

Abstract
Introduction:
Despite apparently complete surgical resection, approximately half of resected early-stage lung cancer patients relapse and die of their disease. Adjuvant chemotherapy reduces this risk by only 5% to 8%. Thus, there is a need for better identifying who benefits from adjuvant therapy, the drivers of relapse, and novel targets in this setting.
Methods:
RNA sequencing and liquid chromatography/liquid chromatography-mass spectrometry proteomics data were generated from 51 surgically resected non-small cell lung tumors with known recurrence status.
Results:
We present a rationale and framework for the incorporation of high-content RNA and protein measurements into integrative biomarkers and show the potential of this approach for predicting risk of recurrence in a group of lung adenocarcinomas. In addition, we characterize the relationship between mRNA and protein measurements in lung adenocarcinoma and show that it is outcome specific.
Conclusions:
Our results suggest that mRNA and protein data possess independent biological and clinical importance, which can be leveraged to create higher-powered expression biomarkers.
Keywords: Lung adenocarcinoma, NSCLC, Biomarkers, Proteomics, Proteogenomics
Introduction
Five-year survival of patients with surgically resected, early-stage lung adenocarcinoma ranges from 50% to 70%, and adjuvant chemotherapy reduces this risk by only a small amount.1 An accurate prediction of the risk of tumor recurrence at the time of surgery could potentially spare patients the toxicity of adjuvant chemotherapy and target other patients for increased therapy and surveillance. Many previous attempts have been made to predict recurrence and prognosticate outcomes after resection of lung adenocarcinomas; however, significant challenges to reproducibility and implementation have prevented the widespread use of these signatures in the clinic.2 To date, there has been no effort to compare and integrate high-content proteomic with transcriptomic approaches in carefully clinically annotated cases of this disease.
In this study, we present an integrative approach combining both transcriptomic and proteomic data. The central hypothesis of this study is that protein and mRNA measurements of lung adenocarcinoma tumors encompass independent information that can be leveraged to discover novel dysregulated genes and integrative clinical biomarkers.
There have been multiple proteogenomics studies in model systems, such as bacteria, yeast, and cell lines.3–5 Initial studies in humans reiterated the poor correlation between mRNA and protein measurements, highlighting the importance of regulation at the post-transcriptional level.6 Recent studies in cell lines have proposed that a greater amount of protein variation can be explained by transcription than previously thought7; however, a picture has emerged of bursts of mRNA transcription creating stable changes in protein expression in response to perturbation.8 In surgically resected tumor samples, the cell states vary from perturbed to steady state, implying that mRNA-protein correlation may vary as well. For example, Wei et al.9 showed that RNA-protein correlation differs between aging and young humans and rhesus macaques. The discovery that mRNA-protein correlation is a phenotype that can be correlated with biological and clinical outcomes necessitates further studies with matched mRNA and protein measurements. Large datasets of matched RNA sequencing (RNAseq) and proteomics results were published by Zhang et al.10 in colorectal and Mertin et al.11 in breast cancer samples of convenience; however, these studies were not designed to explore an integrative clinical biomarker. Recently, Zhang et al.12 published a proteogenomic dataset from high-grade serous ovarian cancer tumors which can be separated into early and late survivors; however, there are no significant differences for mRNA and protein expression between the two groups.
Here, we investigate differential mRNA-protein correlation between recurrent and nonrecurrent lung adenocarcinoma tumors. We then leverage this difference, in combination with differential mRNA and protein abundances to predict lung adenocarcinoma recurrence with matched transcriptomic and proteomic data using a novel supervised classification algorithm.
Materials and Methods
RNAseq Data Collection and Preprocessing
RNA from tumor samples resected at Vanderbilt (Nashville, Tennessee) and MD Anderson (Houston, Texas) was extracted from fresh frozen tissue with Qiagen RNeasy mini kit (Hilden, Germany), converted to a poly-A selected cDNA library, and paired-end sequenced on Illumina HiSeq 2000 (San Diego, California). Raw fastq files were filtered for adapters and low quality, and aligned to University of California-Santa Cruz (UCSC) human genome 19 (hg19) reference genome with TopHat2 using default parameters. Read counts were generated with htseq-count using RefSeq gene definitions.13–15 RNA from tumor samples resected at Washington University (St. Louis, Missouri) was extracted from fresh frozen tissue, converted to a poly-A selected cDNA library with NuGen v2 kit (Ovation, Santa Monica, California) and paired-end sequenced on Illumina HiSeq 2000. Raw fastq files were filtered for adapters and low quality, and aligned to UCSC hg19 reference genome with spliced transcripts alignment to a reference 2-pass method.16,17 Read counts were generated with featureCounts using RefSeq gene definitions. Variants from both RNAseq datasets were extracted with samtools’ mpileup.18,19
The MD Anderson and Vanderbilt Cohort Tumor Tissue Preparation
Formalin-fixed paraffin-embedded (FFPE) tissues of tumor resections collected at Vanderbilt University, MD Anderson, and Washington University (WashU cohort) were used in protein extraction. The Vanderbilt and MD Anderson cohort tissue samples were deparaffnized using sub-x xylene (Surgipath, Richmond, Illinois) followed by rehydration in three ethanol washes as previously described.20 Samples were homogenized in lysis buffer containing trifluoroethanol and 100 mmol/L ammonium bicarbonate at pH 8.0 using Sonic Dismembrator model 100 (Fisher scientific, Pittsburgh, Pennsylvania) at 20 W for 20 second with 30-second intervals. The sonication step was repeated twice, and the samples were stored on ice between sonications. The concentration of the proteins in each lysate was measured using bicinchoninic acid protein assay (Thermo Fisher Pierce, Rockford, Illinois) using the manufacturer’s protocol. A total of 200 μg of lysate was reduced with 20 mmol/L tris(2-carboxyethyl)phosphine (TCEP, Pierce, Rockford, Illinois) and 50 mmol/L DTT (Sigma-Aldrich, St. Louis, Missouri) at 60 °C for 30 minutes followed by alkylation with 100 mmol/L iodoacetamide (Sigma-Aldrich, St. Louis, Missouri) in dark for 20 minutes at room temperature. The concentration of trifluoroethanol was reduced to 10% of the total volume by diluting in 50 mmol/L ammonium bicarbonate. The samples were digested with trypsin (Promega Corporation, Madison, Wisconsin) at a ratio of 1:50 (w:w) overnight at 37 °C followed by acidification with 0.5% trifluoroacetic acid protein digests were frozen at −80 °C and lyophilized to dryness. The samples were re-suspended in high-performance liquid chromatography-grade water with vortexing for 1 minute and desalted using Oasis HLB 96-well μElution plate (30 μm, 5 mg, Waters Corporation, Milford, Massachusetts) as previously described.10
WashU Cohort Tumor Tissue Preparation
The FFPE tumor tissues were deparaffinized in xylene followed by rehydration in ethanol as previously described.21 The tumor tissues were homogenized in a modified lysis buffer containing 0.2% RapiGest (Waters Corporation) in 50 mmol/L ammonium bicarbonate. The lysates were incubated at 105 °C for 30 minutes and stored on ice for 5 minutes. The samples were sonicated using Sonic Dismembrator model 100 (Fisher Scientific) at 20 W for 20 seconds with 30-second intervals. This sonication step was repeated twice, and the samples were incubated at 70 °C for 2 hours. The protein concentration in each lysate was determined by bicinchoninic acid protein assay (Thermo Fisher Pierce) using the manufacturer’s protocol. A total of 100 μg of tissue proteins were reduced with 50 mmol/L DTT at 60 °C for 30 minutes followed by alkylation with 100 mmol/L iodoacetamide in dark at room temperature for 20 minutes. The samples were digested with sequencing grade trypsin (Promega Corporation, Madison, Wisconsin) at a ratio of 1:50 (w:w) and 0.01% ProteaseMax surfactant (Promega Corporation) at 37°C for 3 hours. The samples were acidified with 0.5% trifluoroacetic acid and centrifuged at 14,000 g for 15 minutes. The supernatant was collected and evaporated to dryness in a Speed-Vac concentrator (Thermo Fisher Scientific). The samples were stored in −80 °C until liquid chromatography (LC)/LC-mass spectronomy (MS)/MS analysis.
The Vanderbilt and MD Anderson Cohort Peptide Fractionation by Off-Line High pH Reverse-Phase Chromatography
The samples (n = 44) were reconstituted in 400 μL of 1.0 mol/L triethylammonium bicarbonate at pH 7.5 and injected into the chromatography system. Tryptic peptides were fractionated at high pH reverse-phase XBridge BEH C18 analytical column (250 mm × 4.6 mm, 130 °A, 5 μm, Waters Corporation, Milford, Massachusetts) equipped with an XBridge BEH C18 sentry guard cartridge. The separation was achieved at a flow rate of 0.5 μL/min in 10 mmol/L triethylammonium bicarbonate and water at pH 7.5 (solvent A) and 100% acetonitrile (solvent B). A multistep gradient with three linear gradients were used; from 0 to 5% (solvent) B in 10 minutes, 5% to 35% B in 60 minutes, 35% to 60% B in 15 minutes, and 70% B for 10 minutes before reaching the initial conditions. A total of 60 fractions were collected and recombined into 15 peptide fractions as previously described.10 The samples were evaporated to dryness in a Speed-Vac concentrator and stored in −80 °C until LC MS/MS runs.
The Vanderbilt and MD Anderson Cohort LC-MS/MS Analysis
The protein digests were reconstituted in 50 μL of 2% acetonitrile and 0.1% formic acid. An Eksigent NanoLC 2D pump with an AS1 auto-sampler reverse-phase LC system (Eksigent, Dublin, California) was used for peptide fractionation. A total of 8 μg were injected and separated using 0.1% formic acid (solvent A) and 0.1% formic acid in acetonitrile in a packed capillary tip (Polymicro Technologies, Phoenix, Arizona) containing Jupiter C18 resin (Phenomenex, 5 μm, 300°A) in-line with a solid phase extraction column (packed with the same resin). The gradient was programmed to desalt the samples on the column for 15 minutes at 100% A before separation at a flow rate of 1.5 μL/min. The separation was achieved by changing mobile phase composition from 100% A to 25% B in 50 minutes, 25% to 90% B in 65 minutes and held at 90% for extra 9 minutes. Peptides eluting the column were ionized at 1.45 kV and analyzed with a Thermo Velos Pro dual-pressure linear ion trap mass spectrometer (Thermo Fisher Scientific) by data dependent acquisition. The top five MS/MS scans were acquired for every full MS scan for an m/z range from 400 to 2000. The method was used with an ion transfer tube temperature at 200 °C; S-lens radio frequency 65%; dynamic exclusion with a repeat count 1 and repeat duration of 1 second for an exclusion list size of 50 mass-to-charges; collision-induced dissociation (CID) with normalized collision energy of 30%, q = 0.25, and activation time of 10 ms; and the minimum intensity threshold was set to 1000 counts.
The WashU Cohort LC/LC-MS/MS Analysis
For the analysis of the WashU cohort (17 samples), LC coupled to tandem MS was performed using a Waters nanoacquity two-dimensional (2D) UHPLC system (Waters Corporation) with two reverse-phases interfaced to a Thermo LTQ-Oribitrap Elite hybrid mass spectrometer (Thermo Fisher Scientific, Bremen, Germany). A total of 8 μg of protein digest reconstituted in 100 mM ammonium formate was injected using Acquity UPLC autosampler (Waters Corporation) and the peptides were fractionated online at high pH before analytical separation. The fractionation of peptides was achieved in the first reverse-phase column (Waters BEH C18, 130 °A, 1.7 μm, 300 μm, and 100 mm) at pH 10.0 in buffer A1 (20 mmol/L ammonium formate) by varying the amounts of solvent B1 (100% acetonitrile). The column was equilibrated at 3% B1 (v/v), which was increased to 4.7% (v/v) in 1 minute eluting the first fraction of peptides and decreased back to 3% (v/v) B1 in the next 4 minutes. The column was held at 3% (v/v) B1 during separation at a steady flow rate of 2 μL/min. The solvent % B1 (v/v) was increased from 4.7%, 9.0%, 10.8%, 12.0%, 13.1%, 14.0%, 14.9%, 15.8%, 16.7%, 17.7%, 18.9%, 20.4%, 22.2%, 25.8% and to 65% over 15 fractions. Each fraction eluted from the fractioning column was loaded onto a Waters symmetry C18 trap column (100Å, 5 μm, 180 μm × 20 mm) and desalted at a flow rate of 20 μL/min. The analytical separation was achieved in the second reverse-phase column (Waters HSS T3, C18, 100Å, 1.8 μm, 75 μm × 150 mm) at pH 2.4 which was equilibrated to initial conditions; 95% (v/v) A2 (water with 0.1% formic acid) and 5% (v/v) B2 (acetonitrile with 0.1% formic acid). The subsequent separation was achieved by three linear gradients at 38 °C where the % B2 was increased from 5% to 9% in 3 minutes; 9% to 30% over 44 minutes; 30% to 40% over 5 minutes; and 40% to 85% over 5 minutes at a flow rate of 0.5 μL/min. The column was held at 5% (v/v) B2 from 65 to 70 minutes before reaching initial conditions. The 2D LC was coupled to LTQ-orbitrap Elite via a nanospray Flex ion source (Thermo Fisher Scientific) containing a 30-μm inner-diameter stainless steel emitter (Thermo Fisher Scientific) with spray voltage between 1.7 kV and 1.8 kV. The orbitrap mass spectrometer was operated in data-dependent acquisition mode, where the top 15 MS/MS scans were acquired for every full MS-scan. The full MS-scan was acquired in the orbitrap MS-analyzer with resolution r = 120,000 at m/z 400 for every 107 charges acquired in the ion trap MS-analyzer. This acquisition was set to trigger MS/MS scans for the top 15 most abundant m/z peaks after CID for an automated gain control target value of 5000 charges. The method was programmed with an ion transfer tube temperature at 275 °C; S-lens RF 55%; dynamic exclusion with a repeat count 1 and repeat duration of 15 seconds for exclusion list size of 500 mass-to-charges; CID with normalized collision energy of 35%, q = 0.25 and activation time of 10 ms; the minimum intensity threshold was set to 6000 counts.
Data Processing and Protein Identification
For protein identification, Myrimatch version 2.1.111 was used with a customized RefSeq human database (version 54) and Peptitome version 1.0.42. The raw files generated in Xcaliber software (Thermo Fisher Scientific) for all 15 fractions of each protein digest were used in the peptide identification. The MS/MS spectra were searched with fixed carbamidomethyl modification at cysteine, and variable acetylation at protein N-termini, oxidation of methionines, and deamidation at asparagine and glutamine (only for the WashU cohort data). A maximum of two missed cleavages were allowed for every fully tryptic peptide (proline rule applied) with a minimum peptide length of six amino acids. The data were filtered in IdPicker software version 3.0.504. The proteins present in each sample were identified with a peptide false discovery rate (FDR) of 1% and a protein FDR of 4.45%. Protein groups were filtered to only include proteins with a minimum of two peptides and with spectra required per peptide. For proteogenomic analysis, protein groups identified in each sample were grouped based on the gene group and the respective number of spectral counts for each gene group per patient was recorded.
Normalization and Filtering
Both proteomics and RNAseq datasets were normalized by dividing each patient column by the total number of counts in that column, and then multiplying by 1 million to get counts per million. We then filtered out any genes for which the median across more than half of the patients was 0. Three thousand nine hundred sixty genes were detected at RNA and protein levels in at least one sample in both the Vanderbilt/MD Anderson and WashU cohorts, and after filtering, 2286 genes remained for downstream analysis. We only used features for which there were matching protein and RNA features from the same gene.
Differential Gene Expression and Correlation
We developed a novel method of differential gene expression by comparing the rank median expression of each group and dividing by the total number of genes to get a number between −1 and 1. This method is robust to outliers, simple, and nonparametric. All differential correlation was computed as the absolute value of the difference between RNA-protein Spearman correlation values within each cohort. A cutoff for significance of 0.54 was used. We chose this cutoff by taking the value of correlation or anticorrelation necessary to achieve significance within a single cohort (Spearman ρ > 0.27, estimated p < 0.05) and multiplying by 2, that is, by taking the minimum difference necessary between a significantly correlated and significantly anticorrelated RNA-protein pair.
Construction of the Integrative Biomarker
To create an integrated biomarker of tumor recurrence, we use a model selection approach. For each gene we find a set of models or functions that relate the RNA measurements to the protein measurements in the nonrecurrent and recurrent cohorts. Formally, we define the functions as follows:
| (1) |
Where fR and fNR are the recurrent and nonrecurrent functions and N(0,σR2) and N(0,σNR2) are the normally distributed error terms of the models. After the models are generated on a training set, the likelihood that an expression measurement from a test sample came from a recurrent or nonrecurrent patient is obtained by computing the probability density of the difference between the theoretical and test protein expression values for each model.
To learn the relationship between RNA and protein measurements for each gene, we use L1 trend filtering, which seeks to fit a piecewise linear function to the data. Trend filtering controls for over-fitting with a sparsity term which is optimized using cross-validation. We implemented trend filtering using the R package genlasso. Trend filtering seeks to optimize the following objective function:
| (2) |
Where λ ≥ 0 is the regularization parameter, and D is the second-order difference matrix defined in Kim et al.22 Trend filtering enforces a piecewise linear regression model and the number of knots, or differing slope values, is determined by cross-validation within the training set to optimize the number of kinks given the noisiness of the data.
We compute the overall probability of a patient being recurrent or nonrecurrent using Bayes’ theorem with an uninformative prior and independent genes.
| (3) |
Where Pg,j, Rg,j are the protein and RNA measurements for each gene in the signature for a given patient and M is the number of genes in the signature. To perform feature selection to find the final gene signature, we remove genes that are inaccurate on the training set based on the number of incorrectly predicted log odds ratios for each gene.
Comparison of Individual Versus Integrative Biomarker Results
The integrative biomarker was benchmarked against a method using similar principles, except using only protein or RNA data alone. The individual RNA and protein biomarkers were built using the same operation to build the individual RNA component of the integrative biomarker. First, samples were split into training or testing (here we use leave-one-out cross-validation). For each RNA and protein gene, measurements were first divided into recurrent or nonrecurrent. Next, the validation sample’s RNA or protein values were compared to the distributions of the training recurrent and nonrecurrent samples, and a log likelihood was generated that the validation sample came from either clinical group. Finally, the log likelihoods were combined into a single log likelihood using a naïve Bayesian classifier.
Functional Analysis of Dysregulated Genes
The dysregulated genes identified in this study were examined for enrichments of regulatory factors including RNA binding protein and microRNA binding sites. 5’ and 3’ untranslated region (UTR) coordinates for all available transcripts were downloaded from the UCSC Table Browser for the human genome (hg38).23 UTR exon sequences were extracted for each transcript using the R package BSgenome.Hsapiens.UCSC.hg38.23 Sequence motifs for 178 human RNA-binding Proteins (RBP) binding sites (101 RBPs) were collected from the Catalog of Inferred Sequence Binding Preferences-RNA.24 Each UTR sequence (length L) was scanned for each motif (length M) using a single nucleotide sliding window providing L − M + 1 scores. The maximum score for each transcript was selected as the motif representative score. The set of putative targets for each RBP motif across the whole genome were identified as the set of transcripts with representative scores greater than 90% of the motifs’ theoretical maximum. The set of targets were compare to the dysregulated genes to identify the putative RBP dysregulated targets. The background set of targets were identified as the targets associated with the global set of genes assayed (all genes for which RNA and protein data was available). A hypergeometric test was used to determine whether the dysregulated genes were enriched as targets for each RBP motif.
MicroRNA data was collected from the TargetScan website.25 The human conserved microRNA family targets for were downloaded from the database (214 microRNA families). This provided a list of genomic coordinates for the microRNA binding sites. Using the UCSC liftover tool, the original hg19 binding site coordinates were converted into the hg38 genomic coordinates. Overlap of these sites with transcribed regions provided the set of gene targets for each microRNA family. After identifying the set of dysregulated microRNA targets a hypergeometric test analogous to the RBP analysis was used to calculate the putative enrichment for each of the microRNA targets in the dysregulated gene set. RBP and microRNA motifs with a Benjamini-Hochberg corrected p value < 0.25 were considered significantly enriched.
Results
Proteogenomic Analysis of Surgically Resected NSCLC
We collected fresh frozen and FFPE specimens from 61 patients, half selected for rapid recurrence after surgery, and half selected for long-term (more than 3 years) survival after surgical resection. Forty-four of these patients were recruited at Vanderbilt University and MD Anderson (tissue was processed at Vanderbilt for all samples), and 7 patients were recruited at WashU (Supplementary Fig. 1). The patients were matched for recurrence and adjuvant chemotherapy status (Table 1). RNAseq was performed on the fresh frozen tissues and tandem LC-MS was performed on the FFPE tissues. In total, 5482 and 6581 protein groups were identified in the Vanderbilt and WashU cohorts, respectively. Five thousand two hundred eighty-four and 5,253 of these proteins were matched by gene symbol to their corresponding mRNA in the Vanderbilt and WashU cohorts, respectively. A total of 6577 genes were measured in at least one study, and 3960 genes were quantified in both studies.
Table 1.
Clinical Patient Attributes
| Recurrent (n = 25) | Nonrecurrent (n = 26) | |
|---|---|---|
| Male (%) | 72 | 31 |
| Adjuvant therapy | 9 | 12 |
| No adjuvant therapy | 16 | 14 |
| Stage | ||
| Ia/b | 8/8 | 6/14 |
| IIa/b | 3/4 | 2/1 |
| IIIa/b | 0/1 | 2/1 |
| Collection Site | ||
| Vanderbilt | 17 | 18 |
| MD Anderson | 4 | 5 |
| WashU | 3 | 4 |
RNA-Protein Correlation is Dependent on Tumor Recurrence Status in Lung Adenocarcinoma
We observed high correlation of mRNA measurements across patients, as well as high correlation of protein measurements across patients, indicating that the data generated from each site are suitable to be combined for analysis (Supplementary Fig. 2). Median mRNA-protein Spearman correlations were ρ = 0.07 in the WashU cohort (3004 genes compared) (Fig. 1A) and ρ = 0.17 in the Vanderbilt cohort (4656 genes compared) (Fig. 1B). These values are lower than those found in previous studies conducted in lung cancer (mean Spearman ρ = 0.34 and ρ < 0.4), colon and rectal (mean Spearman ρ = 0.47), breast (mean Pearson r = 0.39), and ovarian cancers (mean Spearman ρ = 0.45).12–14,26–27
Figure 1.

Gene-level mRNA-protein correlation in human lung adenocarcinoma. mRNA protein correlation in Vanderbilt (A) and WashU (B) datasets. (C) Histogram of mRNA-protein correlations within each cohort. The significance of the difference between recurrent and nonrecurrent mRNA-protein correlations was determined by the Wilcoxon rank sum test.
Next, the pathway enrichments for low- and high-correlated genes were studied and found similar trends to those found in previous the cancer genome atlas studies. Interestingly, the mRNA splicing pathway, which is enriched for poor mRNA-protein correlation in colorectal, breast, and ovarian cancers, is enriched for high mRNA-protein correlation in lung adenocarcinoma (Supplementary Table 1). Aberrant splicing has recently been implicated in lung adenocarcinoma, and may contribute to the overall low mRNA-protein correlation seen in this study.28
Prior research has shown that the unexplained protein variability is not solely accounted for by technical noise, but also post-transcriptional regulation.7 As such, we sought to discover genes whose mRNA-protein correlation was dependent on the clinical outcome. mRNA and protein data were matched at the gene level and filtered by expression to obtain a set of 2286 paired RNA and protein measurements per lung adenocarcinoma patient (N = 51, See Materials and Methods for details). Globally, there is a significant difference between the mRNA-protein correlation of all genes in the recurrent group and in the nonrecurrent group (p < 10−16 Wilcoxon Rank sum test) (Fig. 1C). Overall, the genes we investigated were more highly correlated in the nonrecurrent tumors (Fig. 1C).
Synergistic Detection of RNA and Protein Dysregulation
We investigated the gene-level differences in mRNA-protein correlation and abundances with spearman correlation (Fig. 2A) and a 2D differential expression method (Fig. 2B). We show that the mRNA-protein correlation of individual genes can vary greatly between recurrent and nonrecurrent tumors (Fig. 2A). We hypothesized that mRNA-protein correlation itself may contain important information about the state of the cell. Poorly correlated mRNA and protein abundances may reflect post-transcriptional (splicing, microRNA, RNA localization, etc.) and post-translational (phosphorylation, ubiquitination, altered degradation, etc.) regulation. As such, differential correlation can be used to detect dysregulated genes in cancer, and necessitates the collection and analysis of large clinical cohorts with matched mRNA and protein data.
Figure 2.
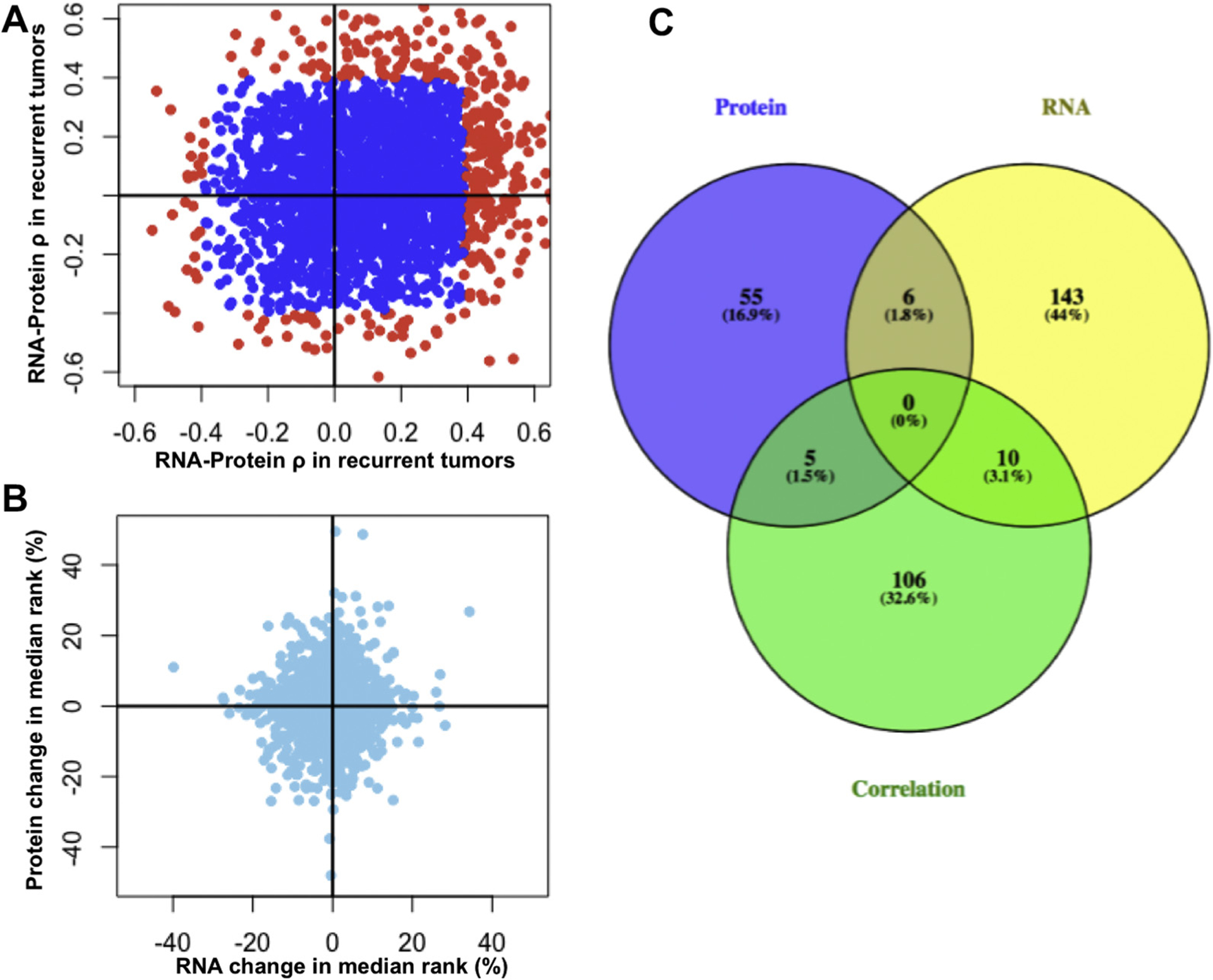
Synergistic discovery of differentially regulated genes using matched RNA and protein abundances. (A) RNA-protein correlations within recurrent and nonrecurrent patient cohorts are shown in a scatterplot. Genes whose RNA-protein abundances are significantly correlated or anticorrelated (uncorrected p value < 0.05) are shown in red. (B) RNA and protein differential expression is shown as the change in median rank abundance between nonrecurrent and recurrent cohorts. Genes are differentially expressed at RNA and protein levels. (C) Overlap of genes differentially expressed at the protein and RNA levels, as well as genes that are differentially correlated. Zero genes displayed simultaneous differential expression at both levels and differential correlation. Please see Supplemental Methods for more information about how differential expression and correlation was computed.
We found that genes can be differentially expressed independently at the mRNA and protein levels (Fig. 2B). Indeed, there is little overlap between genes that are differentially expressed at the mRNA and protein levels, including differential correlation (Fig. 2C) (differential expression p values are reported as uncorrected p values produced by the R package, npSeq, see Supplementary Methods).29 Were we to only use one data type, we would have found 66 differentially expressed proteins or 159 differentially expressed mRNAs; however, the inclusion of both allows us to generate 325 hypotheses of dysregulated genes. The numbers of differentially expressed proteins and mRNAs reported by npSeq are very low due to its stringency; however, we chose this nonparametric approach to minimize the chance of differential expression being driven by outliers. Outlier-driven differential expression is not as useful in biomarker development because it does not capture the behavior of an entire cohort. In addition, we observed high intragroup variability relative to intergroup variability.
We further investigated which genes were most differentially correlated. The most differentially correlated gene, translocase of inner mitochondrial membrane 50 (TIMM50), has highly correlated RNA-protein abundances among nonrecurrent tumors but highly anticorrelated abundances among recurrent tumors (Fig. 3). TIMM50 encodes the protein, Tim50, that is involved in the mitochondrial apoptosis pathway, is upregulated by mutant p53, and its loss induces apoptosis in breast cancer cells.30,31 TIMM50 is weakly differentially expressed at the RNA level, and not differentially expressed at the protein level, such that its discovery as a dysregulated gene in our patient cohort requires the use of both data types.
Figure 3.
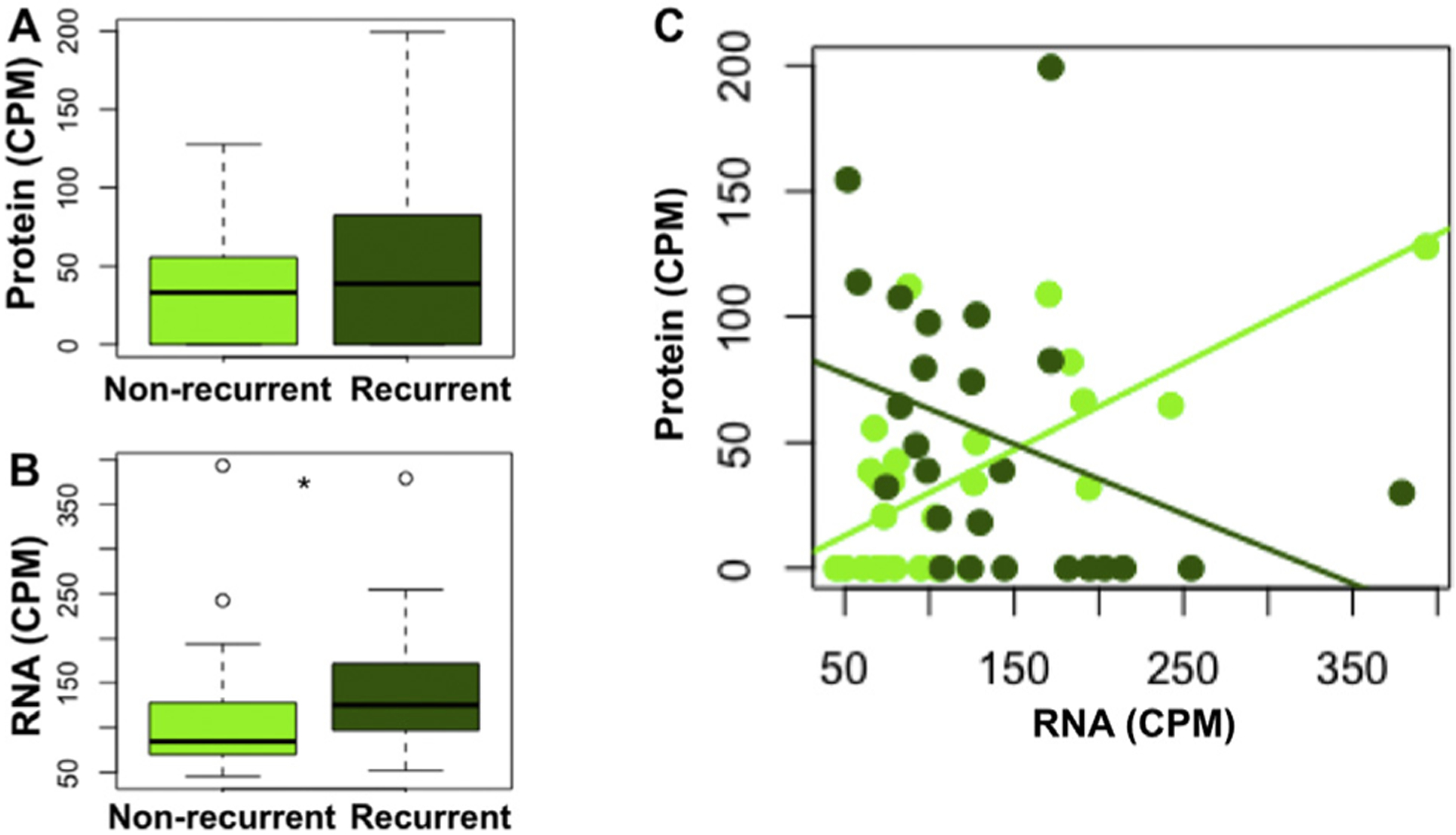
Translocase of inner mitochondrial membrane 50 (Timm50) is differentially correlated between recurrent and nonrecurrent tumors. (A) Timm50 is weakly differentially expressed at the RNA level (p < 0.05), but not differentially expressed at the protein level (B). Timm50 differential RNA-protein correlation between recurrent and nonrecurrent tumors. CPM, counts per million.
To examine whether aberrant post-transcriptional regulation contributed to the poor RNA-protein Spearman correlations in recurrent patients, we search for enriched RBPs and microRNA motifs within these genes. This analysis of 178 RBPs and 214 miRNA family motifs identified no significant enrichment (FDR < 0.25) for post-transcriptional motifs within this gene set (Supplementary Tables 2–4).
Integrating RNA and Protein Abundances for Predicting Tumor Recurrence
We next sought to leverage the RNA and protein data by developing a novel, comprehensive methodology to generate integrative expression biomarkers (Fig. 4). In brief, we separate patients into training and test cohorts, and then further separate the training cohort according to a binary clinical variable (Fig. 4A). In this study, the variable is recurrence status. For each gene, we perform regression using a recently developed machine learning technique, L1 trend filtering, to find a piecewise-linear relationship between RNA and protein abundances in each cohort (Fig. 4A).22 Trend filtering produces a set of piecewise linear equations that seek to balance over- and under-fitting of the model. For instance, if the relationship is highly nonlinear with a high signal-to-noise ratio, then the model will have many knots that closely follow the data. In the case of a highly linear or low signal-to-noise ratio, then there will be no knots, and simple linear regression is performed. The test samples are then compared to the model, and an error is calculated that represents the difference between the model-predicted and test protein values, given the test RNA values (Fig. 4B). Errors are then calculated for each training sample and used to learn parameters for a normal distribution independently for each cohort. p values for the test errors are extracted from recurrent and nonrecurrent distributions and combined to generate a log odds ratio (LOR) for each gene-patient combination (Fig 4C). These LOR values are then summed for all genes included in the signature to generate a final LOR that a tumor will recur or not. (For more details on how genes are included in the final signatures, see Supplemental Methods.)
Figure 4.
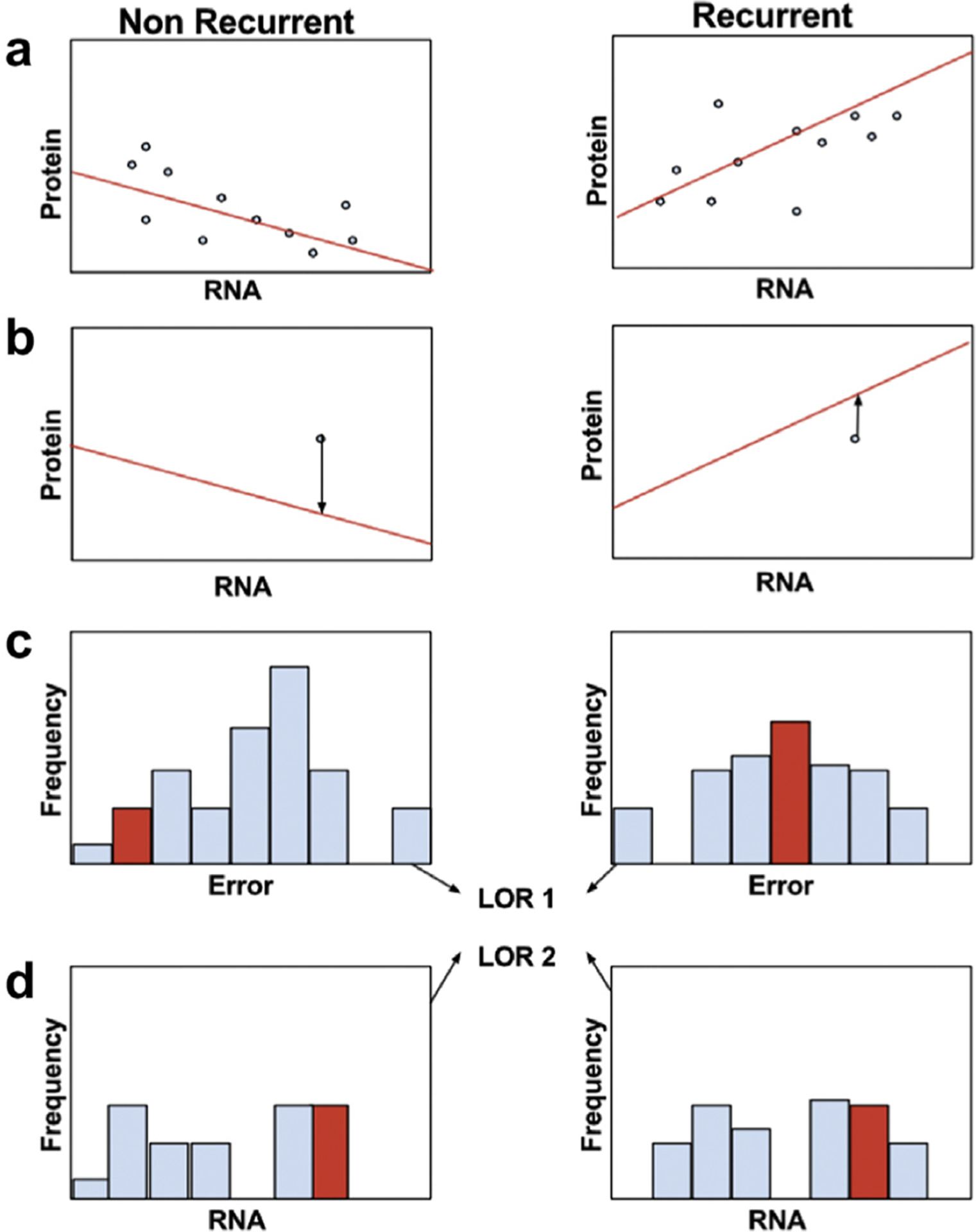
Overview of integrative RNA-protein biomarker discovery pipeline. (A) Patients are divided into their clinical groups; here we use binary recurrence status to group the patients. Regression is then performed using trendfiltering to find a relationship between RNA and protein abundances within each cohort. (B) This model is then used to test a separate test sample or samples. Given a test RNA abundance, the test error is calculated as the difference between the predicted protein value and the test protein value (arrows). (C) The test errors are then compared to the distributions of training errors in each cohort, and a log odds ratio is calculated (LOR1). (D) Because this integrative method does not detect differential RNA abundances in the absence of differential protein abundances, a second log odds ratio is calculated by comparing the test RNA abundances to the training RNA abundances in each cohort (LOR2).
Because our method considers protein as a function of RNA, a gene that has differential RNA expression in the absence of differential protein expression would not be considered as a useful biomarker. We remedy this situation by generating a separate LOR that an RNA measurement was taken from recurrent or nonrecurrent RNA abundance distributions (Fig. 4D). The accuracy of the LORs generated by the integrative or RNA-alone methods are compared on the training set for each gene, and using a simple objective function, the method decides whether to use each gene as an integrative or RNA biomarker.
Using a synthetic dataset, we show that our method is able to simultaneously use changes to protein concentrations, RNA concentrations, and RNA-protein correlations (Supplementary Figs. 3–5). Leave-one-out cross-validation results on our patient cohort are shown in Table 2. Our integrative method was able to correctly predict 36 of 51 (71%) patients’ recurrence status, including 20 of 26 (77%) nonrecurrent patients and 16 of 25 (64%) recurrent patients (Supplementary Fig. 6). This is in contrast to results using protein and RNA expression separately, which collectively had an accuracy of ~ 50%. Interestingly, the majority of prediction errors using our integrative approach of nonrecurrent patients (4 of 6, 67%) came from the misclassification of patients who received chemotherapy. This suggests that our method was able to find tumors that may have recurred without the intervention of adjuvant chemotherapy.
Table 2.
Integrative Biomarker of Recurrence Leave-One-Out Cross Validation Performance
| Dataypes Used | Feature Selection | Total (%) | Nonrecurrent (%) | Recurrent (%) |
|---|---|---|---|---|
| RNA | All | 24 (47) | 12 (46) | 12 (48) |
| Feature Selection | 29 (57) | 13 (50) | 16 (64) | |
| Protein | All | 18 (35) | 10 (38) | 8 (32) |
| Feature Selection | 24 (47) | 12 (46) | 12 (48) | |
| RNA+Protein | All | 19 (37) | 9 (35) | 10 (40) |
| Feature Selection | 25 (49) | 11 (42) | 14 (56) | |
| Integrative | All | 22 (43) | 10 (38) | 12 (48) |
| Feature Selection | 36 (71) | 20 (77) | 16 (64) |
To find genes that best predict patient recurrence status, we include feature selection by evaluating each gene’s performance on the training cohort. The result is a signature generated by each cross-validation test (Supplementary Fig. 7). We evaluated the biological significance of each gene included in a majority of signatures: small ubiquitin-like modifier 1 (SUMO1), pterin-4 alpha-carbinolamine dehydratase 1 (PCBD1), proteasome 26S subunit ATPase 5 (PSMC5), archain 1 (ARCN1), pyrophosphatase 2 (PPA2), and sorcin (SRI) (For a full list of genes included in at least one signature, see Supplementary Table 5). Each of these genes was used as an integrative biomarker, not as an RNA biomarker. Sumo1 is covalently attached to target proteins in a process termed sumoylation. Sumoylation is involved in many cellular responses; most notably, sumoylation of DNA damage response proteins is necessary to repair DNA double-stranded breaks.32 PCBD1 is a dimerization cofactor of HNF1 homeobox A (HNF1A), which has been implicated in numerous cancers.33,34 PSMC5 has proteasomal functions, has been used as a biomarker of radiosensitivity in a lung cancer H460 cell line, and has been identified as a modifier of the transforming growth factor beta transcriptional program.35,36 ARCN1 has been hypothesized to function in vesicle trafficking, and in one study, ARCN1 RNA expression was predictive of survival in surgically resected lung cancer.37,38 PPA2 is a mitochondrial inorganic pyrophosphatase. SRI has been shown to be involved in multidrug resistance in cancer, and protects against mitochondrial apoptosis.39–41 Our integrative biomarker method selected biologically relevant genes to predict lung adenocarcinoma recurrence.
Discussion
In this study, we present a novel comprehensive characterization of 51 lung adenocarcinoma tumors with matched RNA and protein abundance analysis. We further show that the combined analysis of RNA and protein abundances can be used to define candidate biomarkers of recurrence risk for surgically resected lung adenocarcinomas. Although several papers have used RNA data to inform the choice of protein biomarkers, our method is the first, to our knowledge, to integrate RNA and protein expression data into a single signature. In fact, our method can be more broadly implemented to perform supervised learning to predict a binary response variable using any two matched datasets.
There are several limitations of this study. First and foremost, RNAseq data was generated from fresh frozen tissues whereas proteomics data was generated from FFPE tissues. This is a possible explanation for the unusually low RNA-protein correlation. Second, although our integrative biomarker improved upon RNA or protein-based biomarkers for recurrence prediction in our dataset, the accuracy (71%) is too low to be of clinical utility. This result is possibly due to the high intragroup variability observed in our data. Third, these patients do not have matched DNA sequencing data, so a comprehensive catalogue of driver mutations is lacking. Fourth, the majority of recurrent tumors were male (72%) and the majority of nonrecurrent tumors were female (69%). Fifth, our study was not designed with independent training and validation cohorts, so independent validation is necessary to reduce the potential for over-fitting explaining the observed results. A large, publicly available cohort of lung cancer patients with matched RNAseq, proteomics, and clinical data is sorely lacking to aid in the validation of studies such as the one presented here. Ideally, this validation set would be produced solely from fresh frozen tissue obtained from surgical resection and have sufficient numbers of patients with and without tumor progression. Sixth, protein extraction is difficult to perform and can produce variable results, particularly on heterogenous tumors.42 Although our protein extraction methods for the Vanderbilt/MD Anderson and WashU cohorts were highly similar, they were not identical, and could be a source of technical error in our study. Ultimately, independent validation is necessary to show robustness of our findings.
One interesting approach for a future study would be to find combinations of RNA and proteins that are predictive of a clinical or biological outcome that are not necessarily from the same gene. It might be that the expression of one protein as a function of an entirely different RNA, which is possibly noncoding, could be an excellent biomarker. This possibility highlights the fact that our method contextualizes the protein expression within the landscape of RNA expression.
Supplementary Material
Acknowledgments
Financial support was received from the Vanderbilt lung cancer NCI SPORE grant (CA090949). Dr. M. Sharpnack is supported by a National Library of Medicine Fellowship (4T15LM011270).
Disclosure: Dr. M. Sharpnack has received grants from the National Library of Medicine. Dr. Carbone has received personal fees from Genentech/Roche, Bristol-Myers Squibb, and Astra Zeneca. The remaining authors declare no conflict of interest.
Footnotes
Supplementary Data
Note: To access the supplementary material accompanying this article, visit the online version of the Journal of Thoracic Oncology at www.jto.org and https://doi.org/10.1016/j.jtho.2018.06.025.
References
- 1.Winton T, Livingston R, Johnson D, et al. Vinorelbine plus cisplatin vs. observation in resected non-small-cell lung cancer. N Engl J Med. 2005;352:2589–2597. [DOI] [PubMed] [Google Scholar]
- 2.Zhu C-Q, Ding K, Strumpf D, et al. Prognostic and predictive gene signature for adjuvant chemotherapy in resected non-small-cell lung cancer. J Clin Oncol. 2010;28:4417–4424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nie L, Wu G, Zhang W. Correlation between mRNA and protein abundance in Desulfovibrio vulgaris: a multiple regression to identify sources of variations. Biochem Biophys Res Commun. 2006;339:603–610. [DOI] [PubMed] [Google Scholar]
- 4.Marguerat S, Schmidt A, Codlin S, Chen W, Aebersold R, Bahler J. Quantitative analysis of fission yeast transcriptomes and proteomes in proliferating and quiescent cells. Cell. 2012;151:671–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tian Q, Stepaniants SB, Mao M, et al. Integrated genomic and proteomic analyses of gene expression in Mammalian cells. Mol Cell Proteomics. 2004;3:960–969. [DOI] [PubMed] [Google Scholar]
- 6.Chen G, Gharib TG, Huang CC, et al. Discordant protein and mRNA expression in lung adenocarcinomas. Mol Cell Proteomics. 2002;1:304–313. [DOI] [PubMed] [Google Scholar]
- 7.Jingyi B, Li J, Biggin MD. Statistics requantitates the central dogma. Science. 2015;347:1066–1067. [DOI] [PubMed] [Google Scholar]
- 8.Jovanovic M, Rooney MS, Mertins P, et al. Dynamic profiling of the protein life cycle in response to pathogens. Science. 2015;347:1259038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wei YN, Hu HY, Xie GC, et al. Transcript and protein expression decoupling reveals RNA binding proteins and miRNAs as potential modulators of human aging. Genome Biol. 2015;16:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang B, Wang J, Wang X, et al. Proteogenomic characterization of human colon and rectal cancer. Nature. 2014;513:382–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mertins P, Mani DR, Ruggles KV, et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature. 2016;534:55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang H, Liu T, Zhang Z, et al. Integrated proteogenomic characterization of human high-grade serous ovarian cancer. Cell. 2016;166:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL, et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anders S, Pyl PT, Huber W. HTSeq-A python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pruitt KD, Tatusova T, Maglott DR. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007;35:D61–D65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Engström PG, Steijger T, Sipos B, et al. Systematic evaluation of spliced alignment programs for RNA-seq data. Nat. Methods 2013;10:1185–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. [DOI] [PubMed] [Google Scholar]
- 19.Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sprung RW, Brock JWC, Tanksley JP, et al. Equivalence of Protein Inventories Obtained from Formalin-fixed Paraffin-embedded and Frozen Tissue in Multidimensional Liquid Chromatography-Tandem Mass Spectrometry Shotgun Proteomic Analysis. Mol Cell Proteomics. 2002;9:1988–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scicchitano MS, Dalmas DA, Boyce RW, et al. Protein extraction of formalin-fixed, paraffin-embedded tissue enables robust proteomic profiles by mass spectrometry. J. of Histochemistry and Cytochemistry 2009;57:849–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim SJ, Koh K, Boyd S, Gorinevsky D. l1 Trend filtering. SIAM Rev Soc Ind Appl Math. 2009;51:339–360. [Google Scholar]
- 23.Karolchik D, Hinrichs AS, Furey TS, et al. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004;32:D493–D496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ray D, Kazan H, Cook KB, et al. A compendium of RNA-binding motifs for decoding gene regulation. Nature. 2013;499:172–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015;4:1–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stewart PA, Parapatics K, Welsh EA, et al. A pilot proteogenomic study with data integration identifies MCT1 and GLUT1 as prognostic markers in lung adenocarcinoma. PLoS One. 2015;10:e0142162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li L, Wei Y, To C, et al. Integrated omic analysis of lung cancer reveals metabolism proteome signatures with prognostic impact. Nat Commun. 2014;5:5469. [DOI] [PubMed] [Google Scholar]
- 28.Collisson EA, Campbell JD, Brooks AN, et al. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li J, Tibshirani R. Finding consistent patterns: a nonparametric approach for identifying differential expression in RNA-Seq data. Stat Methods Med Res. 2011;22:519–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sankala H, Vaughan C, Wang J, Deb S, Graves PR. Upregulation of the mitochondrial transport protein, Tim50, by mutant p53 contributes to cell growth and chemoresistance. Arch Biochem Biophys. 2011;512: 52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao H, Korn JM, Ferretti S, et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat Med. 2015;21:1318–1325. [DOI] [PubMed] [Google Scholar]
- 32.Eifler K, Vertegaal ACO. SUMOylation-mediated regulation of cell cycle progression and cancer. Trends Biochem Sci. 2015;40:779–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnen G, Kaufman S. Studies on the enzymatic and transcriptional activity of the dimerization cofactor for hepatocyte nuclear factor 1. Proc Natl Acad Sci U S A. 1997;94:13469–13474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoskins JW, Jia J, Flandez M, et al. Transcriptome analysis of pancreatic cancer reveals a tumor suppressor function for HNF1A. Carcinogenesis. 2014;35:2670–2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yim J, Yun HS, Lee SJ, et al. Radiosensitizing effect of PSMC5, a 19S proteasome ATPase, in H460 lung cancer cells. Biochem Biophys Res Commun. 2016;469:94–100. [DOI] [PubMed] [Google Scholar]
- 36.Choy L, Derynck R. The type II transforming growth factor (TGF)-b receptor-interacting protein TRIP-1 acts as a modulator of the TGF-b response. J Biol Chem. 1998;273:31455–31462. [DOI] [PubMed] [Google Scholar]
- 37.Kobayashi H, Nishimura H, Matsumoto K, Yoshida M. Biochemical and Biophysical Research Communications Identification of the determinants of 2-deoxyglucose sensitivity in cancer cells by shRNA library screening. Biochem Biophys Res Commun. 2015;467:121–127. [DOI] [PubMed] [Google Scholar]
- 38.Tomida S, Koshikawa K, Yatabe Y, et al. Gene expression-based, individualized outcome prediction for surgically treated lung cancer patients. Oncogene. 2004;23:5360–5370. [DOI] [PubMed] [Google Scholar]
- 39.Qu Y, Yang Y, Liu B, Xiao W. Comparative proteomic profiling identified sorcin being associated with gemcitabine resistance in non-small cell lung cancer. Med Oncol. 2010;27:1303–1308. [DOI] [PubMed] [Google Scholar]
- 40.Maddalena F, Laudiero G, Piscazzi A, et al. Sorcin induces a drug-resistant phenotype in human colorectal cancer by modulating Ca2+ homeostasis. Cancer Res. 2011;71:7659–7669. [DOI] [PubMed] [Google Scholar]
- 41.Landriscina M, Laudiero G, Maddalena F, et al. Mitochondrial chaperone Trap1 and the calcium binding protein sorcin interact and protect cells against apoptosis induced by antiblastic agents. Cancer Res. 2010;70:6577–6586. [DOI] [PubMed] [Google Scholar]
- 42.Klont F, Bras L, Wolters JC, et al. Assessment of sample preparation bias in mass spectrometry-based proteomics. Anal Chem. 2018;90:5405–5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.