formation and cleavage post MI (Fig. 5C and Supplementary Fig. 6A). In addition, many genes involved in sphingolipid metabolism, inflammation and neutrophil degranulation displayed altered expression following AC modRNA treatment, two days post MI (Fig. 5D and Supplementary Fig. 6C&D). High ceramide levels can lead to neutrophil recruitment54 and activation.55 Analyzing the immune cell population in the infarct area revealed significantly lower neutrophil infiltration into the infarct zone of AC modRNA-treated hearts two days post MI (Fig. 5I), consistent with the post MI altered immune response shown by GO enrichment analysis (Fig. 5D) and lowered expression of the Ngp gene, which is expressed predominantly in neutrophils and promyelocytes56 (Fig. 5E) in these hearts.
Inflammation and immune cell infiltration into infarct cardiac tissue are crucial to healing after MI.57 Completely inhibiting the recruitment of a particular immune cell population, such as neutrophils or macrophages, is detrimental to the heart post MI,58, 59 but reducing the numbers of neutrophils infiltrating the myocardium has been shown to shrink infarct size and promote adaptation to hypoxic stress.60, 61 Administering AC modRNA immediately after MI decreased detrimental neutrophil levels, thereby providing an additional mechanism for improving outcome post MI. It remains unclear how AC modRNA alters the immune cell population in the LV post MI; AC modRNA may influence chemokine expression directly or indirectly.
Additionally, we do not yet know which S1P receptor (1–5) is activated in the heart during cell death rate reduction. The two most abundant receptors in the heart, S1pr1 and 3, increase significantly post MI (Supplementary Table 3). By contrast, S1pr2 levels are low four hours after MI and return to normal by 24 h post MI. The roles of S1pr1 and S1pr3 in cardioprotection are well established,50 but the role of S1p2 in heart function remains unclear.
In addition, AC modRNA’s effects post MI have yet to be investigated in large animals. However, by using hiPS-CMs in a hypoxia chamber, we demonstrated that AC modRNA reduced cell death to control levels in human settings, an effect similar to that reported here for nrCMs (Supplementary Fig. 6).
Overall, our data suggest (as summarized in Fig. 5O) that using AC modRNA to achieve transient overexpression of AC, an essential enzyme in sphingolipid metabolism and cell survival,12 lowers ceramide levels, decreases cardiac cell death and attenuates but does not eliminate detrimental neutrophil infiltration in the LV. Further, these effects improve cardiac function, leading to improved survival after MI in mice.
Supplementary Material
Clinical Perspective.
What is New?
We show that Acid Ceramidase delivery using modified mRNA alters sphingolipid metabolism by hydrolyzing ceramides in the heart post myocardial infarction.
This metabolic change significant reduces cell death, alters immune response by limiting neutrophil infiltration and supports cardiac function.
What are the clinical applications?
Our results suggest that Acid Ceramidase modified mRNA may serve as a cardiac protection target post myocardial infarction.
ACKNOWLEDGEMENTS
The authors acknowledge Jason Kondrat, Sunita DSouza and the Pluripotent Stem Cell Core Facility at the Icahn School of Medicine at Mount Sinai for their help with this manuscript.
FUNDING SOURCES
This work was funded by a cardiology start-up grant and NIH grant R01 HL142768–01 awarded to the Zangi laboratory and by the Genetics and Genomic Sciences budget in support of the Eliyahu laboratory.
Non-standard Abbreviations and Acronyms
- AC
Acid Ceramidase
- Sphk
Sphingosine kinase
- S1P
sphingosine-1-phosphate
- modRNA
modified mRNA
- LV
Left ventricle
- MI
Myocardial infarction
- LAD
Left anterior descending artery
- Luc
Luciferase
- nrCM
Neonatal rat cardiomyocytes
- hiPS
human induce pluripotent stem cells
- AAR
Area at risk
- LVID
Left ventricular internal diameter
- EF
Ejection fraction
- FS
Fractional Shortening
Footnotes
CONFLICT OF INTEREST DISCLOSURES
E.E., L.Z, A.S.V. and Y.H. are Inventors on Provisional Patent Application (MODRNA ENCODING SPHINGOLIPID METABOLIZING PROTEINS TO PROMOTE CELL SURVIVAL) 3710/039P, Filed March 2018, which covers the results in this manuscript.
References
- 1.Spencer FA, Meyer TE, Gore JM and Goldberg RJ. Heterogeneity in the management and outcomes of patients with acute myocardial infarction complicated by heart failure: the National Registry of Myocardial Infarction. Circulation. 2002;105:2605–2610. [DOI] [PubMed] [Google Scholar]
- 2.Hadas Y, Katz MG, Bridges CR and Zangi L. Modified mRNA as a therapeutic tool to induce cardiac regeneration in ischemic heart disease. Wiley Interdiscip Rev Syst Biol Med. 2017;9: wsbm.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lin Z and Pu WT. Strategies for cardiac regeneration and repair. Sci Transl Med. 2014;6:239rv1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meeusen JW, Donato LJ and Jaffe AS. Lipid Biomarkers for Risk Assessment in Acute Coronary Syndromes. Curr Cardiol Rep. 2017;19:48. [DOI] [PubMed] [Google Scholar]
- 5.Yu J, Pan W, Shi R, Yang T, Li Y, Yu G, Bai Y, Schuchman EH, He X and Zhang G. Ceramide is upregulated and associated with mortality in patients with chronic heart failure. Can J Cardiol. 2015;31:357–363. [DOI] [PubMed] [Google Scholar]
- 6.Castro BM, Prieto M and Silva LC. Ceramide: a simple sphingolipid with unique biophysical properties. Prog Lipid Res. 2014;54:53–67. [DOI] [PubMed] [Google Scholar]
- 7.Arana L, Gangoiti P, Ouro A, Trueba M and Gomez-Munoz A. Ceramide and ceramide 1-phosphate in health and disease. Lipids Health Dis. 2010;9:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ji R, Akashi H, Drosatos K, Liao X, Jiang H, Kennel PJ, Brunjes DL, Castillero E, Zhang X, Deng LY, Homma S, George IJ, Takayama H, Naka Y, Goldberg IJ and Schulze PC. Increased de novo ceramide synthesis and accumulation in failing myocardium. JCI Insight. 2017;2:225–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reforgiato MR, Milano G, Fabrias G, Casas J, Gasco P, Paroni R, Samaja M, Ghidoni R, Caretti A and Signorelli P. Inhibition of ceramide de novo synthesis as a postischemic strategy to reduce myocardial reperfusion injury. Basic Res Cardiol. 2016;111:12. [DOI] [PubMed] [Google Scholar]
- 10.Klevstig M, Stahlman M, Lundqvist A, Scharin Tang M, Fogelstrand P, Adiels M, Andersson L, Kolesnick R, Jeppsson A, Boren J and Levin MC. Targeting acid sphingomyelinase reduces cardiac ceramide accumulation in the post-ischemic heart. J Mol Cell Cardiol. 2016;93:69–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eliyahu E, Shtraizent N, Martinuzzi K, Barritt J, He X, Wei H, Chaubal S, Copperman AB and Schuchman EH. Acid ceramidase improves the quality of oocytes and embryos and the outcome of in vitro fertilization. FASEB J. 2010;24:1229–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eliyahu E, Park JH, Shtraizent N, He X and Schuchman EH. Acid ceramidase is a novel factor required for early embryo survival. FASEB J. 2007;21:1403–1409. [DOI] [PubMed] [Google Scholar]
- 13.Koch J, Gartner S, Li CM, Quintern LE, Bernardo K, Levran O, Schnabel D, Desnick RJ, Schuchman EH and Sandhoff K. Molecular cloning and characterization of a full-length complementary DNA encoding human acid ceramidase. Identification Of the first molecular lesion causing Farber disease. J Biol Chem. 1996;271:33110–33115. [DOI] [PubMed] [Google Scholar]
- 14.Bernardo K, Hurwitz R, Zenk T, Desnick RJ, Ferlinz K, Schuchman EH and Sandhoff K. Purification, characterization, and biosynthesis of human acid ceramidase. J Biol Chem. 1995;270:11098–11102. [DOI] [PubMed] [Google Scholar]
- 15.Shtraizent N, Eliyahu E, Park JH, He X, Shalgi R and Schuchman EH. Autoproteolytic cleavage and activation of human acid ceramidase. J Biol Chem. 2008;283:11253–11259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gebai A, Gorelik A, Li Z, Illes K and Nagar B. Structural basis for the activation of acid ceramidase. Nat Commun. 2018;9:1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mann DL. Sphingosine 1-phosphate as a therapeutic target in heart failure: more questions than answers. Circulation. 2012;125:2692–2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Waeber C and Walther T. Sphingosine-1-phosphate as a potential target for the treatment of myocardial infarction. Circ J 2014;78:795–802. [DOI] [PubMed] [Google Scholar]
- 19.Harris CM, Mittelstadt S, Banfor P, Bousquet P, Duignan DB, Gintant G, Hart M, Kim Y and Segreti J. Sphingosine-1-Phosphate (S1P) Lyase Inhibition Causes Increased Cardiac S1P Levels and Bradycardia in Rats. J Pharmacol Exp Ther. 2016;359:151–158. [DOI] [PubMed] [Google Scholar]
- 20.Santos-Gallego CG, Vahl TP, Goliasch G, Picatoste B, Arias T, Ishikawa K, Njerve IU, Sanz J, Narula J, Sengupta PP, Hajjar RJ, Fuster V and Badimon JJ. Sphingosine-1-Phosphate Receptor Agonist Fingolimod Increases Myocardial Salvage and Decreases Adverse Postinfarction Left Ventricular Remodeling in a Porcine Model of Ischemia/Reperfusion. Circulation. 2016;133:954–966. [DOI] [PubMed] [Google Scholar]
- 21.Holland WL, Miller RA, Wang ZV, Sun K, Barth BM, Bui HH, Davis KE, Bikman BT, Halberg N, Rutkowski JM, Wade MR, Tenorio VM, Kuo MS, Brozinick JT, Zhang BB, Birnbaum MJ, Summers SA and Scherer PE. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat Med. 2011;17:55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang CL, Leblond AL, Turner EC, Kumar AH, Martin K, Whelan D, O’Sullivan DM and Caplice NM. Synthetic chemically modified mrna-based delivery of cytoprotective factor promotes early cardiomyocyte survival post-acute myocardial infarction. Mol Pharm. 2015;12:991–996. [DOI] [PubMed] [Google Scholar]
- 23.Turnbull IC, Eltoukhy AA, Fish KM, Nonnenmacher M, Ishikawa K, Chen J, Hajjar RJ, Anderson DG and Costa KD. Myocardial Delivery of Lipidoid Nanoparticle Carrying modRNA Induces Rapid and Transient Expression. Mol Ther. 2016;24:66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zangi L, Lui KO, von Gise A, Ma Q, Ebina W, Ptaszek LM, Spater D, Xu H, Tabebordbar M, Gorbatov R, Sena B, Nahrendorf M, Briscoe DM, Li RA, Wagers AJ, Rossi DJ, Pu WT and Chien KR. Modified mRNA directs the fate of heart progenitor cells and induces vascular regeneration after myocardial infarction. Nat Biotechnol. 2013;31:898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zangi L, Oliveira MS, Ye LY, Ma Q, Sultana N, Hadas Y, Chepurko E, Spater D, Zhou B, Chew WL, Ebina W, Abrial M, Wang QD, Pu WT and Chien KR. Insulin-Like Growth Factor 1 Receptor-Dependent Pathway Drives Epicardial Adipose Tissue Formation After Myocardial Injury. Circulation. 2017;135:59–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kondrat J, Sultana N and Zangi L. Synthesis of Modified mRNA for Myocardial Delivery. Methods Mol Biol. 2017;1521:127–138. [DOI] [PubMed] [Google Scholar]
- 27.Sultana N, Magadum A, Hadas Y, Kondrat J, Singh N, Youssef E, Calderon D, Chepurko E, Dubois N, Hajjar RJ and Zangi L. Optimizing Cardiac Delivery of Modified mRNA. Mol Ther. 2017;25:1306–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Magadum A, Singh N, Kurian AA, Sharkar MTK, Chepurko E and Zangi L. Ablation of a Single N-Glycosylation Site in Human FSTL 1 Induces Cardiomyocyte Proliferation and Cardiac Regeneration. Mol Ther Nucleic Acids. 2018;13:133–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hulsmans M, Sager HB, Roh JD, Valero-Munoz M, Houstis NE, Iwamoto Y, Sun Y, Wilson RM, Wojtkiewicz G, Tricot B, Osborne MT, Hung J, Vinegoni C, Naxerova K, Sosnovik DE, Zile MR, Bradshaw AD, Liao R, Tawakol A, Weissleder R, Rosenzweig A, Swirski FK, Sam F and Nahrendorf M. Cardiac macrophages promote diastolic dysfunction. J Exp Med. 2018;215:423–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Du J, Yuan Z, Ma Z, Song J, Xie X and Chen Y. KEGG-PATH: Kyoto encyclopedia of genes and genomes-based pathway analysis using a path analysis model. Mol Biosyst. 2014;10:2441–2447. [DOI] [PubMed] [Google Scholar]
- 31.Bhabak KP, Kleuser B, Huwiler A and Arenz C. Effective inhibition of acid and neutral ceramidases by novel B-13 and LCL-464 analogues. Bioorg Med Chem. 2013;21:874–882. [DOI] [PubMed] [Google Scholar]
- 32.Pizzirani D, Bach A, Realini N, Armirotti A, Mengatto L, Bauer I, Girotto S, Pagliuca C, De Vivo M, Summa M, Ribeiro A and Piomelli D. Benzoxazolone carboxamides: potent and systemically active inhibitors of intracellular acid ceramidase. Angew Chem Int Ed Engl. 2015;54:485–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rex K, Jeffries S, Brown ML, Carlson T, Coxon A, Fajardo F, Frank B, Gustin D, Kamb A, Kassner PD, Li S, Li Y, Morgenstern K, Plant M, Quon K, Ruefli-Brasse A, Schmidt J, Swearingen E, Walker N, Wang Z, Watson JE, Wickramasinghe D, Wong M, Xu G and Wesche H. Sphingosine kinase activity is not required for tumor cell viability. PLoS One. 2013;8:e68328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu H, Chang DW and Yang X. Interdimer processing and linearity of procaspase-3 activation. A unifying mechanism for the activation of initiator and effector caspases. J Biol Chem. 2005;280:11578–11582. [DOI] [PubMed] [Google Scholar]
- 35.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, McDermott MG, Monteiro CD, Gundersen GW and Ma’ayan A. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44:W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR and Ma’ayan A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Katz MG, Hadas Y, Fargnoli AS, Hadas S, Zangi L and Bridges CR. [Gene Therapy Potential as a Treatment for Heart Failure]. Harefuah. 2018;157:112–116. [PubMed] [Google Scholar]
- 38.Laaksonen R, Ekroos K, Sysi-Aho M, Hilvo M, Vihervaara T, Kauhanen D, Suoniemi M, Hurme R, Marz W, Scharnagl H, Stojakovic T, Vlachopoulou E, Lokki ML, Nieminen MS, Klingenberg R, Matter CM, Hornemann T, Juni P, Rodondi N, Raber L, Windecker S, Gencer B, Pedersen ER, Tell GS, Nygard O, Mach F, Sinisalo J and Luscher TF. Plasma ceramides predict cardiovascular death in patients with stable coronary artery disease and acute coronary syndromes beyond LDL-cholesterol. Eur Heart J. 2016;37:1967–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang DD, Toledo E, Hruby A, Rosner BA, Willett WC, Sun Q, Razquin C, Zheng Y, Ruiz-Canela M, Guasch-Ferre M, Corella D, Gomez-Gracia E, Fiol M, Estruch R, Ros E, Lapetra J, Fito M, Aros F, Serra-Majem L, Lee CH, Clish CB, Liang L, Salas-Salvado J, Martinez-Gonzalez MA and Hu FB. Plasma Ceramides, Mediterranean Diet, and Incident Cardiovascular Disease in the PREDIMED Trial (Prevencion con Dieta Mediterranea). Circulation. 2017;135:2028–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Usta E, Mustafi M, Artunc F, Walker T, Voth V, Aebert H and Ziemer G. The challenge to verify ceramide’s role of apoptosis induction in human cardiomyocytes--a pilot study. J Cardiothorac Surg. 2011;6:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bielawska AE, Shapiro JP, Jiang L, Melkonyan HS, Piot C, Wolfe CL, Tomei LD, Hannun YA and Umansky SR. Ceramide is involved in triggering of cardiomyocyte apoptosis induced by ischemia and reperfusion. Am J Pathol. 1997;151:1257–1263. [PMC free article] [PubMed] [Google Scholar]
- 42.Levkau B Cardiovascular effects of sphingosine-1-phosphate (S1P). Handb Exp Pharmacol. 2013:147–170. [DOI] [PubMed] [Google Scholar]
- 43.Hassler DF and Bell RM. Ceramidases: enzymology and metabolic roles. Adv Lipid Res. 1993;26:49–57. [PubMed] [Google Scholar]
- 44.Siskind LJ, Fluss S, Bui M and Colombini M. Sphingosine forms channels in membranes that differ greatly from those formed by ceramide. J Bioenerg Biomembr. 2005;37:227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Elrick MJ, Fluss S and Colombini M. Sphingosine, a product of ceramide hydrolysis, influences the formation of ceramide channels. Biophys J. 2006;91:1749–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guicciardi ME, Leist M and Gores GJ. Lysosomes in cell death. Oncogene. 2004;23:2881–2890. [DOI] [PubMed] [Google Scholar]
- 47.Benaim G, Pimentel AA, Felibertt P, Mayora A, Colman L, Sojo F, Rojas H and De Sanctis JB. Sphingosine inhibits the sarco(endo)plasmic reticulum Ca(2+)-ATPase (SERCA) activity. Biochem Biophys Res Commun. 2016;473:572–577. [DOI] [PubMed] [Google Scholar]
- 48.del Monte F, Williams E, Lebeche D, Schmidt U, Rosenzweig A, Gwathmey JK, Lewandowski ED and Hajjar RJ. Improvement in survival and cardiac metabolism after gene transfer of sarcoplasmic reticulum Ca(2+)-ATPase in a rat model of heart failure. Circulation. 2001;104:1424–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hajjar RJ, Kang JX, Gwathmey JK and Rosenzweig A. Physiological effects of adenoviral gene transfer of sarcoplasmic reticulum calcium ATPase in isolated rat myocytes. Circulation. 1997;95:423–429. [DOI] [PubMed] [Google Scholar]
- 50.Cannavo A, Liccardo D, Komici K, Corbi G, de Lucia C, Femminella GD, Elia A, Bencivenga L, Ferrara N, Koch WJ, Paolocci N and Rengo G. Sphingosine Kinases and Sphingosine 1-Phosphate Receptors: Signaling and Actions in the Cardiovascular System. Front Pharmacol. 2017;8:556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Duan HF, Wang H, Yi J, Liu HJ, Zhang QW, Li LB, Zhang T, Lu Y, Wu CT and Wang LS. Adenoviral gene transfer of sphingosine kinase 1 protects heart against ischemia/reperfusion-induced injury and attenuates its postischemic failure. Hum Gene Ther. 2007;18:1119–1128. [DOI] [PubMed] [Google Scholar]
- 52.Xia P, Wang L, Moretti PA, Albanese N, Chai F, Pitson SM, D’Andrea RJ, Gamble JR and Vadas MA. Sphingosine kinase interacts with TRAF2 and dissects tumor necrosis factor-alpha signaling. J Biol Chem. 2002;277:7996–8003. [DOI] [PubMed] [Google Scholar]
- 53.Guo X, Yin H, Li L, Chen Y, Li J, Doan J, Steinmetz R and Liu Q. Cardioprotective Role of Tumor Necrosis Factor Receptor-Associated Factor 2 by Suppressing Apoptosis and Necroptosis. Circulation. 2017;136:729–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Teichgraber V, Ulrich M, Endlich N, Riethmuller J, Wilker B, De Oliveira-Munding CC, van Heeckeren AM, Barr ML, von Kurthy G, Schmid KW, Weller M, Tummler B, Lang F, Grassme H, Doring G and Gulbins E. Ceramide accumulation mediates inflammation, cell death and infection susceptibility in cystic fibrosis. Nat Med. 2008;14:382–391. [DOI] [PubMed] [Google Scholar]
- 55.Corriden R, Hollands A, Olson J, Derieux J, Lopez J, Chang JT, Gonzalez DJ and Nizet V. Tamoxifen augments the innate immune function of neutrophils through modulation of intracellular ceramide. Nat Commun. 2015;6:8369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Moscinski LC and Hill B. Molecular cloning of a novel myeloid granule protein. J Cell Biochem. 1995;59:431–442. [DOI] [PubMed] [Google Scholar]
- 57.Liu J, Wang H and Li J. Inflammation and Inflammatory Cells in Myocardial Infarction and Reperfusion Injury: A Double-Edged Sword. Clin Med Insights Cardiol. 2016;10:79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Horckmans M, Ring L, Duchene J, Santovito D, Schloss MJ, Drechsler M, Weber C, Soehnlein O and Steffens S. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur Heart J. 2017;38:187–197. [DOI] [PubMed] [Google Scholar]
- 59.van Amerongen MJ, Harmsen MC, van Rooijen N, Petersen AH and van Luyn MJ. Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am J Pathol. 2007;170:818–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liehn EA, Tuchscheerer N, Kanzler I, Drechsler M, Fraemohs L, Schuh A, Koenen RR, Zander S, Soehnlein O, Hristov M, Grigorescu G, Urs AO, Leabu M, Bucur I, Merx MW, Zernecke A, Ehling J, Gremse F, Lammers T, Kiessling F, Bernhagen J, Schober A and Weber C. Double-edged role of the CXCL12/CXCR4 axis in experimental myocardial infarction. J Am Coll Cardiol. 2011;58:2415–2423. [DOI] [PubMed] [Google Scholar]
- 61.Montecucco F, Lenglet S, Braunersreuther V, Pelli G, Pellieux C, Montessuit C, Lerch R, Deruaz M, Proudfoot AE and Mach F. Single administration of the CXC chemokine-binding protein Evasin-3 during ischemia prevents myocardial reperfusion injury in mice. Arterioscler Thromb Vasc Biol. 2010;30:1371–1377. [DOI] [PubMed] [Google Scholar]
- 62.Bielawski J, Szulc ZM, Hannun YA and Bielawska A. Simultaneous quantitative analysis of bioactive sphingolipids by high-performance liquid chromatography-tandem mass spectrometry. Methods. 2006;39:82–91. [DOI] [PubMed] [Google Scholar]
- 63.Yang L, Soonpaa MH, Adler ED, Roepke TK, Kattman SJ, Kennedy M, Henckaerts E, Bonham K, Abbott GW, Linden RM, Field LJ and Keller GM. Human cardiovascular progenitor cells develop from a KDR+ embryonic-stem-cell-derived population. Nature. 2008;453:524–528. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.