

Abstract
Sonic Hedgehog (SHH) signaling, a developmental pathway promoting lung mesenchymal expansion and differentiation during embryogenesis, has been increasingly recognized as a profibrotic factor in mature lung, where it might contribute to the pathogenesis of lung fibrosis. Pathway inhibition at the level of the downstream Gli transcription factors Gli1 and Gli2 (by GANT61) ameliorates lung fibrosis in the bleomycin model, whereas inhibition proximally at the level of HH ligand (by anti Hh antibody 5E1) or Smo (by GDC-0449) of the canonical pathway does not, implicating Gli1 and/or Gli2 as a key target. The fact that both the Gli1-labelled cell lineage and Gli1 expressing cells expand during fibrosis formation and contribute significantly to the pool of myofibroblasts in the fibrosis scars suggests a fibrogenic role for Gli1. Therefore to further dissect the roles of Gli1 and Gli2 in lung fibrosis we evaluated Gli1 KO and control mice in the bleomycin model. Monitoring of Gli1+/+ (n = 12), Gli1lZ/+ (n = 37) and Gli1lZ/lZ (n = 18) mice did not reveal differences in weight loss or survival. Lung evaluation at the 21-day endpoint did not show differences in lung fibrosis formation (as judged by morphology and trichrome staining), Ashcroft score, lung collagen content, lung weight, BAL protein content or BAL cell differential count. Our data suggest that Gli1 is not required for bleomycin-induced lung fibrosis.
Keywords: Hedgehog signaling, Gli1, bleomycin, myofibroblast, lung fibrosis
Introduction
Sonic Hedgehog (SHH) signaling is one of the key epithelium-derived regulators of lung mesenchyme during lung development, inducing balanced mesenchymal lineage expansion and differentiation.[1–3] Whether the same signals of this pathway contribute to expansion and differentiation of mesenchyme in a pathologic scenario, such as the fibrotic response in lung fibrosis, exemplified by idiopathic pulmonary fibrosis (IPF), remains elusive. However, the profibrotic characteristics of the HH pathway and several important observations render it highly relevant to lung fibrosis. SHH signals from epithelium to mesenchyme, and disturbed epithelial-mesenchymal communication is thought to be central to IPF.[4] SHH signaling is pro-fibrotic: it promotes mesenchymal cell proliferation and differentiation (e.g., to myofibroblasts and bronchial smooth muscle cells) in embryonic lung development,[5,6] postnatal lung development[7] and in pulmonary fibrosis;[8] it also exhibits pro-survival, anti-apoptotic effects, as in vitro studies have shown. Fibroblasts from IPF lungs are more resistant to induction of apoptosis,[9] and we showed prolonged fibroblast survival in a 3D-sphere model in culture.[10] Others have observed aberrant HH pathway expression in IPF,[11,12] in other forms of pulmonary fibrosis,[13,14] and in fibrosis of other organs, such as heart, liver, skin and kidney.[15–18] Furthermore, the Gli1-derived lineage expands during lung fibrosis in the bleomycin model and four other fibrosis models and this lineage contributes to the alpha smooth muscle actin (aSMA)-positive myofibroblast population in fibrosis scars.[19] Together these data indicate a central role for this pathway in fibrosis.
Sonic Hedgehog (Shh), the most widely expressed HH ligand in the developing lung, induces canonical signaling by interacting with its receptor patched 1 (Ptch1), which modulates the Gli transcription factors (Gli1/2/3) and transcription of target genes through the transmembrane protein smoothened (Smo) (reviewed in[20]). Without ligand, Ptch1 inhibits Smo, resulting in transcriptional repressor activity of Gli3. Ligand binding to Ptch1 removes Smo inhibition, promoting transcriptional activator activity of Gli2. Gli1, an obligatory, though redundant, activator of the pathway, provides positive feedback and, as a direct transcriptional target, has proven to be a reliable reporter of HH pathway activity.[21] Ptch1 and hedgehog inhibitory protein (Hhip), also direct transcriptional targets, providing negative feedback. Ptch1 decreases pathway activity through Smo inhibition. Hhip, which is expressed on the cell surface, competes with Ptch1 for SHH ligand.
Several studies that were undertaken to study the role of HH signaling in the experimental model of bleomycin-induced lung fibrosis have drawn attention to the downstream transcription factors Gli1 and Gli2. Targeting upstream molecules of the HH pathway did not alter pulmonary fibrosis, as competition for Shh ligand using the pan-HH antibody 5E1[10,22] and inhibition of Smo with the small molecule GDC-0449 failed to ameliorate lung fibrosis.[23] However, GANT61, a small-molecule inhibitor with binding-specificity to both Gli1 and Gli2[24] significantly reduced the fibrotic lesions in the study of Farrokhi et al.[23] These data suggest, first, a fibrogenic role for Gli1 and/or Gl2 and, second, the possibility of HH pathway activation in a non-canonical manner.[25] Recent work of Salzer et al. revealed that in the CNS Gli1 by itself is critical for neural stem cell remyelination.[26]
Studies to evaluate the role of Gli1 and Gli2 face several obstacles. At present it is not possible to disrupt Gli2 signaling alone and study its effect independent of Gli1 in disease models. First, Gli2−/− mice develop hypoplastic lungs and are not viable.[27] Second, Gli2 loss by itself would affect expression of Gli1 as a target of the canonical HH signaling. Third, while the lungs of Gli2+/− mice appear normal, Gli2+/− mice that are also null for Gli1 develop hypoplastic lungs and 50% of these embryos die at birth.[28] On the other hand, Gli1 is redundant during embryonic and postnatal lung development, as Gli1lZ/lZ mice develop normally into adulthood without any spontaneous phenotype, which makes them suitable to test Gli1-dependence in an appropriate model, where Gli2 expression should remain unimpaired.
Therefore to further dissect the roles of Gli1 and 2 in lung fibrosis we used control, Gli1+/− and Gli1 KO mice in the bleomycin model. We hypothesized that loss of Gli1 alleles will ameliorate lung fibrosis.
Material and methods
Animal experiments
All experimental protocols were authorized by the Institutional Animal Care and Use Committee at NYU Langone Medical Center (New York, NY). Mice harboring Gli1nlacZ knock-in alleles (Swiss Webster background) were genotyped as described.[21] Gli1nlacZ/+ heterozygotes were bred to generate homozygous, heterozygous and wild type off-spring. A total of 67 genotyped littermates (12 Gli1+/+, 37 Gli1lZ/+, and 18 Gli1lZ/lZ mice) of both genders (34 females and 33 males), 8–10 weeks of age, were exposed to endotracheal bleomycin as previously described.[10] In brief, the animals received intraperitoneal anesthetics (Ketamine 80 mg/kg) and Xylazine 6 mg/kg) prior to surgical cutdown and endotracheal administration of 1.125 U/kg bleomycin in 50 μl sterile saline (Sigma-Aldrich, St. Louis, MO) via a 28-gauge syringe. Following recovery the animals were monitored for weight change, activity and well-being every 2–3 days and mortality was documented until lung harvest at day 21.
Lung histology and histochemistry
Lung harvest and processing was performed as previously described.[10] Mice were anesthetized with pentobarbital and exsanguinated. Bronchoalveolar lavage (BAL) was collected immediately for cell count, differential count following the Diff-quick method and protein quantification using BCA assay (Pierce). After right-ventricular perfusion with PBS, the right lung was collected for collagen quantification. The left lung tissue was fixed with 4% PFA in PBS at 25 cm H2O pressure for 15 min at RT, followed by further fixation for 24 h at 4 °C, paraffin embedding and sectioning. Lung sections were stained with Masson’s trichrome to assess collagen distribution. For immunofluorescence-based detection of b-gal and aSMA, sections were de-parafinized, antigen-retrieved with pH6-citrate buffer (DAKO, Carpinteria, CA), followed by blocking and incubation with primary against b-gal (Abcam, ab9361,1:750) and aSMA-FITC (Sigma, F3777, 1:1000), secondary antibodies (Alexa Fluor 555, Molecular Probes, 1:1000) and DAPI nuclear dye. Trichrome- and IF-stained lungs were imaged using a Ni- epifluorescence microscope and NIS-Elements AR software (Nikon Instruments, Inc, Melville, NY).
Ashcroft fibrosis score
The Ashcroft scoring method was performed on Masson-Trichrome-stained lung tissue sections, following the instructions of the original publication.[29] All trichrome stained lung sections were scanned using the Perkin-Elmer multispectral imaging system Vectra3 (Waltham, MA). The obtained lung images were then overlaid with a 10x magnification tile grid using Perkin-Elmer W-Form 2.4.2 software. Tiles covering less than 50% of lung area or containing dominating air-way and/or vascular structures were excluded. Ashcroft scores were assigned to each tile. The average Ashcroft score for each lung was derived by dividing the sum of scores by the number of scored tiles.
Collagen quantification
Right lung collagen was quantified using the hydroxyproline assay as described.[10]
Statistical analysis
Statistical analysis including Kaplan-Meier survival curves was performed using GraphPad Prism 7.0 (GraphPad Software, Inc. La Jolla, CA). Weight data are presented as mean ± standard deviation (SD). Statistical difference between groups were analyzed for parametric data using Student’s t-test. The data for the Ashcroft score, collagen content, lung weight, BAL protein and differential count are presented as individual data points and medians with range. For these non-parametric data sets we used one-way analysis of variance (ANOVA) and the Kruskal-Wallis test to test for statistical significance. A p value greater than 0.05 was considered significant. Our power calculations were based on previous bleomycin experiments, in which we calculated that approximately 10 animals per group were needed to show a 25% difference in collagen content with a power of 90% and a p-value of 0.05.
Results
Gli1+/+, Gli1lZ/+, and Gli1lZ/1Z littermates including both genders were monitored for weight loss and survival during the 21-day study period following bleomcyin injury (Figure 1). Swiss Webster mice show intermediate susceptibility to bleomycin-induced lung injury.[30] There was no significant difference in weight loss between the different genotypes (Figure 1A). Subgroup analysis by gender shows significantly greater weight loss in the female Gli1+/+ animals when compared to Gli1lZ/+ and Gli1lZ/lZ mice (Figure 1B and C). Overall survival was 91% (61/67) and matched the expected mortality of ~10%, which we had established in preliminary dosing experiments for the Swiss-Webster background prior to the study. Survival of all three genotypes was comparable (Figure 1D).
Figure 1.
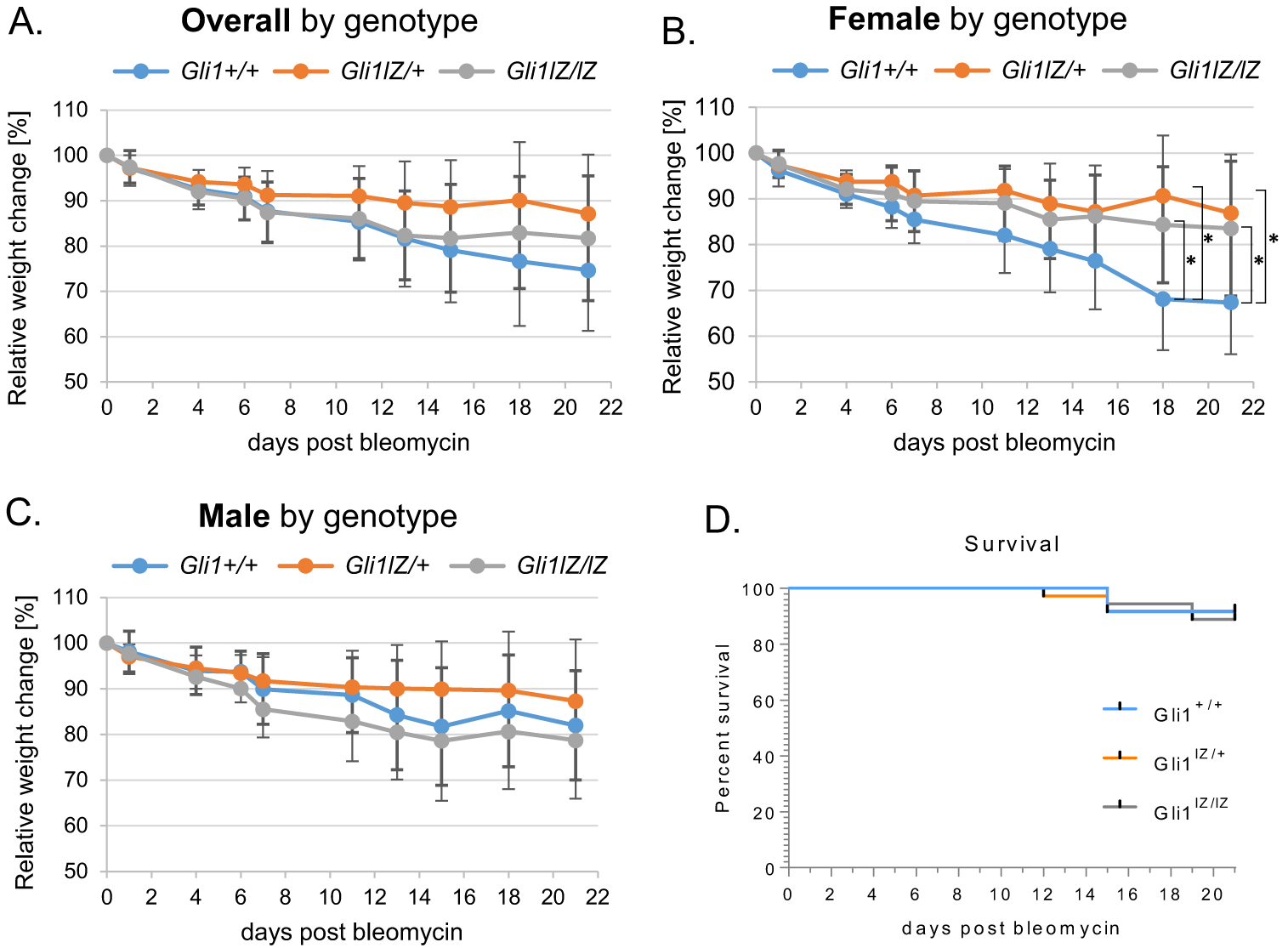
Comparison of bleomycin injury-induced weight loss and survival between Gli1+/+, Gli1lZ/+, and Gli1lZ/lZ mice. (A-C) Relative weight changes in % from original weight at the day of bleo administration were calculated from recorded weights and reported overall by (A) genotype and (B, C) by gender and genotype. (D) Survival curves for each group were calculated from recorded deaths over the 21-day study period. Data are shown as mean and SD of each group.*:p < 0.05.
Evaluation of lung fibrosis was performed 21 days following exposure and included lung morphology, Ashcroft fibrosis score, collagen quantification, lung weight and BAL studies (Figures 2 and 3). Representative low-, intermediate-, and high-magnification images of tri-chrome-stained lungs of the 3 different genotypes show the presence of the typical bleomycin-associated fibrotic changes, which were seen to a significant extent in almost all of the examined lungs despite some variation between mice. Dense fibrosis lesions with significant alveolar-interstitial collagen deposition surrounding stromal cells and inflammatory cell infiltrate, together with alveolar airspace destruction, distortion and bronchiectasis, were not different between Gli1+/+, Gli1lZ/+, and Gli1lZ/lZ mice (Figure 2A). IF-based examination of b-gal protein shows Gli1+ cells in Gli1lZ/+, and Gli1lZ/lZ, but not in Gli1+/+(wild type) lungs, consistent with the presence of the lacZ knock-in allele in the former (Figure 2B). aSMA+ myofibroblasts are present to a similar extent in the fibrosis scars of all three genotypes. Of note, in Gli11Z/+ and Gli1lZ/lZ lungs a significant number of aSMA-positive cells are also Gli1-positive, consistent with our previous findings.[10]
Figure 2.
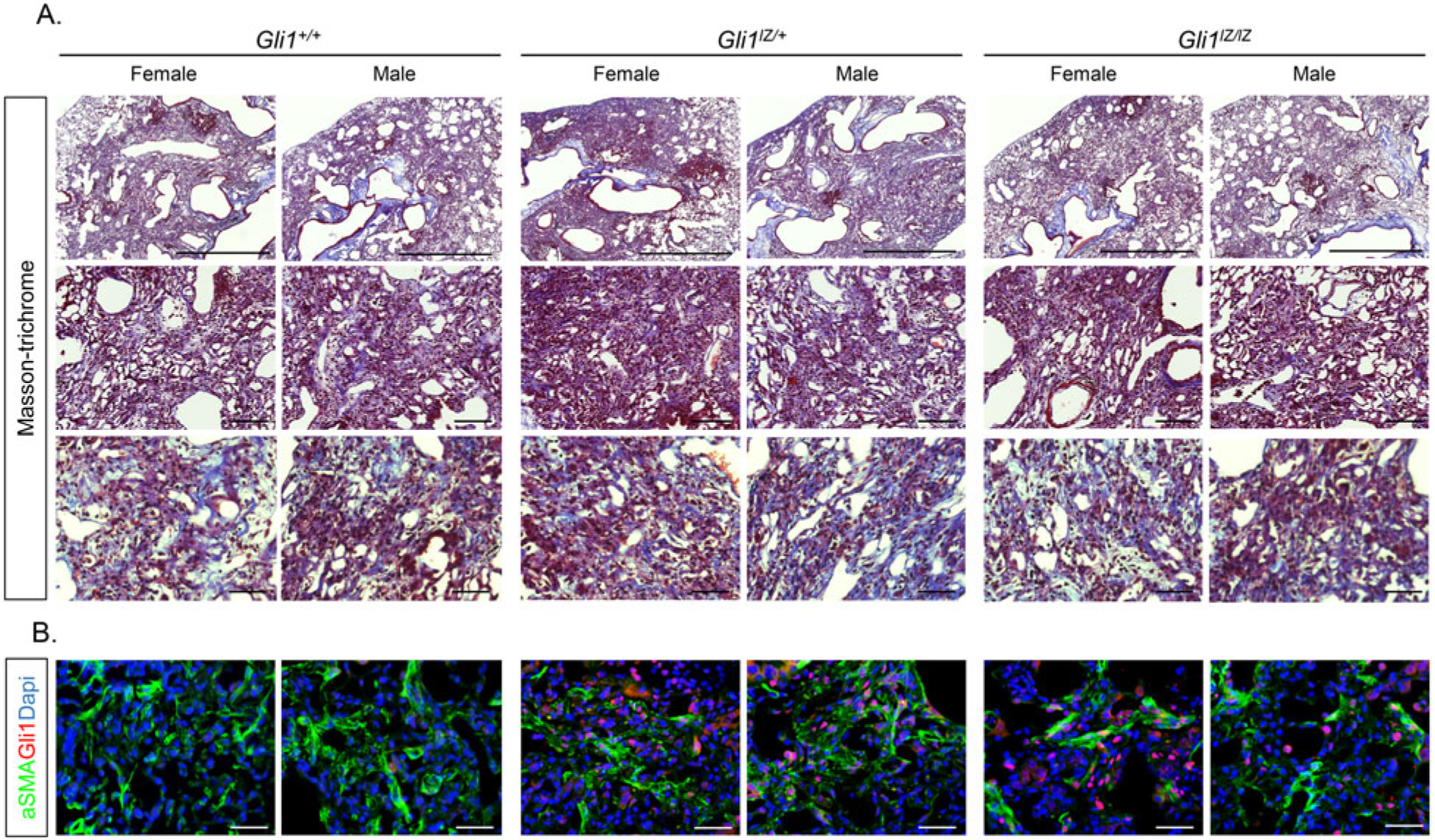
Comparison of morphologic changes in bleomycin-injured Gli1+/+, Gli1lZ/+, and Gli1lZ/lZ lungs. (A) Low-, intermediate-, and high-magnification images of trichrome-stained sections were taken from representative female and male lungs for each genotype. Scale bar upper panel: 250 mm, scale bar middle panel: 100 mm, scale bar lower panel: 50 mm. (B) High-magnification IF images of aSMA (green), Gli1 (red) and DAPI (blue) staining from representative female and male lungs for each genotype. Scale bar: 50 μm.
Figure 3.
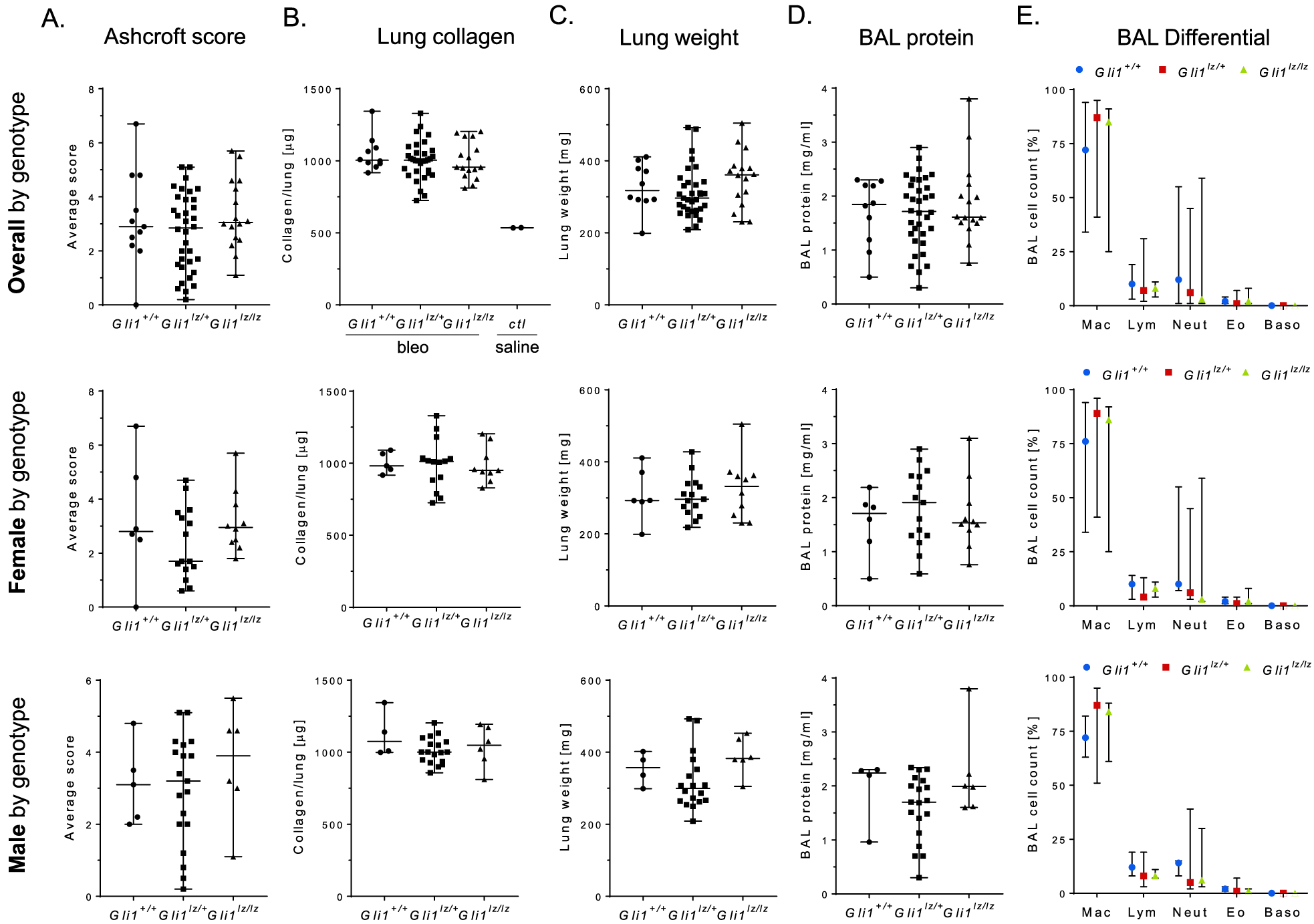
Comparison of quantitative parameters of bleomycin- Gli1+/+, Gli1lZ/+, and Gli1lZ/lZ lungs. (A) Extent of lung fibrosis was assessed using the Ashcroft score method. Average score values for each lung are shown. (B) Lung collagen content was calculated from measured hydroxyproline content and is shown in mg collagen per lung. Bleomycin-treated lungs were compared to each other and to historic saline control (ctl) lungs. (C) Wet lung weight was measured during lung harvest. (D) BAL protein was quantified using a BCA assay. (E) Cell differential counts were performed on Diff-Quik stained cytospin samples. Data are shown as median with range of each group.*: p < 0.05.
We next performed histopathological evaluation of lung fibrosis by applying the established Ashcroft scoring method.[29] Average Ashcroft scores did not significantly differ between Gli1+/+, Gli1lZ/+, and Gli1lZ/lZ lungs (Figure 3A). We also compared lung collagen content and lung weight between the three groups (Figure 3B and C). Collagen content on average was 2-fold higher than in saline-treated historic control lungs, consistent with significant collagen deposition and fibrosis as a result of bleomycin-injury. Collagen content was not significantly different between the genotypes and the same was the case in the gender-based subgroup analysis (Figure 3B). Lung weight was similar in Gli1+/+, Gli1lZ/+, and Gli1lZ/lZ mice (Figure 3C), but, as expected, it was dependent on gender with an average male lung weight of ~340 mg and a female weight of ~310 mg independent of the genotype (data not shown).
Finally, we evaluated BAL protein content, cell count and differential in the three genotypes (Figure 3D and E). BAL protein and total BAL cell count were not significantly between the groups (data not shown). Although the BAL differential cell count for lymphocytosis and neutrophilia varied to a certain extent, the three groups did not differ significantly in their inflammatory cell composition in the fibrosed lungs.
Discussion
In this study we carried out a large-scale experiment using a genetic approach to determine the role of Gli1 in bleomycin-induced fibrosis. Despite the large number of animals and inclusion of both genders in a bleomycin-susceptible mouse cohort we were unable to discern a difference following increasing loss of Gli1 alleles.
There are several reasons why the redundance of Gli1 in the bleomycin model comes as a surprising result. First, we and others have shown that there is an increased number of Gli1+ cells localized in the fibrosis areas of bleomycin-treated lungs and that many of them are aSMA-expressing myofibroblasts, one of the key characteristics of pulmonary fibrosis.[19,23,30] In the same experiment using the Gli1lZ/+ reporter, we also showed increased numbers of Gli1+ cells in fibrosis-adjacent areas, raising the possibility of pathway activation in the pool of Gli2+ cells located in the alveolar interstitial septa of uninjured lungs. Farrokhi and colleagues described both Gli1+;aSMA+ and Gli2+;aSMA+ double-positive cells in the fibrotic scars of C57Bl6 wild-type mice.[23] Kramann et al., who used the lineage tracer Gli1creERT2;R26RtdTom, showed that the Gli1-labeled cell population expands during bleomycin-induced lung fibrosis and contributes significantly to the aSMA+ myofibroblast population in fibrosis scars.[19] Of note, they also detected the same phenomenon in fibrosis models of the kidney, heart, liver. Furthermore, genetic ablation of Gli1 cell lineage using diphtheria-toxin receptor methodology ameliorated kidney and heart fibrosis,[19] implicating an important functional contribution of Gli1-expressing cells to the fibrosis process.
Second, if Gli1 is a central element impacting fibrosis induction and is not dependent on canonical HH pathway input, as suggested by Farrokhi’s results and our prior study,[10,23] one would still expect blockage of this non-canonical signaling axis when Gli1 is knocked out. As Gli1−/− mice develop normally into adulthood, its relevance for mesenchymal cell function could well be questioned. However, canonical HH pathway inhibition during embryonic and postnatal lung development decreased both total lung Gli1 gene expression and the number of Gli1+ cells, and these findings were accompanied by effects on key mesenchymal functions such as proliferation, myofibroblast and bronchial smooth muscle cell differentiation, and overall lung structure.[5,7]
One possible explanation for our results is that Gli2 rather than Gli1 contributes to the regulation of mesenchymal cell function in adult lung. Alveolar Gli2+ cells are more numerous than Gli1+ cells. Therefore, inhibition of Gli2 might affect a greater number of cells, even if it does not include the whole Gli2+ cell population. The significance of this idea is supported by a recent study of Wang and colleagues, in which Gli2-labelled cells were sorted into Gli1high and Gli1low groups, which anatomically localized to small air-ways and to alveoli, respectively, and were able to show that over-activation of Gli2 that included distal Gli1low cells using the SmoM2 allele resulted in loss of alveoli and enlarged airspace size.[31] Therefore it is conceivable that inhibition of the same population could shift the balance towards mesenchymal expansion and extracellular matrix deposition and maybe even spontaneous fibrosis formation. Altering the Gli2 cell pool in Wang’s study was also accompanied by shifted expression of genes that regulate alveolar stem cell regeneration and suggests a role of Gli2 in stem cell maintenance, a link that has been observed in the mammary gland.[32] In light of this idea, the inhibitory effect of GANT61 on bleomycin-induced lung fibrosis was most likely due to its inhibitory effect on Gli2 rather than on Gli1.[23] However, the concern of pharmacologic inhibitors has always been that they might exert nonspecific effects that could affect the outcome, in this case fibrosis. Therefore, we tried to circumvent this problem by using a genetic approach of Gli1 allelic loss for our study. Of note, there is a Gli2flox conditional allele available for targeted Gli2 deletion,[33] however, given the essential requirement of Gli2 during embryonic lung development,[27,28] this approach in homozygote mice might require restriction of Cre recombination to either a lung cell population or to a postnatal time point.
One of the limitations of our study is that we used an outbred background, Swiss-Webster, of intermediate bleomycin-susceptibility while the inbred C57Bl6 background would be most the most susceptible lung fibrosis. However, based on the extent of fibrosis we observed, we believe that there was sufficient fibrosis formation to reliably detect a difference between the different genotypes if present.
In the future we need to more closely evaluate the requirement for Gli2 in pulmonary fibrosis, as Gli2 appears to be a promising target and inhibitors that specifically target Gli2 could be used to target pulmonary fibrosis and IPF.
Acknowledgments
We would like to thank the following funding entities for their support: the NIH/NHLBI (K08-HL128842)(M.C.K) and (5R21-HL104455)(J.S.M.), NIH/NCI (P30-CA016087) (C.A.L.), the Stony Wold-Herbert Foundation NYC (M.C.K), the NYU Weissmann Scholarship program (M.C.K), and the Will Rogers Foundation (M.C.K, J.S.M). We also thank the members of the Experimental Pathology Research laboratory, which is partially supported by the Cancer Center Support Grant P30CA016087.
Funding
National Heart, Lung, and Blood Institute; Stony Wold-Herbert Fund; National Cancer Institute.
Footnotes
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- [1].Maeda Y, Dave V, Whitsett JA. Transcriptional control of lung morphogenesis. Physiol Rev. 2007; 87(1):219–244. doi: 10.1152/physrev.00028.2006. [DOI] [PubMed] [Google Scholar]
- [2].Hines EA, Sun X. Tissue crosstalk in lung development. J Cell Biochem. 2014;115(9):1469–1477. doi: 10.1002/jcb.24811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Shannon JM, Hyatt BA. Epithelial-mesenchymal interactions in the developing lung. Annu Rev Physiol. 2004;66(1):625–645. doi: 10.1146/annurev.-physiol.66.032102.135749. [DOI] [PubMed] [Google Scholar]
- [4].Sakai N, Tager AM. Fibrosis of two: Epithelial cell-fibroblast interactions in pulmonary fibrosis. Biochim Biophys Acta. 2013;1832(7):911–921. doi: 10.1016/j.bbadis.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Miller L-AD, Wert SE, Clark JC, et al. Role of Sonic hedgehog in patterning of tracheal-bronchial cartilage and the peripheral lung. Dev Dyn. 2004;231(1): 57–71. doi: 10.1002/dvdy.20105. [DOI] [PubMed] [Google Scholar]
- [6].Li C, Li M, Li S, et al. Progenitors of secondary crest myofibroblasts are developmentally committed in early lung mesoderm. Stem Cells. 2015;33(3): 999–1012. doi: 10.1002/stem.1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kugler MC, Loomis CA, Zhao Z, Cushman JC, Liu L, Munger JS. Sonic Hedgehog signaling regulates myofibroblast function during alveolar septum formation in murine postnatal lung. Am J Respir Cell Mol Biol. 2017;57(3):280–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hu B, Liu J, Wu Z, et al. Reemergence of Hedgehog mediates epithelial-mesenchymal crosstalk in pulmonary fibrosis. Am J Respir Cell Mol Biol. 2014;52(4): 418–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lozano Bolanos A, Milla CM, Lira JC, et al. Role of Sonic Hedgehog in idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2012;303(11): L978–L990. [DOI] [PubMed] [Google Scholar]
- [10].Liu L, Kugler MC, Loomis CA, et al. Hedgehog signaling in neonatal and adult lung. Am J Respir Cell Mol Biol. 2013;48(6):703–710. doi: 10.1165/rcmb.2012-0347OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Selman M, Pardo A, Kaminski N. Idiopathic pulmonary fibrosis: aberrant recapitulation of developmental programs? PLoS Med. 2008;5(3):e62. doi: 10.1371/journal.pmed.0050062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Cigna N, Farrokhi Moshai E, Brayer S, et al. The Hedgehog system machinery controls transforming growth factor-beta-dependent myofibroblastic differentiation in humans: involvement in idiopathic pulmonary fibrosis. Am J Pathol. 2012;181(6): 2126–2137. doi: 10.1016/j.ajpath.2012.08.019. [DOI] [PubMed] [Google Scholar]
- [13].Coon DR, Roberts DJ, Loscertales M, et al. Differential epithelial expression of SHH and FOXF1 in usual and nonspecific interstitial pneumonia. Exp Mol Pathol. 2006;80(2):119–123. doi: 10.1016/j.yexmp.2005.12.003. [DOI] [PubMed] [Google Scholar]
- [14].Stewart GA, Hoyne GF, Ahmad SA, et al. Expression of the developmental Sonic hedgehog (Shh) signalling pathway is up-regulated in chronic lung fibrosis and the Shh receptor patched 1 is present in circulating T lymphocytes. 2003;199(4):488–495. doi: 10.1002/path.1295. [DOI] [PubMed] [Google Scholar]
- [15].Fabian SL, Penchev RR, St-Jacques B, et al. Hedgehog-Gli pathway activation during kidney fibrosis. Am J Pathol. 2012;180(4):1441–1453. doi: 10.1016/j.ajpath.2011.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Horn A, Palumbo K, Cordazzo C, et al. Hedgehog signaling controls fibroblast activation and tissue fibrosis in systemic sclerosis. Arthritis Rheum. 2012; 64(8):2724–2733. doi: 10.1002/art.34444. [DOI] [PubMed] [Google Scholar]
- [17].Omenetti A, Porrello A, Jung Y, et al. Hedgehog signaling regulates epithelial-mesenchymal transition during biliary fibrosis in rodents and humans. J Clin Invest. 2008;118(10):3331–3342. doi: 10.1172/JCI35875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bijlsma MF, Leenders PJA, Janssen BJA, et al. Endogenous hedgehog expression contributes to myocardial ischemia-reperfusion-induced injury. Exp Biol Med (Maywood). 2008;233(8) :989–996. doi: 10.3181/0711-RM-307. [DOI] [PubMed] [Google Scholar]
- [19].Kramann R, Schneider RK, DiRocco DP, et al. Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell. 2015;16(1):51–66. doi: 10.1016/j.stem.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kugler MC, Joyner AL, Loomis CA, Munger JS. Sonic Hedgehog signaling in the lung - From development to disease. Am J Respir Cell Mol Biol. 2015; 52(1):1–13. doi: 10.1165/rcmb.2014-0132TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bai CB, Auerbach W, Lee JS, et al. Gli2, but not Gli1, is required for initial Shh signaling and ectopic activation of the Shh pathway. Development. 2002; 129(20):4753–4761. [DOI] [PubMed] [Google Scholar]
- [22].Ericson J, Morton S, Kawakami A, et al. Two critical periods of Sonic Hedgehog signaling required for the specification of motor neuron identity. Cell 1996; 87(4):661–673. doi: 10.1016/S0092-8674(00)81386-0. [DOI] [PubMed] [Google Scholar]
- [23].Farrokhi Moshai E, Wémeau-Stervinou L, Cigna N. Targeting the Hedgehog-Gli pathway inhibits Bleomycin-induced lung fibrosis in mice. Am J Respir Cell Mol Biol. 2014;51(1):11–25. [DOI] [PubMed] [Google Scholar]
- [24].Lauth M, Bergstrom A, Shimokawa T, et al. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc Natl Acad Sci U S A. 2007;104(20):8455–8460. doi: 10.1073/pnas.0609699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Jenkins D Hedgehog signalling: emerging evidence for non-canonical pathways. Cell Signal. 2009;21(7): 1023–1034. [DOI] [PubMed] [Google Scholar]
- [26].Samanta J, Grund EM, Silva HM, et al. Inhibition of Gli1 mobilizes endogenous neural stem cells for remyelination. Nature 2015;526(7573):448–452. doi: 10.1038/nature14957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Motoyama J, Liu J, Mo R, et al. Essential function of Gli2 and Gli3 in the formation of lung, trachea and oesophagus. Nat Genet. 1998;20(1):54–57. doi: 10.1038/1711. [DOI] [PubMed] [Google Scholar]
- [28].Park HL, Bai C, Platt KA, et al. Mouse Gli1 mutants are viable but have defects in SHH signaling in combination with a Gli2 mutation. Development. 2000; 127(8):1593–1605. [DOI] [PubMed] [Google Scholar]
- [29].Ashcroft T, Simpson JM, Timbrell V. Simple method of estimating severity of pulmonary fibrosis on a numerical scale. J Clin Pathol. 1988;41(4):467–470. doi: 10.1136/jcp.41.4.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Schrier DJ, Kunkel RG, Phan SH. The role of strain variation in murine bleomycin-induced pulmonary fibrosis. Am Rev Respir Dis. 1983;127(1):63–66. doi: 10.1164/arrd.1983.127.1.63. [DOI] [PubMed] [Google Scholar]
- [31].Wang C, de Mochel NSR, Christenson SA, et al. Expansion of hedgehog disrupts mesenchymal identity and induces emphysema phenotype. J Clin Invest. 2018;128(10):4343–4358. doi: 10.1172/JCI99435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zhao C, Cai S, Shin K. Stromal Gli2 activity coordinates a niche signaling program for mammary epithelial stem cells. Science. 2017;356(6335):eaal3485. [DOI] [PubMed] [Google Scholar]
- [33].Corrales JD. The level of sonic hedgehog signaling regulates the complexity of cerebellar foliation. Development 2006;133(9):1811–1821. doi: 10.1242/dev.02351. [DOI] [PubMed] [Google Scholar]