

Abstract
The necrosome is a protein complex required for signaling in cells that results in necroptosis, which is also dependent on tumor necrosis factor receptor (TNF-R) signaling. TNFα promotes necroptosis, and its expression is facilitated by mitogen-activated protein (MAP) kinase–activated protein kinase 2 (MK2) but is inhibited by the RNA-binding protein tristetraprolin (TTP, encoded by the Zfp36 gene). We have stimulated murine macrophages from WT, MyD88−/−, Trif−/−, MyD88−/−Trif−/−, MK2−/−, and Zfp36−/− mice with graded doses of lipopolysaccharide (LPS) and various inhibitors to evaluate the role of various genes in Toll-like receptor 4 (TLR4)–induced necroptosis. Necrosome signaling, cytokine production, and cell death were evaluated by immunoblotting, ELISA, and cell death assays, respectively. We observed that during TLR4 signaling, necrosome activation is mediated through the adaptor proteins MyD88 and TRIF, and this is inhibited by MK2. In the absence of MK2-mediated necrosome activation, lipopolysaccharide-induced TNFα expression was drastically reduced, but MK2-deficient cells became highly sensitive to necroptosis even at low TNFα levels. In contrast, during tonic TLR4 signaling, WT cells did not undergo necroptosis, even when MK2 was disabled. Of note, necroptosis occurred only in the absence of TTP and was mediated by the expression of TNFα and activation of JUN N-terminal kinase (JNK). These results reveal that TTP plays an important role in inhibiting TNFα/JNK-induced necrosome signaling and resultant cytotoxicity.
Keywords: inflammation, cell death, Toll-like receptor 4 (TLR4), necrosis, necrotic death, cytokine, tristetraprolin, ZFP36 ring finger protein (ZFP36), necrosome
Introduction
Toll-like receptor stimulation results in the activation of NF-κB, interferon regulatory factor, and MAPK2 pathways and consequent expression of inflammatory cytokines and chemokines (1, 2). The expression of cytokines such as TNFα is modulated through posttranscriptional mechanisms. MAP kinase-activated protein kinase 2 (MK2) and the RNA-binding protein tristetraprolin (TTP) have opposite impact on TNFα expression as Mk2−/− cells express reduced levels of TNFα in response to TLR signaling (3), whereas TTP-deficient cells express high levels of TNFα that is associated with inflammation and autoimmunity (4, 5). The p38MAPK/MK2 pathway stabilizes TNFα mRNA and stimulates its translation in part by inactivating (phosphorylating) TTP, which leads to an impairment in the binding of TTP to the AU-rich elements of TNFα-mRNA (3, 6–10).
Engagement of the TNF-R has been reported to promote NF-κB signaling, and has been shown to be facilitated by the receptor interacting protein kinase-1 (RipK1) (11). Cellular inhibitor of apoptosis proteins (cIAPs) maintain RipK1 in this prosurvival complex (12). Upon degradation of cIAPs through second mitochondrial inhibitor of apoptosis (SMAC), RipK1 undergoes ubiquitin editing and participates in two cell death platforms: the ripoptosome and the necrosome (13, 14). The ripoptosome complex consists of RipK1, RipK3, FADD, and caspase-8, which induces apoptotic cell death (15). When caspase-8 activity is inhibited, it allows further interaction of the complex with RipK3 and MLKL (called the necrosome complex), which results in phosphorylation and consequent trimerization of MLKL and cell death by necroptosis (13, 16–21).
In addition to TNF-R engagement, TLR signaling in the absence of caspase activity results in the assembly of an necroptosis (22–24) The pathological role of necroptosis has been revealed in various chronic diseases (25–33). Recently, it was reported that the treatment of myeloid leukemic cells with SMAC mimetic failed to induce TNFα expression and ripoptosome induced cell death unless MK2 was inhibited (34). Furthermore, MK2 was shown to induce an inhibitory phosphorylation at Ser-321 on RipK1 in myeloid leukemic cells that restricted SMAC mimetic–induced cell death (34–37). However, this enhancement of cell death by the inhibition of MK2 was not dependent on MLKL or RipK3 but required the kinase function of RipK1 (34).
Here we evaluated the impact of MK2 and TTP in macrophages, and we demonstrate that TTP inhibits necrosome activation during tonic TLR signaling through the inhibition of TNFα and JNK expression/activation.
Results
MyD88 and TRIF signaling promote necroptosis of macrophages
Necrosome signaling is induced in cells by engagement of TLRs or cytokine receptors (TNF-R/IFNAR) signaling in the absence of caspase-8 activity (17, 38). We stimulated macrophages with various concentrations of the TLR4 ligand LPS in the presence of a high concentration of the pan-caspase inhibitor zVAD-fmk (50 μm) to identify the relative concentration of LPS required to induce cell death by necroptosis. To confirm the mechanism of cell death as necroptosis, we treated cells with necrostatin-1 (Nec-1) to inhibit the kinase region of RipK1 and consequent RipK1–RipK3 interaction. Our results indicate that a minimal concentration of 200 pg/ml of LPS is required to induce necroptosis of macrophages (Fig. 1A). Furthermore, at 10 ng/ml of LPS, 50% cell death of macrophages occurred by 6 h post stimulation, whereas at 0.1 ng/ml LPS, 50% cell death occurred at 24 h (Fig. 1B). Necroptosis was not induced when the concentration of LPS was reduced to 0.01 ng/ml. This suggests that higher concentrations of LPS accelerate the timing of necroptosis. We stimulated primary macrophages with high (100 ng/ml) (Fig. 1, A, C–I) or low (1 ng/ml) (Fig. S1, A–F) concentration of LPS, and/or zVAD-fmk (50 μm) and measured cell death at 24 h. Both at high and low concentration of LPS, cell death depended on RipK3 and was rescued by the RipK3 inhibitor GSK872 (Fig. 1C and Fig. S1A). Whereas Ifnar1−/− macrophages showed resistance against necroptosis at both LPS concentrations (Fig. 1C and Fig. S1A), Tnfr2−/− macrophages showed protection only in response to stimulation with low concentration of LPS (Fig. S1A). Thus, higher concentration of LPS shifts the dependence of cell death less toward TNFR2 and more toward IFN-R. There was no impact of TNFR1 on necroptosis. Although we have previously reported that TRIF-signaling promotes necroptosis of macrophages (38), our results indicate that resistance against necroptosis induced by high or low concentration of LPS occurs only when both MyD88 and TRIF-signaling pathways are disabled (Fig. 1, D and E and Fig. S1B). Combined deficiency in MyD88 and Trif compromised necrosome signaling in macrophages as revealed by lack of Ser-166 phosphorylation of RipK1 and undetectable Ser-345 phosphorylation of MLKL (Fig. 1F). Furthermore, the total phosphorylation of RipK1 (upper band) was undetectable in MyD88−/−Trif−/− macrophages (Fig. 1F). We further observed that the phosphorylation of RipK3 was reduced in Trif−/− but completely inhibited in MyD88−/−Trif−/− macrophages (Fig. 1G). This suggests that both TLR4 adaptor proteins synergize to induce necroptosis with TRIF playing a more dominant role over MyD88. We observed that MyD88 promoted the expression of TNFα whereas TRIF induced IFN-I. The expression of both cytokines was inhibited only in macrophages with double deficiency of TRIF and MyD88 (Fig. 1H and Fig. S1C).
Figure 1.

TRIF and MyD88 synergize to induce necroptosis in macrophages during stimulation with high dose of LPS. A and B, bone marrow–derived macrophages (BMMs) were treated with zVAD-fmk (50 μm) and different concentrations of LPS, and cell viability was evaluated by MTT assay at 24 h (A) or at various time intervals post stimulation (B). C–J, BMMs were generated from WT or various knockout mice indicated in the figure and treated with LPS (100 ng/ml) and zVAD-fmk (50 μm). Cell viability was evaluated by MTT assay (24 h) (C and J), alamarBlue assay (24 h) (D), or at 18 h by Zombie Yellow assay (E). Cell extracts were collected at various time intervals and the expression/activation of various proteins evaluated by Western blotting (F and G). Production of TNFα (ELISA) and IFN-I (bioassay) was measured in cell supernatants collected at 24 h (H). A graphic version of results is shown in panel I. Cells were also treated with LPS and zVAD-fmk as mentioned above and p38MAPK inhibitor (LY2228820, 4 μm) or MK2 inhibitor (PF3644022, 5 μm) (J), and cell death was evaluated at 24 h by MTT assay. Representative data of one experiment of three similar experiments are shown. Graphs show the percentage of viable cells ± S.D. relative to controls. Each experiment was repeated three times. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
Interestingly, the activation of p38MAPK was reduced in MyD88−/− and Trif−/− macrophages but was undetectable in MyD88−/−Trif−/− macrophages (Fig. 1F). Thus, despite little p38MAPK phosphorylation in MyD88−/−Trif−/− macrophages, there was poor necroptosis of macrophages. This result is surprising because p38MAPK pathway has been recently shown to induce an inhibitory phosphorylation of RipK1 and inhibit cell death (35–37). Because we observed that the phosphorylation of RipK1 (Ser-166)-dependent necroptosis was also inhibited in MyD88−/−Trif−/− macrophages, this result suggests that the inhibition of the p38MAPK-induced inhibitory phosphorylation of RipK1 does not necessarily drive the cells toward necroptosis in the absence of TNFα and IFN-I.
Because p38MAPK and the downstream kinase MK2 promotes TNFα expression by macrophages following LPS treatment (3), we evaluated the impact of p38MAPK inhibitor (LY2228820, 4 μm) or MK2 inhibitor (PF3644022, 5 μm) on necroptosis induced by high (Fig. 1I) or low (Fig. S1D) concentrations of LPS. There was no appreciable increase in necroptosis when the p38MAPK pathway was disabled. Similar results were observed with Mk2−/− macrophages (Fig. 1I and Fig. S1E). Total phosphorylation of RipK1 (upper band) was inhibited in Mk2−/− cells and there was a slight increase in the Ser-345 phosphorylation of MLKL at earlier time period (Fig. S1F).
MK2 promotes TNFα expression but impairs the sensitivity of cells to necroptosis at lower levels of TNFα
Whereas induction of necroptosis in macrophages requires high concentrations of zVAD-fmk (>25 μm) (Fig. 2A), lower concentrations are needed to inhibit caspase activity (39). We therefore revised our experimental model and treated cells with LPS (1 ng/ml) and a reduced concentration of zVAD-fmk (10 μm) that does not induce significant necroptosis (Fig. 2A) to determine whether the inhibition of p38MAPK induces their sensitivity to cell death under these conditions. Our results indicate that the inhibition of p38MAPK makes the cells significantly susceptible to cell death (Fig. 2B), which correlates with Ser-345 phosphorylation of MLKL (Fig. 2C). We observed that the inhibition of p38MAPK accelerated the kinetics and magnitude of necroptosis (Fig. 2D) compared with the cells that did not receive the p38MAPK inhibitor (Fig. 1B). Induction of cell death by the inhibition of p38MAPK was still dependent on RipK3, TNFα (Fig. 2E), and MLKL (Fig. 2F) further confirming the mechanism of cell death to be necroptosis. In the absence of the p38MAPK inhibitor, there was no detectable cell death in WT, RipK3−/−, or TNFα−/− macrophages upon treatment with LPS (1 ng/ml) and low level of zVAD-fmk (10 μm) (Fig. S2A). Similar results were obtained with the MK2 inhibitor (Fig. 2G). To further confirm the role of MK2, we tested the cell death of Mk2−/− macrophages and observed that the deficiency of MK2 promotes significantly increased cell death when cells are treated with reduced zVAD-fmk concentration (Fig. 2H). Furthermore, Mk2−/− macrophages displayed an absence of overall RipK1 phosphorylation (upper band), and an increase in the stimulatory phosphorylation of RipK1 (Ser-166) and that was associated with phosphorylation of RipK3 and MLKL (Fig. 2I). We observed that when necroptosis was induced by treatment of cells with LPS (1 ng/ml) + zVAD-fmk (10 μm) + p38MAPK inhibitor (4 μm), cell death was mainly dependent on TRIF at 6 h post stimulation of cells (Fig. S2B). However, at 24 h post stimulation, complete rescue of cell death was observed only when both MyD88 and TRIF were disabled (Fig. 2J and Fig. S2C), suggesting that both adaptor proteins synergize to promote necroptosis under these conditions.
Figure 2.

p38MAPKpathway inhibits necrosome signaling in macrophages only at reduced concentration of LPS. A, BMMs were treated with LPS (1 ng/ml) and different concentrations of zVAD-fmk. B–L, BMMs were generated from WT and various knockout mice indicated in the figure and treated with LPS (1 ng/ml) (B, C, E–L) or various concentrations of LPS (D), and a reduced concentration (10 μm) of zVAD-fmk. M–P, a different pan-caspase inhibitor (Q-VD-OPh, 50 μm) (M) and two caspase-8 inhibitors (zIETD-fmk, 50 μm) (M-O) and Emricasan (EMR, 10 μm) (P) were used in some experiments. Cell death was evaluated at 24 h by MTT assay (A, B, E–H, L–N), CCK8 assay (D, P) or at 18 h by Zombie Yellow assay (J). Cell extracts were collected at various time intervals and the expression/activation of various proteins evaluated by Western blotting (C, I, O). Expression of TNFα was measured in the supernatants collected at various time intervals after stimulation of WT or Mk2−/− macrophages with LPS (1 ng/ml) + zVAD-fmk (10 μm) (K). Tnfα−/− macrophages were stimulated with LPS (1 ng/ml) + zVAD-fmk (10 μm) and different concentrations of recombinant TNFα in the presence or absence of p38MAPK/MK2 inhibitors, and cell death was evaluated by MTT assay at 24 h (L). Panel L shows asymmetric sigmoidal curve fit analysis of experimental data shown in Fig. S2E. Graphs show the percentage of viable cells ± S.D. relative to controls. Representative data of one experiment of three similar experiments are shown. Each experiment was repeated three times. ***, p < 0.001; ****, p < 0.0001.
We observed that the expression of TNFα was potently reduced in Mk2−/− cells in response to stimulation with LPS (Fig. S2D) or LPS+zVAD-fmk (Fig. 2K), although cell death occurring in the absence of MK2 is TNFα dependent (Fig. 2G). To reconcile this paradox, we treated Tnfa−/− macrophages with LPS (1 ng/ml)+zVAD-fmk (10 μm) in the presence of increasing concentrations of recombinant TNFα. Our results indicate that the inhibition of MK2 or p38MAPK makes macrophages highly sensitive (>100-fold) to minimal concentrations of TNFα required to induce cell death (Fig. 2L and Fig. S2E). These results suggest that the inhibition of p38MAPK/MK2 signaling increases the sensitivity of macrophages to necroptosis induced by very low levels of TNFα.
p38MAPK restricts necroptosis induced by the inhibition of caspase-8
We observed that when we use higher concentration (50 μm) of the pan-caspase inhibitor zVAD-fmk, potent necroptosis is induced in macrophages, and addition of the p38MAPK inhibitor has negligible impact (Fig. 1J). However, when the concentration of zVAD-fmk is lowered to 10 μm, then necroptosis is induced only when the p38MAPK pathway is inhibited (Fig. 2B). Thus, the use of higher zVAD-fmk concentration masks the important role of p38MAPK in the inhibition of necrosome signaling. Interestingly, treatment of macrophages with LPS (1 ng/ml) and the specific caspase-8 inhibitor (zIETD-fmk, 50 μm) or another pan-caspase inhibitor (Q-VD-OPh, 50 μm) did not induce necroptosis (Fig. 2M). These results suggest that zVAD-fmk is uniquely able to induce necroptosis. Interestingly, we observed that zIETD-fmk does induce necroptosis of macrophages only when p38MAPK is inhibited (Fig. 2, M and N). In addition to zIETD-fmk, treatment of cells with Q-VD-OPh resulted in cell death only upon co-treatment with LPS and p38MAPK inhibitor (Fig. 2M). Because the zIETD-fmk induced potent necrosome signaling in macrophages only when p38MAPK/MK2 signaling was inhibited, this suggests that zIETD-fmk was functional as a caspase-8 inhibitor. Furthermore, treatment of macrophages with LPS+ zIETD-fmk+p38MAPK inhibitor resulted in the phosphorylation of MLKL (Fig. 2O).
It has been suggested that zVAD-fmk treatment results in necroptosis, because it blocks caspase-8–cFLIPs heterodimer better than the zIETD-fmk or Q-VD-OPh (40, 41). We therefore used another specific caspase-8 inhibitor, Emricasan, which has increased specificity toward caspase-8 and blocks cFLIP-caspase-8 heterodimers similar to zVAD-fmk (42). The treatment of cells with LPS (1 ng/ml)+Emricasan (10 μm) resulted in cell death, which was enhanced by co-treatment with p38MAPK inhibitor (Fig. 2P). Cell death was rescued by GSK872, indicating the mechanism of cell death to be necroptosis. Additional experiments indicated that when the concentration of Emricasan was reduced to 1 μm, cell death was undetectable in macrophages unless p38MAPK pathway was inhibited (Fig. S2F). It has been shown that the phosphorylation of RipK1 by TAK1 governs the induction of apoptosis or necroptosis (43). We observed that the inhibition of TAK1 results in enhanced cell death of macrophages, and that was comparable to what was observed with the p38MAPK inhibitor (Fig. S2G).
TTP inhibits necrosome activation during tonic TLR4 signaling
Because TTP (Zfp36) is a downstream target of MK2 (MAPKAP2), we evaluated the role of TTP in necroptosis of macrophages. Our results indicate that when Zfp-36−/− macrophages are stimulated with 1 ng/ml of LPS and 25 μm zVAD-fmk, they undergo significantly more necroptosis (Fig. S3). We reasoned that this regulation of necroptosis by TTP may become more apparent at even more reduced concentrations of LPS. We observed that when cells were stimulated with very low dose of LPS (0.01 ng/ml), WT macrophages failed to undergo cell death, whereas Zfp36-deficient macrophages displayed a slight increase in cell death (Fig. 3, A–C). Interestingly, the inhibition of p38MAPK in WT cells did not induce any detectable cell death at low LPS concentration, whereas the inhibition of p38MAPK induced significant cell death in Zfp36−/− macrophages (Fig. 3, D–G). This induction of cell death in Zfp36−/− macrophages was because of necroptosis because the inhibition of RipK3 by GSK872 rescued cell death. Interestingly, the potent activation of necrosome, as revealed by Ser-345 phosphorylation of MLKL and Ser-166 phosphorylation of RipK1, was observed only when p38MAPK pathway was inhibited in Zfp36−/− cells (Fig. 3H).
Figure 3.

TTP regulates necrosome activation in response to tonic TLR4 signaling. A–J, WT and TTP (Zfp-36)−/− macrophages were stimulated with LPS (0.01 ng/ml) and zVAD-fmk (10 μm). D–J, in some experiments, cells were also co-treated with inhibitors against p38MAPK (LY2228820, 4 μm) (D–I) or MK2 (PF3644022, 5 μm) (J). Cell death was evaluated at 24 h by MTT assay (A, D, and J), by Zombie Yellow assay (B, C, E, and F), or by propidium iodide/Hoechst staining (G). Western blot analysis was performed on cell extracts collected at various time intervals (H and I). WT and TTP (Zfp-36)−/− macrophages were stimulated with LPS (0.01 ng/ml) and Emricasan (EMR) (500 nm) ± inhibitor against p38MAPK (LY2228820, 4 μm), and cell death was evaluated 24 h later by CCK8 assay (K). Representative data of one experiment of three similar experiments are shown. Graphs show the percentage of viable cells ± S.D. relative to controls. Each experiment was repeated three times. *, p < 0.05; ***, p < 0.001; ****, p < 0.0001.
Because RipK1 has been shown to have cell death–independent role in promoting cytokine synthesis through ERK1/2 and NF-κB activation (44), and TTP has been reported to promote degradation of various signaling proteins in the NF-κB pathway (45), we evaluated the activation of NF-κB by Western blotting. There was an increase in the activation of NF-κB in Zfp36−/− macrophages (Fig. 3I). Comparable to the results obtained with the inhibitor of p38MAPK, the inhibition of MK2 also resulted in cell death in Zfp36−/− cells (Fig. 3J). The inhibition of p38MAPK pathway also resulted in enhanced cell death in Zfp36−/− cells when cells were treated with low concentration of LPS and the specific inhibitor of caspase-8 (Emricasan) (42) (Fig. 3K). We observed that the inhibition of TAK1 or p38MAPK resulted in comparable enhancement in the induction of necroptosis in Zfp36−/− macrophages (Fig. S4A). The inhibition of p38MAPK resulted in enhanced RipK3 phosphorylation in Zfp36−/− macrophages (Fig. S4B).
The inhibition of necrosome signaling by TTP occurs through abrogation of both TNFα expression and JNK activation
Because regulation of TNFα mRNA stability is critically dependent on TTP (3), we evaluated the impact of TTP and p38MAPK pathway on TNFα expression in response to tonic LPS stimulation. At a highly reduced concentration of LPS (0.01 ng/ml) and zVAD-fmk (10 μm), the TNFα production was undetectable in WT cells but was significantly induced in Zfp36−/− cells particularly after inactivation of the p38MAPK pathway (Fig. 4, A and B). Necroptosis of Zfp36−/− macrophages, following stimulation with low level of LPS (10 pg/ml), was partially inhibited by blocking TNFR-I and completely rescued by blocking TNFR-II (Fig. 4, C–F). This suggests that TTP blocks necrosome signaling through regulation of TNFR signaling. Addition of exogenous TNFα to cells instead of LPS resulted in induction of necroptosis in both WT cells and Zfp36−/− macrophages to the same degree (Fig. 4G), which correlated with the Ser-166 phosphorylation of RipK1 and Ser-345 phosphorylation of MLKL (Fig. 4H). This is in contrast to the situation with LPS treatment where cell death and necrosome activation was only observed in Zfp36−/− macrophages (Fig. 3, D–H). These results suggest that the difference in cell death observed in WT and Zfp36−/− macrophages following LPS treatment must be due selective induction/maintenance of TNFα expression in Zfp36−/− macrophages.
Figure 4.

The inhibition of necroptosis by Zfp-36 depends on TNFR signaling. A–H, WT and Zfp36−/−macrophages were stimulated with LPS (0.01 ng/ml) and zVAD-fmk (10 μm). Some cells were also co-treated with inhibitors against p38MAPK (LY2228820, 4 μm) (A, C, and D–H), MK2 (PF3644022, 5 μm) (B) or with antibodies against TNFR1 (25 μg/ml) (C and F) or TNFR2 (25 μg/ml) (D and F). Expression of TNFα was measured by ELISA in the supernatants collected at 6 h (A and B). Cell death was evaluated at 24 h by alamarBlue assay (C, D, and G), at 18 h by Zombie Yellow assay (E and F). Western blotting was performed on cell extracts collected at various time intervals (H). Representative data of one experiment of three similar experiments are shown. Graphs show the percentage of viable cells ± S.D. relative to controls. Each experiment was repeated three times. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
We observed that there was an increase in the expression of cIAP1/2, JNK1/2, and pERK1/2 in Zfp36−/− cells (Fig. 5A). Activation of JNK1/2 was also detected only in Zfp36−/− cells upon the inhibition of the p38MAPK pathway (Fig. 5A). Knowing that JNK1/2 signaling has been shown to promote necroptosis (46), we treated macrophages with JNK1/2 inhibitor (SP60012, 50 μm) and observed a total abrogation of TNFα expression (Fig. 5B), and rescue of cell death in Zfp36−/− macrophages (Fig. 5C).
Figure 5.
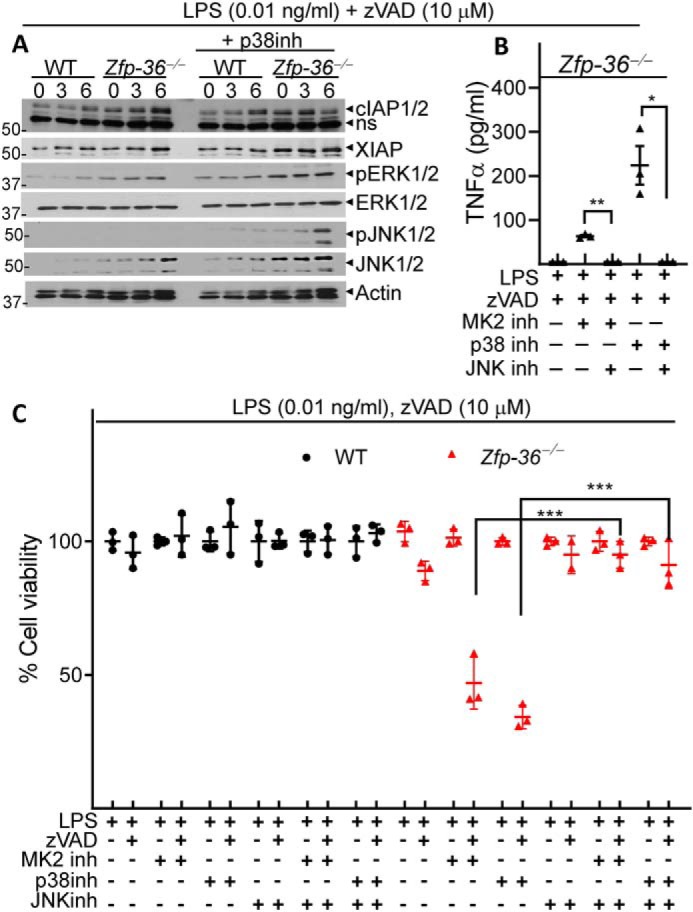
The inhibition of necroptosis by Zfp-36 depends on JNK signaling. A–C, WT and Zfp36−/− macrophages were stimulated with LPS (0.01 ng/ml) and zVAD-fmk (10 μm). Some cells were also co-treated with inhibitors against p38MAPK (LY2228820, 4 μm) (A–C), or MK2 (PF3644022, 5 μm) (B and C), or JNK1/2 (SP60012, 50 μm) (B and C). Cell death was evaluated at 24 h by alamarBlue assay (A, C) and the expression of TNFα was measured in the supernatants at 6 h post stimulation (B). Representative data of one experiment of three similar experiments are shown. Graphs show the percentage of viable cells ± S.D. relative to controls. Each experiment was repeated three times. ***, p < 0.001.
Thus, these results indicate that when necrosome activation is induced by very high concentrations of both LPS and the pan-caspase inhibitor zVAD-fmk, the impact of p38MAPK/MK2 signaling on necroptosis is barely appreciable. The inhibitory effect of p38MAPK/MK2 on necrosome signaling becomes apparent only when the concentrations of LPS and zVAD-fmk are reduced to low levels. Because TTP degrades TNFα transcripts (3), we predict that this results in complete abrogation of TNFα when cells are stimulated with low doses of LPS, and consequently, no necrosome signaling ensues. However, abrogation of TTP is not sufficient to switch the cells to necroptosis because MK2 still promotes the inhibitory phosphorylation of RipK1. Thus, disabling both the TTP and MK2 pathways is required to promote the necrosome activation of macrophages (Fig. 6).
Figure 6.
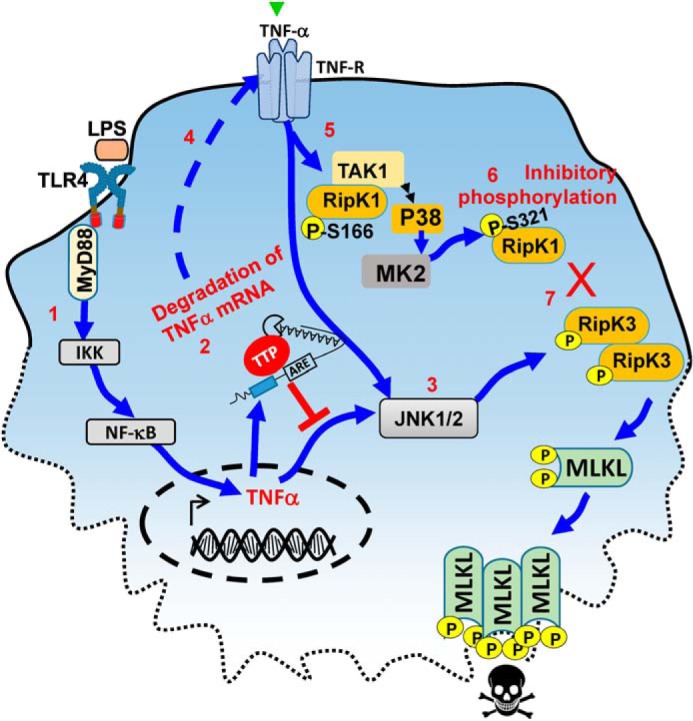
Regulation of necrosome signaling by TTP. Tonic TLR4 engagement in macrophages leads to expression of TNFα through Myd88 signaling (1). TNFα mRNA is rapidly degraded by TTP through recognition of AU-rich regions (2). As a consequence, less TNFα is available for TNF-receptor signaling and activation of JNK1/2 (3). Small levels of TNFα bind to the TNF-receptor (4) which is sufficient to induce phosphorylation of TAK1, P38, MK2, and Ser-166–RipK1 (5). MK2 induces an inhibitory Ser-321 phosphorylation of RipK1 (6). As a consequence, RipK3 fails to phosphorylate MLKL and mediate necroptosis (7). TTP regulates the level of TNFα which is the critical first step in promoting the activation of RipK1.
Discussion
TLR signaling of myeloid cells is a key driver of inflammatory response that facilitates the control of pathogens (1). Rupture of cells as a result of inflammatory cell death pathways results in the release of DAMPs and further amplification of the inflammatory response, which can lead to impairment in host survival (28). The triad of RipK1, caspase-8, and RipK3 maintains homeostasis, and an imbalance in the expression/function of any one of these proteins leads to host fatality because of over-activation of the others (47–50). Thus, regulation of cell death pathways is critical for maintenance of homeostasis. Although caspase-8 is considered a bona fide regulator of necrosome signaling, additional regulatory mechanisms of necrosome signaling have been recently revealed (24, 34–37). In this report we show that TTP plays a key role in inhibiting necrosome signaling of macrophages.
Our results indicate that during necrosome signaling of WT macrophages, p38MAPK/MK2 signaling does not impact necroptosis of cells unless the concentration of zVAD-fmk is reduced or caspase-8 inhibitor is specifically used. Although caspase-8 is a potent inhibitor of necrosome signaling in fibroblasts, specific inhibition of caspase-8 by zIETD-fmk does not result in necroptosis of macrophages even at high concentrations (24). Although it was considered that the concentration of the caspase-8 inhibitor used (50 μm) may not be sufficient to inhibit caspase-8, it inhibited necrosome signaling when p38MAPK/MK2 signaling was also inhibited, suggesting that the caspase-8 inhibitor is functional at that concentration. It is currently not clear why necroptosis in macrophages can be induced by the pan-caspase inhibitor zVAD-fmk but not by the caspase-8 inhibitor zIETD-fmk (24) or the other pan-caspase inhibitor, Q-VD-OPh. Because cFLIP is an endogenous inhibitor of caspase-8 (40) which interacts with caspase-8 as a heterodimer (41), it is conceivable that zVAD-fmk uniquely inhibits the caspase-8–cFLIPs heterodimer better than the zIETD or Q-VD-fmk.
We have reported previously that during necrosome signaling in macrophages, TLR4-engagement induces the phosphorylation of RipK1 at Thr-235 and Ser-313 (51). Recently, a new inhibitory phosphorylation of RipK1 was reported at Ser-321 in myeloid leukemia cells following treatment with SMAC mimetics, and that was mediated by MK2 (34–37). However, this enhancement of leukemic cell death that was induced by the inhibition of MK2 was not dependent on MLKL or RipK3 but required the kinase function of RipK1 (34). The role of MK2 in necroptosis was evaluated following treatment of MEFs with TNFα+SMAC-mimetic+zVAD-fmk, and this was reported to be enhanced by the inhibition of MK2 (37). Although these studies did not evaluate the impact of MK2 in TLR4-induced necrosome signaling of macrophages, we did not observe any significant impact of MK2 when cells were stimulated with traditional doses of LPS (1 or 100 ng/ml) and zVAD-fmk (50 μm) required to induce necroptosis of macrophages. Rather, we observed that necroptosis was induced only when cells were stimulated with very low doses of LPS (1 ng/ml) and zVAD-fmk (10 μm), and the p38MAPK pathway was inhibited. Furthermore, our results also indicate that the inhibition of the p38MAPK pathway enhances necroptosis induced by TNFα+zVAD-fmk; however, this was not impacted by Zfp-36 (Fig. 4G). We have revealed that the role of Zfp-36 is mainly related to expression of TNFα when TLR4 signaling is at tonic levels. Under these conditions, the inhibition of p38MAPK pathway by itself fails to induce necroptosis in WT cells (Fig. 3D). We believe that signaling mechanisms that operate at very low levels of stimulants are physiologically more relevant as they may exercise a greater impact on homeostasis.
We have previously reported that the higher molecular weight band of RipK1 is the phosphorylated version of this protein (38). Because this phosphorylation of RipK1 was inhibited in MK2-deficient cells, it appears that the predominant phosphorylation of RipK1 in response to TLR signaling is inhibitory in nature and is mediated by MK2. The knockdown of RipK1 has been shown to enhance necroptosis of human monocytic THP1 cells (52), suggesting that RipK1 scaffold may function more as an inhibitor rather than activator of necroptosis. Interestingly, disabling p38MAPK/MK2 results in amplification of the stimulatory RipK1 phosphorylation (Ser-166) while completely obliterating the inhibitory phosphorylation, and this dichotomy becomes more pronounced when necrosome stimulation is reduced, particularly when TTP is also disabled. It has been shown that direct phosphorylation of RipK1 at Ser-321 by MK2 suppresses RipK1 Ser-166 autophosphorylation and inhibits its ability to bind FADD/caspase-8 and induce RipK1-kinase–dependent apoptosis and necroptosis (37).
We have reported that IFNAR1 signaling (53), and particularly the downstream transcription factor complex ISGF3, is required for necrosome activation in macrophages (38). Recently, TNF-R2 signaling has been shown to be required for necroptosis of macrophages that depends on IFNAR1 signaling (54). This explains why necrosome signaling depends on IFNAR1 and TNFR2 particularly when a relatively reduced dose of LPS (1 ng/ml) is used. It is not clear why necroptosis becomes less dependent on TNFR2 when the concentration of LPS is increased further to 100 ng/ml. An interesting paradox that we have revealed here is that during necrosome signaling the levels of TNFα are substantially reduced in MK2-deficient macrophages, yet these cells become highly susceptible to TNFα-dependent necroptosis. We have shown that MK2 deficiency results in increased sensitivity of macrophages to necroptosis at highly reduced TNFα concentrations.
Our results reveal that the p38MAPK and TTP pathways synergize to regulate necrosome signaling, and this synergistic regulation of necrosome signaling achieves a greater significance when the concentrations of LPS and zVAD-fmk are reduced. According to our model, TTP inhibits TNFα expression when the TLR4 signaling is very low, and TNFα is required for necrosome signaling. The inhibition of TTP does not enhance necroptosis because MK2 is still able to induce an inhibitory phosphorylation on RipK1 and hence inhibit necroptosis. Thus, disabling both TTP and MK2 results in necroptosis.
It has been previously reported that transfection of the human embryonic kidney cell line HEK293 with a TTP and XIAP-expressing plasmid results in maintenance of RipK1 expression through degradation of XIAP and cIAP2 causing increased cell death (55). Our Western blots did not reveal any impact of TTP on the expression of RipK1 (Fig. 5, A and B), although we did observe increased expression of cIAP1/2 and XIAP in Zfp36-deficient macrophages. However, IAPs inhibit ripoptosome-induced cell death (15, 39), without having any impact on necrosome signaling and necroptosis (56). It remains to be seen whether the increased expression of XIAP in Zfp36-deficient macrophages results in reduced ripoptosome-induced cell death following treatment with a SMAC mimetic. In contrast to the effect of RipK1 in ripoptosome signaling, RipK1 has been shown to inhibit necrosome signaling (52). Similar to necrosome signaling, TTP was recently shown to inhibit the expression of NLRP3 inflammasome-mediated cell death (57). Thus, we believe that the regulation of TNFα expression by TTP and induction of the inhibitory phosphorylation on RipK1 by MK2 result in synergistic inhibition of necrosome signaling in macrophages (Fig. 6). Both, Zfp-36- and MK2-deficient mice have major phenotypic impact in vivo (3, 4). We expect that necrosome signaling is dysregulated and facilitates the effects observed in the inflammatory response in these mice.
Experimental procedures
Mice
C57BL/6J (stock no. 0664), TNF-R1−/− (stock no. 03242), TNF-R2−/− (stock no. 02620), TNFα−/− (stock no. 05540), Trif-mutant (stock no. 05037), and MyD88−/− (stock no. 09088) were obtained from The Jackson Laboratory (Bar Harbor, ME). RipK3−/− were obtained from Dr. Vishva Dixit (Genentech, South San Francisco, California). Homozygous mating strategy was used to breed the mice mentioned above. Heterozygous breeding strategy was used to maintain Zfp-36−/− and MK2−/− mice because of poor breeding associated with homozygous breeding. When using macrophages from Zfp-36−/− and MK2−/− mice, macrophages from littermate +/+ mice were used as WT controls. Mice were maintained at the animal facility of the University of Ottawa, Faculty of Medicine. All procedures were approved by University of Ottawa Animal Care Committee.
Macrophages and cell cultures
Macrophages (BMM) were generated from the bone marrow of the appropriate mouse strain after differentiating bone marrow cells with M-CSF (5 ng/ml) for 7–12 days. Cells were cultured in RPMI + 8% FBS. Cells were plated in 96- or 24-well plates in RPMI+8%FBS and various reagents added after overnight incubation. At various time intervals, cell death and activation of various proteins were evaluated.
Cell death assays
Cell viability was measured by various approaches. For colorimetric assay of cell death measurement, MTT was added to cells at a final concentration of 0.5 mg/ml and incubated at 37 °C for 2 h. Cells were lysed by adding 5 mm HCl in isopropyl alcohol and vigorously pipette up and down to solubilize the MTT crystals. Using Emax plate reader, optical density values were obtained by measuring absorption at 570 nm with a reference wavelength of 650 nm. The data were normalized to the corresponding stimulated control (i.e. LPS+zVAD-fmk treated cells were normalized to LPS treated cells whereas TNFα+zVAD-fmk treated cells were normalized to TNFα). In some experiments cell viability was measured by CCK8 assay (Enzo Life Sciences, Cat#ALX-850–039-K101). Optical density was measured at 450 nm and normalized to the corresponding stimulation control.
As an alternative colorimetric assay, alamarBlue® (cat. no. R7017, Sigma) was used at a final concentration of 0.04% dye (Resazurin) and fluorescence was measured (excitation 530 nm, emission 590 nm).
Cell death was also evaluated by immunofluorescence following staining with Hoëchst 33342 (2.5 μg/ml; Invitrogen) and propidium iodide (1:10 dilution; BD Pharmingen, 550825). Cells were stained in RPMI lacking phenol for 25–30 min. to distinguish live and dead cells. A Zeiss AxioObserver D1 microscope and the AxioVision Rel. 4.8 program were utilized to capture and analyze images. Cell death was also evaluated by flow cytometry following staining of cells with Zombie YellowTM (BioLegend, San Diego, CA). Zombie YellowTM solution (1:100 in PBS) was added into each well and the plate was incubated at room temperature, in the dark, for 15–30 min before washing with 100 μl FACS buffer (PBS, 1% BSA, 1 mm EDTA) once. Samples were fixed in 1% paraformaldehyde and acquired on LSR Fortessa Flow cytometer.
Western blotting
Cells were washed with cold PBS and were directly lysed in 1% SDS lysis buffer with 1% β-mercaptoethanol and transferred to 1.5 ml Eppendorf tubes. Samples were boiled instantly at 96 °C and frozen at −20 °C for future use. Lysates were run on polyacrylamide gel, based on the size of the proteins of interest. Gels were typically loaded with 25 μl of lysate and run at 135 V for 75 min. The transfer was run at 100 V for an hour. Once transferred, 5% skim milk or 5% BSA was used for 1 h to block the membrane. Primary antibodies diluted in appropriate blocking buffer (5% milk or BSA), were added to the membrane and incubated overnight on a shaker at 4 °C. The following antibodies were used: Mouse anti-Ripk1 (BD Biosciences, 610459), rabbit anti–phospho-RipK1 Ser-166 (Cell Signaling Technology, 31122), rabbit anti-RipK3 (ProSci Inc., 2283), rat anti-MLKL monoclonal (MABC604, EMD Millipore, Billerica, MA), rabbit anti-phospho MLKL(S345) monoclonal (D6E3G, Cell signaling, CA), mouse anti-β-actin (sc-81178, Santa Cruz, TX), rabbit anti p38MAPK (Cell Signaling Technology, 8690), rabbit anti-p-p38 (Cell Signaling Technology, 4511), anti-mouse MK2 (Cell Signaling Technology, 3042), anti-mouse p-MK2 (Cell Signaling Technology, 3007), rabbit anti-mouse FLIP (Abcam, 8421), mouse anti-mouse FADD (Enzo, ADI-AMM-212-E), rabbit anti-mouse P65 (Cell Signaling Technology, 8242), rabbit anti-mouse phospho (Ser-536) P65 (Cell Signaling Technology, 3033), rabbit anti-mouse XIAP (Cell Signaling Technology, 14334), Rabbit anti-mouse ERK (Cell Signaling Technology, 4695), rabbit anti-mouse phospho (T202/Y204) ERK (Cell Signaling Technology, 4370), rabbit anti JNK (Cell Signaling Technology, 9252), rabbit anti-mouse phospho (Thr-183/Tyr-185) JNK (Cell Signaling Technology, 9251), and rabbit anti-mouse cIAP1/2 (Cyclex, CY-P1041). Results were quantified using ImageJ software to perform the densitometric analysis.
Reagents and inhibitors
p38MAPK inhibitor (LY2228820, S4279) and MK2 inhibitor (PF3644022, S1494) were obtained from Selleck Chemicals (Houston, TX). GSK872 (GLXC-03990) was obtained from Glixx (Southborough, MA). Necrostatin-1 (Nec-1 9037), ultrapure LPS (Escherichia coli 0111:B4, (L4524) and MTT (M5655) were obtained from Sigma-Aldrich). zVAD-FMK (A1902) was obtained from ApexBio (Houston, TX). Caspase 8 inhibitor (C8I) (zIETD-fmk, 1064–20C) was obtained from BioVision (Milpitas, CA). JNK inhibitor (SP60012) was obtained from Sigma-Aldrich (S5567). TAK1 inhibitor ((5Z)-7-oxozeaenol) was obtained from Tocris (3604). Emricasan was obtained from Selleck Chemicals (S7775). Recombinant mouse TNFα (410-MT) was obtained from R&D Systems (Minneapolis, MN).
Cytokine analysis
Production of TNFα was measured in supernatants collected at 6 h by BD Biosciences optEIA ELISA kit (eBioscience). The expression of IFN-I was measured using a reporter cell line. ISRE-L929 cells were seeded at 5 × 104 cells per well in a 96-well tissue culture plate and incubated at 37 °C with 40 μl of culture supernatants for 4 h. Using the luciferase assay system kit (Promega, E1500), the luminescence was measured by a Molecular Devices Emax plate reader and data were analyzed by SoftMax Pro and Graphpad Prism 6.
Statistical analyses
Statistical analysis was determined by Student's t test using GraphPad Prism 8 software (La Jolla, California).
Author contributions
A. A. data curation; A. A., N. A. A., and S. H. investigation; A. A. and N. A. A. methodology; N. A. A. validation; N. A. A., S. H., M. G., P. J. B., and S. S. writing-review and editing; D. J. S., C. T., E. S. A., M. G., P. J. B., and S. S. resources; D. J. S., C. T., and S. S. writing-original draft; S. S. conceptualization; S. S. software; S. S. supervision; S. S. project administration.
Supplementary Material
This work was supported by Natural Sciences and Engineering Research Council (NSERC) Grant RGPIN-2017-03836 and by Canadian Institutes of Health Research (CIHR) Grant 375862 (to S. S.). This work was also supported by the Intramural Research Program of the NIEHS, National Institutes of Health (to P. J. B. and D. J. S.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S4.
- MAPK
- mitogen-activated protein kinase
- TTP
- tristetraprolin
- TNF-R
- tumor necrosis factor receptor
- cIAP
- cellular inhibitor of apoptosis protein
- SMAC
- second mitochondrial activator of caspases
- TLR
- Toll-like receptor 4
- JNK
- c-Jun N-terminal kinase
- LPS
- lipopolysaccharide
- BMM
- bone marrow–derived macrophages
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide.
References
- 1. Brodsky I. E., and Medzhitov R. (2009) Targeting of immune signalling networks by bacterial pathogens. Nat. Cell Biol. 11, 521–526 10.1038/ncb0509-521 [DOI] [PubMed] [Google Scholar]
- 2. Eckmann L., and Kagnoff M. F. (2001) Cytokines in host defense against Salmonella. Microbes. Infect. 3, 1191–1200 10.1016/S1286-4579(01)01479-4 [DOI] [PubMed] [Google Scholar]
- 3. Kotlyarov A., Neininger A., Schubert C., Eckert R., Birchmeier C., Volk H. D., and Gaestel M. (1999) MAPKAP kinase 2 is essential for LPS-induced TNF-α biosynthesis. Nat. Cell Biol. 1, 94–97 10.1038/10061 [DOI] [PubMed] [Google Scholar]
- 4. Taylor G. A., Carballo E., Lee D. M., Lai W. S., Thompson M. J., Patel D. D., Schenkman D. I., Gilkeson G. S., Broxmeyer H. E., Haynes B. F., and Blackshear P. J. (1996) A pathogenetic role for TNFα in the syndrome of cachexia, arthritis, and autoimmunity resulting from tristetraprolin (TTP) deficiency. Immunity 4, 445–454 10.1016/S1074-7613(00)80411-2 [DOI] [PubMed] [Google Scholar]
- 5. Carballo E., Lai W. S., and Blackshear P. J. (1998) Feedback inhibition of macrophage tumor necrosis factor-α production by tristetraprolin. Science 281, 1001–1005 10.1126/science.281.5379.1001 [DOI] [PubMed] [Google Scholar]
- 6. Hitti E., Iakovleva T., Brook M., Deppenmeier S., Gruber A. D., Radzioch D., Clark A. R., Blackshear P. J., Kotlyarov A., and Gaestel M. (2006) Mitogen-activated protein kinase-activated protein kinase 2 regulates tumor necrosis factor mRNA stability and translation mainly by altering tristetraprolin expression, stability, and binding to adenine/uridine-rich element. Mol. Cell Biol. 26, 2399–2407 10.1128/MCB.26.6.2399-2407.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Neininger A., Kontoyiannis D., Kotlyarov A., Winzen R., Eckert R., Volk H. D., Holtmann H., Kollias G., and Gaestel M. (2002) MK2 targets AU-rich elements and regulates biosynthesis of tumor necrosis factor and interleukin-6 independently at different post-transcriptional levels. J. Biol. Chem. 277, 3065–3068 10.1074/jbc.C100685200 [DOI] [PubMed] [Google Scholar]
- 8. Sabio G., and Davis R. J. (2014) TNF and MAP kinase signalling pathways. Semin. Immunol. 26, 237–245 10.1016/j.smim.2014.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Arthur J. S., and Ley S. C. (2013) Mitogen-activated protein kinases in innate immunity. Nat. Rev. Immunol. 13, 679–692 10.1038/nri3495 [DOI] [PubMed] [Google Scholar]
- 10. Carballo E., Cao H., Lai W. S., Kennington E. A., Campbell D., and Blackshear P. J. (2001) Decreased sensitivity of tristetraprolin-deficient cells to p38 inhibitors suggests the involvement of tristetraprolin in the p38 signaling pathway. J. Biol. Chem. 276, 42580–42587 10.1074/jbc.M104953200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Meylan E., Burns K., Hofmann K., Blancheteau V., Martinon F., Kelliher M., and Tschopp J. (2004) RIP1 is an essential mediator of Toll-like receptor 3-induced NF-κB activation. Nat. Immunol. 5, 503–507 10.1038/ni1061 [DOI] [PubMed] [Google Scholar]
- 12. Conte D., Holcik M., Lefebvre C. A., LaCasse E., Picketts D. J., Wright K. E., and Korneluk R. G. (2006) Inhibitor of apoptosis protein cIAP2 is essential for lipopolysaccharide-induced macrophage survival. Mol. Cell Biol. 26, 699–708 10.1128/MCB.26.2.699-708.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Degterev A., Huang Z., Boyce M., Li Y., Jagtap P., Mizushima N., Cuny G. D., Mitchison T. J., Moskowitz M. A., and Yuan J. (2005) Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 1, 112–119 10.1038/nchembio711 [DOI] [PubMed] [Google Scholar]
- 14. Pasparakis M., and Vandenabeele P. (2015) Necroptosis and its role in inflammation. Nature 517, 311–320 10.1038/nature14191 [DOI] [PubMed] [Google Scholar]
- 15. Tenev T., Bianchi K., Darding M., Broemer M., Langlais C., Wallberg F., Zachariou A., Lopez J., MacFarlane M., Cain K., and Meier P. (2011) The ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol. Cell 43, 432–448 10.1016/j.molcel.2011.06.006 [DOI] [PubMed] [Google Scholar]
- 16. Cho Y. S., Challa S., Moquin D., Genga R., Ray T. D., Guildford M., and Chan F. K. (2009) Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137, 1112–1123 10.1016/j.cell.2009.05.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Christofferson D. E., and Yuan J. (2010) Necroptosis as an alternative form of programmed cell death. Curr. Opin. Cell Biol. 22, 263–268 10.1016/j.ceb.2009.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Galluzzi L., and Kroemer G. (2008) Necroptosis: A specialized pathway of programmed necrosis. Cell 135, 1161–1163 10.1016/j.cell.2008.12.004 [DOI] [PubMed] [Google Scholar]
- 19. Festjens N., Vanden Berghe T., and Vandenabeele P. (2006) Necrosis, a well-orchestrated form of cell demise: Signalling cascades, important mediators and concomitant immune response. Biochim. Biophys. Acta 1757, 1371–1387 10.1016/j.bbabio.2006.06.014 [DOI] [PubMed] [Google Scholar]
- 20. Hitomi J., Christofferson D. E., Ng A., Yao J., Degterev A., Xavier R. J., and Yuan J. (2008) Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell 135, 1311–1323 10.1016/j.cell.2008.10.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Declercq W., Vanden Berghe T., and Vandenabeele P. (2009) RIP kinases at the crossroads of cell death and survival. Cell 138, 229–232 10.1016/j.cell.2009.07.006 [DOI] [PubMed] [Google Scholar]
- 22. He S., Liang Y., Shao F., and Wang X. (2011) Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc. Natl. Acad. Sci. U.S.A. 108, 20054–20059 10.1073/pnas.1116302108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McComb S., Cheung H. H., Korneluk R. G., Wang S., Krishnan L., and Sad S. (2012) cIAP1 and cIAP2 limit macrophage necroptosis by inhibiting Rip1 and Rip3 activation. Cell Death Differ. 19, 1791–1801 10.1038/cdd.2012.59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McComb S., Shutinoski B., Thurston S., Cessford E., Kumar K., and Sad S. (2014) Cathepsins limit macrophage necroptosis through cleavage of Rip1 kinase. J. Immunol. 192, 5671–5678 10.4049/jimmunol.1303380 [DOI] [PubMed] [Google Scholar]
- 25. Kaczmarek A., Vandenabeele P., and Krysko D. V. (2013) Necroptosis: The release of damage-associated molecular patterns and its physiological relevance. Immunity 38, 209–223 10.1016/j.immuni.2013.02.003 [DOI] [PubMed] [Google Scholar]
- 26. Vandenabeele P., Galluzzi L., Vanden Berghe T., and Kroemer G. (2010) Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 11, 700–714 10.1038/nrm2970 [DOI] [PubMed] [Google Scholar]
- 27. Günther C., Martini E., Wittkopf N., Amann K., Weigmann B., Neumann H., Waldner M. J., Hedrick S. M., Tenzer S., Neurath M. F., and Becker C. (2011) Caspase-8 regulates TNF-α-induced epithelial necroptosis and terminal ileitis. Nature 477, 335–339 10.1038/nature10400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Duprez L., Takahashi N., Van Hauwermeiren F., Vandendriessche B., Goossens V., Vanden Berghe T., Declercq W., Libert C., Cauwels A., and Vandenabeele P. (2011) RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity 35, 908–918 10.1016/j.immuni.2011.09.020 [DOI] [PubMed] [Google Scholar]
- 29. Ito Y., Ofengeim D., Najafov A., Das S., Saberi S., Li Y., Hitomi J., Zhu H., Chen H., Mayo L., Geng J., Amin P., DeWitt J. P., Mookhtiar A. K., Florez M., et al. (2016) RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science 353, 603–608 10.1126/science.aaf6803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Karunakaran D., Geoffrion M., Wei L., Gan W., Richards L., Shangari P., DeKemp E. M., Beanlands R. A., Perisic L., Maegdefessel L., Hedin U., Sad S., Guo L., Kolodgie F. D., Virmani R., Ruddy T., and Rayner K. J. (2016) Targeting macrophage necroptosis for therapeutic and diagnostic interventions in atherosclerosis. Sci. Adv. 2, e1600224 10.1126/sciadv.1600224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ofengeim D., Ito Y., Najafov A., Zhang Y., Shan B., DeWitt J. P., Ye J., Zhang X., Chang A., Vakifahmetoglu-Norberg H., Geng J., Py B., Zhou W., Amin P., Berlink Lima J., Qi C., Yu Q., Trapp B., and Yuan J. (2015) Activation of necroptosis in multiple sclerosis. Cell Rep. 10, 1836–1849 10.1016/j.celrep.2015.02.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Berger S. B., Kasparcova V., Hoffman S., Swift B., Dare L., Schaeffer M., Capriotti C., Cook M., Finger J., Hughes-Earle A., Harris P. A., Kaiser W. J., Mocarski E. S., Bertin J., and Gough P. J. (2014) Cutting edge: RIP1 kinase activity is dispensable for normal development but is a key regulator of inflammation in SHARPIN-deficient mice. J. Immunol. 192, 5476–5480 10.4049/jimmunol.1400499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang W., Marinis J. M., Beal A. M., Savadkar S., Wu Y., Khan M., Taunk P. S., Wu N., Su W., Wu J., Ahsan A., Kurz E., Chen T., Yaboh I., Li F., et al. (2018) RIP1 kinase drives macrophage-mediated adaptive immune tolerance in pancreatic cancer. Cancer Cell 34, 757–774.e7 10.1016/j.ccell.2018.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lalaoui N., Hänggi K., Brumatti G., Chau D., Nguyen N. Y., Vasilikos L., Spilgies L. M., Heckmann D. A., Ma C., Ghisi M., Salmon J. M., Matthews G. M., de Valle E., Moujalled D. M., Menon M. B., et al. (2016) Targeting p38 or MK2 enhances the anti-leukemic activity of Smac-mimetics. Cancer Cell 29, 145–158 10.1016/j.ccell.2016.01.006 [DOI] [PubMed] [Google Scholar]
- 35. Menon M. B., Gropengießer J., Fischer J., Novikova L., Deuretzbacher A., Lafera J., Schimmeck H., Czymmeck N., Ronkina N., Kotlyarov A., Aepfelbacher M., Gaestel M., and Ruckdeschel K. (2017) p38MAPK/MK2-dependent phosphorylation controls cytotoxic RIPK1 signalling in inflammation and infection. Nat. Cell Biol. 19, 1248–1259 10.1038/ncb3614 [DOI] [PubMed] [Google Scholar]
- 36. Dondelinger Y., Delanghe T., Rojas-Rivera D., Priem D., Delvaeye T., Bruggeman I., Van Herreweghe F., Vandenabeele P., and Bertrand M. J. M. (2017) MK2 phosphorylation of RIPK1 regulates TNF-mediated cell death. Nat. Cell Biol. 19, 1237–1247 10.1038/ncb3608 [DOI] [PubMed] [Google Scholar]
- 37. Jaco I., Annibaldi A., Lalaoui N., Wilson R., Tenev T., Laurien L., Kim C., Jamal K., Wicky John S., Liccardi G., Chau D., Murphy J. M., Brumatti G., Feltham R., Pasparakis M., Silke J., and Meier P. (2017) MK2 phosphorylates RIPK1 to prevent TNF-induced cell death. Mol. Cell 66, 698–710.e5 10.1016/j.molcel.2017.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McComb S., Cessford E., Alturki N. A., Joseph J., Shutinoski B., Startek J. B., Gamero A. M., Mossman K. L., and Sad S. (2014) Type-I interferon signaling through ISGF3 complex is required for sustained Rip3 activation and necroptosis in macrophages. Proc. Natl. Acad. Sci. U.S.A. 111, E3206–E3213 10.1073/pnas.1407068111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rijal D., Ariana A., Wight A., Kim K., Alturki N. A., Aamir Z., Ametepe E. S., Korneluk R. G., Tiedje C., Menon M. B., Gaestel M., McComb S., and Sad S. (2018) Differentiated macrophages acquire a pro-inflammatory and cell death-resistant phenotype due to increasing XIAP and p38-mediated inhibition of RipK1. J. Biol. Chem. 293, 11913–11927 10.1074/jbc.RA118.003614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hughes M. A., Powley I. R., Jukes-Jones R., Horn S., Feoktistova M., Fairall L., Schwabe J. W., Leverkus M., Cain K., and MacFarlane M. (2016) Co-operative and hierarchical binding of c-FLIP and caspase-8: A unified model defines how c-FLIP isoforms differentially control cell fate. Mol. Cell 61, 834–849 10.1016/j.molcel.2016.02.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Oberst A., Dillon C. P., Weinlich R., McCormick L. L., Fitzgerald P., Pop C., Hakem R., Salvesen G. S., and Green D. R. (2011) Catalytic activity of the caspase-8-FLIPL complex inhibits RIPK3-dependent necrosis. Nature 471, 363–367 10.1038/nature09852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Brumatti G., Ma C., Lalaoui N., Nguyen N. Y., Navarro M., Tanzer M. C., Richmond J., Ghisi M., Salmon J. M., Silke N., Pomilio G., Glaser S. P., de Valle E., Gugasyan R., Gurthridge M. A., et al. (2016) The caspase-8 inhibitor Emricasan combines with the SMAC mimetic birinapant to induce necroptosis and treat acute myeloid leukemia. Sci. Transl. Med. 8, 339ra69 10.1126/scitranslmed.aad3099 [DOI] [PubMed] [Google Scholar]
- 43. Geng J., Ito Y., Shi L., Amin P., Chu J., Ouchida A. T., Mookhtiar A. K., Zhao H., Xu D., Shan B., Najafov A., Gao G., Akira S., and Yuan J. (2017) Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nat. Commun. 8, 359 10.1038/s41467-017-00406-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Najjar M., Saleh D., Zelic M., Nogusa S., Shah S., Tai A., Finger J. N., Polykratis A., Gough P. J., Bertin J., Whalen M., Pasparakis M., Balachandran S., Kelliher M., Poltorak A., and Degterev A. (2016) RIPK1 and RIPK3 kinases promote cell-death-independent inflammation by Toll-like receptor 4. Immunity 45, 46–59 10.1016/j.immuni.2016.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tiedje C., Diaz-Muñoz M. D., Trulley P., Ahlfors H., Laaß K., Blackshear P. J., Turner M., and Gaestel M. (2016) The RNA-binding protein TTP is a global post-transcriptional regulator of feedback control in inflammation. Nucleic Acids Res. 44, 7418–7440 10.1093/nar/gkw474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cao M., Chen F., Xie N., Cao M. Y., Chen P., Lou Q., Zhao Y., He C., Zhang S., Song X., Sun Y., Zhu W., Mou L., Luan S., and Gao H. (2018) c-Jun N-terminal kinases differentially regulate TNF- and TLRs-mediated necroptosis through their kinase-dependent and -independent activities. Cell Death Dis. 9, 1140 10.1038/s41419-018-1189-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kaiser W. J., Upton J. W., Long A. B., Livingston-Rosanoff D., Daley-Bauer L. P., Hakem R., Caspary T., and Mocarski E. S. (2011) RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 471, 368–372 10.1038/nature09857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dillon C. P., Weinlich R., Rodriguez D. A., Cripps J. G., Quarato G., Gurung P., Verbist K. C., Brewer T. L., Llambi F., Gong Y. N., Janke L. J., Kelliher M. A., Kanneganti T. D., and Green D. R. (2014) RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell 157, 1189–1202 10.1016/j.cell.2014.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dillon C. P., Oberst A., Weinlich R., Janke L. J., Kang T. B., Ben-Moshe T., Mak T. W., Wallach D., and Green D. R. (2012) Survival function of the FADD-CASPASE-8-cFLIPL complex. Cell Rep. 1, 401–407 10.1016/j.celrep.2012.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Newton K., Dugger D. L., Wickliffe K. E., Kapoor N., de Almagro M. C., Vucic D., Komuves L., Ferrando R. E., French D. M., Webster J., Roose-Girma M., Warming S., and Dixit V. M. (2014) Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science 343, 1357–1360 10.1126/science.1249361 [DOI] [PubMed] [Google Scholar]
- 51. Shutinoski B., Alturki N. A., Rijal D., Bertin J., Gough P. J., Schlossmacher M. G., and Sad S. (2016) K45A mutation of RIPK1 results in poor necroptosis and cytokine signaling in macrophages, which impacts inflammatory responses in vivo. Cell Death Differ. 23, 1628–1637 10.1038/cdd.2016.51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kearney C. J., Cullen S. P., Clancy D., and Martin S. J. (2014) RIPK1 can function as an inhibitor rather than an initiator of RIPK3-dependent necroptosis. FEBS J. 281, 4921–4934 10.1111/febs.13034 [DOI] [PubMed] [Google Scholar]
- 53. Robinson N., McComb S., Mulligan R., Dudani R., Krishnan L., and Sad S. (2012) Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat. Immunol. 13, 954–962 10.1038/ni.2397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Legarda D., Justus S. J., Ang R. L., Rikhi N., Li W., Moran T. M., Zhang J., Mizoguchi E., Zelic M., Kelliher M. A., Blander J. M., and Ting A. T. (2016) CYLD proteolysis protects macrophages from TNF-mediated auto-necroptosis induced by LPS and licensed by type I IFN. Cell Rep. 15, 2449–2461 10.1016/j.celrep.2016.05.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Selmi T., Alecci C., dell'Aquila M., Montorsi L., Martello A., Guizzetti F., Volpi N., Parenti S., Ferrari S., Salomoni P., Grande A., and Zanocco-Marani T. (2015) ZFP36 stabilizes RIP1 via degradation of XIAP and cIAP2 thereby promoting ripoptosome assembly. BMC Cancer 15, 357 10.1186/s12885-015-1388-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Alturki N. A., McComb S., Ariana A., Rijal D., Korneluk R. G., Sun S.-C., Alnemri E., and Sad S. (2018) Triad3a induces the degradation of early necrosome to limit RipK1-dependent cytokine production and necroptosis. Cell Death Dis. 9, 592 10.1038/s41419-018-0672-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Haneklaus M., O'Neil J. D., Clark A. R., Masters S. L., and O'Neill L. A. J. (2017) The RNA-binding protein tristetraprolin (TTP) is a critical negative regulator of the NLRP3 inflammasome. J. Biol. Chem. 292, 6869–6881 10.1074/jbc.M116.772947 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.