

Abstract
In the United States and other westernized nations, CVDs are the leading cause of death in adults over 65 years of age. Large artery stiffness and endothelial dysfunction are increased with age and age-associated arterial dysfunction is an important antecedent of CVDs. One age-associated change that may contribute to vascular dysfunction and CVD risk is an increase in the number of resident senescent cells in the vasculature. Senescent cells display a pro-oxidant, pro-inflammatory phenotype known as the SASP. However, the mechanisms that drive the SASP and the vascular aging phenotype remain elusive. A putative mechanism is the involvement of oxidative stress and inflammation in telomere function. Telomeres are the end caps of chromosomes which are maintained by a six-protein complex known as shelterin. Disruption of shelterin can uncap telomeres and induce cellular senescence. Accordingly, in this review, we propose that oxidative stress and inflammation disrupt shelterin in vascular cells, driving telomere dysfunction and that this mechanism may be responsible for the induction of SASP. The proposed mechanisms may represent some of the initial changes that lead to vascular dysfunction in advanced age.
Keywords: aging, cardiovascular disease, endothelium, inflammation, large artery stiffness, oxidative stress, senescence, shelterin, telomere
1 |. INTRODUCTION
In the United States, CVDs are the leading cause of death in adults over 65 years of age.1 Also, individuals diagnosed with diseases of accelerated aging, such as progeria syndromes, are most likely to die of cardiovascular complications.2 This suggests that the cardiovascular system is highly susceptible to the changes that occur with advancing age. With normal aging, arterial function begins to decline and the majority of cardiovascular diseases are preceded by arterial dysfunction.3 For example, large artery stiffness increases and endothelial dependent dilation decreases, independent of any overt CVDs or traditional cardiovascular risk factors.4–7 These findings suggest that the fundamental processes that occur with aging induce arterial dysfunction and precede the development of CVDs.
Mechanisms underlying age-related arterial dysfunction including chronic inflammation and elevated oxidative stress have been reviewed in detail by us and others.8,9 However, the local changes within the vasculature that drive and perpetuate the pro-inflammatory and pro-oxidant vascular phenotype remain elusive. While the consequences of these processes have been well characterized, identifying a root cause may allow for improved countermeasures to reverse or prevent age-related arterial dysfunction. The goal of this review is to look at prospective mechanisms that induce the pro-inflammatory and pro-oxidant phenotype that suppresses arterial function in older adults, specifically alterations in telomeres (chromosome ends) and telomere associated proteins (shelterin) and resultant senescence (permanent cell cycle arrest).
2 |. CELL SENESCENCE IN VASCULAR AGING
One hypothesis for the mechanism underlying the changes in arterial phenotype that occur with aging is an increase in the number of resident senescent cells.10,11 Senescence is defined as permanent cell cycle arrest, and is thought to act as a tumor suppressor mechanism, preventing cells from passing on damaged DNA to daughter cells.12,13 Most eukaryotic mitotic cells have a limited replicative capacity (there are exceptions, for example, cancer cells), whereby cells become senescent after numerous divisions in vivo or successive passages in vitro, a process known as replicative senescence.14,15 Oxidative stress,16 strong mitogenic signaling,17 DNA damage,18 and telomere dysfunction can lead to premature senescence through the downstream activation of the tumor suppressor proteins, including p53, and subsequent transcription of p21, and the p16 phosphorylation of pRB.13 This process is known as stress-induced senescence.
Senescent cells display a SASP that impacts the local tissue milieu. Senescent cells act in both an autocrine and paracrine manner by upregulating inflammatory mediators, such as chemokines and cytokines, and by the production of ROS.13,19 Therefore, the accumulation of senescent cells within the vasculature could act as a positive feedback loop, with senescent cells secreting chemokines that recruit immune cells, further increasing cytokine and ROS production and perpetuating a pro-inflammatory and pro-oxidant phenotype in the tissue (Figure 1C,D). Furthermore, these senescent cells could push surrounding cells toward senescence by impacting their ability to effectively cap telomeres, while simultaneously reducing vascular function. Indeed, early observations showed that markers of senescent cells were found in atherosclerotic plaques but not in non-atherogenic arteries.20 More recently, we have demonstrated that arteries from older healthy adults and hypertensive patients have greater expression of p21, a marker of senescence, as well as the SASP pro-inflammatory phenotype.21 Likewise, endothelial cells sampled from healthy older adults have greater expression of the senescence markers p21 and p16, both of which are correlated with blunted endothelial function.22 In contrast, older adults that perform habitual exercise (a vasoprotective lifestyle) have normalized endothelial senescent markers and endothelial function.22 Lastly, in preclinical rodent models, drugs that remove senescent cells, known as senolytics, improve arterial function in older mice (3 months of treatment starting at 24 months).23 Therefore, senescent cells are a viable candidate for explaining the upregulation and maintenance of both inflammation and oxidative stress that are seen in aged vessels (Figure 1). Further elucidation of the mechanisms by which arterial cells are driven to senescence is of great importance to better understand vascular aging.
FIGURE 1.
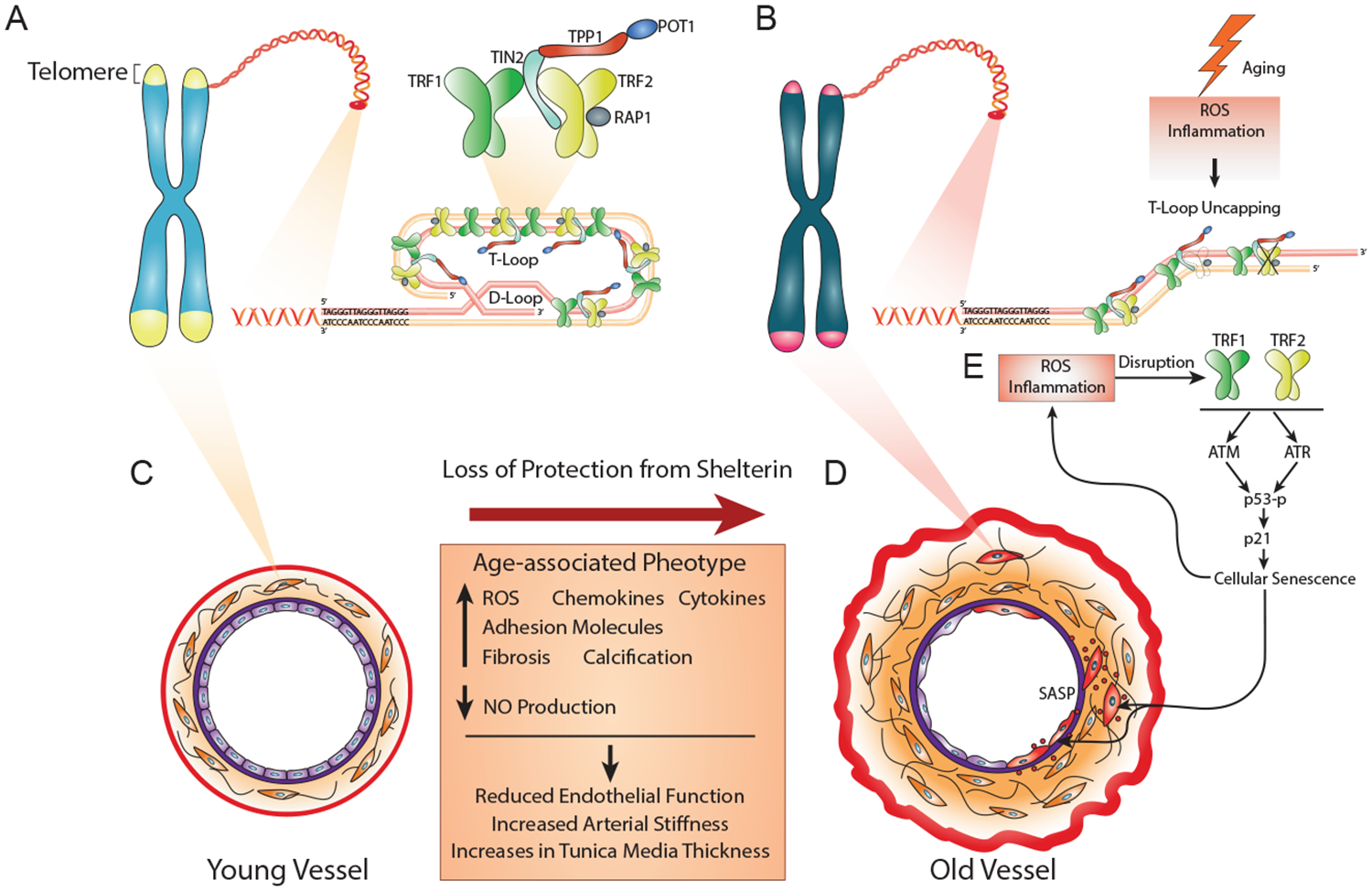
Telomere Dysfunction and Shelterin Disruption Lead to Senescence and Age-related Vascular Phenotype. Telomeres are the endcaps of chromosomes comprised of TTAGGG repeats. The repeat sequence serves as a binding substrate for a six subunit protein complex known as the shelterin complex. The six shelterin proteins include TRF1 and TRF2, and POT1. TRF1 and TRF2 bind double stranded telomeric DNA, whereas POT1 localizes to single stranded regions. Both TRF1 and TRF2 are found as homodimers, and are critical for recruiting the other shelterin proteins, TRF1-TIN2, RAP1, and TPP1. Shelterin regulates the invasion of the single stranded 3′ overhang at the end of the telomere into the duplex telomeric DNA, forming what is known as a t-loop effectively inhibiting the activation of the DNA damage response. In response to double stranded DNA breaks or exposed telomeres (loss or disruption of TRF2), ATM kinase is activated, whereas in response to single stranded DNA (loss or disruption of POT1), ATR kinase is activated. Activation of both ATM and ATR leads to the phosphorylation of the tumor suppressor protein p53, and subsequent transcription of the p21, leading to cellular senescence (permanent cell cycle arrest). In arteries disruption of TRF1 and TRF2 lead to senescence, and could be induced directly by ROS or indirectly by inflammation. Senescent cells display what is known as a SASP, that increases production of ROS and inflammation. With aging, an increase in the number of resident senescent cells represents one possible explanation for the aged arterial phenotype, as it is characterized by increased ROS and inflammation that could further uncap telomeres or induce DNA damage, forming a positive feedback loop. Thus, upregulating chemokines, cytokines, and adhesion molecules, as well as fibrosis and calcification, reducing production of NO. Collectively, these changes reduce endothelial dependent dilation, and increase arterial stiffness and tunica media thickness
Here, we will discuss recent findings in the vasculature that indicate telomere dysfunction may be a causal factor in age-related senescence and arterial dysfunction. Furthermore, we will speculate on the possible mechanisms responsible for changes in telomere regulation that activate the DNA damage response (DDR) pathways that are responsible for the induction of cellular senescence. These mechanisms include inflammation and oxidative stress, as well as other factors which alter telomeric DNA and its associated proteins that lead to the age-related changes in the vasculature that precede CVDs.
3 |. TELOMERES AND SHELTERIN
Telomeres are the ends of eukaryotic chromosomes and are made up of non-coding DNA comprised of hexanucleotide TTAGGG repeats (Figure 1A).24 Telomeres serve a functional role, solving two critical problems within DNA. The first is known as the end-protection problem, which occurs due to the linear nature of DNA.25 The ends of chromosomes resemble damaged DNA, which has negative consequences for cell fate as recognition of these chromosome ends as a DNA break will activate the DDR, whereby the cell will attempt to repair the DNA. Furthermore, this DDR can lead to cell cycle arrest, as it involves the activation of either the ATM or ATR kinase pathways that ultimately up-regulate p53-mediated transcription of p21, leading to cellular senescence.25
Solving this problem is the role of a six subunit protein complex known as shelterin that modulates the structure of telomeres and inhibits the DDR.25 Shelterin mediates the invasion of a single stranded 3′ overhang into the duplex telomeric DNA, effectively tucking away the chromosome ends. This caps the telomere by forming what is known as a t-loop.25 Shelterin is critical for maintaining the t-loop and preventing the activation of the DDR that would otherwise induce cellular senescence.25 Three of the shelterin proteins directly bind to the TTAGGG repeats within the telomeres, including TRF1 and TRF2, and POT1. TRF1 and TRF2 bind double stranded telomeric DNA, whereas POT1 localizes to single stranded regions.26 Both TRF1 and TRF2 are found as homodimers and are critical for recruiting the other shelterin proteins including TIN2, RAP1, and TPP1 (Figure 1A).26
Each shelterin protein has a unique role in telomere function. TRF2 and POT1 act to repress activation of two DDR pathways that signal cells to become senescent: the ATM kinase pathway and the ATR kinase pathway, respectively.25 ATM is activated in response to DNA ends, thus TRF2 is able to prevent ATM activation as a result of its role in forming the t-loop or through direct inhibition.25,26 ATR is activated in response to single stranded overhanging DNA, but binding of POT1 prevents the recruitment of the factors necessary for activation.26 TRF1, on the other hand, is involved in the regulation of telomere length27 and is also critical for telomere replication, as it prevents stalling of the replication fork.28 In total, the shelterin complex acts to regulate telomere homeostasis and form the t-loop, thereby “capping” telomeres and preventing inappropriate activation of the DDR that could lead to cellular senescence or other negative cellular consequences (Figure 1B). While it is known that all of the shelterin components play a role in telomere maintenance, for the purpose of this review we will focus on TRF1 and TRF2 due to lack of evidence in the other shelterin components and the critical nature of these specific shelterin components for telomere t-loop stability.
Telomeres solve a second problem known as the end-replication problem. This occurs during DNA replication: DNA polymerases are unable to fully replicate the nucleotide sequence on the 3′ of the template strand, causing a loss of nucleotides on the 5′ end of the new strand of DNA.29 Because telomeres contain non-coding DNA they serve as a protective boundary against damage and loss of important genetic information contained within coding DNA.30 However, the loss of telomeric DNA with successive cell divisions ultimately causes telomeres to shorten until they have reached a critical length and can no longer form a t-loop, at which time the cells become senescent.31
It is possible that telomere attrition could lead to the presence within tissue of several ECs or VSMCs that have critically short telomeres, and that these cells could be responsible for the presence of SASP in old vessels. This would most likely be proximal to vascular branches and bifurcations, as the turbulent blood flow in these regions may increase cellular proliferation rates in comparison to laminar flow found in non-branched regions.32 However, telomere shortening per se likely is not the sole cause of cellular senescence. Our laboratory has previously shown evidence of telomere uncapping in arteries from older adults and ECs isolated from aged mice that were independent of telomere length and correlated with classic DNA damage signaling. This indicates that a disruption of the t-loop or shelterin is a more likely mechanism of cellular senescence.21,33 In total, the recognition of double stranded or single stranded DNA by the DDR machinery can lead to the senescent phenotype found in aged arteries. In addition to telomere attrition and uncapping, several structural properties of telomeres also make them likely sites of DNA damage in comparison to other heterochromatic or intergenic regions within chromosomes.
4 |. TELOMERE DYSFUNCTION: AN INITIAL STEP FOR VASCULAR AGING
In clinical studies, correlations have been made between leukocyte telomere length and age-related chronic diseases, with telomere length of leukocytes regarded as a reliable marker for prognosis in cancer and type 2 diabetes, although it varies between individuals.34,35 However, in the setting of coronary artery disease, leukocyte telomere length is a poor predictor of clinical outcome and is independent of conventional vascular risk factors.36 While the telomere length of leukocytes is typically shorter with aging and age-related chronic disease, this could be due to chronic inflammation induction of leukocyte proliferation and not advanced age per se. Therefore, it may have value as a biomarker, but it is difficult to conclude that shorter telomeres in leukocytes directly contribute to vascular aging or CVD. Still, in mice, the accumulation of critically short telomeres in vascular tissue resulting from telomerase deficiency is sufficient to induce endothelial dysfunction,37 whereas ectopic expression of telomerase in human VSMCs both lengthens telomeres and increases plaque stability via greater cap stability and a normalized VSMC phenotype in atherosclerosis.20 Still, TERT not only extends the telomere, but also regulates eNOS expression in human microvessels at translational and posttranslational levels as well as prevents oxidative stress via reduced mitochondrial ROS and improved endothelial function.38 Therefore, while these findings indicate that telomere shortening is associated with senescence, vascular aging, and CVD, it is unclear whether telomere shortening is sufficient to be the root cause of the senescent cells and vascular dysfunction found in the aged vasculature.
Interestingly, unlike telomere length, telomere uncapping was found to strongly correlate to markers of p53/p21-associated senescence in arteries from healthy young and old individuals.21 Furthermore, in comparison with healthy old controls, age-matched hypertensive individuals have greater p53/p21-dependent senescence and telomere uncapping, whereas arterial mean telomere length was not significantly different between groups.39 These findings indicate that telomere uncapping plays a vital role in the activation of p53/p21 that leads to senescence in arteries independent of telomere shortening. Thus, a better understanding of how aging impacts telomere protection may inform strategies to prevent or reduce the age-associated senescent burden in arteries.
It is well established that TRF2 is important for maintaining the t-loop structure, as telomere uncapping can be caused by disruption or loss of TRF2 expression.40 Thus, alterations in the regulation of TRF2 with aging could uncap telomeres and lead to senescence that participates in CVD pathology. Several findings support this hypothesis including the observation that the senescent VSMCs that accumulate in human atherosclerotic plaques have decreased expression of TRF2 and increased DNA damage.41 Similarly, a mouse model of VSMC-specific TRF2 dysfunction displays accelerated atherogenesis.41 Conversely, overexpression of TRF2 reduces DNA damage and prevents senescence in VSMCs in vitro.41 Furthermore, by employing a transgenic mouse model of inducible TRF2 deletion, our group has recently demonstrated that telomere uncapping caused by deletion of TRF2 gene induces dysfunction in both large and small arteries. This included impaired endothelium dependent dilation, as well as increased systolic blood pressure and oxidative stress,33 indicating a critical role for TRF2 in vascular function. The role of TRF1, another shelterin complex component, is to protect against DNA damage and replicative stress. Interestingly, ECs isolated from patients with severe coronary artery disease had a greater accumulation of senescent ECs with lower TRF1 protein levels as well as higher telomere oxidation.42 This clinical finding is supported by in vitro data in which serial passaging of HUVECs reduces TRF1 expression, whereas overexpression of TRF1 in serially passaged HUVECs, a cell culture model of aging, partially reduces telomere-associated DNA damage foci (markers of double stranded DNA breaks)43 and the SASP.44 Notably, it was found that telomere dysfunction and the SASP occur prior to replicative senescence.44 Therefore, telomere dysfunction may contribute to the early changes that promote senescence in ECs or VSMCs that induce vascular dysfunction and lead to CVD development. Thus, protecting telomere integrity by repairing normal function of the shelterin complex holds promise as a strategy to control/prevent cell senescence associated with CVD.
5 |. FUNCTIONAL REGULATION OF VASCULAR AGEING WITH SHELTERIN
As previously discussed, shelterin is critical for maintaining telomere integrity and evidence suggests that it plays a vital role in vascular function. However, important unanswered questions remain, including how changes in the regulation of shelterin could lead to age-related arterial dysfunction. Oxidative stress and inflammation are important contributors to age-related CVD by triggering EC dysfunction and increasing arterial stiffness, as well as by increasing the deposition of ECM.8 As mentioned earlier in this review, there exists a positive feedback loop between inflammation/oxidative stress and telomere dysfunction that acts to potentiate vascular dysfunction. For example, telomere damage or attrition can signal cellular senescence and induce the SASP and subsequent inflammation and ROS generation, ROS can also damage to telomeres, leading to cellular senescence that perpetuates this vicious cycle. Thus, senescent cells represent a plausible candidate mechanism underlying the upregulation of both inflammation and oxidative stress in aged vessels. In the following sections, we will explore the possible connection between oxidative stress, inflammation, cellular senescence, and telomere structure and function (Figure 1).
5.1 |. Oxidative stress
Oxidative stress, defined as an abundance of ROS above that which can be handled by antioxidant defenses, has been well addressed in the pathogenesis of various age-related CVDs.45 Vascular cells, specifically ECs, are continually exposed to numerous ROS generating stimuli in the circulating plasma, including immune cells, lipids and glucose, as well as stimuli that increase endogenous ROS production such as oscillatory shear stress.46,47 Since ECs are particularly long-lived cells,48 the accumulation of oxidative damage over the life of the cell is likely. This will lead to damage in both genomic and mitochondrial DNA that can further impair the redox balance and result in impaired vascular function (reviewed in detail elsewhere49,50).
Telomeric DNA is comprised of TTAGGG repeats and is therefore guanine-rich. This makes it particularly susceptible to oxidation because guanine has the lowest redox potential of any of the bases within DNA.51 Oxidative guanine damage has a critical influence on the telomere integrity.52 Thus, telomeres within vascular cells represent a likely site of damage due to oxidative stress. One potential consequence of telomere oxidation is a reduction in the amount of bound TRF1 and TRF2, as even a single oxidized guanine reduces binding of these shelterin components by >50%.53,54 In contrast, overexpression of TRF2 reduces oxidative damage at telomeres in human fibroblasts.55 Therefore, it is possible that vascular cells exposed to oxidative stress may be more likely to become senescent because of a decrease in TRF1 and TRF2 binding that could uncap telomeres. However, currently, only a few studies in ECs or VSMCs have directly connected oxidative stress with telomere dysfunction and the shelterin complex. In one such study, VSMCs that overexpressed TRF2 had reduced DNA damage induced by an oxidative reagent as well as an accelerated DNA repair process.41 In ECs, the findings are less clear. In HUVECs exposed to H2O2, there was an increase in TRF1 but no change in TRF2, despite senescence-associated β-gal staining indicating that the number of senescent ECs increased.56 Taken together, these studies suggest that although the shelterin proteins may help to modulate the effects of oxidative stress on DNA damage, their expression may not be regulated by the presence of ROS.56 Still, it should be noted that the H2O2 levels used in this study appear to be higher than that which may be present in normal physiological conditions, so extrapolation of these findings to the in vivo condition is limited.56 In summary, telomeres within the vasculature are likely sites of oxidative damage which can disrupt the binding of shelterin or alter expression necessary for preventing cellular senescence.
5.2 |. Inflammation
There is extensive evidence of vascular remodeling and endothelial dysfunction due to the chronic low-grade inflammation that occurs in advanced age (reviewed in detail elsewhere8,57). This is characterized by greater circulating levels of pro-inflammatory cytokines and chemokines, as well as increased expression of adhesion molecules in the vascular wall,19 and the accumulation of immune cells in the adventitia.58 In addition to immune-cell derived cytokines, senescent cells within the arteries may contribute to the pro-inflammatory environment via the SASP and lead to a persistent activation of DDR and ROS production. This could impact adjacent cells, leading to their cell cycle arrest and a further increase in the senescent burden. Indeed, senescent human fibroblasts produce ROS sufficient to cause DNA damage in neighboring cells.59 Interestingly, eliminating the extracellular ROS using antioxidants significantly diminishes DNA damage foci formation in neighboring cells.59 In addition, successively passaged human fibroblast cells display increased mitochondrial ROS, which was thought to be a consequence of SASP-induced inflammation rather than primary senescence.60 Thus, inflammatory processes originating in non-senescent cells, infiltrating immune cells, or senescent cells within the vessel can lead to increases in ROS and cytokine production that will perpetuate/enhance the local pro-inflammatory environment and contribute to dysfunction in the artery. Understanding these mechanisms and the regulation of these inflammation pathways may be critical to the development of strategies to limit the senescent burden and improve vascular function with aging.
NF-κB, is a canonical pro-inflammatory transcription factor that plays a central role in regulation of the SASP with advancing age.61 Upregulation of inflammation via the NF-κB pathway may increase production of ROS in vascular cells62 that stimulate SASP-associated inflammation via the sequelae of events from telomere damage to uncapping to the induction of senescence. For example, transgenic activation of NF-kB in mice leads to chronic inflammation and an increase in the number of telomere induced foci in fibroblasts.63 Similar to our findings in aged and hypertensive adults,21,39 telomere dysfunction in this model was not associated with the shortened telomere length, but was associated with oxidative damage marker (4-HNE) and double stranded DNA damage marker γ-H2AX.63 Similarly, our group has demonstrated that telomere uncapping in human arteries is associated with the increased abundance of SASP factors such as IL-6, IL-8 and MCP-1.21 However, further research is warranted to determine if upregulation of inflammatory pathways directly induces telomere uncapping or indirectly impacts telomere function in a ROS-dependent manner. Furthermore, there is a paucity of in vivo data that elucidates whether the upregulation of ROS and inflammation in the setting of age-related telomere uncapping is required for SASP feedback regulation in senescent ECs.
5.3 |. Senescence barriers: p53/p21 pathway
Senescence associated cell cycle arrest has been shown to depend on p53 and pRB, although the relative contribution of them is variable depending on the cell type.64 In comparison to pRB, which is involved in mitogenic stress and chromatin disruption, the p53 pathway is predominantly activated by DNA damage and telomere dysfunction.8 Furthermore, data from human fibroblasts may provide insight into possible mechanisms for the regulation of senescence. In response to double stranded DNA breaks, TRF2 can be rapidly phosphorylated and lose its ability to bind telomeric DNA. This process is recognized as DNA damage by the DDR machinery, leading to activation of the ATM kinase pathway.65 Phosphorylated ATM can further phosphorylate and activate p53 which transcribes p21 to enforce cellular senescence, establishing TRF2-ATM-p53 as a downstream signal of DDR. Additionally, p53 can reduce TRF2 protein levels, which occurs via Siah1 mediated ubiquitin-proteosomal degradation.66 This indicates that when telomeres lose the protection of TRF2, the ATM-p53 signal accentuates the DDR by further repressing TRF2 protein levels. Conversely, extra-telomeric TRF2 has the ability to directly repress p21 transcription by occupying the p21 promoter,67 potentially acting to inhibit cellular senescence. Interestingly, VSMCs from atherosclerotic plaques display increased abundance of Siah-1a and p16, whereas levels of TRF2 increased in cells lacking p53.41 However, in ECs the relationship between TRF2 and p53/p21 is still largely unknown. Taken together, these findings demonstrate that while TRF2 acts to repress the DDR, activation of these pathways can accentuate DNA damage signaling and cellular senescence by reducing TRF2 levels.
6 |. PROSPECTIVE: TARGETING SHELTERIN AS A NOVEL ANTI-AGING STRATEGY
As targeting removal of senescent cells with senolytics has been shown to improve the age-related phenotypes and vascular pathology seen in old rodents,23 several recent clinical trials have been initiated in age-related diseases or other diseases characterized by telomere dysfunction and an abundance of senescent cells.68,69 Thus, a new frontier in drug development has emerged with the ultimate goal being prevention and treatment of premature senescence by developing novel drugs aimed at maintaining shelterin function and telomere homeostasis as well as at preventing inappropriate activation of DDR pathways. Future studies examining the effect of modulating TRF1 and TRF2 to improve age-related vascular dysfunction are warranted, as a number of studies have indicated that both TRF1 and TRF2 can be modified at the post-translational level (reviewed in detail elswhere70). Presently, there are no known compounds that can directly regulate TRF1 or TRF2 expression/activation directly; however, interactions of these telomeric proteins with non-telomeric signaling pathways may provide a novel means to manipulate their activities. For example, TRF2 expression can be controlled by the Wnt/β-catenin pathway, which is critical in stem cell pluripotency and cell fate specification during embryonic development and tumorgenesis.71 Similarly, TRF1 is known to interact with Akt, a ubiquitous serine threonine protein kinase involved in metabolism and growth factor signaling. Indeed, PI3Kα, a downstream target of Akt, has been recently identified to regulate TRF1 protein expression.72 Further studies elucidating these potential shelterin regulators may allow for small molecule compounds which may help improve shelterin function, reduce senescence in old arteries or CVDs thereby improving function.
7 |. CONCLUSIONS
In this short review, we have provided a brief overview of the functional role of cellular senescence and telomere function in vascular function in advanced age and disease. Furthermore, we have outlined a potential role for alterations in the shelterin complex and oxidation of telomeric DNA and the resultant telomere uncapping and DNA damage response in the mechanisms of the age-related vascular phenotypes. The expanding interest in this field will hopefully help to accelerate the development of novel pathways or compounds that can modify the expression/activity of shelterin complex or alleviate DNA damage signaling specific to these insults without altering necessary and beneficial tumor suppressor pathways. Overall, this area is vastly understudied and has immense potential not only in arterial aging but also in treatment of CVD.
Funding information
National Institute on Aging, Grant/Award Number: AG045339, AG050238 and AG053131
Abbreviations:
- CVDs
cardiovascular diseases
- DDR
DNA damage response
- EC
endothelial cells
- ECM
extracellular matrix
- ECs
endothelial cells
- HUVEC
human umbilical vein endothelial cells
- HUVECs
human umbilical vein endothelial cells
- NF- κB
nuclear factor kappa B
- NO
nitric oxide
- p16
cyclin dependent kinase inhibitor 2A
- p21
cyclin-dependent kinase inhibitor 1
- POT1
protection of telomeres-1
- PRB
retinoblastoma protein
- RAP1
Ras-related protein-1
- ROS
reactive oxygen species
- SASP
senescence associated secretory phenotype
- SASP
senescence associated secretory phenotype
- Siah1
Siah E3 Ubiquitin Protein Ligase 1
- TERT
telomerase reverse transcriptase
- TIN2
TRF1-interacting protein 2
- TPP1
tripeptidyl peptidase 1
- TRF1
telomere repeat binding factor-1
- TRF2
telomere repeat binding factor-2
- VSMC
vascular smooth muscle cells
REFERENCES
- 1.Heron M. Deaths: leading causes for 2011. Natl Vital Stat Rep. 2015;64(7):1–96. [PubMed] [Google Scholar]
- 2.Capell BC, Collins FS, Nabel EG. Mechanisms of cardiovascular disease in accelerated aging syndromes. Circ Res. 2007;101(1): 13–26. [DOI] [PubMed] [Google Scholar]
- 3.Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: part I: aging arteries: a “set up” for vascular disease. Circulation. 2003;107(1):139–146. [DOI] [PubMed] [Google Scholar]
- 4.Lakatta EG, Wang M, Najjar SS. Arterial aging and subclinical arterial disease are fundamentally intertwined at macroscopic and molecular levels. Med Clin North Am. 2009;93(3):583–604, Table of Contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seals DR, Jablonski KL, Donato AJ. Aging and vascular endothelial function in humans. Clin Sci (Lond). 2011;120(9):357–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee HY, Oh BH. Aging and arterial stiffness. Circ J. 2010;74(11):2257–2262. [DOI] [PubMed] [Google Scholar]
- 7.Celermajer DS, Sorensen KE, Spiegelhalter DJ, Georgakopoulos D, Robinson J, Deanfield JE. Aging is associated with endothelial dysfunction in healthy men years before the age-related decline in women. J Am Coll Cardiol. 1994;24(2):471–476. [DOI] [PubMed] [Google Scholar]
- 8.Donato AJ, Morgan RG, Walker AE, Lesniewski LA. Cellular and molecular biology of aging endothelial cells. J Mol Cell Cardiol. 2015;89(Pt B):122–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nowak KL, Rossman MJ, Chonchol M, Seals DR. Strategies for achieving healthy vascular aging. Hypertension. 2018;71(3):389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Minamino T, Komuro I. Vascular cell senescence: contribution to atherosclerosis. Circ Res. 2007;100(1):15–26. [DOI] [PubMed] [Google Scholar]
- 11.Minamino T, Miyauchi H, Yoshida T, Tateno K, Kunieda T, Komuro I. Vascular cell senescence and vascular aging. J Mol Cell Cardiol. 2004;36(2):175–183. [DOI] [PubMed] [Google Scholar]
- 12.Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8(9):729–740. [DOI] [PubMed] [Google Scholar]
- 14.Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 2005;120(4):513–522. [DOI] [PubMed] [Google Scholar]
- 15.Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621. [DOI] [PubMed] [Google Scholar]
- 16.Parrinello S, Samper E, Krtolica A, Goldstein J, Melov S, Campisi J. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol. 2003;5(8):741–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88(5):593–602. [DOI] [PubMed] [Google Scholar]
- 18.Di Leonardo A, Linke SP, Clarkin K, Wahl GM. DNA damage triggers a prolonged p53-dependent G1 arrest and long-term induction of Cip1 in normal human fibroblasts. Genes Dev. 1994;8(21):2540–2551. [DOI] [PubMed] [Google Scholar]
- 19.Pantsulaia I, Ciszewski WM, Niewiarowska J. Senescent endothelial cells: potential modulators of immunosenescence and ageing. Ageing Res Rev. 2016;29:13–25. [DOI] [PubMed] [Google Scholar]
- 20.Matthews C, Gorenne I, Scott S, et al. Vascular smooth muscle cells undergo telomere-based senescence in human atherosclerosis: effects of telomerase and oxidative stress. Circ Res. 2006;99(2):156–164. [DOI] [PubMed] [Google Scholar]
- 21.Morgan RG, Ives SJ, Lesniewski LA, et al. Age-related telomere uncapping is associated with cellular senescence and inflammation independent of telomere shortening in human arteries. Am J Physiol Heart Circ Physiol. 2013;305(2):H251–H258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rossman MJ, Kaplon RE, Hill SD, et al. Endothelial cell senescence with aging in healthy humans: prevention by habitual exercise and relation to vascular endothelial function. Am J Physiol Heart Circ Physiol. 2017;313(5):H890–H895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roos CM, Zhang B, Palmer AK, et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell. 2016;15(5):973–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moyzis RK, Buckingham JM, Cram LS, et al. A highly conserved repetitive DNA sequence, (TTAGGG)n, present at the telomeres of human chromosomes. Proc Natl Acad Sci USA. 1988;85(18):6622–6626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Lange T. How telomeres solve the end-protection problem. Science. 2009;326(5955):948–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lazzerini-Denchi E, Sfeir A. Stop pulling my strings - what telomeres taught us about the DNA damage response. Nat Rev Mol Cell Biol. 2016;17(6):364–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Steensel B, de Lange T. Control of telomere length by the human telomeric protein TRF1. Nature. 1997;385(6618):740–743. [DOI] [PubMed] [Google Scholar]
- 28.Sfeir A, Kosiyatrakul ST, Hockemeyer D, et al. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell. 2009;138(1):90–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Levy MZ, Allsopp RC, Futcher AB, Greider CW, Harley CB. Telomere end-replication problem and cell aging. J Mol Biol. 1992;225(4):951–960. [DOI] [PubMed] [Google Scholar]
- 30.Shay JW, Wright WE. Telomeres and telomerase in normal and cancer stem cells. FEBS Lett. 2010;584(17):3819–3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gilson E, Geli V. How telomeres are replicated. Nat Rev Mol Cell Biol. 2007;8(10):825–838. [DOI] [PubMed] [Google Scholar]
- 32.Corey DM, Rinkevich Y, Weissman IL. Dynamic patterns of clonal evolution in tumor vasculature underlie alterations in lymphocyte-endothelial recognition to foster tumor immune escape. Can Res. 2016;76(6):1348–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Machin D, Dobson P, Ghaffari S, Walker A, Lesniewski L, Donato A. Age-related telomere uncapping occurs independent of telomere shortening in mouse endothelial cells. FASEB J 2015; 29(1_supplement):642. [Google Scholar]
- 34.Wang J, Dong X, Cao L, et al. Association between telomere length and diabetes mellitus: a meta-analysis. J Int Med Res. 2016;44(6):1156–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu X, Qu K, Pang Q, Wang Z, Zhou Y, Liu C. Association between telomere length and survival in cancer patients: a meta-analysis and review of literature. Front Med. 2016;10(2):191–203. [DOI] [PubMed] [Google Scholar]
- 36.Haycock PC, Heydon EE, Kaptoge S, Butterworth AS, Thompson A, Willeit P. Leucocyte telomere length and risk of cardiovascular disease: systematic review and meta-analysis. BMJ. 2014;349:g4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bhayadia R, Schmidt BM, Melk A, Homme M. Senescence-induced oxidative stress causes endothelial dysfunction. J Gerontol A Biol Sci Med Sci. 2016;71(2):161–169. [DOI] [PubMed] [Google Scholar]
- 38.Beyer AM, Freed JK, Durand MJ, et al. Critical role for telomerase in the mechanism of flow-mediated dilation in the human microcirculation. Circ Res. 2016;118(5):856–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morgan RG, Ives SJ, Walker AE, et al. Role of arterial telomere dysfunction in hypertension: relative contributions of telomere shortening and telomere uncapping. J Hypertens. 2014;32(6):1293–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Griffith JD, Comeau L, Rosenfield S, et al. Mammalian telomeres end in a large duplex loop. Cell. 1999;97(4):503–514. [DOI] [PubMed] [Google Scholar]
- 41.Wang J, Uryga AK, Reinhold J, et al. Vascular smooth muscle cell senescence promotes atherosclerosis and features of plaque vulnerability. Circulation. 2015;132(20):1909–1919. [DOI] [PubMed] [Google Scholar]
- 42.Voghel G, Thorin-Trescases N, Farhat N, et al. Cellular senescence in endothelial cells from atherosclerotic patients is accelerated by oxidative stress associated with cardiovascular risk factors. Mech Ageing Dev. 2007;128(11–12):662–671. [DOI] [PubMed] [Google Scholar]
- 43.Rothkamm K, Barnard S, Moquet J, Ellender M, Rana Z, Burdak-Rothkamm S. DNA damage foci: meaning and significance. Environ Mol Mutagen. 2015;56(6):491–504. [DOI] [PubMed] [Google Scholar]
- 44.Hohensinner PJ, Kaun C, Buchberger E, et al. Age intrinsic loss of telomere protection via TRF1 reduction in endothelial cells. Biochem Biophys Acta. 2016;1863(2):360–367. [DOI] [PubMed] [Google Scholar]
- 45.Guzik TJ, Touyz RM. Oxidative stress, inflammation, and vascular aging in hypertension. Hypertension. 2017;70(4):660–667. [DOI] [PubMed] [Google Scholar]
- 46.Wung BS, Cheng JJ, Hsieh HJ, Shyy YJ, Wang DL. Cyclic strain-induced monocyte chemotactic protein-1 gene expression in endothelial cells involves reactive oxygen species activation of activator protein 1. Circ Res. 1997;81(1):1–7. [DOI] [PubMed] [Google Scholar]
- 47.Gray SP, Di Marco E, Okabe J, et al. NADPH oxidase 1 plays a key role in diabetes mellitus-accelerated atherosclerosis. Circulation. 2013;127(18):1888–1902. [DOI] [PubMed] [Google Scholar]
- 48.Schwartz SM, Benditt EP. Aortic endothelial cell replication. I. Effects of age and hypertension in the rat. Circ Res. 1977;41(2):248–255. [DOI] [PubMed] [Google Scholar]
- 49.Incalza MA, D’Oria R, Natalicchio A, Perrini S, Laviola L, Giorgino F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul Pharmacol. 2018;100:1–19. [DOI] [PubMed] [Google Scholar]
- 50.Li H, Horke S, Forstermann U. Vascular oxidative stress, nitric oxide and atherosclerosis. Atherosclerosis. 2014;237(1):208–219. [DOI] [PubMed] [Google Scholar]
- 51.Fleming AM, Burrows CJ. G-quadruplex folds of the human telomere sequence alter the site reactivity and reaction pathway of guanine oxidation compared to duplex DNA. Chem Res Toxicol. 2013;26(4):593–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang Z, Rhee DB, Lu J, et al. Characterization of oxidative guanine damage and repair in mammalian telomeres. PLoS Genet. 2010;6(5):e1000951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Opresko PL, Fan J, Danzy S, Wilson DM 3rd, Bohr VA. Oxidative damage in telomeric DNA disrupts recognition by TRF1 and TRF2. Nucleic Acids Res. 2005;33(4):1230–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rhee DB, Ghosh A, Lu J, Bohr VA, Liu Y. Factors that influence telomeric oxidative base damage and repair by DNA glycosylase OGG1. DNA Repair. 2011;10(1):34–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Richter T, Saretzki G, Nelson G, Melcher M, Olijslagers S, von Zglinicki T. TRF2 overexpression diminishes repair of telomeric single-strand breaks and accelerates telomere shortening in human fibroblasts. Mech Ageing Dev. 2007;128(4):340–345. [DOI] [PubMed] [Google Scholar]
- 56.Maeda T, Guan JZ, Koyanagi M, Makino N. Telomerase activity and telomere length distribution in vascular endothelial cells in a short-term culture under the presence of hydrogen peroxide. Geriatr Gerontol Int. 2013;13(3):774–782. [DOI] [PubMed] [Google Scholar]
- 57.Wang M, Shah AM. Age-associated pro-inflammatory remodeling and functional phenotype in the heart and large arteries. J Mol Cell Cardiol. 2015;83:101–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moos MP, John N, Grabner R, et al. The lamina adventitia is the major site of immune cell accumulation in standard chow-fed apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2005;25(11):2386–2391. [DOI] [PubMed] [Google Scholar]
- 59.Nelson G, Wordsworth J, Wang C, et al. A senescent cell bystander effect: senescence-induced senescence. Aging Cell. 2012;11(2):345–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lawless C, Jurk D, Gillespie CS, et al. A stochastic step model of replicative senescence explains ROS production rate in ageing cell populations. PLoS ONE. 2012;7(2):e32117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Donato AJ, Pierce GL, Lesniewski LA, Seals DR. Role of NFkappaB in age-related vascular endothelial dysfunction in humans. Aging. 2009;1(8):678–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Foncea R, Carvajal C, Almarza C, Leighton F. Endothelial cell oxidative stress and signal transduction. Biol Res. 2000;33(2):89–96. [DOI] [PubMed] [Google Scholar]
- 63.Jurk D, Wilson C, Passos JF, et al. Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nat Commun. 2014;2:4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tanaka H, Mendonca MS, Bradshaw PS, et al. DNA damage-induced phosphorylation of the human telomere-associated protein TRF2. Proc Natl Acad Sci USA. 2005;102(43):15539–15544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fujita K, Horikawa I, Mondal AM, et al. Positive feedback between p53 and TRF2 during telomere-damage signalling and cellular senescence. Nat Cell Biol. 2010;12(12):1205–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hussain T, Saha D, Purohit G, et al. Transcription regulation of CDKN1A (p21/CIP1/WAF1) by TRF2 is epigenetically controlled through the REST repressor complex. Sci Rep. 2017;7(1):11541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Garcia-Calzon S, Zalba G, Ruiz-Canela M, et al. Dietary inflammatory index and telomere length in subjects with a high cardiovascular disease risk from the PREDIMED-NAVARRA study: cross-sectional and longitudinal analyses over 5 y. Am J Clin Nutr. 2015;102(4):897–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Salvador L, Singaravelu G, Harley CB, Flom P, Suram A, Raffaele JM. A natural product telomerase activator lengthens telomeres in humans: a randomized, double blind, and placebo controlled study. Rejuvenation Res. 2016;19(6):478–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Walker JR, Zhu XD. Post-translational modifications of TRF1 and TRF2 and their roles in telomere maintenance. Mech Ageing Dev. 2012;133(6):421–434. [DOI] [PubMed] [Google Scholar]
- 71.Diala I, Wagner N, Magdinier F, et al. Telomere protection and TRF2 expression are enhanced by the canonical Wnt signalling pathway. EMBO Rep. 2013;14(4):356–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mendez-Pertuz M, Martinez P, Blanco-Aparicio C, et al. Modulation of telomere protection by the PI3K/AKT pathway. Nat Commun. 2017;8(1):1278. [DOI] [PMC free article] [PubMed] [Google Scholar]