

Abstract
Cell senescence is one of the most important processes determining cell fate and is involved in many pathophysiological conditions, including cancer, neurodegenerative diseases, and other aging-associated diseases. It has recently been discovered that the E3 ubiquitin ligase STIP1 homology and U-box–containing protein 1 (STUB1 or CHIP) is up-regulated during the senescence of human fibroblasts and modulates cell senescence. However, the molecular mechanism underlying STUB1-controlled senescence is not clear. Here, using affinity purification and MS-based analysis, we discovered that STUB1 binds to brain and muscle ARNT-like 1 (BMAL1, also called aryl hydrocarbon receptor nuclear translocator–like protein 1 (ARNTL)). Through biochemical experiments, we confirmed the STUB1-BMAL1 interaction, identified their interaction domains, and revealed that STUB1 overexpression down-regulates BMAL1 protein levels through STUB1's enzymatic activity and that STUB1 knockdown increases BMAL1 levels. Further experiments disclosed that STUB1 enhances BMAL1 degradation, which is abolished upon proteasome inhibition. Moreover, we found that STUB1 promotes the formation of Lys-48–linked polyubiquitin chains on BMAL1, facilitating its proteasomal degradation. Interestingly, we also discovered that oxidative stress promotes STUB1 nuclear translocation and enhances its co-localization with BMAL1. STUB1 expression attenuates hydrogen peroxide–induced cell senescence, indicated by a reduced signal in senescence-associated β-gal staining and decreased protein levels of two cell senescence markers, p53 and p21. BMAL1 knockdown diminishes this effect, and BMAL1 overexpression abolishes STUB1's effect on cell senescence. In summary, the results of our work reveal that the E3 ubiquitin ligase STUB1 ubiquitinates and degrades its substrate BMAL1 and thereby alleviates hydrogen peroxide–induced cell senescence.
Keywords: E3 ubiquitin ligase; senescence; ubiquitylation (ubiquitination); protein degradation; hydrogen peroxide; brain and muscle ARNT-like 1 (BMAL1, ARNTL, MOP3); cell cycle regulation; circadian clock; proteasome; STIP1 homology and U-box-containing protein 1 (STUB1)
Introduction
Senescence is an irreversible form of cell-cycle arrest with specific morphologic, molecular, and functional characteristics (1). Certain types of stress, including oxidative stress, can cause cellular senescence (2). Reactive oxygen species–induced cell senescence is involved in the response to DNA damage, epigenetic regulation, and activation of tumor suppression pathway. The hallmarks for senescent cells are reduced proliferative Ki-67 protein level, enhanced activity of senescence-associated β-gal (SA-β-Gal)3 (3), and elevated expression of cell cycle inhibitors, such as p21, and tumor suppressors, such as p53 and retinoblastoma protein pRB (4, 5). The p53-p21 signaling pathway has been implicated in the initiation of cell senescence, and their high expression could be considered as an indicator for cell senescence (5).
Cell senescence can also be accelerated by the activation of oncogenes, such as ras (6, 7), or the inhibition of important biological pathways, including the ubiquitin-proteasome system (UPS) (8). In the UPS, besides E1 activating enzymes and E2 conjugating enzymes, E3 ubiquitin ligases are the major enzymes that determine substrate specificity (9), leading to the modulation of specific signaling pathways through the ubiquitination of downstream targets. Different types of ubiquitination, such as monoubiquitination and polyubiquitination, alter the biological functions of the modified substrates (10–12). Most notably, the Lys-48–linked polyubiquitin chains are recognized by the 26S proteasome for subsequent degradation of the modified substrates (10). Several E3 ubiquitin ligases, such as MDM2 and SCFFBXO46, have been found to modulate cell senescence through degrading specific substrates (13, 14).
STIP1 homology and U-box–containing protein 1 STUB1 (also called C terminus of HSP70-interacting protein (CHIP)) possesses chaperone activity and U-box–dependent E3 ubiquitin ligase activity (15, 16). This protein plays essential roles in protein quality control by coupling the molecular chaperone machinery with the UPS (17). It has been found that STUB1 regulates the ubiquitination of diverse substrates, including endonuclease G (18), expanded polyglutamine proteins (19), FOXP3 (20), RIPK3 (21), SMAD3 (22), tau (23), unfolded proteins (24), and oxidized proteins (25). Thus, STUB1 mediates a variety of biological processes, including the regulatory T-cell function, necroptosis, TGF-β signaling, unfolding protein response, and stress response, although not all of the modified substrates are degraded by the 26S proteasome in these processes. It has been discovered that STUB1 protein level is up-regulated in senescent human fibroblasts (26), whereas Stub1 knockout mice have reduced life span (27) and STUB1 silencing induces premature senescence (25). However, the exact molecular mechanism by which STUB1 regulates cell senescence is still not completely clear.
Brain and muscle ARNT-like 1 (BMAL1; also called aryl hydrocarbon receptor nuclear translocator–like protein 1 (ARNTL) or basic-helix-loop-helix-PAS protein MOP3) is one of the master regulators for the circadian clock, and its knockout in mice completely disrupts the rhythmic behavior in constant darkness (28). Dysregulation of the circadian clock is associated with premature aging in Bmal1 knockout mice (29). Many enzymes in the UPS interact with BMAL1 and regulate its ubiquitination and degradation (30–33). However, the E3 ubiquitin ligases in the upstream signaling pathways of BMAL1 and their roles in the regulation of cell senescence have not been explored.
In this work, using MS and biochemical approaches, we identify an E3 ubiquitin ligase, STUB1, which interacts with BMAL1 and regulates its stability, ubiquitination, and degradation. We further use SA-β-Gal staining and immunoblotting of cell senescence markers to demonstrate that STUB1 attenuates hydrogen peroxide–induced senescence in HEK293T cells. Detailed mechanistic studies reveal that this regulation is mediated by BMAL1 and that restoration of BMAL1 protein level abolishes the effect of STUB1 on cell senescence. Our work reveals a novel molecular mechanism by which STUB1 regulates hydrogen peroxide–induced cell senescence.
Results
MS analysis identifies STUB1 as a BMAL1-interacting partner
It has been discovered that BMAL1 regulates the transcription activity of the cell senescence marker p53 and its downstream targets (34, 35). Therefore, to further explore potential upstream modulators for cell senescence, we carried out immunoprecipitation and MS analyses for BMAL1-interacting partners using a procedure described previously (31). MS analyses of FLAG immunoprecipitates from cell lysates resulting from HEK293T cells expressing FLAG-BMAL1 identified the E3 ubiquitin ligase STUB1 as a BMAL1-interacting protein. Two biological MS replicates detected 14 and 5 peptide spectrum matches (PSMs) of STUB1 in the experimental samples, but no peptides were identified in the control samples (PSMs = 0). The sequence coverage of STUB1 was 27.39% (Fig. 1A). In total, eight different tryptic peptides plus one containing an oxidized methionine were derived from STUB1 (Fig. 1B and Fig. S1). The Δmass was less than 1 ppm, the charge state was 2, and Xcorr was larger than 2.5 for all of the identified peptides. The annotated MS/MS spectrum of one representative peptide showed clear b- and y-ions. These data indicate the confident identification of STUB1 as a BMAL1 binding partner.
Figure 1.

Immunoprecipitation and MS analysis identified STUB1 as an interacting partner of BMAL1. A, PSMs for BMAL1 and STUB1 identified from two biological replicates of MS analyses for the FLAG immunoprecipitates of cell lysates from HEK293T cells expressing control or FLAG-BMAL1 plasmid, respectively. The number of peptides, sequence coverage, and PSMs for BMAL1 and STUB1 were provided. B, information for the MS-identified tryptic peptides derived from STUB1 in two biological replicates. The peptide sequence, charge state (z), MH+, Δmass, Xcorr, and amino acid position of the peptides derived from STUB1 were provided. C, a representative annotated MS/MS spectrum of an identified STUB1 tryptic peptide. The amino acid sequence, MH+, charge state (z), and Δmass of the identified peptide were also shown along with the annotated b- and y-ions.
Biochemical validation of the interaction between STUB1 and BMAL1
Next we used biochemical approaches to validate the discovery obtained from MS analyses. We expressed either FLAG-BMAL1 or Myc-STUB1 in HEK293T cells and purified them using FLAG affinity gel or Myc magnetic beads, respectively. Western blotting of immunoprecipitates demonstrated the presence of endogenous STUB1 or BMAL1 in the experimental samples but not in the control samples (Fig. 2, A and B). Furthermore, immunoprecipitation of endogenous BMAL1 in HEK293T cell lysate with an anti-BMAL1 antibody demonstrated that BMAL1 and STUB1 interact endogenously (Fig. 2C). These results confirmed the interaction between BMAL1 and STUB1 biochemically, validating the MS results.
Figure 2.

STUB1 preferably interacts with BMAL1 over CLOCK. A and B, STUB1 co-immunoprecipitates BMAL1 (A) and BMAL1 co-immunoprecipitates STUB1 (B). HEK293T cells were transfected with pcDNA3.1, Myc-STUB1, or FLAG-BMAL1 plasmid using PEI transfection reagent for 48 h. STUB1 or BMAL1 was immunoprecipitated (IP) with Myc or FLAG antibodies, respectively. C, STUB1 interacts with BMAL1 endogenously. Endogenous BMAL1 in cell lysate from HEK293T cells was immunoprecipitated with an anti-BMAL1 antibody. IgG was used as a control. D, STUB1 interacts with CLOCK. HEK293T cells were transfected with HA-CLOCK plasmid and then split into two plates for the transfection of control or Myc-STUB1 plasmid for 48 h, respectively. STUB1 was immunoprecipitated with an anti-Myc antibody. HSP70 and HSP90β were used as positive controls for their interaction with STUB1. E, STUB1 preferably interacts with BMAL1 over CLOCK. HEK293T cells were co-transfected with HA-CLOCK and Myc-STUB1 plasmids and then split into two plates for the transfection of control or FLAG-BMAL1 plasmid, respectively. STUB1 was immunoprecipitated with anti-Myc magnetic beads. F, BMAL1 knockdown enhances the interaction between STUB1 and CLOCK. HEK293T cells were co-transfected with HA-CLOCK and Myc-STUB1 plasmids and then split into two plates for the transfection of control or shBMAL1 plasmid, respectively. CLOCK was immunoprecipitated with anti-HA magnetic beads. The cell lysates and immunoprecipitates in A–F were immunoblotted with the indicated antibodies.
STUB1 preferentially interacts with BMAL1 over CLOCK
Because BMAL1 forms a heterodimer with CLOCK (36), we next asked whether STUB1 could also interact with CLOCK. To do so, we transfected HA-CLOCK plasmid along with the control or Myc-STUB1 plasmid and then carried out immunoprecipitation. The Western blotting images showed that STUB1 could also co-immunoprecipitate CLOCK (Fig. 2D). Here, HSP70 and HSP90β were used as positive controls because it was previously reported that they interacted with STUB1 and could modulate its biological function (24, 37). To examine whether BMAL1 could affect the interaction between STUB1 and CLOCK, we performed the same experiment as described above in the presence or absence of BMAL1. The immunoprecipitation and Western blotting results indicated that STUB1 did not interact with CLOCK any more when BMAL1 was expressed (Fig. 2E), suggesting that BMAL1 might disrupt the interaction between STUB1 and CLOCK through the competitive interaction. However, the interaction between STUB1 and HSP70 or HSP90β was not affected by BMAL1 (Fig. 2E). To further verify the effect of BMAL1 on the interaction between STUB1 and CLOCK, we expressed Myc-STUB1 and HA-CLOCK and then knocked down BMAL1 in HEK293T cells. Immunoprecipitation of HA-COCK and immunoblotting experiments indicated that BMAL1 depletion slightly increased STUB1-CLOCK interaction (Fig. 2F).
The STUB1 U-box domain interacts with BMAL1 and the BMAL1 middle domain interacts with STUB1
Next, we conducted two experiments to determine the interaction domains between STUB1 and BMAL1. First, pulldown of FLAG-BMAL1 from cell lysate with GST-tagged STUB1 and its truncations expressed in Escherichia coli found that the full-length STUB1, ΔTPR and U-box truncations interact with BMAL1, but other domains do not (Fig. 3, A and B), indicating that U-box domain interacts with BMAL1. Second, STUB1 was present in the immunoprecipitates of FLAG-tagged BMAL1 and its truncations but not of the negative control (Fig. 3, C and D). All BMAL1 truncations used in our experiments contain the middle domain (amino acids 143–325, PAS-A and the disorder 1 region). Therefore, the above data suggest that the middle domain of BMAL1 interacts with STUB1.
Figure 3.

BMAL1 interacts with the STUB1 U-box domain and STUB1 interacts with the BMAL1 middle domain. A, illustration of domains for the GST-tagged full-length STUB1 and its truncations. B, BMAL1 preferably interacts with the STUB1 U-box domain. GST-tagged STUB1 and its truncations were expressed in E. coli, purified with GSH-agarose beads, incubated with cell lysate from HEK293T cells expressing FLAG-BMAL1, washed, and eluted by heating with 2× SDS loading buffer. The eluates were stained with Ponceau S or immunoblotted (WB) with an anti-FLAG antibody. C, illustration of domains for the FLAG-tagged full-length BMAL1 and its truncations. D, STUB1 interacts with the BMAL1 middle domain. FLAG-tagged full-length BMAL1 and its truncations were co-transfected with Myc-STUB1 into HEK293T cells. Cells were lysed, and BMAL1 and its truncations were immunoprecipitated with an anti-FLAG antibody. Cell lysates and immunoprecipitates were immunoblotted with the indicated antibodies.
STUB1 down-regulates BMAL1 through its E3 ligase activity
STUB1 mediates the ubiquitination and proteasomal degradation of many substrates, including FOXP3, SMAD3, and RIPK3 (20–22). Therefore, we first tested whether STUB1 could also down-regulate BMAL1. The endogenous BMAL1 protein was significantly reduced after expressing Myc-STUB1 (Fig. 4A). Gradient increase of the transfected STUB1 led to a progressive reduction of BMAL1 (Fig. 4B). Next, we asked whether the enzymatic activity of STUB1 is required for this regulation. We transfected the control, WT, or catalytically inactive H260Q STUB1 plasmid (20) into HEK293T cells. Immunoblotting of endogenous BMAL1 in cell lysates showed that the H260Q mutant did not affect the BMAL1 protein level, although the WT STUB1 unambiguously reduced BMAL1 (Fig. 4C), indicating that the enzymatic activity is required for this regulation. Similar results were also obtained when we examined the effect of STUB1 on CLOCK protein level (Fig. S2).
Figure 4.

The E3 ligase activity is required for the STUB1-mediated BMAL1 reduction. A, STUB1 expression down-regulates BMAL1 protein. HEK293T cells were transfected with the control or Myc-STUB1 plasmid, respectively. Cell lysates were immunoblotted for endogenous BMAL1, Myc-STUB1, and β-tubulin. The experiments were carried out three times, and means ± S.D. (error bars) were plotted with GraphPad Prism. p value was calculated using Student's t test. **, p < 0.01. B, gradient-increased transfection of STUB1 gradually reduces BMAL1 protein level. HEK293T cells were transfected with different amounts of the control or Myc-STUB1 plasmid in 6-well plates. Cell lysates were immunoblotted for endogenous BMAL1, Myc-STUB1, and β-tubulin. The experiments were carried out three times, and means ± S.D. were plotted with GraphPad Prism. Student's t test was used: *, p < 0.05; ns, not significant. C, the regulation of BMAL1 by STUB1 requires its E3 ligase activity. HEK293T cells were transfected with the control, WT STUB1, or H260Q STUB1 (inactive mutant) plasmid, and cell lysates were immunoblotted. Student's t test was performed for the data from triplicates, and means ± S.D. were plotted. ***, p < 0.001; ns, not significant. D, STUB1 knockdown increases BMAL1 protein level. HEK293T cells were transfected with control or STUB1 specific siRNAs, respectively, for 48 h using Lipofectamine 2000 transfection reagent. Cell lysates were immunoblotted with the indicated antibodies. The experiments were performed in triplicates, and means ± S.D. were plotted. Student's t test was used: ***, p < 0.001.
To further validate the regulation of BMAL1 by STUB1, we transfected the control or STUB1-specific siRNAs, respectively, into HEK293T cells using Lipofectamine 2000 transfection reagent. Immunoblotting of cell lysates clearly demonstrated the increase of BMAL1 protein level after STUB1 knockdown (Fig. 4D). Taken together, our experiments demonstrated the regulation of BMAL1 by STUB1.
Proteasome inhibition abolishes the effect of STUB1 on BMAL1
Next, we asked whether the regulation of BMAL1 by STUB1 worked through the UPS. To test this, we first expressed FLAG-BMAL1 with the control or Myc-STUB1 in HEK293T cells, split the cells, and examined the BMAL1 protein level at different time points after the addition of cycloheximide (CHX), which blocks protein synthesis. The result showed that STUB1 significantly increased the BMAL1 degradation rate (Fig. 5A). Second, we expressed the control or Myc-STUB1 plasmid, split the cells, and treated them with DMSO or MG132 (a proteasome inhibitor), respectively. Similar to the above result, STUB1 significantly reduced BMAL1 protein level in the DMSO-treated samples. However, after the addition of MG132, the BMAL1 protein level was no longer regulated by STUB1 (Fig. 5B). These data indicate that STUB1 mediates BMAL1 degradation most probably through the UPS.
Figure 5.

STUB1 mediates the BMAL1 protein level through the ubiquitin-proteasome system. A, STUB1 enhances BMAL1 degradation. HEK293T cells were transfected with FLAG-BMAL1 plasmid and the control or Myc-STUB1 plasmid. Cells were split into 6-well plates, and at 48 h post-transfection, cells were treated with CHX (200 μg/ml) for the indicated time. Cell lysates were immunoblotted with the indicated antibodies. Experiments were repeated three times, and means ± S.D. (error bars) were plotted. Two-way analysis of variance was used: **, p < 0.01. Note that the amount of plasmid used for transfection was slightly adjusted to ensure that the FLAG-BMAL1 protein level at the zero time point was similar in the absence or presence of Myc-STUB1. B, MG132 eliminates the effect of STUB1 on BMAL1 protein level. HEK293T cells were transfected with a control or Myc-STUB1 plasmid, respectively, and at 48 h after transfection, cells were treated with DMSO or MG132 (10 μm) for 12 h. The resulting cell lysates were immunoblotted. The experiments were repeated three times, and means ± S.D. were plotted. Student's t test was used: *, p < 0.05; ns, not significant. C, STUB1 increases BMAL1 ubiquitination. HEK293T cells were first transfected with FLAG-BMAL1 plasmid and divided into four plates 6 h after transfection. The cells were again transfected with a control, WT, or H260Q STUB1 plasmid for 36 h. Cells were treated with DMSO or MG132 (20 μm) for 6 h. FLAG-BMAL1 was immunoprecipitated with FLAG M2 affinity gel. The cell lysates and immunoprecipitates were immunoblotted. D, STUB1 promotes the formation of the Lys-48–linked polyubiquitin chains on BMAL1. HEK293T cells were first transfected with FLAG-BMAL1 and Myc-STUB1 and then split into four plates 6 h after transfection. Cells were again transfected with a control, WT, K48R, or K63R ubiquitin (Ub) plasmid, and at 36 h after the second transfection, cells were treated with MG132 (10 μm) for 12 h. The cell lysates and FLAG immunoprecipitates were immunoblotted with the indicated antibodies.
STUB1 promotes the formation of Lys-48–linked polyubiquitin chains on BMAL1
To further test whether STUB1 enhances the BMAL1 ubiquitination, we carried out two more experiments. In the first experiment, we expressed FLAG-BMAL1 in HEK293T cells and then divided the cells into four plates. Cells were further transfected with the control, WT, or H260Q STUB1 plasmid and treated with DMSO or MG132. BMAL1 was immunoprecipitated with FLAG affinity beads, and the resulting immunoprecipitates were immunoblotted with an anti-ubiquitin antibody. The results clearly showed the increase of BMAL1 ubiquitination upon the expression of WT STUB1 but not the H260Q mutant, indicating that the E3 ligase activity is required for STUB1 to modulate BMAL1 ubiquitination (Fig. 5C), which is consistent with the above discovery that WT STUB1 but not the H260Q mutant reduces BMAL1 protein level (Fig. 4C). In the second experiment, HEK293T cells were transfected with FLAG-BMAL1 and Myc-STUB1 plasmids, divided into four plates, and transfected again with control, WT, K48R, or K63R HA-ubiquitin plasmid followed by MG132 treatment. The anti-HA (ubiquitin) immunoblotting of FLAG immunoprecipitates showed the significant decrease of BMAL1 ubiquitination when the K48R ubiquitin was expressed (Fig. 5D), suggesting that STUB1 promotes the formation of Lys-48–linked polyubiquitin chains on BMAL1. Taken together with previous results, these data indicate that STUB1 mediates BMAL1 ubiquitination for proteasomal degradation.
STUB1 attenuates cell senescence through down-regulating BMAL1
It has been discovered that STUB1 is up-regulated in senescent human fibroblasts (26) and modulates the senescence in HFL-1 human lung fibroblast cells (25). Moreover, both STUB1 and BMAL1 regulate the cell senescence markers p53 and p21. Therefore, we sought to test whether STUB1 modulates cell senescence through regulating BMAL1. First, we examined the effect of hydrogen peroxide (H2O2) on the protein level of p53 and p21. The immunoblotting results showed that H2O2 increased the expression of p53 and p21 in HEK293T cells, indicating the occurrence of cell senescence. Their expression level reached a maximum at 400 μm H2O2 (Fig. S3). Therefore, in the following experiments, we used this concentration to treat cells to induce cell senescence. Next, we examined the effect of H2O2 on the localization of STUB1 and BMAL1. To do so, we expressed Myc-STUB1 and FLAG-BMAL1 in HEK293 cells for immunofluorescence experiments (Fig. 6A). In the mock PBS treatment, the majority of STUB1 is located in the cytoplasm, and only a small fraction of STUB1 is localized in the nucleus. Upon H2O2 treatment, the majority of STUB1 is translocated to the nucleus. However, under both experimental conditions, BMAL1 is mainly localized in the nucleus, although it is a nucleocytoplasmic shuttling protein (38). This experiment also demonstrates that H2O2 enhances the co-localization of STUB1 and BMAL1. Next, we used SA-β-Gal staining (3) and immunoblotting of p53 and p21 to evaluate the effect of STUB1 on the regulation of H2O2–induced cell senescence. When HEK293T cells were transfected with STUB1 plasmid and treated with H2O2, cell senescence was significantly reduced when compared with the mock transfection, demonstrated by the decrease of SA-β-Gal–positive cells and by the reduction of p53 and p21 (Fig. 6, B and C).
Figure 6.

Hydrogen peroxide enhances STUB1 nuclear localization, and STUB1 reduces cell senescence by down-regulating BMAL1. A, H2O2 elevates STUB1 nuclear translocation and promotes the co-localization between STUB1 and BMAL1. HEK293 cells were first transfected with Myc-STUB1 and FLAG-BMAL1, split into two plates, and treated with PBS or 400 μm H2O2 for 24 h. Cells were fixed and incubated with anti-Myc and anti-FLAG antibodies for immunofluorescence measurement. Scale bar, 5 μm. B and C, STUB1 attenuates H2O2-induced cell senescence. HEK293T cells were first transfected with the control or Myc-STUB1 plasmid and then treated with 400 μm H2O2 for 24 h. Half of the cells were stained with an SA-β-Gal staining kit (B), and the other half were used for immunoblotting (C). Images were taken under a microscope, and the senescent cells (blue cells) were counted. Three images were taken for quantification (means ± S.D. (error bars)). Scale bar, 50 μm. Student's t test was used: ***, p < 0.001. The experiments were repeated in triplicates, and similar results were obtained. D and E, BMAL1 knockdown almost completely eliminates the effect of STUB1 on hydrogen peroxide–induced cell senescence. HEK293T cells were first transfected with the control or BMAL1-specific shRNA, and then each was split into two plates for the transfection of the control or Myc-STUB1 plasmid. Cells were treated and processed as described in B and C. Scale bar, 50 μm. Student's t test was used: *, p < 0.05; **, p < 0.01; ***, p < 0.001. F and G, BMAL1 expression abolishes the effect of STUB1 on cell senescence. HEK293T cells were transfected with the control or Myc-STUB1 plasmid with or without FLAG-BMAL1 plasmid. Cells were treated and processed as described in B and C. Scale bar, 50 μm. Student's t test was used: **, p < 0.01; ***, p < 0.001; ns, not significant.
To further examine whether this reduction worked through the down-regulation of BMAL1 by STUB1, we used shBMAL1 to knock down endogenous BMAL1 and then transfected the control or STUB1 plasmid. The SA-β-Gal staining and immunoblotting experiments showed that after BMAL1 knockdown, the effect of STUB1 on the percentage of SA-β-Gal–positive cells or the relative amount of p53 and p21 was significantly altered (Fig. 6, D and E). These data unambiguously indicate that STUB1 attenuates H2O2-induced cell senescence at least partially through BMAL1. To further validate this conclusion, we restored the BMAL1 protein level through transfecting BMAL1 plasmid into the STUB1-expressing cells. The SA-β-Gal staining and Western blotting analyses clearly indicated that STUB1 reduced cell senescence and further restoration of BMAL1 abolished the effect of STUB1 on cell senescence (Fig. 6, F and G). Taken together, these data further support the idea that STUB1 regulates cell senescence through down-regulating BMAL1.
Discussion
In this work, using affinity purification and MS analysis, we identified STUB1 as a BMAL1 interactor and found that it reduced BMAL1 stability and promoted its ubiquitin-mediated degradation. In a model cell line, we further discovered that STUB1 expression attenuated hydrogen peroxide–induced cell senescence through down-regulating BMAL1, demonstrated by the reduced SA-β-Gal–positive cells and by the decreased cell senescence markers p53 and p21. Knockdown of BMAL1 and restoration of BMAL1 almost completely abolished the effect of STUB1 on cell senescence, indicating that the reduction of BMAL1 by STUB1 is the primary mechanism for the decreased senescence mediated by STUB1. Therefore, our work identified a new regulator and a new molecular mechanism for the regulation of hydrogen peroxide–induced cell senescence (Fig. 7).
Figure 7.
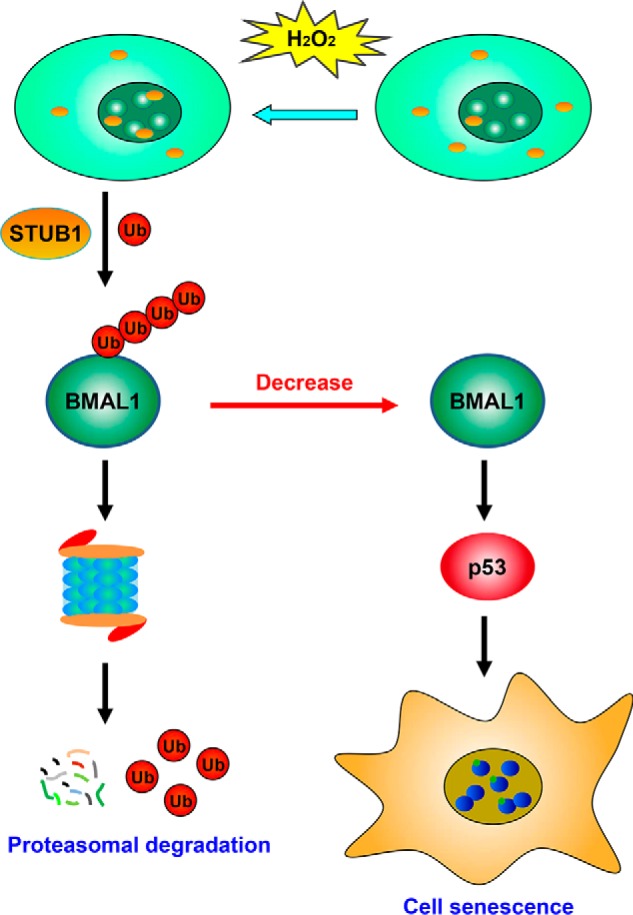
Proposed model for the regulation of cell senescence by STUB1. Upon hydrogen peroxide treatment, STUB1 is translocated to the nucleus, promotes the ubiquitination and proteasomal degradation of BMAL1, decreases the BMAL1 protein level, and further reduces the expression of two cell senescence marker proteins, p53 and p21, leading to the attenuated cell senescence induced by hydrogen peroxide.
Using biochemical approaches, we not only discovered that STUB1 interacted with BMAL1 but also revealed that STUB1 could bind to the BMAL1-interacting partner CLOCK. However, co-expression of BMAL1 and CLOCK eliminated the interaction between STUB1 and CLOCK. Moreover, we found that the interaction between STUB1 and BMAL1 or CLOCK led to the reduction of both proteins in the absence of the other binding partner, and the E3 ligase activity of STUB1 was required for this reduction. These data also suggest that the use of HEK293T cells, which do not express CLOCK protein, might facilitate the identification of STUB1 as a BMAL1 binding partner through immunoprecipitation and MS analysis.
It has been reported that the percentage of SA-β-Gal–positive cells is lower but not statistically significant in Bmal1 knockout mouse embryo fibroblast cells when treated with a low concentration of hydrogen peroxide (39). Our results showed that treatment with a higher concentration of hydrogen peroxide in BMAL1 knockdown cells resulted in a significant reduction of senescent cells. It should be noted that it might also be possible that different cell lines have distinct effects upon BMAL1 knockdown and oxidative stress.
In our experiments, we found that STUB1 expression reduces BMAL1, p53, and p21 protein level, which is consistent with the previous discovery that BMAL1 activates the transcription of p53 and its downstream genes (34, 35). It has been reported that STUB1 reduces p53 protein level during cellular senescence and that proteasome inhibition prevents its reduction (26). However, it is unknown whether this regulation occurs directly through the ubiquitination of p53 by STUB1 even though STUB1 can ubiquitinate p53 in cooperation with E2 UBCH5B and HSC70 (40). Our experiments might suggest a possible alternative mechanism for the regulation of p53 by STUB1 during cell senescence. In this mechanism, STUB1 down-regulates BMAL1 through promoting its ubiquitination and proteasomal degradation, thus leading to the reduced BMAL1 transcription activity and p53 expression. It is interesting to know how the interaction between BMAL1 and STUB1 is regulates. Our experiments revealed that oxidative stress promoted the STUB1 nuclear translocation and enhances its co-localization with BMAL1, although the molecular mechanism for STUB1 nuclear translocation is unknown. Therefore, we expect that other signals that trigger the translocation of STUB1 to the nucleus would most probably enhance their interaction and promote the STUB1-mediated ubiquitination and degradation of BMAL1. It would also be valuable to discover other physiological and pathological conditions that regulate this interaction because cell senescence is involved in many diseases (41).
It has been reported that STUB1 regulates the ubiquitination and degradation of many proteins, such as FOXP3, SMAD3, and RIPK3 (20–22), and thus executes its biological functions. Here, using shRNA to knockdown BMAL1, we also demonstrated that STUB1 may down-regulate p53 and its downstream target p21, at least partially, through the regulation of the ubiquitination and degradation of BMAL1. This is in accordance with our finding that STUB1 attenuates cell senescence through down-regulating BMAL1, supported by the alteration of p53 and p21 protein level. Several possible reasons may explain the role of BMAL1 but not other STUB1 substrates in cell senescence. First, some STUB1 substrates, such as CLOCK, may not be expressed in our model cell line. Second, other proteins mediated by STUB1 might not exhibit a profound effect on the modulation of cell senescence under our experimental conditions. Third, BMAL1 might be the most significant influencing player in mediating H2O2-induced cell senescence in the cell line we used here. However, we cannot rule out the possibility that different cell lines might have distinct signaling pathways modulating cell senescence.
It should be mentioned that STUB1 exhibits protective roles in oxidative stress–induced cell death through down-regulating endonuclease G in cancer cells and primary rat cortical neurons (18). Here we found that STUB1 protected cells from senescence through the ubiquitination and degradation of BMAL1. These results suggest that STUB1 exhibits its protective roles through different downstream targets in different cells and biological processes. Indeed, the protective function of STUB1 has also been associated with the neurodegenerative diseases (42), by targeting the misfolded proteins, such as expanded polyglutamine proteins, premature cystic fibrosis transmembrane conductance regulator, α-synuclein, and tau, for proteasomal degradation (19, 23, 24).
BMAL1 is one of the master regulators in maintaining the integrity of the circadian clock, the roughly 24-h wake and sleep cycle. Genetic knockout of Bmal1 in mice completely disrupts the circadian rhythm in constant darkness (28). Modulating BMAL1 by E2, E3, and deubiquitinase may affect the mouse circadian rhythm or cellular clock behaviors (30–33). Because our experiments were carried out in HEK293T cells, which are deficient in CLOCK, we did not test the effect of STUB1 on cellular circadian behavior. A different cell line should be used if one wants to explore this behavior. However, it should be noted that analysis of gene expression in a circadian database (CircaDB, RRID:SCR_018078) (43) did not detect a profound and significant oscillation of Stub1 mRNA in major mouse tissues. In addition, several important functions of BMAL1, such as regulation of protein synthesis (44) and metabolism (45, 46), were not explored in this work. Because STUB1 profoundly down-regulates BMAL1, it might also participate in the regulation of these processes.
In summary, our work discovered a new upstream regulator, an E3 ubiquitin ligase, STUB1, for BMAL1 and elucidated the underlying regulatory molecular mechanism. We also discovered that STUB1 attenuates hydrogen peroxide–induced cell senescence through down-regulating BMAL1. Because BMAL1 has a more profound effect on many biological processes, this regulation might have diverse impacts on aspects other than cell senescence.
Experimental procedures
Materials
The HEK293T cell line was from American Type Culture Collection. Antibodies were from the following companies: FLAG M2 affinity gel for immunoprecipitation from Sigma; anti-Myc and anti-HA magnetic beads from Bimake (Houston, TX); STUB1 and p21 antibodies from ProteinTech Group (Wuhan, Hubei, China); BMAL1, HA, and ubiquitin from Santa Cruz Biotechnology, Inc.; FLAG, Myc, and glyceraldehyde-3-phosphate dehydrogenase from HuaAn Biotechnology (Hangzhou, Zhejiang, China); α-tubulin, β-tubulin, and β-actin from Vazyme Biotech Co. (Nanjing, Jiangsu, China); HSP70, HSP90β, and p53 from Cohesion Biosciences (Suzhou, Jiangsu, China); horseradish peroxidase–labeled secondary antibodies from Beyotime Biotechnology (Haimen, Jiangsu, China); and Alexa Fluor 594 goat anti-mouse IgG and Alexa Fluor 488 goat anti-rabbit IgG from Thermo Fisher Scientific (Waltham, MA). FLAG peptide (DYKDDDDK) was synthesized by ChinaPeptides (Hangzhou, Zhejiang, China); CHX was from Sigma and MG132 was from Selleck (Houston, TX); reagents for SA-β-Gal staining were ordered from Beyotime Biotechnology; and H2O2 was from Lingfeng Chemical Reagent Co., Ltd. (Shanghai, China).
Plasmids
Myc-tagged WT STUB1 and catalytically inactive H260Q mutant plasmids (20) were kindly provided by Dr. Bin Li (Institut Pasteur of Shanghai, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences). Plasmids expressing GST-STUB1 and its truncations in E. coli were provided by Dr. Zijie Chang (Tsinghua University). FLAG-BMAL1 and HA-CLOCK plasmids (47) were gifts from Dr. Ying Xu, and WT, K48R, and K63R HA-ubiquitin plasmids were from Dr. Xinliang Mao (Soochow University). FLAG-BMAL1 truncations were from a previous study (31).
Cell culture
High-glucose Dulbecco's modified Eagle's medium (Gibco), supplemented with 10% fetal bovine serum (Lonsera, Shanghai, China), 1% penicillin, and streptomycin (Gibco), was used to culture HEK293T cells in a humidified 37 °C incubator containing 5% CO2.
Transfection
Control (catalog no. 160818) and STUB1 siRNAs were synthesized by RiboBio Co. (Guangzhou, Guangdong, China). The siSTUB1 sequences were as follows: #1, 5′-AUACAUGGCAGAUAUGGAUTT-3′ (sense) and 5′-AUCCAUAUCUGCCAUGUAUTT-3′ (antisense); #2, 5′-AACAGGCACUUGCUGACUGTT-3′ (sense) and 5′-CAGUCAGCAAGUGCCUGUUTT-3′ (antisense). Control or STUB1 siRNAs (25 pmol) were used to transfect HEK293T cells in 12-well plates using Lipofectamine 2000 transfection reagent in a similar way as described previously (48). pLKO.1-shBMAL1 was constructed using the primers CCGGGCAGAATGTCATAGGCAAGTTCTCGAGAACTTGCCTATGACATTCTGCTTTTTG (forward) and AATTCAAAAAGCAGAATGTCATAGGCAAGTTCTCGAGAACTTGCCTATGACATTCTGC (reverse) according to a method published previously (49). HEK293T cells were transfected with shBMAL1 alone or with the indicated plasmids using polyethyleneimide (PEI) (Sigma) transfection reagent. Control shRNA or pcDNA3.1 was used to balance the total amount of plasmids transfected in each sample. Fresh medium was replaced 6 h after transfection. Cells were cultured for the subsequent experiments.
Immunoprecipitation
Cells were washed twice with ice-cold PBS and incubated with the RIPA lysis buffer (150 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% Triton X-100, 0.1% SDS, and 1 mm EDTA) containing 10% glycerol and fresh protease inhibitor (Selleck) for 30 min on ice. After brief sonication, the mixture was centrifuged at 16,000 × g (4 °C) for 10 min to obtain the cell lysate.
FLAG-BMAL1 was immunoprecipitated using a method reported previously (50). In short, prewashed FLAG M2 affinity gel (20 μl) was mixed with cell lysate. After overnight incubation at 4 °C, the affinity gel was washed six times with RIPA buffer. Protein was eluted twice with 40 μl of 200 μg/ml FLAG peptide in TBST (TBS with 0.1% Tween 20). For the purification of Myc-tagged STUB1 and its interacting proteins, cell lysates were incubated with prewashed anti-Myc magnetic beads overnight at 4 °C, and RIPA buffer was used to wash the beads six times. SDS sample loading buffer (60 μl) was added to the beads and heated at 98 °C for 10 min to elute the proteins. HA-tagged CLOCK and its interacting partners were purified by incubating the cell lysate with prewashed anti-HA magnetic beads for 6 h, and the beads were then washed with RIPA buffer three times. SDS sample loading buffer (60 μl) was added to the beads and heated at 98 °C for 10 min to elute proteins. For the immunoprecipitation of endogenous BMAL1, HEK293T cell lysate was precleaned with protein A/G-agarose beads, incubated with IgG and anti-BMAL1 antibody (0.8 μg) overnight, and again incubated with protein A/G-agarose beads (40 μl slurry) for 3 h. The protein A/G beads were washed three times with RIPA buffer, and proteins were eluted with SDS sample loading buffer by heating at 98 °C for 10 min.
GST pulldown
GST-tagged proteins were expressed in E. coli and purified with GSH-agarose beads according to a protocol published previously (51). The GSH-agarose beads with purified GST-tagged proteins were incubated with cell lysate obtained from HEK293T cells expressing FLAG-BMAL1 and washed with RIPA buffer three times. The proteins were eluted by heating the beads with 2× SDS loading buffer and separated by SDS-PAGE. Proteins were transferred to polyvinylidene difluoride membrane and stained with Ponceau S before performing immunoblotting analysis.
CHX and MG132 treatment
For the CHX treatment experiment, HEK293T cells were transfected with FLAG-BMAL1 plasmid and pcDNA3.1 or Myc-STUB1 plasmid using PEI transfection reagent. Cells were then divided equally into 6-well plates. At 48 h after transfection, cells were treated with CHX (200 μg/ml) for different time periods. For proteasome inhibition experiments, HEK293T cells were transfected with pcDNA3.1 or Myc-STUB1 plasmid and then divided into two sets of plates. Cells were treated with DMSO or MG132, respectively, at the indicated concentration and time. Cells were washed with ice-cold PBS and processed as described above for subsequent experiments.
Immunoblotting
Immunoblotting experiments were performed as reported previously (52). In short, samples (cell lysates or immunoprecipitates) were mixed with SDS sample loading buffer, heated for 10 min at 98 °C, centrifuged for 10 min at 16,000 × g, and loaded on SDS-PAGE for separation, and proteins were transferred to polyvinylidene difluoride membrane (Millipore, Burlington, MA). Double-distilled water was used to briefly wash the membrane, which was then incubated with 5% nonfat milk for 1 h. Primary antibodies with a proper dilution were used to incubate the membrane for 1–2 h at room temperature, followed by washing three times (each for 10 min) with TBST on a plate shaker. Then the membrane was incubated with secondary antibodies, followed by washing with TBST. The membrane was incubated with hypersensitive ECL chemiluminescence reagents (NCM Biotech) for 5 min. A ChemiDoc MP or Tanon 5200 chemiluminescent imaging system was used to visualize the target protein bands and to record chemiluminescent signal. ImageJ was used for densitometry quantification.
Immunofluorescence
HEK293 cells were transfected with Myc-STUB1 and FLAG-BMAL1 plasmids for 24 h and treated with PBS or H2O2 (400 μm) for 24 h, cells were washed with PBS, fixed with pre-chilled 4% paraformaldehyde for 15 min, and then permeabilized with 0.1% Triton X-100 for 15 min at room temperature. Cells were further blocked with 5% BSA for 1 h at room temperature, incubated with an anti-FLAG mouse mAb (1:300 dilution) or anti-Myc rabbit polyclonal antibody (1:300 dilution) at 4 °C overnight. Cells were washed three times (5 min each) with TBST and stained again with Alexa Fluor 594 goat anti-mouse IgG and Alexa Fluor 488 goat anti-rabbit IgG (1:300 dilution) for 2 h at room temperature in the dark. Cells were washed three times with TBST (5 min each), stained with DAPI (1:10,000 dilution) for 15 min at room temperature. Images were captured using a laser confocal microscope (LSM 710, Carl Zeiss) with Plan-Apochromat ×63/1.40 numerical aperture oil DICM27 objective.
SA-β-Gal
An SA-β-Gal staining kit was used to analyze the senescent cells. Cells were fixed with 4% paraformaldehyde for 15 min and then washed three times with PBS. Cells were then incubated with staining solution (a mixture of 10 μl of solution A, 10 μl of solution B, 930 μl of solution C, and 50 μl of X-Gal solution) for 14 h in a nonhumidified incubator at 37 °C. A microscope was used to record the images, and the blue-staining cells were counted as senescent cells.
Statistical analysis
All of the data were plotted as mean ± S.D. using GraphPad Prism. Student's t test or two-way analysis of variance was used for statistical analyses.
Author contributions
K. U., S. C., J. L., X. W., Q. L., Y. Z., Y. L., Z. H., and G. X. conceptualization; K. U., S. C., J. L., X. W., Q. L., and Y. Z. data curation; K. U. formal analysis; K. U., S. C., J. L., X. W., Q. L., and Y. Z. investigation; K. U., Z. H., and G. X. writing-original draft; K. U., Y. L., Z. H., and G. X. writing-review and editing; G. X. supervision; Z. H. and G. X. funding acquisition.
Supplementary Material
Acknowledgments
We are grateful to Dr. Bin Li (Institut Pasteur of Shanghai, Chinese Academy of Sciences) and Dr. Zijie Chang (Tsinghua University) for the STUB1 plasmids, Dr. Ying Xu (Soochow University) for the FLAG-BMAL1 and HA-CLOCK plasmids, and Dr. Xinliang Mao (Soochow University) for HA-ubiquitin and its mutant plasmids. We also thank Xiafang Xu and Dr. Xiaogang Jiang for assistance during the cell senescence experiments.
This work was supported by National Key R&D Program of China Grant 2019YFA0800400, National Natural Science Foundation of China Grants 31670833 and 31700722, Suzhou Bureau of Science and Technology (Basic Research in Medical and Health Sciences) Grant SYS201718, Postgraduate Research and Practice Innovation Program of Jiangsu Province Grant KYCX17_2040, Jiangsu Key Laboratory of Neuropsychiatric Diseases Grant BM2013003, National Center for International Research Grant 2017B01012, and a project funded by the Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S3.
- SA-β-Gal
- senescence-associated β-gal
- HEK
- human embryonic kidney
- UPS
- ubiquitin-proteasome system
- PSM
- peptide spectrum match
- CHX
- cycloheximide
- PEI
- polyethyleneimide
- RIPA
- radioimmune precipitation assay.
References
- 1. Hernandez-Segura A., Nehme J., and Demaria M. (2018) Hallmarks of cellular senescence. Trends Cell Biol. 28, 436–453 10.1016/j.tcb.2018.02.001 [DOI] [PubMed] [Google Scholar]
- 2. Chen Q. M., Bartholomew J. C., Campisi J., Acosta M., Reagan J. D., and Ames B. N. (1998) Molecular analysis of H2O2-induced senescent-like growth arrest in normal human fibroblasts: p53 and Rb control G1 arrest but not cell replication. Biochem. J. 332, 43–50 10.1042/bj3320043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dimri G. P., Lee X., Basile G., Acosta M., Scott G., Roskelley C., Medrano E. E., Linskens M., Rubelj I., and Pereira-Smith O. (1995) A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. U.S.A. 92, 9363–9367 10.1073/pnas.92.20.9363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shay J. W., Pereira-Smith O. M., and Wright W. E. (1991) A role for both RB and p53 in the regulation of human cellular senescence. Exp. Cell Res. 196, 33–39 10.1016/0014-4827(91)90453-2 [DOI] [PubMed] [Google Scholar]
- 5. Mavrogonatou E., Pratsinis H., and Kletsas D. (2019) The role of senescence in cancer development. Semin. Cancer Biol. 10.1016/j.semcancer.2019.06.018 [DOI] [PubMed] [Google Scholar]
- 6. Serrano M., Lin A. W., McCurrach M. E., Beach D., and Lowe S. W. (1997) Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88, 593–602 10.1016/S0092-8674(00)81902-9 [DOI] [PubMed] [Google Scholar]
- 7. Zhang Y., and Yang J. M. (2011) The impact of cellular senescence in cancer therapy: is it true or not? Acta Pharmacol. Sin. 32, 1199–1207 10.1038/aps.2011.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chondrogianni N., Stratford F. L., Trougakos I. P., Friguet B., Rivett A. J., and Gonos E. S. (2003) Central role of the proteasome in senescence and survival of human fibroblasts: induction of a senescence-like phenotype upon its inhibition and resistance to stress upon its activation. J. Biol. Chem. 278, 28026–28037 10.1074/jbc.M301048200 [DOI] [PubMed] [Google Scholar]
- 9. Xu G., and Jaffrey S. R. (2013) Comprehensive profiling of protein ubiquitination for drug discovery. Curr. Pharm. Des. 19, 3315–3328 10.2174/13816128113199990305 [DOI] [PubMed] [Google Scholar]
- 10. Wilkinson K. D., Tashayev V. L., O'Connor L. B., Larsen C. N., Kasperek E., and Pickart C. M. (1995) Metabolism of the polyubiquitin degradation signal: structure, mechanism, and role of isopeptidase T. Biochemistry 34, 14535–14546 10.1021/bi00044a032 [DOI] [PubMed] [Google Scholar]
- 11. Deng L., Wang C., Spencer E., Yang L., Braun A., You J., Slaughter C., Pickart C., and Chen Z. J. (2000) Activation of the IκB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 103, 351–361 10.1016/S0092-8674(00)00126-4 [DOI] [PubMed] [Google Scholar]
- 12. Mukhopadhyay D., and Riezman H. (2007) Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science 315, 201–205 10.1126/science.1127085 [DOI] [PubMed] [Google Scholar]
- 13. Choppara S., Ganga S., Manne R., Dutta P., Singh S., and Santra M. K. (2018) The SCFFBXO46 ubiquitin ligase complex mediates degradation of the tumor suppressor FBXO31 and thereby prevents premature cellular senescence. J. Biol. Chem. 293, 16291–16306 10.1074/jbc.RA118.005354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu B., Yi J., Yang X., Liu L., Lou X., Zhang Z., Qi H., Wang Z., Zou J., Zhu W. G., Gu W., and Luo J. (2019) MDM2-mediated degradation of WRN promotes cellular senescence in a p53-independent manner. Oncogene 38, 2501–2515 10.1038/s41388-018-0605-5 [DOI] [PubMed] [Google Scholar]
- 15. Ballinger C. A., Connell P., Wu Y., Hu Z., Thompson L. J., Yin L. Y., and Patterson C. (1999) Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol. Cell. Biol. 19, 4535–4545 10.1128/MCB.19.6.4535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rosser M. F., Washburn E., Muchowski P. J., Patterson C., and Cyr D. M. (2007) Chaperone functions of the E3 ubiquitin ligase CHIP. J. Biol. Chem. 282, 22267–22277 10.1074/jbc.M700513200 [DOI] [PubMed] [Google Scholar]
- 17. VanPelt J., and Page R. C. (2017) Unraveling the CHIP:Hsp70 complex as an information processor for protein quality control. Biochim. Biophys. Acta 1865, 133–141 10.1016/j.bbapap.2016.11.005 [DOI] [PubMed] [Google Scholar]
- 18. Lee J. S., Seo T. W., Yi J. H., Shin K. S., and Yoo S. J. (2013) CHIP has a protective role against oxidative stress-induced cell death through specific regulation of endonuclease G. Cell Death Dis. 4, e666 10.1038/cddis.2013.181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jana N. R., Dikshit P., Goswami A., Kotliarova S., Murata S., Tanaka K., and Nukina N. (2005) Co-chaperone CHIP associates with expanded polyglutamine protein and promotes their degradation by proteasomes. J. Biol. Chem. 280, 11635–11640 10.1074/jbc.M412042200 [DOI] [PubMed] [Google Scholar]
- 20. Chen Z., Barbi J., Bu S., Yang H. Y., Li Z., Gao Y., Jinasena D., Fu J., Lin F., Chen C., Zhang J., Yu N., Li X., Shan Z., Nie J., et al. (2013) The ubiquitin ligase Stub1 negatively modulates regulatory T cell suppressive activity by promoting degradation of the transcription factor Foxp3. Immunity 39, 272–285 10.1016/j.immuni.2013.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Seo J., Lee E. W., Sung H., Seong D., Dondelinger Y., Shin J., Jeong M., Lee H. K., Kim J. H., Han S. Y., Lee C., Seong J. K., Vandenabeele P., and Song J. (2016) CHIP controls necroptosis through ubiquitylation- and lysosome-dependent degradation of RIPK3. Nat. Cell Biol. 18, 291–302 10.1038/ncb3314 [DOI] [PubMed] [Google Scholar]
- 22. Xin H., Xu X., Li L., Ning H., Rong Y., Shang Y., Wang Y., Fu X. Y., and Chang Z. (2005) CHIP controls the sensitivity of transforming growth factor-β signaling by modulating the basal level of Smad3 through ubiquitin-mediated degradation. J. Biol. Chem. 280, 20842–20850 10.1074/jbc.M412275200 [DOI] [PubMed] [Google Scholar]
- 23. Hatakeyama S., Matsumoto M., Kamura T., Murayama M., Chui D. H., Planel E., Takahashi R., Nakayama K. I., and Takashima A. (2004) U-box protein carboxyl terminus of Hsc70-interacting protein (CHIP) mediates poly-ubiquitylation preferentially on four-repeat Tau and is involved in neurodegeneration of tauopathy. J. Neurochem. 91, 299–307 10.1111/j.1471-4159.2004.02713.x [DOI] [PubMed] [Google Scholar]
- 24. Murata S., Minami Y., Minami M., Chiba T., and Tanaka K. (2001) CHIP is a chaperone-dependent E3 ligase that ubiquitylates unfolded protein. EMBO Rep. 2, 1133–1138 10.1093/embo-reports/kve246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sisoula C., and Gonos E. S. (2011) CHIP E3 ligase regulates mammalian senescence by modulating the levels of oxidized proteins. Mech. Ageing Dev. 132, 269–272 10.1016/j.mad.2011.04.003 [DOI] [PubMed] [Google Scholar]
- 26. Sisoula C., Trachana V., Patterson C., and Gonos E. S. (2011) CHIP-dependent p53 regulation occurs specifically during cellular senescence. Free Radic. Biol. Med. 50, 157–165 10.1016/j.freeradbiomed.2010.10.701 [DOI] [PubMed] [Google Scholar]
- 27. Min J. N., Whaley R. A., Sharpless N. E., Lockyer P., Portbury A. L., and Patterson C. (2008) CHIP deficiency decreases longevity, with accelerated aging phenotypes accompanied by altered protein quality control. Mol. Cell. Biol. 28, 4018–4025 10.1128/MCB.00296-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bunger M. K., Wilsbacher L. D., Moran S. M., Clendenin C., Radcliffe L. A., Hogenesch J. B., Simon M. C., Takahashi J. S., and Bradfield C. A. (2000) Mop3 is an essential component of the master circadian pacemaker in mammals. Cell 103, 1009–1017 10.1016/S0092-8674(00)00205-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Khapre R. V., Kondratova A. A., Susova O., and Kondratov R. V. (2011) Circadian clock protein BMAL1 regulates cellular senescence in vivo. Cell Cycle 10, 4162–4169 10.4161/cc.10.23.18381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gossan N. C., Zhang F., Guo B., Jin D., Yoshitane H., Yao A., Glossop N., Zhang Y. Q., Fukada Y., and Meng Q. J. (2014) The E3 ubiquitin ligase UBE3A is an integral component of the molecular circadian clock through regulating the BMAL1 transcription factor. Nucleic Acids Res. 42, 5765–5775 10.1093/nar/gku225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen S., Yang J., Zhang Y., Duan C., Liu Q., Huang Z., Xu Y., Zhou L., and Xu G. (2018) Ubiquitin-conjugating enzyme UBE2O regulates cellular clock function by promoting the degradation of the transcription factor BMAL1. J. Biol. Chem. 293, 11296–11309 10.1074/jbc.RA117.001432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen S., Yang J., Yang L., Zhang Y., Zhou L., Liu Q., Duan C., Mieres C. A., Zhou G., and Xu G. (2018) Ubiquitin ligase TRAF2 attenuates the transcriptional activity of the core clock protein BMAL1 and affects the maximal Per1 mRNA level of the circadian clock in cells. FEBS J 285, 2987–3001 10.1111/febs.14595 [DOI] [PubMed] [Google Scholar]
- 33. Zhang Y., Duan C., Yang J., Chen S., Liu Q., Zhou L., Huang Z., Xu Y., and Xu G. (2018) Deubiquitinating enzyme USP9X regulates cellular clock function by modulating the ubiquitination and degradation of a core circadian protein BMAL1. Biochem. J. 475, 1507–1522 10.1042/BCJ20180005 [DOI] [PubMed] [Google Scholar]
- 34. Mullenders J., Fabius A. W., Madiredjo M., Bernards R., and Beijersbergen R. L. (2009) A large scale shRNA barcode screen identifies the circadian clock component ARNTL as putative regulator of the p53 tumor suppressor pathway. PLoS One 4, e4798 10.1371/journal.pone.0004798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jiang W., Zhao S., Jiang X., Zhang E., Hu G., Hu B., Zheng P., Xiao J., Lu Z., Lu Y., Ni J., Chen C., Wang X., Yang L., and Wan R. (2016) The circadian clock gene Bmal1 acts as a potential anti-oncogene in pancreatic cancer by activating the p53 tumor suppressor pathway. Cancer Lett. 371, 314–325 10.1016/j.canlet.2015.12.002 [DOI] [PubMed] [Google Scholar]
- 36. Darlington T. K., Wager-Smith K., Ceriani M. F., Staknis D., Gekakis N., Steeves T. D., Weitz C. J., Takahashi J. S., and Kay S. A. (1998) Closing the circadian loop: CLOCK-induced transcription of its own inhibitors per and tim. Science 280, 1599–1603 10.1126/science.280.5369.1599 [DOI] [PubMed] [Google Scholar]
- 37. Shang Y., Xu X., Duan X., Guo J., Wang Y., Ren F., He D., and Chang Z. (2014) Hsp70 and Hsp90 oppositely regulate TGF-β signaling through CHIP/Stub1. Biochem. Biophys. Res. Commun. 446, 387–392 10.1016/j.bbrc.2014.02.124 [DOI] [PubMed] [Google Scholar]
- 38. Kwon I., Lee J., Chang S. H., Jung N. C., Lee B. J., Son G. H., Kim K., and Lee K. H. (2006) BMAL1 shuttling controls transactivation and degradation of the CLOCK/BMAL1 heterodimer. Mol. Cell. Biol. 26, 7318–7330 10.1128/MCB.00337-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nakahata Y., Yasukawa S., Khaidizar F. D., Shimba S., Matsui T., and Bessho Y. (2018) Bmal1-deficient mouse fibroblast cells do not provide premature cellular senescence in vitro. Chronobiol. Int. 35, 730–738 10.1080/07420528.2018.1430038 [DOI] [PubMed] [Google Scholar]
- 40. Esser C., Scheffner M., and Höhfeld J. (2005) The chaperone-associated ubiquitin ligase CHIP is able to target p53 for proteasomal degradation. J. Biol. Chem. 280, 27443–27448 10.1074/jbc.M501574200 [DOI] [PubMed] [Google Scholar]
- 41. Rhinn M., Ritschka B., and Keyes W. M. (2019) Cellular senescence in development, regeneration and disease. Development 146, dev151837 10.1242/dev.151837 [DOI] [PubMed] [Google Scholar]
- 42. Dickey C. A., Patterson C., Dickson D., and Petrucelli L. (2007) Brain CHIP: removing the culprits in neurodegenerative disease. Trends Mol. Med. 13, 32–38 10.1016/j.molmed.2006.11.003 [DOI] [PubMed] [Google Scholar]
- 43. Pizarro A., Hayer K., Lahens N. F., and Hogenesch J. B. (2013) CircaDB: a database of mammalian circadian gene expression profiles. Nucleic Acids Res. 41, D1009–D1013 10.1093/nar/gks1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lipton J. O., Yuan E. D., Boyle L. M., Ebrahimi-Fakhari D., Kwiatkowski E., Nathan A., Güttler T., Davis F., Asara J. M., and Sahin M. (2015) The circadian protein BMAL1 regulates translation in response to S6K1-mediated phosphorylation. Cell 161, 1138–1151 10.1016/j.cell.2015.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shimba S., Ishii N., Ohta Y., Ohno T., Watabe Y., Hayashi M., Wada T., Aoyagi T., and Tezuka M. (2005) Brain and muscle Arnt-like protein-1 (BMAL1), a component of the molecular clock, regulates adipogenesis. Proc. Natl. Acad. Sci. U.S.A. 102, 12071–12076 10.1073/pnas.0502383102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hatanaka F., Matsubara C., Myung J., Yoritaka T., Kamimura N., Tsutsumi S., Kanai A., Suzuki Y., Sassone-Corsi P., Aburatani H., Sugano S., and Takumi T. (2010) Genome-wide profiling of the core clock protein BMAL1 targets reveals a strict relationship with metabolism. Mol. Cell. Biol. 30, 5636–5648 10.1128/MCB.00781-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shi G., Xie P., Qu Z., Zhang Z., Dong Z., An Y., Xing L., Liu Z., Dong Y., Xu G., Yang L., Liu Y., and Xu Y. (2016) Distinct roles of HDAC3 in the core circadian negative feedback loop are critical for clock function. Cell Rep. 14, 823–834 10.1016/j.celrep.2015.12.076 [DOI] [PubMed] [Google Scholar]
- 48. Fang L. M., Li B., Guan J. J., Xu H. D., Shen G. H., Gao Q. G., and Qin Z. H. (2017) Transcription factor EB is involved in autophagy-mediated chemoresistance to doxorubicin in human cancer cells. Acta Pharmacol. Sin. 38, 1305–1316 10.1038/aps.2017.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang Q. Q., Wang W. J., Li J., Yang N., Chen G., Wang Z., and Liang Z. Q. (2015) Cathepsin L suppression increases the radiosensitivity of human glioma U251 cells via G2/M cell cycle arrest and DNA damage. Acta Pharmacol. Sin. 36, 1113–1125 10.1038/aps.2015.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhou Y., Xiong L., Zhang Y., Yu R., Jiang X., and Xu G. (2016) Quantitative proteomics identifies myoferlin as a novel regulator of A disintegrin and metalloproteinase 12 in HeLa cells. J. Proteomics 148, 94–104 10.1016/j.jprot.2016.07.015 [DOI] [PubMed] [Google Scholar]
- 51. Ma Q., Hu Q. S., Xu R. J., Zhen X. C., and Wang G. H. (2015) Protease Omi facilitates neurite outgrowth in mouse neuroblastoma N2a cells by cleaving transcription factor E2F1. Acta Pharmacol. Sin. 36, 966–975 10.1038/aps.2015.48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hou X.-O., Si J.-M., Ren H.-G., Chen D., Wang H.-F., Ying Z., Hu Q.-S., Gao F., and Wang G.-H. (2015) Parkin represses 6-hydroxydopamine-induced apoptosis via stabilizing scaffold protein p62 in PC12 cells. Acta Pharmacol. Sin. 36, 1300–1307 10.1038/aps.2015.54 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.